

Summary
Cells control dynamic transitions in transcript levels by regulating transcription, processing and/or degradation through an integrated regulatory strategy. Here, we combine RNA metabolic labeling, rRNA-depleted RNA-seq, and DRiLL, a novel computational framework, to quantify the level, editing sites, and transcription, processing and degradation rates of each transcript at a splice junction resolution during the LPS response of mouse dendritic cells. Four key regulatory strategies, dominated by RNA transcription changes, generate most temporal gene expression patterns. Non-canonical strategies that also employ dynamic posttranscriptional regulation control only a minority of genes, but provide unique signal processing features. We validate Tristetraprolin (TTP) as a major regulator of RNA degradation in one non-canonical strategy. Applying DRiLL to the regulation of non-coding RNAs and to zebrafish embryogenesis demonstrates its broad utility. Our study provides a new quantitative approach to discover transcriptional and post-transcriptional events that control dynamic changes in transcript levels using RNA-Seq data.
Introduction
Dynamic changes in transcript levels are tightly regulated by the interplay of RNA transcription, processing and degradation. Cells can produce complex dynamic mRNA patterns by changing one or more of these rates (Figure 1a). For example, either increasing transcription or decreasing splicing or degradation rates can yield a similar temporal mRNA profile (Figure 1a, red). Compensatory changes in two (or more) of these rates can also leave the mRNA levels unchanged and thus diminish or obscure regulatory transitions, say if decreased processing counteracts increased transcription (Figure 1b). However, most studies only measure mRNA levels and tacitly focus on transcriptional regulation, excluding changes in RNA degradation or processing from consideration.
Figure 1. Dynamic transitions in mature RNA levels can arise from changes in transcription, processing, or degradation.
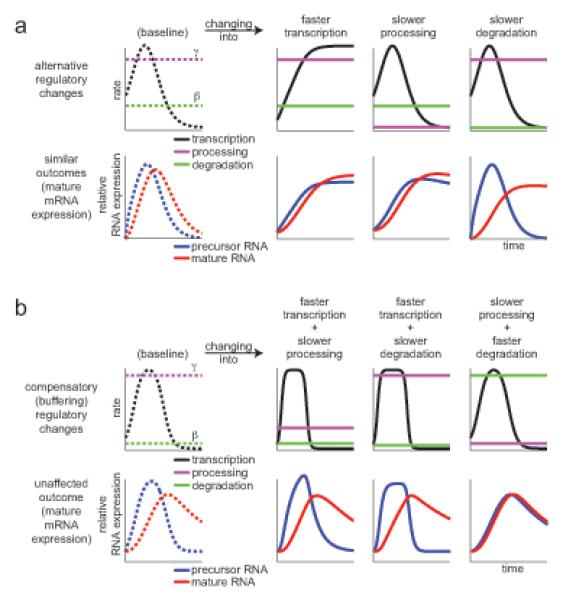
(a) Different regulatory changes can lead to a similar mRNA temporal expression profile. Top: transcription (black, RNA/min), processing (magenta, 1/min) and degradation rates (green, 1/min). Bottom: precursor (blue) and mature (red) RNA expression levels. Left (dashed lines): baseline reference expression (dashed lines). Three columns (solid lines): changes in each of three possible rates, lead to the same new ,mRNA profile (solid red, bottom). (b) Compensatory changes in two of three rates (rows as in (a)) leave mRNA levels (red, bottom) unchanged. Left column (dashed lines): reference expression; three columns (solid lines): changes from reference in two of three possible rates; mRNA levels (red, bottom) do not change vs. baseline.
The many possible regulatory strategies raise important questions. How does each regulatory strategy combine a transcript’s transcription, processing and degradation rates to generate its expression pattern? Are genes with similar temporal mRNA profiles controlled by the same strategy? If not, what function do different strategies serve if their outcome (mRNA profile) is seemingly the same? Does local variation in transcription or splicing rates along a transcript’s length regulate its expression? These questions are not fully understood, even for specific transcripts.
Technical and computational challenges have limited the availability of genome-wide dynamic data on RNA transcription, processing and degradation. Methods for measuring RNA regulation rates in vivo typically require severe manipulations (Audibert et al., 2002; Core et al., 2008; Shalem et al., 2008; Singh and Padgett, 2009), impacting physiological relevance. Fractionation-based methods (Churchman and Weissman, 2011; Pandya-Jones et al., 2013) may be impacted by non-specific RNA binding and co-precipitating proteins. Recently, several studies (Dolken et al., 2008; Eser et al., 2013; Rabani et al., 2011; Windhager et al., 2012) used short pulses of 4-thiouridine (4sU) RNA labeling to isolate newly-transcribed RNA and determine RNA kinetics during dynamic responses. Although most focused on RNA transcription and degradation, this strategy was also applied for RNA processing (Rabani et al., 2011; Windhager et al., 2012), albeit not dynamically. Moreover, although the excision rate of particular introns was described (Audibert et al., 2002; Singh and Padgett, 2009), there is no large-scale data on intron specific processing rates, and few studies that measured RNA processing intermediates (Pandya-Jones et al., 2013; Rabani et al., 2011; Windhager et al., 2012; Zeisel et al., 2011) had insufficient resolution to study individual introns. Consequently, RNA-Seq analysis tools (Katz et al., 2010; Trapnell et al., 2009; Wang et al., 2008) are optimized for mature transcripts, but not unstable precursors.
Here, we generate a high-resolution map of the transcriptome in response to lipopolysaccharide (LPS) stimulation in mouse immune dendritic cells (DCs). We combine high resolution sequencing of rRNA-depleted and of metabolically labeled RNA and a novel computational modeling approach (DRiLL) to quantify (1) precursor and mature RNA levels at a splice junction resolution from rRNA-depleted sequencing counts, (2) kinetic rates of RNA transcription, processing and degradation from metabolic labeling data, and (3) reliable RNA editing sites by detecting local differences in base composition between recently transcribed and overall RNA. Four regulatory strategies generate most (65%) expression patterns through changes in RNA transcription; non-canonical strategies with a dynamic posttranscriptional component affect a minority (35%) of genes and provide unique signal processing features. Finally, we apply DRiLL to the early zebrafish transcriptome and to the regulation of unstable non-coding RNAs, establishing its general utility.
Results
A high-resolution map of the temporal response of mouse DCs to LPS
To monitor the relative regulatory contributions of RNA transcription, processing and degradation, we sampled RNA from mouse DCs every 15 minutes, for the first 3 hours of their response to LPS (Figure 2a, Experimental Procedures), following a short (10 minute) metabolic labeling pulse with 4sU preceding the sampled time point. We isolated RNA from each sample in two ways: (1) RNA depleted of rRNA (RNA-Total) to measure total RNA regardless of its transcription time; (2) 4sU-labeled RNA (RNA-4sU) that captures primarily RNA transcribed during the 10 minute labeling pulse and is thus enriched for short-lived transcripts, including mRNA precursors and processing intermediates. We deeply sequenced each sample (80-200 million paired-end 101 base reads per sample, Table S1, Experimental Procedures). Although any time point is measured only once, we analyzed them jointly to minimize biases in any one sample.
Figure 2. DRiLL infers the abundance and kinetics of precursor and mature transcripts at single junction resolution.

(a) A high-resolution map of the temporal LPS response. Orange: 4sU pulse and 4sU-RNA. Dark brown: sampled RNA; light brown: rRNA-depleted Total RNA; blue, red: inferred precursor and mature levels, respectively; black, purple, green: estimated rates of RNA transcription, processing, and degradation, respectively. (b) Binomial model. Counts of sequencing reads that are located on exons, introns or the junctions between them (grayscale, dark to light) are used to infer, for each splicing junction, the abundance of transcripts with an unspliced precursor (P, blue) and mature junction (M, red), in either RNA-Total (solid) or RNA-4sU (dashed) samples. (c) Kinetic model. Transcription makes a precursor (P, blue) of the junction (at some temporally changing rate α, black), and that product (P) is processed (at rate γ, purple, constant or temporally changing) into a mature transcript (M, red). Degradation (at rate β, green, constant or temporally changing) eliminates the mature (M) junction. Comparing the kinetic model estimates of P and M to their levels as inferred by the binomial model (red and blue, respectively), the model fits the kinetic parameters of a junction. See also Figures S1, S7 and Table S1.
A model-based approach quantifies the abundance and kinetics of precursor and mature transcripts at single junction resolution
We developed DRiLL (Dynamic RNA Lifecycle), a novel computational scheme to quantify transcript abundance and kinetic rates at the level of individual splice junctions in precursor and mature transcripts (Figure 2b-c, Figure S1a-b). DRiLL consists of two consecutive modules.
First, a binomial model (Figure 2b, Experimental Procedures) uses RNA-Seq counts to infer, for each splicing junction, the abundance of transcripts with an unspliced junction (precursor transcripts, P) and those with a fully spliced junction (mature transcripts, M), and, when appropriate, distinguish the relative abundance of several mature isoforms (M1, M2, … Mk) that arise from a single precursor. Inference relies on separating the different sequencing reads that span an annotated junction by their location on exons, introns or the junctions between them. It applies independently to each RNA-Seq sample, and thus is applicable to any deeply sequenced RNA, but is most appropriate for rRNA-depleted samples (see Discussion).
Second, a dynamic model uses the estimated abundance of the precursor and mature junctions from different RNA populations to infer each transcript’s kinetic parameters: transcription, splicing and degradation rates (Figure 2c, Experimental Procedures). In this model, transcription (α) produces a primary precursor of the junction that is subsequently processed by splicing into a mature junction, and ultimately degraded. A precursor processing rate (γ) represents the junction’s half-life at its unprocessed form, and a mature degradation rate (β) models the mature junction’s half-life, balancing RNA processing and decay. While degradation is expected to be uniform across a transcript, because we model each junction separately, its ‘degradation rate’ reflects a local stability that is affected both by its own maturation and by the mature transcript’s decay. For example, if one junction within a transcript is spliced much faster than others, its ‘degradation rate’ is lower than that of other junctions, simply because it starts its life as a ‘mature junction’ earlier, while the rest of the transcript is still being processed (see below). Reliable estimation of kinetic parameters usually requires high-resolution temporal data of total and metabolically labeled RNA, but less data suffices under certain conditions (see below).
Large-scale analysis of RNA regulatory rates at per-junction resolution
We first used DRiLL to quantify junction-specific kinetic rates for transcription, processing and degradation of the top 10% of expressed genes with highest coverage (1,128 genes, encompassing 9% of all annotated junctions; Experimental Procedures, Figure S2a-b) and hence can be analyzed most precisely (98% with tight confidence intervals).
DRiLL’s inferred expression levels and rates were reproducible and accurate by several tests (Figure S1, Experimental Procedures). The transcription rates of an unprocessed junction range from tens of seconds to tens of minutes per junction (median of 4.1 min/junction, Figure 3a), well in line with recent measurements of RNA polymerase elongation rates in human HeLa cells (Fuchs et al., 2014). The half-life of a precursor junction, reflecting its splicing rate, ranges from fractions of minutes to an hour (median of 14.0 minutes, Figure 3b), and agrees with few measured individual intron splicing rates in human (Singh and Padgett, 2009) and mouse (Audibert et al., 2002). The mature junction’s half-life, reflecting the stability of the processed junction, ranges more widely from a few minutes to a few hours (86.1 minutes median, Figure 3c), and is typically longer than the precursor junction’s half-life (p<1.7×10−33, KS-test; median difference of 55 minutes, Figure S2c). Significant dynamic changes in processing and/or degradation rates are evident in 15% of junctions, and are also faster on average (Figure S2d-f).
Figure 3. Genome wide kinetic rates at per-junction resolution.
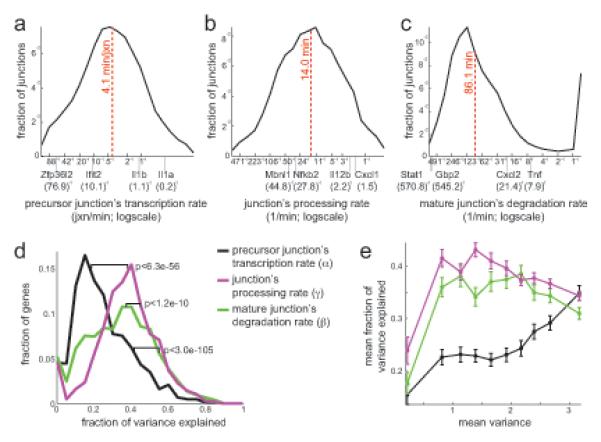
(a-c) Distribution of junction kinetic rates (x-axis, logscale) predicted for 10,351 substantially expressed junctions (fraction of junctions, y-axis). Example transcripts and half-life values in minutes are marked. Dashed line: median. (a) Precursor junction transcription rates (jxn/min, x-axis, logscale). (b) Junction processing rate (1/min, x-axis, logscale). (c) Mature junction degradation rate (1/min, x-axis, logscale). (d) Distribution of the fraction of the variance between a gene’s junctions that is explained by differences in transcription (black), processing (purple) or degradation (green) rates, in 1,693 genes with ≥2 junctions (fraction, y-axis). P-values: KS-test. (e) The mean fraction (y-axis) of the variance between a gene’s junctions that is explained by differences in its transcription (black), processing (purple) or degradation (green) rates, estimated in each of 10 quantiles of genes (x-axis) partitioned by mean variance between junctions. Error bars: standard error. See also Figures S2, S3.
Differential processing efficiency is a major source of intra-transcript variation
Junctions from the same transcript are generally regulated jointly, and thus have highly similar levels and associated rates compared to those on separate transcripts (p < 8×10−111, KS-test), especially when comparing adjacent junctions (p < 5×10−250, KS test; Figure S2h). Indeed, internal transcript differences account for only 40% of overall variation (Figure S2g), and contribute less to the total variance in our data than differences between transcripts.
Nevertheless, local events can give rise to variation in the level of different splicing junctions within a transcript. Transcriptional pausing or length differences can lead to changes in transcription rates between junctions (e.g., compare jxn 1 and 6 in Tcfec, Figure S3a), while differences in splicing efficiency between junctions would result in different half-lives of their individual precursor (e.g., jxn 1 and 3 in Cxcl2, Figure S3b) or mature (e.g., jxn 9 and 10 in Zc3hav1, Figure S3c) forms, with some junctions spliced long before the rest of the transcript matures (e.g., jxn 3 in Il12b, Figure S3d).
Globally, half-life differences explain most (75%) of the internal variation between junctions of the same transcript (37% by precursor and 38% by mature junction’s half-life differences; Figure 3d), supporting differential splicing efficiency as a main source of internal transcript variability. Local differences in transcription explain only 25% of the variation, but their contribution is more prominent for transcripts with high internal variation (35% in the most variable transcripts quantile; Figure 3e). Indeed, correlation between individual junctions’ and whole-transcripts’ rates (see below) is higher when comparing transcription rates (r=0.6) than for processing (r=0.38) or degradation (r=0.47) rates (Figure S4a).
Gene specific regulatory rates in the dynamic RNA life cycle
We systematically studied dynamic RNA regulation in top 70% of annotated transcripts, (7,872 transcripts, spanning 76% of all junctions; Experimental Procedures) with at least a minimal coverage of their exons, introns and junctions. Given the high similarity in expression of junctions within most transcripts, we took the median abundance across all junctions in a transcript as representative of the gene’s dynamics, and estimated from that the transcript’s overall kinetics.
Faster (top 20-30%) transcription, processing and degradation rates are typically associated with shorter transcripts with fewer (7 on average) and shorter (29kb overall length on average) exons and introns, while slower rates (low 20-30%) are associated with longer transcripts with a larger number (13 on average) of longer (54kb overall length on average) exons and introns (Table S2). Very slow processing rates (top 10%) are associated with alternatively spliced transcripts, but unexpectedly also with short transcripts and short introns. Finally, transcription and processing rates are more highly correlated to each other (Figure S4b,c; r = 0.61, p<1×10−40) than either is with degradation rates (r=0.48 and 0.47 respectively; p<1×10−40), consistent with a coherent regulatory coordination between the two biosynthesis steps (transcription and processing).
Most genes are regulated by transcription-dominated canonical strategies
To understand how the regulatory steps are coordinated, we examined which regulatory strategies (Figure 1a) are predominantly used in the DC response. We clustered the genes into 22 groups based on their kinetic parameters (Figure 4a). The genes in each group use rates in a similar way to shape the dynamics of their final mature product (Figure 4a, Figure S5a), and therefore share the same ‘regulatory strategy’. There are four temporal categories of mRNA profiles: transiently induced (groups 1-4), up regulated (groups 5-11), transiently repressed (groups 12-15) and down regulated (groups 16-21). More than half the genes in each category (Figure 4b) employ a single strategy, thus most patterns (65%) arise from just one of four strategies.
Figure 4. Regulatory strategies that generate dynamic mRNA profiles.

(a) Dynamics of transcription (left), processing (middle) and degradation (right) kinetic rates predicted by the kinetic model and ,mRNA levels inferred by the binomial model, relative to unstimulated (t0) control (white: t0, red: 2 fold above t0, blue: 2 fold below t0; logscale), for each of 7,872 expressed genes (rows) during 3 hours of the response (columns). Genes are divided into 22 groups (solid black lines), in four modes of mRNA regulation (dashed black lines, from top to bottom): transiently up, up regulated, transiently down and down regulated. (b) Fraction of genes (y-axis) using canonical (light gray) or non-canonical (dark gray) strategies in each of the four modes (x-axis). Fraction of genes within each mode is marked. (c) Canonical regulatory strategies. Typical transcription (α, black), processing (γ, purple) and degradation (β, green) rates of canonical strategies in each of the four modes. (d) Example genes (name on top, group in brackets) from canonical and non-canonical strategies. Right plots: t0-relative expression (y-axis) of a gene’s precursor (blue) and mature (red) RNA inferred by the binomial model for RNA-total (solid) and RNA-4sU (dashed). Left plots: kinetic parameters of a gene (relative to rate at t0, y-axis): transcription (black), processing (dashed purple) and degradation (dashed green). See also Figures S4, S5, S6 and Tables S2, S5.
All four predominant regulatory strategies (Figure 4c) combine dynamic changes in transcription with temporally-constant processing and degradation rates (these rates change in only 4.3% and 10% of genes respectively, below). Genes with transiently induced mature RNA are enriched for inflammatory signaling proteins (e.g., Tnf) and transcription factors (e.g., Nfkb), and typically (group 3; 70%) arise from transient increases in transcription rates, combined with fast (constant) processing rates (e.g., Ifrd, Figure 4d, Figure S5b-e). Up regulated genes are enriched for viral and interferon response genes, and typically (groups 9-10, 62%) arise from an increase in transcription rate combined with constantly fast processing and constantly slow degradation rates (e.g., Cpeb4, Figure 4d, Figure S5b-e). The transcriptional increase is commonly (44%) a “production overshoot” as previously reported in macrophages (Zeisel et al., 2011): strong transcriptional induction (transcription fold-change is at least twice as high as that of mRNA levels) that contributes to a fast accumulation of the mature transcript, and can either be transient (clusters 5 and 8) or persistent (clusters 9 and 10) in our time scope. Transiently repressed genes are generally enriched for housekeeping genes, with canonical group 14 (71%) also specifically enriched for mitochondrial and vesicular genes. All are canonically regulated at two stages (e.g., Atp6v, Figure 4d): Initially, there is little to no new transcription, and fast degradation eliminates pre-existing mRNAs; subsequently, transcription increases rapidly and mRNAs accumulate again, albeit with a temporal delay. This expression profile replaces old transcripts with new ones, rather than accumulating on top of them. Finally, down regulated genes are enriched for proliferation and cell cycle factors (e.g., EGFR signaling), and generally (groups 19 and 21; 53%) arise from a decrease in transcription rates combined with (constant) slow processing and fast degradation rates (e.g., Coro1a, Figure 4d, Figure S5b-e). Slow processing rates either delay the effect of increases in transcription on final mRNA levels (in transiently repressed cluster 13), or buffer them such that they do not manifest in mRNA levels during our temporal span (in down-regulated clusters 16, 18 and 20).
Alternative regulatory strategies use dynamic regulation at multiple steps to generate similar expression patterns with unique functionalities
A minority of genes (35%) follow different regulatory strategies that often involve a dynamically regulated posttranscriptional component but seemingly result in the same mRNA patterns as canonical strategies. Regulation of mRNA through both transcription and RNA processing/degradation forms a feed-forward loop (FFL) (Figure 5a) that we use in simulation studies (Experimental Procedures) to compare with matching canonical strategies, to determine the function of the posttranscriptional component.
Figure 5. Simulations suggest functional role of alternative regulatory strategies.

(a) Canonical and alternative strategies. Top to bottom: simple regulatory strategy where only transcription rates change dynamically (red arrow); incoherent feed-forward loop (FFL) regulation of mRNA expression with additional temporal changes in degradation rates (dashed red arrow, temporally delayed); incoherent FFL regulation of precursor expression with additional temporal changes in processing (dashed red arrow, temporally delayed); a double incoherent FFL with temporal changes in transcription, processing and degradation rates. (b-e) Comparing simple (dashed lines) and alternative (solid lines) strategies: a double incoherent FFL (b), a precursor incoherent FFL (c), and a mature RNA incoherent FFL (d). Top: temporal (x-axis, minutes) precursor (blue) and mature (red) RNA expression (y-axis) by either strategy. Bottom: temporal (x-axis) transcription (black), processing (purple) and degradation (green) rate changes relative to unstimulated cells by a simple (top) or alternative (bottom) strategy. See also Figure S5.
Among transiently induced genes, those in cluster 2 (enriched for inflammatory response genes) quickly reach a maximal transcription rate and maintain it, while their mRNA levels increase and decrease more gradually, because of dynamic regulation of both processing and degradation rates (e.g., Zfp36, Figure 4d). As transcription increases, the precursor accumulates due to an initially low processing rate, but once processing rate increases, mRNA peaks quickly and very highly. Next, degradation rate increases result in quick removal of these transcripts. The canonical regulatory strategy in cluster 3 generates an apparently similar mRNA dynamics through regulation of transcription rates alone, while processing and degradation remain constant (e.g., Ifrd, Figure 4d). However, important differences between those groups suggest a functional role for the alternative strategy. First, genes in cluster 2 have a much higher maximal expression than those in cluster 3 (3-fold higher median, Figure S5b-e), possibly due to a prolonged period of maximal transcription (Figure 5b). Furthermore, simulations suggest that a coupled increase in transcription, processing and degradation rates maintains the same peak expression level even for noisy signals, whereas if only transcription is regulated, peak expression is much lower when the signal is noisy (Figure S5f). While both clusters 2 and 4 display a delayed increase in degradation following transcriptional induction, which produces sharp peaks of RNA levels as previously described (Rabani et al., 2011), our current analysis reveals that processing rates increase in cluster 2 but decrease in cluster 4 and contribute to the shutoff rather than the onset phase.
Among up-regulated genes, transcription rates of cluster 11 genes steadily increase, but their processing rate is also dynamically regulated. At the beginning of the response a low processing rate allows the unprocessed transcripts to accumulate (e.g., Eif2ak2, Figure 4d). Subsequent increase in both transcription and processing rates results in a faster accumulation of the mature mRNA to higher levels (2-fold on average, Figure S5b-e) and a faster predicted future shutoff (Figure 5c) than the canonical strategy of clusters 9 and 10. This regulatory strategy is more sensitive to expression noise (Figure S5g), which might explain why it is not implemented for the lower expressed, and thus noisier, genes in clusters 9 and 10. Another alternative strategy, in clusters 5 and 8, gives rise to a very similar mRNA pattern: instead of a steady increase in transcription, an early short burst of high transcription rates levels off to a moderate rate (a transient “production overshoot”, e.g., Plat, Figure 4d). Coupled with constantly slow degradation rates, mRNAs that are relatively long lived accumulate quickly.
Transiently repressed genes in clusters 12, 13 and 15 have dynamically regulated transcription and degradation rates. Unlike the canonical strategy (cluster 14) where degradation rate is constantly high (e.g., Atp6v, Figure 4d), here degradation rate is only high initially, contributing to eliminating all existing transcripts, and then slows down (e.g., Xrn1, Figure 4d). The reduced degradation rate is combined with increased transcription rate and leads to RNA accumulation and eventually to a higher steady state expression compared to the canonical strategy (2-fold on average, Figure S5b-e, Figure 5d). Similarly, in the down-regulated genes of clusters 16 and 17 both transcription and degradation rates decrease (e.g., Mbnl1, Figure 4d), resulting in a slower decrease of mRNA levels and higher steady state mRNA levels (Figure 5e) compared to the canonical strategy (clusters 19 and 21). Cluster 16 is enriched for many housekeeping genes, which cells must maintain even if at lower levels. This strategy lowers the energetic price of expression, but at the cost of slower regulation.
Predicting molecular regulatory mechanisms by integration of RNA and protein life cycle data
To explore some of the molecular mechanisms governing distinct regulatory strategies, we analyzed the clusters (Figure 4a) for their correlation to changes in putative regulatory proteins with a known RNA binding activity as measured by pulsed SILAC proteomics (Jovanovic et al., personal communication). Seven RNA binding proteins were each highly correlated (r>0.98) to changes in RNA degradation or processing rates of at least 10 transcripts.
Several lines of evidence support a role for one of these candidates, Tristetraprolin (TTP, Zfp36) in regulating changes in RNA degradation rate in DCs. First, TTP is a known regulator of RNA stability (Brooks and Blackshear, 2013) of several key immune genes (Lai et al., 2006), and responds to many LPS-activated signaling pathways (Lai et al., 2006). Second, the consensus ARE heptamer (UAUUUAU) associated with RNA destabilization by TTP (Lai et al., 2006) is present in 3’UTRs of 58/109 genes (p < 2.3×10−13) with a predicted increase in degradation rates. Finally, consistent with TTP’s known auto-regulatory role (Brooks and Blackshear, 2013), TTP’s RNA degradation rate increases in the LPS response (Figure 4d), as does that of its most well established target, Tnf (Carballo et al., 1998) (data not shown).
TTP is required for up-regulation of degradation rates in transiently induced genes
To test the hypothesis that TTP regulates RNA degradation rates during the LPS response, we measured RNA levels and transcription rates in DCs derived from either normal (WT) or from homozygous TTP knockout mice (TTP-KO) every 15 minutes along a 3 hour time course of their response to LPS (Figure 6a). We used the nCounter to measure each of 267 signature immune genes (Experimental Procedures). Transcripts regulated by TTP should demonstrate a changed degradation rate between WT and TTP-KO cells. To identify and quantify these changes we used a novel molecular model of a trans regulator of mRNA degradation (here, TTP; Figure 6b; Experimental Procedures).
Figure 6. TTP as a regulator of dynamic RNA degradation rates.

(a) Method overview. 4sU (orange) and total (brown) RNA are sampled from DCs from wildtype (WT, light gray) and TTP-KO (dark gray) mice, following LPS stimulation and short (10 minute) metabolic labeling pulses, and quantified for a 267 transcript signature by the nCounter. (b) Kinetic model of factor induced RNA degradation. Gene X is transcribed at rate α (black) that differs in WT (dotted) or R-KO (solid) cells, and is degraded either at basal rate β1 (dark green) from the unbound state (XFree), or through factor-mediated (R, yellow, commonly an RBP) degradation (rate β2, light green) from the bound state (XR), in either WT (dotted) or R-KO (solid, inactive) cells. The regulator’s association and dissociation constants (kb, kd) determine the binding efficiency (Km). We optimize the parameters per gene by comparing the model predictions (bottom, RNA-Total: brown; RNA-4sU: orange) to the nCounter measurements. (c) 36 predicted TTP targets. Rows: Genes (left; red: known TTP targets). Left heatmap: estimated WT degradation profiles (relative rate; red: high; blue: low) at 13 time points (columns). Right heatmap: predicted 1/Km (binding affinity, left column) and β2 (factor induced degradation, right column). (d) Predicted levels of the active regulator protein (solid yellow), TTP protein levels measured in WT cells (dashed yellow; average of two replicates), and TTP RNA levels in WT (dashed red) and TTP-KO (solid red) cells. (f) Mean ratio of predicted transcription rate (WT vs. TTP-KO rate; y-axis; logscale) over time (x-axis) for 36 predicted TTP targets (black) and non-targets (gray). Error bars: standard error. See also Table S4.
TTP is predicted by the model to regulate the degradation of 36 transcripts within our ‘signature set’: in WT cells, degradation of these transcripts increases at 60-90 minutes post stimulation, but in TTP-KO cells they have only minimal changes of degradation (Figure 6c). These include 7/11 known TTP targets (Brooks and Blackshear, 2013) (p<10−4), and are enriched (17/36 targets, p<5.3×10−11) with upregulated degradation rates from RNA-Seq data (above). Furthermore, our model’s estimated regulator activity function agrees with measured (Jovanovic et al., personal communication) changes in TTP protein levels (Figure 6d), our predicted Km values negatively correlate (Spearman ρ = −0.21, p < 7.7×10−4) with known TTP binding preferences (Brooks and Blackshear, 2013), and our estimated Hill coefficient (n = 2.9) that suggests that TTP binds cooperatively, is consistent with previous studies (Brooks and Blackshear, 2013), and with enrichment of targets for multiple occurrences of the TTP binding pentamer (14/36 targets, p<3.0×10−5). Our analysis suggests that TTP may also independently affect its targets’ transcription, since transcription rates of the 36 predicted TTP targets significantly decrease in TT-KO cells (3-fold vs. 1.3-fold on average for non-targets, Figure 6e). This is likely an indirect effect, at either the transcription or processing level.
Revealing reliable RNA editing sites in noncoding portions of LPS response transcripts
We used our data to identify other steps in the RNA life cycle, such as RNA editing events, whose detection by high throughput sequencing (Danecek et al., 2012; Li et al., 2009; Li et al., 2011; Neeman et al., 2006) raised substantial debates (Kleinman and Majewski, 2012; Lin et al., 2012; Pickrell et al., 2012) due to difficulties to computationally control for the current error rates in RNA-seq. As an alternative (Figure 7a, Experimental Procedures), rather than comparing to DNA sequences, we compared base changes between two RNA-seq experiments: RNA-4sU-Seq and RNA-total-Seq, expecting that editing changes will be more prominent in RNA-total than in newly transcribed RNA-4sU, but that error-prone positions will be equally affected in both samples.
Figure 7. High resolution metabolic labeling can reliably detect RNA editing.
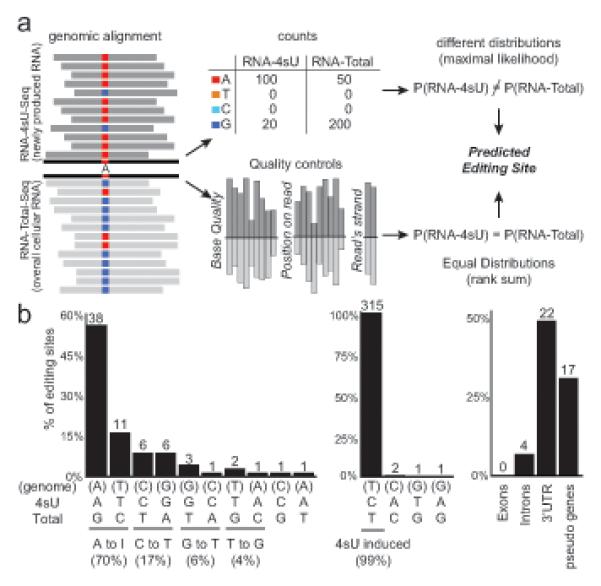
(a) Method for detecting editing sites. We search for positions where the distribution of sequenced nucleotides is different in RNA-4sU-Seq (dark gray, top) and RNA-total-Seq (light gray, bottom) using maximum likelihood estimation (top row), and also require that other measures associated with base quality distribute evenly between the two samples (bottom row). (b) Distribution of predicted editing sites (% of sites, y-axis). Left: nucleotide changes in RNA-total (editing sites, nucleotide changes on x-axis; top: genomic base, middle: RNA-4sU base, bottom: RNA-total base), middle: nucleotide changes in RNA-4sU data (4sU induced base changes), right: distinct annotations associated with RNA-total nucleotide changes. Number of sites is marked. See also Table S3.
We found 70 editing sites in 43 loci across the DCs transcriptome (Table S3), a substantially lower number than estimates in human (Li et al., 2009), and supported them by several lines of evidence. First, a lower editing level is expected in mouse in the absence of primate-specific Alu repeats (Neeman et al., 2006). Second, as an internal positive control, nucleotide changes called in newly transcribed RNA-4sU are almost exclusively (315/319) C to T modifications that are known (Hafner et al., 2010) to arise when sequencing 4sU residues. Conversely, predicted edits (Figure 7b) are mostly (61/70) known deaminations: either A to I (38 A/G changes and 11 complement T/C changes, which likely arise from sequencing strand biases), or C to U (6 C/T changes and 6 complement G/A changes). Surrounding sequences are enriched for forming stem-loop structures with an upstream sequence (p<5.7×10−12), but not with a downstream sequence, consistent with the known binding preference of adenine-deaminase (ADAR). Third, none of the edited sites affects an annotated protein sequence (Figure 7b), while many sites (17/43) are associated with annotated and putative pseudogenes (e.g., Taldo1, Psme2b), which often contain multiple edited positions (8/17); this is consistent with a postulation that editing controls the expression of many transposable elements in human (Neeman et al., 2006). Finally, mass-spectrometry detected 18 peptides that match a reading frame within one of the putative pseudogenes (within the intron of the gene Ccrn4l) and confirmed a predicted G/U editing that changes a Valine residue into a Leucine. All other sites are located in non-protein coding portions of expressed genes (22/43 in 3’UTRs, 4/43 in introns), and potentially contribute to their posttranscriptional regulation.
Applying DRiLL to diverse systems and non-coding transcripts demonstrates its general utility
To demonstrate DRiLL’s wide applicability, we used it to examine the regulation of non-coding RNAs in our system, and of maternally deposited vs. zygotic transcripts in early embryogenesis.
First, we used DRiLL to dissect the regulation of unstable non-coding regulatory RNAs in DCs (Figure S6). Although both eRNAs (Kaikkonen et al., 2013) (Figure S6a) and lincRNAs (Carpenter et al., 2013) (Figure S6b) were implicated in key innate immune functions, including in DCs, their regulation has not been extensively studied in any system, since they are usually lowly expressed (Carninci et al., 2005). Applying DRiLL to these newly annotated non-coding transcripts in our data, we found that eRNAs are transcribed at a very high rate, but are also very quickly degraded. Conversely, lincRNAs are transcribed and processed at comparable rates to protein coding genes, but are significantly less stable (Figure S6c-i). This could help explain how lincRNAs are both lowly expressed and tissue specific (Cabili et al., 2011).
Second, we used DRiLL to analyze transcriptome dynamics during early zebrafish embryogenesis (Figure S7). Embryos initially rely on maternally provided mRNAs and only activate zygotic (embryonic) transcription ~3 hours post fertilization (hpf) (Lee et al., 2014; Schier, 2007). Using rRNA-depleted RNA-Seq data (Lee et al., 2013), DRiLL distinguished maternal from zygotic mRNAs (Figure S7a-b); using polyA+ RNA-Seq (Pauli et al., 2012), DRiLL estimated the onset time and rate of decay of maternally provided messages (Figure S7c-e). We find two major waves of degradation of maternal messages: immediately after fertilization (0-1 hpf, 18%) or after the maternal-to-zygotic (MZT) transition (3-5 hpf, 47%). Post-MZT decaying mRNAs are degraded faster than early decaying mRNAs (KS-test p < 10−29, Figure S7e), and are selectively enriched in their 3’UTR for seed sequences for miR-430 (p < 8.3×10−11), a microRNA involved in the degradation of maternal mRNAs (Giraldez et al., 2006). This suggests that different degradation pathways are active before vs. after MZT. Indeed, early (2-4 hpf) polyA tail lengths of maternal mRNAs (as measured in (Subtelny et al., 2014)) correlate to their ribosomal occupancy (as measured in (Chew et al., 2013), Figure S7f), but later (4-6 hpf) lengths are correlated with mRNA stability (Figure S7g). These findings support and extend the idea (Subtelny et al., 2014) that zebrafish post-transcriptional mechanisms change from a maternally derived control over mRNA translation into a zygotic regulation of mRNA stability.
Discussion
We present a novel approach (Figure 2) that combines high-resolution RNA labeling and sequencing with advanced computational modeling (DRiLL) and uses it to study the regulatory strategies that generate temporal RNA levels during the LPS response.
Quantitative dissection of the RNA life cycle in dynamic responses
DRiLL uses RNA-seq data to predict the frequency of mature and alternative transcripts and of their unstable precursors and processing intermediates, which are mostly disregarded in other transcriptome analysis tools (Katz et al., 2010; Trapnell et al., 2009; Wang et al., 2008). As rRNA depletion becomes increasingly popular, especially when RNA quality is low (Adiconis et al., 2013), DRiLL will help researchers to explore transcriptomes at unprecedented depth and resolution. When temporal metabolic labeling data is also available, DRiLL further predicts kinetic transcription, processing and degradation rates, both between transcripts and within transcripts (per-junction). This can be extended to other aspects of the RNA life cycle, such as RNA editing. DRiLL is broadly applicable, as we demonstrated for unstable lncRNA and for maternally provided mRNAs in zebrafish embryogenesis. Our genomic portal www.broadinstitute.org/rnalifecycle) provides the scientific community with ready access to our analysis and tools.
Although the levels quantified by DRiLL are reproducible and reliable by several tests, they can be impacted by noise and biases in sequencing data, variations in coverage along genes and considering paired-end reads as independent observations. Introns retention in mRNAs can lead to further inconsistencies between junctions. Simplifying assumptions of the kinetic model (e.g., that global RNA levels in cells remain constant upon LPS stimulation, or that individual junctions are independently regulated) would affect our estimated rates, but would not change the ranking between genes (as all estimates will be similarly affected by such global events). Using a likelihood ratio test to select between constant and dynamic rate models also reduces DRiLL’s sensitivity to detect changes in lowly expressed genes.
Key principles of temporal RNA regulation in mammalian cells
We determined the key regulatory strategies that DCs implement to generate their mRNA outputs (Figure 4), demonstrating how similar or correlated mRNA profiles are generated in distinct ways, and hypothesized on their possible distinct functional utility (Figure 5). Our extensive dataset can be further combined with other genome-scale data in this system. For example, decreased degradation rates in down-regulated cluster 16 counteract transcriptional repression and increase the new steady state levels. Pulsed SILAC measurements of Cluster 16 proteins (Jovanovic et al., personal communication) also show an increase in their translation rate upon LPS stimulation, possibly as a second level of post-transcriptional buffering of their transcriptional repression.
Our work provides new and effective quantitative tools to study RNA dynamics at both transcript and per junction resolution from RNA-Seq data, and generates a unique view of the different kinetic strategies that cells use to coordinate transcriptional and posttranscriptional events and regulate transcript levels during a dynamic response.
Experimental Procedures
DCs culture and sample collection
DCs culture and treatment, RNA sample collection, and 4sU labeled RNA isolation were done as described in (Rabani et al., 2011), with the following modifications. We added 4sU to a 500 μM final concentration for 10 minutes before RNA collection. For RNA-Seq, 10 μg total RNA from each sample was depleted of rRNA by RiboZero (Epicenter), a 100 ng aliquot was kept for sequencing, and 4sU purification was done for the remainder of the sample.
RNA-seq, read mapping and annotation
RNA-Seq libraries were constructed by dUTP second strand protocol (Levin et al., 2010), sequenced by Illumina HiSeq2000 with paired-end, 101bp reads (Table S2). We align reads to the mouse reference genome (NCBI37/mm9) using TopHat (Trapnell et al., 2009) with default parameters. We use polyA+ RNA-seq data (Garber et al., 2012) to reconstruct mRNA annotations with Trinity (Grabherr et al., 2011) and Cufflinks (Trapnell et al., 2010), and collect all annotated mouse transcripts (Refseq and UCSC genes NCBI37/mm9 (Rhead et al., 2010)) that matched a reconstructed transcript.
nCounter sample preparation and data processing
nCounter sample preparation, capture and analysis were done as described in (Rabani et al., 2011), with the following modifications. Our code set (Table S4) detects 246 signature LPS transcripts (Amit et al., 2009) and 21 control genes with constant basal expression levels (9 of which used for normalization), via a probe that matches their exon sequence (captures their pre-mRNA and multiple mature mRNA isoforms). For 30/246 transcripts, we had a second probe that matches their intron sequence and captures their precursor.
Substantially expressed genes
We define a splicing junction as substantially expressed if all its exons, introns and the junctions between them have normalized counts (RPKM) sums (of all times and all RNA-Total or RNA-4sU samples) above their respective thresholds (10% or 70% substantially expressed genes). We take all genes with at least one substantially expressed junction.
Precursor and mature RNA abundance
We count sequencing reads that span an annotated junction by their location on either exons, introns or the junctions between them, and use these counts to quantify, for each splicing junction, the abundance of transcripts with an unspliced junction (precursor) and those with a fully spliced junction (mature). We use a binomial model in which the frequency of precursor and mature RNA directly relates to the probability of observing a given number of reads at each location, considering the depth of the sequencing library and the genomic lengths. We use derivative-free methods (’Neadler-Mead simplex algorithm’ as implemented in Matlab) to find the expression levels that are most likely to generate these read counts. We extend this to annotated alternative splicing and predict the relative abundance of several mature isoforms that arise from a single precursor junction. We apply this to the dynamic sequencing data of all substantially expressed junctions, independently for each RNA-Seq sample.
Quantifying transcript kinetics
Our kinetic system model describes the time evolution of a junction’s precursor (P) and mature (M1, …, Mn) mRNA by its transcription (α), degradation (β) and processing (γ) rates:
We use gradient descent optimization to find the model parameters (□ = [□, □,□, X0]) that minimize the difference between the kinetic model predictions of precursor and mature transcripts levels to their direct estimates from RNA-Seq by the binomial model (above). We compare four alternative hypotheses in which rates are either constant or change over time through a likelihood ratio test to identify genes in which dynamic changes in one or both rates significantly (P < 0.01) contribute to temporal changes in overall RNA levels, and assign them with a time-dependent, rather than a constant, rate function. We apply this model using all temporal total and 4sU RNA levels (26 samples) either per an entire transcript or per a specific junction.
Model fit, reproducibility and accuracy
We use a goodness of fit test (χ2 test) with the null hypothesis that the data is governed by the estimated binomial model, and find minimal discrepancy (p < 0.01) with sequencing counts in >70% of our data, with mostly tight confidence intervals. Spearman correlation to an independent biological replicate set of nCounter measurements confirms reproducibility (r>0.73). As expected, shorter-lived precursors are enriched in RNA-4sU samples, and mature junctions in RNA-Total and RNA-polyA samples. The rate predictions are robust to normal additive error (estimated from genome-wide data), with a tight fit (p < 0.01, χ2 test) in >90% of junctions. Confidence intervals are tight (least square error) by bootstrapping for 15 representative examples. Model predictions fit well to two unseen test datasets: polyA+-RNA-Seq and nCounter data, taken at times within and beyond the scope of our training set. Predicted rates are significantly correlated with earlier predictions (Rabani et al., 2011) (degradation: r=0.39; processing: 0.23), despite different time scale, resolution and modeling.
Functional Enrichments
We test enrichment using a hypergeometric p-value (for binary features) or the KS test (for numerical features) and a 5% False Discovery Rate (FDR) across all tested annotations or all ‘substantially expressed’ genes, respectively (Table S5). We calculate functional enrichments of rates by splitting all rates (at all times and all genes) into 10 quintiles, assigning the most abundant quintile (across times) per gene and using hypergeometric p-value.
Clustering
We first cluster (k-means clustering as implemented in Matlab) a subset of 17% highly expressed genes (1,305 genes) after standardizing its log2(expression) and/or log2(rate) temporal data. We iteratively increase the number of clusters as long as none of the clusters has less than 2% of the genes. We assign each of the other genes into the same cluster of the gene in the initial subset with which it has a maximal Pearson correlation.
Simulation studies
We simulate expression data using our kinetic model and characteristic kinetic parameters of RNA transcription, processing and degradation rates. All rate functions are modeled as step functions with a basal rate and an active rate that is used only when an external signal exists. Input signal is modeled by a binary (0/1) function, and noise is introduced by random changes to its values. We simulate a temporal delay of processing and degradation response to an external signal by switching to the active rate only when precursor RNA levels (for processing) or mature RNA levels (for degradation) exceed a predefined threshold.
Factor-dependent RNA degradation
We model factor-dependent regulation of RNA degradation by a non-linear Hill function with two constants: basal degradation of the unbound transcript (β1) and factor-mediated degradation (β2) that also depends on the unbound regulator’s concentration. We fit two alternative models to both the WT and KO measurements, and use a likelihood ratio test (P < 0.01) to select (for each gene) between the null hypothesis of a constant, factor-independent, degradation rate (β2=0 in WT and KO), and a dynamic, factor-dependent regulation (β2>0 in WT only).
RNA editing
We search for edited positions with a different distribution of sequenced nucleotides between RNA-total and RNA-4sU (maximum likelihood test), but an equal distribution of base quality (Wilcoxon Rank-Sum test), location on read (Wilcoxon Rank-Sum test) and strand assignment of read (Fischer exact test).
Supplementary Material
Acknowledgements
We thank the Broad Genomics Platform for sequencing and C. de Boer for comments on the manuscript. Work was supported by MIT Albert Memorial and Xerox-MIT fellowships (MR), SNSF for advanced researchers and Marie Sklodowska-Curie IOF fellowships (MJ), NIH R01GM056211 (AFS), CEGS (1P50HG006193-01, NH, IA, AR), and Pioneer Award (DP1OD003958-01, AR), HHMI (AR), BSF (NF), ERC (309788, IA; 340712 NF), ISF (1782/11, IA), I-CORE (IA, NF), and the Intramural Research Program of the NIH, NIEHS (PJB).
RNA-Seq data and processed files are available at the Gene Expression Omnibus (GSE56977).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adiconis X, Borges-Rivera D, Satija R, DeLuca DS, Busby MA, Berlin AM, Sivachenko A, Thompson DA, Wysoker A, Fennell T, et al. Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nature methods. 2013;10:623–629. doi: 10.1038/nmeth.2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amit I, Garber M, Chevrier N, Leite AP, Donner Y, Eisenhaure T, Guttman M, Grenier JK, Li W, Zuk O, et al. Unbiased reconstruction of a mammalian transcriptional network mediating pathogen responses. Science. 2009;326:257–263. doi: 10.1126/science.1179050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audibert A, Weil D, Dautry F. In vivo kinetics of mRNA splicing and transport in mammalian cells. Molecular and cellular biology. 2002;22:6706–6718. doi: 10.1128/MCB.22.19.6706-6718.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks SA, Blackshear PJ. Tristetraprolin (TTP): interactions with mRNA and proteins, and current thoughts on mechanisms of action. Biochimica et biophysica acta. 2013;1829:666–679. doi: 10.1016/j.bbagrm.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes & development. 2011;25:1915–1927. doi: 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carballo E, Lai WS, Blackshear PJ. Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science. 1998;281:1001–1005. doi: 10.1126/science.281.5379.1001. [DOI] [PubMed] [Google Scholar]
- Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, et al. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–1563. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- Carpenter S, Aiello D, Atianand MK, Ricci EP, Gandhi P, Hall LL, Byron M, Monks B, Henry-Bezy M, Lawrence JB, et al. A long noncoding RNA mediates both activation and repression of immune response genes. Science. 2013;341:789–792. doi: 10.1126/science.1240925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew GL, Pauli A, Rinn JL, Regev A, Schier AF, Valen E. Ribosome profiling reveals resemblance between long non-coding RNAs and 5' leaders of coding RNAs. Development. 2013;140:2828–2834. doi: 10.1242/dev.098343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchman LS, Weissman JS. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature. 2011;469:368–373. doi: 10.1038/nature09652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–1848. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Nellaker C, McIntyre RE, Buendia-Buendia JE, Bumpstead S, Ponting CP, Flint J, Durbin R, Keane TM, Adams DJ. High levels of RNA-editing site conservation amongst 15 laboratory mouse strains. Genome biology. 2012;13:26. doi: 10.1186/gb-2012-13-4-r26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolken L, Ruzsics Z, Radle B, Friedel CC, Zimmer R, Mages J, Hoffmann R, Dickinson P, Forster T, Ghazal P, et al. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. Rna. 2008;14:1959–1972. doi: 10.1261/rna.1136108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eser P, Demel C, Maier KC, Schwalb B, Pirkl N, Martin DE, Cramer P, Tresch A. Periodic mRNA synthesis and degradation co-operate during cell cycle gene expression. Molecular systems biology. 2013;10:717. doi: 10.1002/msb.134886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs G, Voichek Y, Benjamin S, Gilad S, Amit I, Oren M. 4sUDRB-seq: measuring genomewide transcriptional elongation rates and initiation frequencies within cells. Genome biology. 2014;15:R69. doi: 10.1186/gb-2014-15-5-r69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber M, Yosef N, Goren A, Raychowdhury R, Thielke A, Guttman M, Robinson J, Minie B, Chevrier N, Itzhaki Z, et al. A high-throughput chromatin immunoprecipitation approach reveals principles of dynamic gene regulation in mammals. Molecular cell. 2012;47:810–822. doi: 10.1016/j.molcel.2012.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraldez AJ, Mishima Y, Rihel J, Grocock RJ, Van Dongen S, Inoue K, Enright AJ, Schier AF. Zebrafish MiR-430 promotes deadenylation and clearance of maternal mRNAs. Science. 2006;312:75–79. doi: 10.1126/science.1122689. [DOI] [PubMed] [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature biotechnology. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jr., Jungkamp AC, Munschauer M, et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaikkonen MU, Spann NJ, Heinz S, Romanoski CE, Allison KA, Stender JD, Chun HB, Tough DF, Prinjha RK, Benner C, et al. Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Molecular cell. 2013;51:310–325. doi: 10.1016/j.molcel.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz Y, Wang ET, Airoldi EM, Burge CB. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nature methods. 2010;7:1009–1015. doi: 10.1038/nmeth.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinman CL, Majewski J. Comment on "Widespread RNA and DNA sequence differences in the human transcriptome". Science. 2012;335:1302. doi: 10.1126/science.1209658. author reply 1302. [DOI] [PubMed] [Google Scholar]
- Lai WS, Parker JS, Grissom SF, Stumpo DJ, Blackshear PJ. Novel mRNA targets for tristetraprolin (TTP) identified by global analysis of stabilized transcripts in TTP-deficient fibroblasts. Molecular and cellular biology. 2006;26:9196–9208. doi: 10.1128/MCB.00945-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MT, Bonneau AR, Giraldez AJ. Zygotic Genome Activation During the Maternal-to-Zygotic Transition. Annual review of cell and developmental biology. 2014 doi: 10.1146/annurev-cellbio-100913-013027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MT, Bonneau AR, Takacs CM, Bazzini AA, DiVito KR, Fleming ES, Giraldez AJ. Nanog, Pou5f1 and SoxB1 activate zygotic gene expression during the maternal-to-zygotic transition. Nature. 2013;503:360–364. doi: 10.1038/nature12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin JZ, Yassour M, Adiconis X, Nusbaum C, Thompson DA, Friedman N, Gnirke A, Regev A. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nature methods. 2010;7:709–715. doi: 10.1038/nmeth.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JB, Levanon EY, Yoon JK, Aach J, Xie B, Leproust E, Zhang K, Gao Y, Church GM. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science. 2009;324:1210–1213. doi: 10.1126/science.1170995. [DOI] [PubMed] [Google Scholar]
- Li M, Wang IX, Li Y, Bruzel A, Richards AL, Toung JM, Cheung VG. Widespread RNA and DNA sequence differences in the human transcriptome. Science. 2011;333:53–58. doi: 10.1126/science.1207018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Piskol R, Tan MH, Li JB. Comment on "Widespread RNA and DNA sequence differences in the human transcriptome". Science. 2012;335:1302. doi: 10.1126/science.1210419. author reply 1302. [DOI] [PubMed] [Google Scholar]
- Neeman Y, Levanon EY, Jantsch MF, Eisenberg E. RNA editing level in the mouse is determined by the genomic repeat repertoire. Rna. 2006;12:1802–1809. doi: 10.1261/rna.165106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya-Jones A, Bhatt DM, Lin CH, Tong AJ, Smale ST, Black DL. Splicing kinetics and transcript release from the chromatin compartment limit the rate of Lipid A-induced gene expression. Rna. 2013;19:811–827. doi: 10.1261/rna.039081.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauli A, Valen E, Lin MF, Garber M, Vastenhouw NL, Levin JZ, Fan L, Sandelin A, Rinn JL, Regev A, et al. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome research. 2012;22:577–591. doi: 10.1101/gr.133009.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Gilad Y, Pritchard JK. Comment on "Widespread RNA and DNA sequence differences in the human transcriptome". Science. 2012;335:1302. doi: 10.1126/science.1210484. author reply 1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabani M, Levin JZ, Fan L, Adiconis X, Raychowdhury R, Garber M, Gnirke A, Nusbaum C, Hacohen N, Friedman N, et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nature biotechnology. 2011;29:436–442. doi: 10.1038/nbt.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhead B, Karolchik D, Kuhn RM, Hinrichs AS, Zweig AS, Fujita PA, Diekhans M, Smith KE, Rosenbloom KR, Raney BJ, et al. The UCSC Genome Browser database: update 2010. Nucleic acids research. 2010;38:D613–619. doi: 10.1093/nar/gkp939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schier AF. The maternal-zygotic transition: death and birth of RNAs. Science. 2007;316:406–407. doi: 10.1126/science.1140693. [DOI] [PubMed] [Google Scholar]
- Shalem O, Dahan O, Levo M, Martinez MR, Furman I, Segal E, Pilpel Y. Transient transcriptional responses to stress are generated by opposing effects of mRNA production and degradation. Molecular systems biology. 2008;4:223. doi: 10.1038/msb.2008.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, Padgett RA. Rates of in situ transcription and splicing in large human genes. Nature structural & molecular biology. 2009;16:1128–1133. doi: 10.1038/nsmb.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subtelny AO, Eichhorn SW, Chen GR, Sive H, Bartel DP. Poly(A)-tail profiling reveals an embryonic switch in translational control. Nature. 2014;508:66–71. doi: 10.1038/nature13007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature biotechnology. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windhager L, Bonfert T, Burger K, Ruzsics Z, Krebs S, Kaufmann S, Malterer G, L'Hernault A, Schilhabel M, Schreiber S, et al. Ultrashort and progressive 4sU-tagging reveals key characteristics of RNA processing at nucleotide resolution. Genome research. 2012;22:2031–2042. doi: 10.1101/gr.131847.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A, Kostler WJ, Molotski N, Tsai JM, Krauthgamer R, Jacob-Hirsch J, Rechavi G, Soen Y, Jung S, Yarden Y, et al. Coupled pre-mRNA and mRNA dynamics unveil operational strategies underlying transcriptional responses to stimuli. Molecular systems biology. 2011;7:529. doi: 10.1038/msb.2011.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.