

Summary
Ras GTPases regulate intracellular signaling involved in cell proliferation. Elevated Ras signaling activity has been associated with human cancers. Ras activation is catalyzed by guanine-nucleotide exchange factors (GEFs), of which SOS1 is a major member that transduces receptor tyrosine kinase signaling to Ras. We have developed a rational approach coupling virtual screening with experimental screening in identifying small-molecule inhibitors targeting the catalytic site of SOS1 and SOS1-regulated Ras activity. A lead inhibitor, NSC-658497, is found to bind to SOS1, competitively suppresses SOS1-Ras interaction, and dose-dependently inhibits SOS1 GEF activity. Mutagenesis and structure-activity relationship studies map the NSC-658497 site of action to the SOS1 catalytic site, and define the chemical moieties in the inhibitor essential for the activity. NSC-658497 showed dose-dependent efficacy in inhibiting Ras, downstream signaling activities, and associated cell proliferation. These studies establish a proof of principle for rational design of small-molecule inhibitors targeting Ras GEF enzymatic activity.
Keywords: Drug Discovery, Rational Design, GTPase signaling, Ras, Cancer
Introduction
H-,N-,K-Ras are the founding members of the Ras superfamily of small-GTPases, and are known to transduce signals from various cellular receptors including receptor tyrosine kinases, G-protein coupled receptors, and cytokine receptors, to engage in multiple intracellular signaling pathways leading to cell proliferation, differentiation, survival, and gene expression (Buday and Downward, 2008; Rojas et al., 2011; Vigil et al., 2010). Gain of function mutations of H-,N-, K-Ras and components of Ras signaling (e.g. EGFR, BCR-Abl, MEK, PI3K, and AKT) have been identified in a myriad of human cancers (Karnoub and Weinberg, 2008; Pylayeva-Gupta et al., 2011). In addition, aberrant Ras signaling is involved in several developmental disorders, known as the Cardio-Facio-Cutaneous (CFC) diseases (i.e. NF-1, Costello syndrome, and Noonan syndrome) (Cox and Der, 2010). Ras activity in cells is regulated by two classes of enzymes that control its guanine nucleotide bound states – the activators, guanine nucleotide exchange factors (GEFs), that stimulate the exchange of Ras bound GDP for GTP and enable Ras to interact with their effectors leading to downstream signaling, and the inactivators, GTPase-activating proteins (GAPs), that stimulate GTP-hydrolysis and switch off Ras signaling (Buday and Downward, 2008; Pierre et al., 2011; Rojas et al., 2011; Vigil et al., 2010).
There are three subfamilies of Ras GEFs, i.e. SOS, Ras-GRF, and Ras-GRP, that are expressed in different cell-types (Rojas et al., 2011). In particular, SOS1 is a ubiquitously expressed multi-domain enzyme with a REM domain and a Cdc25 catalytic domain essential for Ras GTPase activation and signaling (Pierre et al., 2011; Rojas et al., 2011). The REM and Cdc25 domains contain two distinct sites for Ras binding - a catalytic site and an allosteric site. The catalytic site binds GDP-bound Ras and stimulates GDP/GTP exchange on Ras (Margarit et al., 2003; Sondermann et al., 2004). Gain of function SOS1 mutations have been identified in patients with the developmental disorder Noonan syndrome (Roberts et al., 2007; Tartaglia et al., 2007) and genetic disorder hereditary gingival fibromatosis type 1 (Hart et al., 2002; Jang et al., 2007), and SOS1 has been shown to be differentially expressed in prostate and breast cancer samples (Field et al., 2012; Timofeeva et al., 2009). In addition, SOS1 has been recognized as a critical nodal mediating signal flow from receptor tyrosine kinases to Ras and downstream ERK and PI3K cascades.
To date, the Ras signaling module has been difficult to target; most strategies targeting Ras signaling have been geared toward downstream effectors (e.g. Raf, MEK, PI3K, and AKT) (Vigil et al., 2010) or post-translational modification events (e.g. farnesyltransferases) (Appels et al., 2005; Hara et al., 1993; James et al., 1993). In recent years, there have been several efforts that show limited success in suppressing Ras signaling by targeting either the GEF or effector binding site on Ras, and by designing SOS1-Ras interface peptidomimetics derived from SOS1 (Hocker et al., 2013; Patgiri et al., 2011; Schopel et al., 2013; Zimmermann et al., 2013). More recently, studies in high-throughput screening and fragment-based screening of chemical libraries to disrupt Ras signaling (Maurer et al., 2012; Ostrem et al., 2013; Sun et al., 2012) have produced interesting leads of various small molecules affecting Ras activity in cells, including a small molecule activator of SOS1 that binds to a pocket of SOS1 aside from the Ras catalytic pocket which potentiates SOS1-mediated Ras activation (Burns et al., 2014). However, it remains to be seen if targeting of the catalytic site of SOS1 that directly disrupts SOS1 catalysis and binding of Ras could yield useful small molecule inhibitors, which may present a more “druggable” strategy in the pursuit of rational inhibition of the crucial Ras signaling nodal.
Here we have carried out a virtual screen of small molecule inhibitors targeting the Ras interactive catalytic site of SOS1 based upon structural information of the SOS1-Ras complex (Sondermann et al., 2004), followed by an experimental validation in SOS1-catalyzed Ras GEF reaction. We have identified a class of synthetic compounds that recognize the catalytic pocket of SOS1, and characterized them as effective inhibitors of Ras signaling in cells. The studies establish a new targeting approach for “Rasopthapies” that can be useful for the development of future therapeutics.
Results
Targeting the Ras Catalytic Site of SOS1 by Virtual and Experimental Screening
Crystal structures of the Ras-bound SOS1 complex, as well as other resolved structures of SOS1 and its complexes, provide structural data to guide the rational design of inhibitors of SOS1-Ras interactions (Sondermann et al., 2004). Here, the Ras-bound structure (PDB ID: 1XD2) was used to carry out a virtual screen for chemicals targeting the SOS1 pocket that interacts with Ras (Figure 1A). The Ras switch II region binding site in the catalytic binding region on SOS1 (Figure 1B) contains a hydrophobic pocket formed by residues W809, T829, H911, and K939 that appears to be suitable for binding of a small molecule (Figure 1B). Therefore, we hypothesized that there may exist small molecules that can bind in this pocket and interfere with the interaction between SOS1 and Ras.
Figure 1. Rational design and identification of small molecule inhibitors of SOS1.

(A) The crystal structure of H-Ras in complex with the catalytic site of SOS1 (REM and Cdc25 domains) (PDB ID: 1XD2, chains C and B) (Sondermann et al., 2004). SOS1 and Ras are depicted in green and orange, respectively. The targeted pocket on SOS1 is highlighted with magenta. (B) The structure of SOS1 (PDB ID: 1XD2, chain C) with the amino acid residues that interact with H-Ras shown in magenta, and the residues surrounding the H-Ras Switch II binding site labeled in white. The approximate position of the simulation box for virtual screening is indicated by the red rectangle. Both the top and side views of the H-Ras interacting surface on SOS1 are depicted. (C) Distribution of predicted binding affinities (defined as negative log of the median Ki obtained in multiple runs of docking simulations, and binned into intervals on the x-axis) in subsequent iterations of a multistage virtual screening protocol as described in the SI Experimental Procedures. The first, second, and third steps of the docking protocol are indicated by the blue, red, and green histograms, respectively. The top candidate compounds were ranked using a combination of −log(Ki) and entropy of clustering of docking poses (shown in the insert). The position of our top hit compound, NSC-658497, is indicated by a star (*). Further filtering as described in the SI Experimental Procedures resulted in the selection of 36 top candidate compounds for experimental testing. (D) The overall scheme of the computational and experimental screening approach. 118,500 compounds from the NCI synthetic chemical collection were docked in silico as described in panel C against the catalytic site on SOS1. 135 compounds were identified and 36 top candidate compounds were selected and ordered from the NCI for experimental testing as described in the flow-chart and in the SI Experimental Procedures. As a result of the experimental dissociation assay screen at a dose of 100 μM, 2 hit compounds were identified. (E) 100 μM NSC-674954 (●) partially inhibited 50 nM SOS1-cat (●) mediated GDP/GTP nucleotide exchange upon 2 μM H-Ras (aa. 1-166) (●) in the BODIPY-FL-GDP dissociation assay. (F) 100 μM NSC-658497 (●) completely abrogated 50 nM SOS1-cat (●) mediated GDP/GTP nucleotide exchange upon 2 μM H-Ras (aa. 1-166) (●) in the BODIPY-FL-GDP dissociation assay. Data in E and F are expressed as percent change of relative fluorescence units normalized to the initial time point over 15 minutes. Data in E and F were measured in triplicate and represent the mean of N = 3 experiments.
Using a subset of 118,500 small-molecules from the NCI/DTP Open Chemical Repository, a multistage docking protocol was adopted to identify top hits for experimental screening and validation. In the first step, a set of 30,000 candidates were selected using a limited sampling. This set was subsequently reduced to a set of top 3,000 hits with improved sampling, and further re-ranked using extensive sampling in docking simulations (Figure 1C–D) (Biesiada et al., 2011). Top hits with relatively high predicted binding affinity and consistent binding to a specific site in a dominant pose within the simulation box, thus resulting in low entropy of clustering poses obtained in multiple docking runs, were combined and clustered by their structural similarities (Figure 1C). This resulted in a set of 135 candidate chemicals, of which 36 chemical compounds were selected for experimental screening based upon additional filtering involving an assessment of drug-like properties, similarity to classes of compounds often identified in virtual screening as false positives, and availability of compounds from the NCI/DTP Open Chemical Repository (Figure 1C–D and Table S1). For experimental screening, a fluorescence-based guanine nucleotide exchange assay utilizing a BODIPY-fluorescein (FL) labeled GDP nucleotide was refined based upon previous studies (Figure S1) (Evelyn et al., 2009; Lenzen et al., 1998; Lenzen et al., 1995; McEwen et al., 2001, 2002). The REM-Cdc25 domains of SOS1 and the H-Ras protein with c-terminal 21 amino acid truncation were expressed as histidine-tagged proteins in Escherichia coli and purified. The set of 36 compounds were initially screened at a concentration of 100 μM for their ability to inhibit SOS1 catalyzed BODIPY-FL GDP nucleotide dissociation from H-Ras in exchange for GTP (Figure 1D and Figure S2). Two hit compounds, NSC-674954 and NSC-658497, as partial and complete inhibitors at 100 μM, respectively, of SOS1 catalyzed Ras GEF reaction were identified (Figure 1E–F and Figure S2). The more active chemical inhibitor, NSC-658497, was selected for further characterizations.
Biochemical Characterization of NSC-658497 as an Inhibitor of SOS1
To validate NSC-658497 as an inhibitor of SOS1 catalytic activity, two complementary GEF reaction assays were performed in the presence or absence of the chemical. First, NSC-658497 was found to inhibit SOS1 catalyzed BODIPY-FL GDP nucleotide dissociation from H-Ras in exchange for GTP in a dose-dependent manner (Figure 2A). Secondly, NSC-658497 inhibited SOS1 catalyzed BODIPY-texas red (TR) GTP loading of H-Ras dose-dependently (Figure 2B). NSC-658497 also conformed to our prediction of disrupting the SOS1-Ras interaction in blocking the binding of SOS1-cat to H-Ras competitively in a microscale thermophoresis assay (Figure 2D) and a glutathione-s-transferase-tagged H-Ras pull-down assay (Figure S3A). Direct titration of NSC-658497 to SOS1 revealed that it directly bound to SOS1 with a low micromolar affinity (Kd - 7.0 μM), but not to H-Ras (Figure 2D and Figure S3B). To further rule out potential artifacts of spectroscopic interference, UV-Vis absorbance spectrum of NSC-658497 (Figure S4) was measured to confirm that NSC-658497 does not show absorption at any of the wavelengths used for the fluorescence-based GEF or binding assays. Taken together, these biochemical results validate that NSC-658497 is an effective SOS1 inhibitor in interfering with SOS1-catalyzed Ras GEF reaction.
Figure 2. Biochemical validation of NSC-658497 as an inhibitor of SOS1.
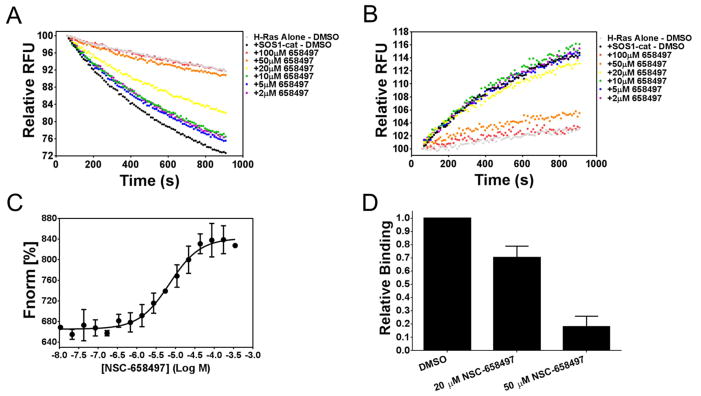
(A) Dose-dependent inhibition of 50 nM SOS1-cat (●) mediated GDP/GTP nucleotide exchange upon 2 μM H-Ras (●) in the BODIPY-FL-GDP dissociation assay at the indicated concentrations of NSC-658497. (B) Dose-dependent inhibition of 100 nM SOS1-cat (●) mediated GDP/GTP nucleotide exchange upon 2 μM H-Ras (●) in the BODIPY-TR-GTP loading assay at the indicated concentrations of NSC-658497. (C) Label-free microscale thermophoresis analysis of NSC-658497 direct binding to SOS1-cat. NSC-658497 was titrated between 0.01 and 350 μM to a constant amount of purified his6-SOS1-cat (500 nM) resulting in a Kd of −5.153 ± 0.076 Log M (7.04 μM). (D) Competition of microscale thermophoresis binding was performed in which NSC-658497 dose-dependently inhibited titration of purified his6-H-Ras (aa. 1-166) (0.4 – 100 μM) binding to NT-647 cysteine-labeled purified his6-SOS1-cat (50 nM) (bar graph). Data is expressed as relative binding and represents normalized binding values to the DMSO control at a non-saturating concentration H-Ras (12.5 μM). Data in A and B are expressed as percent change of relative fluorescence units normalized to the initial time point over 15 minutes. Data in A and B were measured in triplicate and represent the mean of N = 3 experiments. Data in C and D represent the mean ± SEM of N= 3 experiments.
Mutagenesis of SOS1 and Structure-activity Relationship of NSC-658497
To map the site of action for NSC-658497, alanine scanning mutagenesis of the SOS1 residues predicted to be involved in binding to NSC-658497 or Ras were carried out by mutating fourteen residues in the SOS1 catalytic site to alanine one at a time. Out of these fourteen single point mutants, four (I825A, T828A, T829A, and Y912A) completely abrogated binding to NSC-658497 (Figure 3A). The lack of binding activity was not likely due to improper protein folding as three of the mutants remained catalytically active toward Ras (Figure S3C). The fourth mutant, T829A, was catalytically dead, but is known to be required for interaction with H-Ras (Boriack-Sjodin et al., 1998). Interestingly, three of these four mutants (I825A, T828A, and T829A) mapped to a hydrophobic cavity in the catalytic site of SOS1 involved in Ras switch II recognition as predicted by computational docking studies (Figure 3A–B). Two other mutants, W809A and K814A, showed an enhanced binding to NSC-658497, likely due to a relieved steric hindrance and creation of a deeper pocket for accommodating NSC-658497 (Figure 3A), while H911A and K939A displayed only a slight reduction in binding, possibly due to being substituted by water molecules (Figure 3A). Taken together, these mutagenesis studies suggest that NSC-658497 binds to the catalytic site of SOS1 involved in interaction with the Ras switch II region (Figure 3B).
Figure 3. Mapping the site of action on SOS1 for NSC-658497.

(A) Identification of the site of action of NSC-658497 on SOS1-cat through mutagenesis studies. (right panel) Single alanine mutants of both the catalytic and allosteric sites of SOS1-cat were generated as described in the SI Experimental Procedures. Microscale thermophoresis analysis of NSC-658497 direct binding to SOS1-cat was performed. NSC-658497 was titrated between 0.006 and 100 μM to a constant amount of NT-647 amine-labeled purified his6-SOS1-cat (100 nM). Dissociation constants (Kd) were determined by nonlinear regression as described in the Experimental Procedures and expressed as a bar graph. Key interacting residues are indicated by the red, blue, and magenta boxes on the bar graph. (left panel) A structural view of the site of action for NSC-658497 in the catalytic site of SOS1. Key residues in SOS1 that interact with NSC-658497 based upon the mutagenesis studies are labeled in white and highlighted in red, blue, or magenta. (B) (left panel) A structural depiction of the interaction of the switch II region of H-Ras bound to the docking pocket of NSC-658497 in the catalytic site of SOS1. (right panel) in silico docking model of NSC-658497 bound to the catalytic site of SOS1 based upon docking simulations, mutagenesis and SAR data. Data in A is expressed as a bar graph of relative binding affinities normalized to the binding affinity of wild-type SOS1-cat. In panel A, if Kd was unable to be determined due to lack of a binding event, data is expressed as 0 in the bar graph. Data in A represent the mean ± SEM of N = 3 experiments.
To further understand the structure-activity relationship of the SOS1 inhibitor, a series of structural analogs of NSC-658497 containing the rhodanine or analogous hydantoin core moieties were examined by the SOS1 catalyzed BODIPY-FL GDP dissociation guanine nucleotide exchange reaction of Ras (Figure 4). Consistent with the mutagenesis data, alterations of the benzopyran moiety, which maps to the hydrophobic cavity in the catalytic site of SOS1, yielded significant changes in inhibitory potency. Elimination of the benzene ring while retaining the same pyran substitutions in Compound A1 (IC50 – 10.8 μM) led to a slight increase in potency. Retention of the dicarbonyl structure lacking a ring structure, as in Compound A2, caused a three-fold decrease in activity, while saturation of the double bond linker completely abrogated activity as seen in Compound A3. Furthermore, replacement of the benzopyran moiety with simple phenyl systems or removal of the benzopyran moiety (Compounds B1-4) resulted in a complete loss of activity (Figure 4 and Table S2). The overall similar activity seen with variance of the nitrophenyl moiety across analogs C1-C5 is consistent with attributing the activity to the benzofuran modifications.
Figure 4. Structure activity relationship study of NSC-658497 structural analogs.

NSC-658497 and related structural analogs were tested for their ability to dose-dependently inhibit 50 nM SOS1-cat mediated GDP/GTP nucleotide exchange upon 2 μM H-Ras (aa. 1-166) in the BODIPYFL-GDP dissociation assay. (Bottom panel) IC50s are depicted in the table and were calculated based upon a single time point of 900 seconds and nonlinear regression analysis (Prism 6; GraphPad Software Inc.). (Top panel) The NSC-658497 analogs were grouped into three groups (A, B, and C) based on their chemical structure as depicted in the schematic. IC50 values represent the mean ± SEM of N = 3 experiments.
Alteration of the nitrophenyl group in NSC-658497, which may interact with the polar residue Y912 in SOS1, modestly reduced activity. Deletion of the nitro function entirely as in Compound C1 led to a three-fold decrease in activity. Additions of simple hydroxyl (Compound C2) or methoxy (Compound C3) substituents at the ortho position had no effect upon the three-fold loss of activity. Adding both the hydroxyl and methoxy substituents, as in compound C5, at the ortho and meta positions, respectively, led to slight improvements in activity, but did not affect the level of NSC-658497 activity. Dimethoxy substitutions at the meta and para positions led to further loss of activity as seen in Compound C4 (Figure 4 and Table S1). These structure-activity relationship results are consistent with the mutagenesis data in identifying both the pyran and nitrophenyl moieties of NSC-658497 to be preferential for binding to SOS1.
NSC-658497 Inhibits Ras Signaling and Proliferation of Murine Fibroblasts
To assess whether NSC-658497 is active in inhibiting Ras activity and signaling in cells, NIH 3T3 mouse fibroblast cells were serum-starved and treated with varying concentrations of NSC-658497. Subsequently, the cells were stimulated with epidermal growth factor (EGF) to activate the EGF receptor (EGFR) - SOS1 - Ras signaling (Buday and Downward, 2008; Pierre et al., 2011). As shown in Figures 5A–B, NSC-658497 dose-dependently inhibited EGF-stimulated Ras, but not EGFR activation. Ras activation is known to signal downstream through the RAF-MEK signaling axis (Karnoub and Weinberg, 2008; Pierre et al., 2011; Vigil et al., 2010) to activate ERK1/2 or through the PI3K signaling axis to activate AKT (Boulbes et al., 2010; Jacinto et al., 2006; Karnoub and Weinberg, 2008; Vigil et al., 2010). Concomitant to Ras inhibition, NSC-658497 dose-dependently inhibited the EGF activated, Ras downstream targets ERK1/2 and AKT (Figure 5B). NSC-658497 appears to be selective for SOS1-Ras signaling as it did not affect H-Ras related R-Ras activity (Figure 5A) known to be independent of EGF-SOS1 signaling (Buday and Downward, 2008; Ohba et al., 2000; Tian and Feig, 2001). Additionally, NSC-658497 did not affect activity of the more distantly related Rho GTPase, Rac1, in cells (Figure S5A), further supporting the selectivity of NSC-658497.
Figure 5. NSC-658497 inhibits Ras signaling and proliferation of mouse fibroblast cells.

(A–B) NIH/3T3 cells were serum-starved overnight and pre-treated with the indicated concentrations of NSC-658497 for 2 hours. Subsequently, the cells were stimulated with 50 ng/mL of EGF for 5 minutes and were subjected to GST-Raf1 effector domain pull-down experiments or downstream Ras signaling western blotting experiments. The activation of pan-Ras and R-Ras, along with the activation of phospho-ERK1/2, phospho-AKT, and phospho-EGFR were analyzed by western blotting as described in the SI Experimental Procedures. (C) NIH/3T3 cells were grown in the presence of DMSO or indicated concentrations of NSC-658497 over three days. Every 24 hours the proliferation of the NIH/3T3 cells were measured by MTS assay as described in the Experimental Procedures. (D) After overnight starvation and 2 hour pre-treatment with the indicated concentrations of NSC-658497, cells were subjected to downstream Ras signaling western blotting for phospho-ERK1/2 and phospho-AKT activation as described in the SI Experimental Procedures. Wild-type (WT) MEF cells (E) and H-RasG12V transduced MEF cells (G) were subjected to a 2 day proliferation assay. MEF cells were grown in the presence of DMSO or indicated concentrations of NSC-658497 for 2 days. Then, proliferation was measured by MTS assay as described in the Experimental Procedures. (F,H) WT-MEF and H-RasG12V-MEF cells were serum-starved overnight, and then treated with the indicated concentrations of NSC-658497 for 2 hours. After compound treatment, the cells were subjected to phospho-ERK1/2 and phospho-AKT activation western blotting as described in the SI Experimental Procedures. Data in A, B, and D are representative of N = 3 experiments. Data in C were measured in triplicate and represent the mean ± SEM. Data in C is plotted as fold change in growth using Day 0 as the baseline. Data in E and G were performed in triplicate and represent the mean ± SEM. Data in E and G are plotted as fold change in growth using Day 0 as the baseline. Data in F and H are representative of N = 3 experiments. Statistical t test analyses were performed using GraphPad Prism 6 on Data in E (10 μM NSC-658497: p-value = 0.039; 20 μM NSC-658497: p-value = 0.047; 40 μM NSC-658497: p-value = 0.011).
The Ras-MEK-ERK1/2 and Ras-PI3K-AKT signaling axes are critical for cell proliferation (Karnoub and Weinberg, 2008; Pierre et al., 2011; Vigil et al., 2010). NSC-658497 dose-dependently inhibited cell proliferation in parallel with its inhibitory activity for the Ras downstream targets ERK1/2 and AKT in both NIH 3T3 fibroblasts and Hela cells (Figure 5C–D and Figure S5B). Additionally, the effect of NSC-658497 appeared reversible in inhibiting downstream ERK1/2 activity (Figure S5C). To examine the specificity of NSC-658497 in suppressing SOS1-Ras signaling, mouse embryonic fibroblasts overexpressing an active H-Ras mutant (G12V) known to be GTP-hydrolysis deficient and can signal independently of SOS1-mediated Ras activation (Pylayeva-Gupta et al., 2011) were generated (Stengel and Zheng, 2012). While NSC-658497 inhibited Ras signaling and proliferation of wild-type MEF cells in a dose-dependent manner (Figure 5E–F), it did not affect the proliferation or signaling of the H-Ras mutant cells (Figure 5G–H). Consistent with these results, NSC-658497 dose-dependently suppressed Ras signaling mediated by the overexpression of an active SOS1 mutant (W729L), originally identified in Noonan’s Syndrome (Tartaglia et al., 2007), in human embryonic kidney cells (Figure S5D). Taken together, these results suggest that NSC-658497 is specific for SOS1-Ras mediated cell functions.
NSC-658497 Inhibits Ras Signaling and Proliferation of Cancer Cells
SOS1 has been shown to be differentially expressed in prostate and breast cancer patient populations (Field et al., 2012; Timofeeva et al., 2009) and is required for Ras signaling and cell proliferation of prostate cancer cells (Timofeeva et al., 2009). We next tested the ability of NSC-658497 to inhibit DU-145 and PC-3 prostate cancer cell proliferation and Ras signaling. As shown in Figure 6, NSC-658497 dose-dependently inhibited Ras-GTP activity and the downstream p-ERK1/2 and p-Akt activities, as well as the proliferation, of these cells, similar to that by a SOS1-specific siRNA knockdown approach (Timofeeva et al., 2009).
Figure 6. NSC-658497 inhibits Ras signaling and proliferation of prostate cancer cells.

(A, C) DU-145 and PC-3 prostate cancer cells were subjected to a 3 day proliferation assay. PC-3 and DU-145 cells were grown in the presence of DMSO or the indicated concentrations of NSC-658497 for 3 days. Every 24 hours proliferation was measured by MTS assay as described in the Experimental Procedures. (B, D) DU-145 and PC-3 cells were serum-starved overnight, and then treated with the indicated concentrations of NSC-658497 for 2 hours. GST-Raf1 effector domain pull-down experiments for pan-Ras and downstream Ras signaling western blot experiments for phospho-ERK1/2 and phospho-AKT activation were performed as described in the SI Experimental Procedures. Data in A and C were performed in triplicate and represent the mean ± SEM. Data in A and C are plotted as relative growth by normalizing the data to Day 0. Data in B and D are representative of N = 3 experiments.
As a component of the NCI chemical library, NSC-658497 has been tested in the NCI-60 DTP human tumor cell line screen (http://dtp.nci.nih.gov). The DTP/NCI database shows that NSC-658497 was most sensitive on HOP-92 non-small cell lung cancer cells and OVCAR-3 ovarian cancer cells comparing with A549 non-small cell lung cancer and OVAR-5 ovarian cancer cells, respectively (Figure S5E). Interestingly, the HOP-92 and OVCAR-3 cells contain wild-type K-Ras status, while the A549 and OVCAR-5 cells contain oncogenic K-Ras mutations. These results are consistent with the MEF cell data suggesting a selectivity of NSC-658497 in inhibiting the growth of cells bearing wild type Ras vs. oncogenic mutant Ras (Figure 5C). NSC-658497 could be useful for selectively inhibiting proliferation of prostate, lung and ovarian cancer cells that show elevated Ras activity but do not contain oncogenic Ras mutations.
Discussion
Exploration for small molecule inhibitors of protein-protein interactions to dissect cell signaling pathways and as leads for therapeutic development has become increasingly attractive (Bosco et al., 2012). Multiple strategies can be employed to identify such candidate inhibitors, including non-biased high-throughput screening or structure based in silico screening (Evelyn et al., 2009; Evelyn et al., 2007; Gao et al., 2004). In recent years, there has been a renewed effort at targeting Ras GTPases that were once considered “undruggable”. In particular, Rac and Rho inhibitors, NSC-23766 and Rhosin, were identified that target the GEF interactive pockets in the small GTPase Rac1 and RhoA, respectively (Gao et al., 2004; Shang et al., 2012). In related studies, fragment-based high-throughput screening has been used to identify chemical fragments of small-molecules that bind to the GEF interaction surface of K-Ras, and as a consequence disrupting SOS-mediated Ras signaling (Maurer et al., 2012; Sun et al., 2012). In addition, several recent studies targeting oncogenic K-Ras GEF and effector binding have been carried out using high-throughput screening and fragment-based screening of cysteine-modifying small-molecule fragments, respectively (Ostrem et al., 2013; Zimmermann et al., 2013). Intriguingly, one such study yielded a small-molecule activator, not inhibitor, of Ras activity in vitro (Burns et al., 2014). It should be noted, however, that this class of small-molecules appears to inhibit, rather than activate, Ras downstream signaling events in cells (Burns et al., 2014). Another study has used a SOS1 derived peptide to disrupt SOS1-Ras interaction and subsequent Ras signaling, but its utility in cells remains unclear (Patgiri et al., 2011).
Conceptually, the activator enzymes, i.e. GEFs, of small GTPases represent attractive targets in manipulating their substrates, Ras GTPases, activities because they typically contain deeper, more druggable catalytic pockets that are critical for maintaining the activities of individual Ras family members. Our recent studies in rational targeting of Rho GEFs by small molecule inhibitors have provided an experimental basis for pursuing inhibitors with a similar mode of action against Ras GEF (Shang et al., 2013). In the present work, structural information on SOS1 and its complexes (Sondermann et al., 2004) serve as starting points to perform a virtual screen using the NCI chemical library. We select a hydrophobic cavity and nearby polar residues in the SOS1 catalytic domain that form an oblong bowl for Ras binding as the target for small molecule docking (Boriack-Sjodin et al., 1998; Hall et al., 2001). Multistage virtual screening against this site generates a short list of potential candidates that were subjected to a subsequent low-throughput experimental screen using the GEF reaction assay. As a result, a lead chemical inhibitor of SOS1 GEF activity, NSC-658497, was identified. Biochemical analysis of this lead compound, confirmed that it bound to SOS1 directly with a binding constant in the micromolar range, and it can competitively interfere with the SOS1-Ras interaction. Subsequent mutagenesis mapping and SAR analysis of the NSC-658497 structure indicate that the site of action in SOS1 likely includes the hydrophobic residue I825 and the surrounding polar Y912 and T829 residues that are critical for binding to the switch II region of Ras, whereas the aromatic benzopyran and the polar nitrophenyl moieties of NSC-658497 are critically involved in the inhibitory activity. Given its hydrophobicity, the benzopyran moiety may bind to the hydrophobic pocket of the SOS1 site thus serving as an anchor within the catalytic site of SOS1, while the polar nitrophenyl moiety may be involved in interactions outside of the hydrophobic core of the SOS1 site. Future co-crystal studies of the lead inhibitor-SOS1 complex will hopefully corroborate with these experimental data. Additionally, since most key residues identified in the SOS1 site of action are conserved in SOS2 (Figure S6A), it is likely that NSC-658497 could act upon both SOS isoforms. Conversely, because the key residues in the targeted pocket of SO1 are not well conserved in the other Ras GEFs, i.e. RasGRF1 and RasGRP1 (Figure S6B–D), it is possible that NSC-658497 does not work on these Ras activators. These predictions will need further experimental testing to verify.
In cells, NSC-658497 shows a dose-dependent inhibition of H-Ras activation through the well-established EGF-SOS1-Ras cascade but not the EGF-SOS1 signaling independent R-Ras activity (Buday and Downward, 2008; Ohba et al., 2000; Tian and Feig, 2001). It potently suppresses the Ras downstream p-ERK and p-Akt activities and the associated cell proliferation without affecting upstream EGFR signaling or active mutant Ras driven cell signaling. The inhibitory activity is not restricted to fibroblasts, as the inhibitor is equally functional in HeLa cells and prostate cancer cells. Interestingly, NSC-658497 mimics the effects of SOS1 knockdown in the prostate DU-145 cells where SOS1 is overexpressed and dominant in supporting Ras activity and cell growth (Timofeeva et al., 2009). Among the cancer cell lines tested in the NCI repository (http://dtp.nci.nih.gov), NSC-658497 appears to show a selective inhibition of growth of lung and ovarian cancer cells bearing WT K-Ras alleles, not the oncogenic K-Ras activating mutations, suggesting that SOS1 is a potential target for patient groups with cancers overexpressing SOS1 as a biomarker. Additionally, there has been increasing interest in targeting oncogenic Ras in active K-Ras mutant driven cancers. Recent studies suggest that SOS1 may play a role in oncogenic K-Ras tumorigenesis in pancreatic, colon, and breast cancer models dependent upon wild-type H- and N-Ras through an autocrine loop (Grabocka et al., 2014; Hocker et al., 2013; Jeng et al., 2012). It will be interesting to see if the SOS1 inhibitor could also have an advantage in targeting oncogenic K-Ras-driven tumors both in in vitro cell and in vivo mouse models, or whether the SOS1-H/N-Ras dependent proliferation of K-Ras driven tumors is limited to certain cancer types.
In summary, we have employed structural information and a functional biochemical GEF reaction assay to rationally design and identify a lead class of small molecule inhibitors of the RasGEF, SOS1. Our studies of the lead inhibitor, NSC-658497, establish a new approach of rational targeting of Rasopathies that Ras activating enzymes, such as SOS1, may serve as useful targets for future development of anti-cancer therapeutics.
Significance
Ras GTPases are well-known regulators of cell growth and survival activities critical for the development and progression of multiple human cancers. The activators of Ras, RasGEFs, are attractive targets in Ras pathway-driven tumors due to their potential “druggability” as enzymes catalyzing the Ras activation reaction. This study uses structural information of a key RasGEF, SOS1, to perform a virtual screen to identify small-molecule inhibitors that suppress SOS1-catalyzed Ras activation and disrupt the SOS1-Ras interaction. Our lead small-molecule inhibitor identified in this study binds to the catalytic site on SOS1 with a low micromolar affinity. Additionally, our lead small-molecule inhibitor suppresses Ras activation and downstream signaling in cells, along with preventing the growth of cancer cells dependent upon Ras activity. We present a new approach of targeting Rasopathies and a novel class of small-molecule inhibitors of SOS1 useful as both biological tools and for future therapeutic development.
Experimental Procedures
Plasmids, Cell Lines, and Reagents
Details for plasmid generation, cell lines used, and reagents used are described in the SI Experimental Procedures.
Computational Virtual Screening and SAR Analysis
Virtual Screening was performed to identify candidate molecules that could disrupt the interaction between SOS1 and H-Ras, by targeting the SOS1 interaction interface with H-Ras (“catalytic site”). The docking simulations for the virtual screening were performed as described in the SI Experimental Procedures using a subset of 118,500 drug-like synthetic compounds from the NCI/DTP Open Chemical Repository (http://dtp.cancer.gov).
Protein Purifications
Details for protein purifications are described in the SI Experimental Procedures.
Microscale Thermophoresis (MST) Binding Assays
Microscale thermophoresis is a biophysical technique that measures the motion of molecules along microscopic temperature gradients. Fluorescent-labeled molecules or intrinsic fluorescence of molecules are used to monitor their movement along these temperature gradients. Changes in a molecule’s hydration shell, charge, or size will affect a molecule’s movement along these temperature gradients. These changes are utilized to determine binding events (Wienken et al., 2010).
For label-free analysis, 500 nM his6-SOS1-cat was incubated with indicated concentrations of NSC-658497 in binding buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.1% Tween-20, and 3.5% DMSO) at room temperature for 30 minutes. Subsequently, samples were loaded into hydrophilic capillaries (NanoTemper Technologies, Munich, Germany) and binding was measured with a Monolith NT.Label Free reader (NanoTemper Technologies) based upon intrinsic tryptophan fluorescence of the SOS1-cat protein.
For labeled analysis, both purified his6-SOS1-cat (wild-type and mutant) and c-terminal truncated his6-H-Ras were labeled with the red NT-647 amine-reactive dye using N-hydroxy succinimide-ester chemistry using a protein labeling kit (NanoTemper Technologies). Indicated concentrations of NSC-658497 were incubated with NT-647-labeled SOS1cat(100 nM) or H-Ras(100 nM) in binding buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM MgCl2, 0.1% Triton X-100, and 2% DMSO) at room temperature in the dark for 30 minutes. For competition binding experiments, purified his6-SOS1-cat was labeled with the red NT-647 cysteine-reactive dye using a protein labeling kit (NanoTemper Technologies). Indicated concentrations of purified his6-H-Ras (aa. 1-166) were incubated with NT-647-labeled SOS1-cat (50 nM) in the presence of indicated concentrations of NSC-658497 in binding buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM MgCl2, 0.1% Tween-20, and 2% DMSO) at room temperature in the dark for 30 minutes. Subsequently, samples were loaded into hydrophilic capillaries (NanoTemper Technologies) and binding was measured with a Monolith NT.115 reader (NanoTemper Technologies). Binding data were analyzed using Thermophoresis or Hot/Cold analysis as described previously (Jerabek-Willemsen et al., 2011). Data were normalized to either ΔFnorm[%] (10*(Fnorm(bound) – Fnorm(unbound))) or Fraction bound (ΔFnorm[%]/amplitude) (Jerabek-Willemsen et al., 2011). Dissociation constants (Kds) of the binding data were calculated using nonlinear regression analysis (Prism 6; GraphPad Software Inc., La Jolla, CA).
Western Blot Analysis
Details for the western blot analysis is described in the SI Experimental Procedures.
Guanine Nucleotide Exchange Reaction Assays
For the GDP dissociation assay, purified his6-H-Ras (aa. 1-166) was pre-loaded with BODIPY-FL-GDP for 1 hour at room temperature (Stochiometric Ratio – 4 protein:1 labeled nucleotide). Exchange buffer (EB) (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM MgCl2, 0.01% NP-40 Alternative, 1 mM DTT) was added to wells of a 96-well plate (Corning; cat.#: 3915) for both the GDP-dissociation and GTP-loading assays. 82 μl and 85 μl of EB were added to wells containing H-Ras alone for the GDP-dissociation and GTP-loading assays, respectively. 73 μl and 76 μl of EB were added to wells containing both H-Ras and SOS1-cat for the GDP-dissociation and GTP-loading assays, respectively. For both assays, 9 μl of purified his6-SOS1-cat (50 nM in GDP-dissociation assay, and 100 nM in GTP-loading assay) was added to appropriate wells. For the GDP-dissociation assay, 10 μl of 50-fold excess of GTP (100 μM – final) was added to each well. Subsequently, in both assays DMSO and indicated concentrations of NSC-658497 were added to the wells (1% DMSO in final concentration) and the plate was briefly shaken for 30 seconds and incubated for 10 minutes at room temperature. In the GDP-dissociation assay, 8 μl of his6-H-Ras (aa. 1-166) bound to Bodipy-FL-GDP (2 μM final concentration) was added to each well. In the GTP-loading assay, 2.5-fold excess BODIPY-TR-GTP (5 μM final concentration) was added to each well. The plates were read continuously for 15 minutes on an EnVision plate reader (Perkin-Elmer) with the excitation at 485 nm, emission at 535 nm in the GDP-dissociation assay, and the excitation at 560 nm, emission at 635 nm in the GTP-loading assay.
MTS Cell Proliferation Assay
MTS cell proliferation assay was carried out as per the manufacturer’s instructions (Promega, Madison, WI; #G3580) starting with 2,000 cells (H-Ras-G12V MEF cells) or 1,000 cells (wild-type MEF, NIH/3T3, HeLa, DU-145, or PC-3 cells) per well, in triplicate per sample. Briefly 100 μl cell suspension aliquots containing 2,000 cells or 1,000 cells were added to every well of a 96-well plate (Corning, Corning, NY) and cultured 24 hours. Then, cells were treated with indicated concentrations of NSC-658497 or DMSO for 24, 48, or 72 hours. Subsequently, the medium was changed to 100 μl of complete DMEM medium plus 20 μl of MTS substrate (Promega). The metabolic activity of the cells were analyzed by absorbance change at 490 nm with a VMax Kinetic ELISA microplate reader (Molecular Devices, Sunnyvale, CA) after a 2 hour incubation at 37°C and 5% CO2.
Supplementary Material
Highlights.
Translation of the mechanism of Ras regulation into a new Ras targeting approach;
Structure based rational design of small-molecule inhibitors of SOS1;
A proof of principle of inhibiting the catalytic activity of the Ras GEF, SOS1;
Lead chemical inhibitors as useful tools against Rasopathies.
Acknowledgments
We thank Dr. Thomas Schubert (Regensburg, Germany) for performing a label-free MST binding assay. We thank Dr. James Mulloy (Cincinnati Children’s Hospital Medical Center, Division of Experimental Hematology and Cancer Biology) for providing the HeLa cells, and Dr. Zhongyun Dong (University of Cincinnati, Department of Internal Medicine) for providing the prostate cancer cell lines. This work was supported in part by National Institutes of Health Grants.
Footnotes
Author Contributions
C.R.E. designed research, performed research, analyzed data, wrote paper, and edited paper. X.D. performed research. J.B performed research. W.L.B. analyzed data and edited paper. J.M. designed research, contributed computational tools, analyzed computational data, and edited paper. Y.Z. designed research, wrote paper, and edited paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Appels NM, Beijnen JH, Schellens JH. Development of farnesyl transferase inhibitors: a review. The oncologist. 2005;10:565–578. doi: 10.1634/theoncologist.10-8-565. [DOI] [PubMed] [Google Scholar]
- Biesiada J, Porollo A, Velayutham P, Kouril M, Meller J. Survey of public domain software for docking simulations and virtual screening. Human genomics. 2011;5:497–505. doi: 10.1186/1479-7364-5-5-497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boriack-Sjodin PA, Margarit SM, Bar-Sagi D, Kuriyan J. The structural basis of the activation of Ras by Sos. Nature. 1998;394:337–343. doi: 10.1038/28548. [DOI] [PubMed] [Google Scholar]
- Bosco EE, Kumar S, Marchioni F, Biesiada J, Kordos M, Szczur K, Meller J, Seibel W, Mizrahi A, Pick E, et al. Rational design of small molecule inhibitors targeting the Rac GTPase-p67(phox) signaling axis in inflammation. Chemistry & biology. 2012;19:228–242. doi: 10.1016/j.chembiol.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulbes D, Chen CH, Shaikenov T, Agarwal NK, Peterson TR, Addona TA, Keshishian H, Carr SA, Magnuson MA, Sabatini DM, et al. Rictor phosphorylation on the Thr-1135 site does not require mammalian target of rapamycin complex 2. Molecular cancer research: MCR. 2010;8:896–906. doi: 10.1158/1541-7786.MCR-09-0409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buday L, Downward J. Many faces of Ras activation. Biochimica et biophysica acta. 2008;1786:178–187. doi: 10.1016/j.bbcan.2008.05.001. [DOI] [PubMed] [Google Scholar]
- Burns MC, Sun Q, Daniels RN, Camper D, Kennedy JP, Phan J, Olejniczak ET, Lee T, Waterson AG, Rossanese OW, et al. Approach for targeting Ras with small molecules that activate SOS-mediated nucleotide exchange. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:3401–3406. doi: 10.1073/pnas.1315798111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox AD, Der CJ. Ras history: The saga continues. Small GTPases. 2010;1:2–27. doi: 10.4161/sgtp.1.1.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evelyn CR, Ferng T, Rojas RJ, Larsen MJ, Sondek J, Neubig RR. High-throughput screening for small-molecule inhibitors of LARG-stimulated RhoA nucleotide binding via a novel fluorescence polarization assay. Journal of biomolecular screening. 2009;14:161–172. doi: 10.1177/1087057108328761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evelyn CR, Wade SM, Wang Q, Wu M, Iniguez-Lluhi JA, Merajver SD, Neubig RR. CCG-1423: a small-molecule inhibitor of RhoA transcriptional signaling. Molecular cancer therapeutics. 2007;6:2249–2260. doi: 10.1158/1535-7163.MCT-06-0782. [DOI] [PubMed] [Google Scholar]
- Field LA, Love B, Deyarmin B, Hooke JA, Shriver CD, Ellsworth RE. Identification of differentially expressed genes in breast tumors from African American compared with Caucasian women. Cancer. 2012;118:1334–1344. doi: 10.1002/cncr.26405. [DOI] [PubMed] [Google Scholar]
- Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:7618–7623. doi: 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabocka E, Pylayeva-Gupta Y, Jones MJ, Lubkov V, Yemanaberhan E, Taylor L, Jeng HH, Bar-Sagi D. Wild-type H- and N-Ras promote mutant K-Ras-driven tumorigenesis by modulating the DNA damage response. Cancer cell. 2014;25:243–256. doi: 10.1016/j.ccr.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BE, Yang SS, Boriack-Sjodin PA, Kuriyan J, Bar-Sagi D. Structure-based mutagenesis reveals distinct functions for Ras switch 1 and switch 2 in Sos-catalyzed guanine nucleotide exchange. The Journal of biological chemistry. 2001;276:27629–27637. doi: 10.1074/jbc.M101727200. [DOI] [PubMed] [Google Scholar]
- Hara M, Akasaka K, Akinaga S, Okabe M, Nakano H, Gomez R, Wood D, Uh M, Tamanoi F. Identification of Ras farnesyltransferase inhibitors by microbial screening. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:2281–2285. doi: 10.1073/pnas.90.6.2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart TC, Zhang Y, Gorry MC, Hart PS, Cooper M, Marazita ML, Marks JM, Cortelli JR, Pallos D. A mutation in the SOS1 gene causes hereditary gingival fibromatosis type 1. American journal of human genetics. 2002;70:943–954. doi: 10.1086/339689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocker HJ, Cho KJ, Chen CY, Rambahal N, Sagineedu SR, Shaari K, Stanslas J, Hancock JF, Gorfe AA. Andrographolide derivatives inhibit guanine nucleotide exchange and abrogate oncogenic Ras function. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:10201–10206. doi: 10.1073/pnas.1300016110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin JJ, Shoichet BK. ZINC--a free database of commercially available compounds for virtual screening. Journal of chemical information and modeling. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. ZINC: a free tool to discover chemistry for biology. Journal of chemical information and modeling. 2012;52:1757–1768. doi: 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- James GL, Goldstein JL, Brown MS, Rawson TE, Somers TC, McDowell RS, Crowley CW, Lucas BK, Levinson AD, Marsters JC., Jr Benzodiazepine peptidomimetics: potent inhibitors of Ras farnesylation in animal cells. Science. 1993;260:1937–1942. doi: 10.1126/science.8316834. [DOI] [PubMed] [Google Scholar]
- Jang SI, Lee EJ, Hart PS, Ramaswami M, Pallos D, Hart TC. Germ line gain of function with SOS1 mutation in hereditary gingival fibromatosis. The Journal of biological chemistry. 2007;282:20245–20255. doi: 10.1074/jbc.M701609200. [DOI] [PubMed] [Google Scholar]
- Jeng HH, Taylor LJ, Bar-Sagi D. Sos-mediated cross-activation of wild-type Ras by oncogenic Ras is essential for tumorigenesis. Nature communications. 2012;3:1168. doi: 10.1038/ncomms2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerabek-Willemsen M, Wienken CJ, Braun D, Baaske P, Duhr S. Molecular interaction studies using microscale thermophoresis. Assay and drug development technologies. 2011;9:342–353. doi: 10.1089/adt.2011.0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nature reviews Molecular cell biology. 2008;9:517–531. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenzen C, Cool RH, Prinz H, Kuhlmann J, Wittinghofer A. Kinetic analysis by fluorescence of the interaction between Ras and the catalytic domain of the guanine nucleotide exchange factor Cdc25Mm. Biochemistry. 1998;37:7420–7430. doi: 10.1021/bi972621j. [DOI] [PubMed] [Google Scholar]
- Lenzen C, Cool RH, Wittinghofer A. Analysis of intrinsic and CDC25-stimulated guanine nucleotide exchange of p21ras-nucleotide complexes by fluorescence measurements. Methods in enzymology. 1995;255:95–109. doi: 10.1016/s0076-6879(95)55012-7. [DOI] [PubMed] [Google Scholar]
- Margarit SM, Sondermann H, Hall BE, Nagar B, Hoelz A, Pirruccello M, Bar-Sagi D, Kuriyan J. Structural evidence for feedback activation by Ras.GTP of the Ras-specific nucleotide exchange factor SOS. Cell. 2003;112:685–695. doi: 10.1016/s0092-8674(03)00149-1. [DOI] [PubMed] [Google Scholar]
- Maurer T, Garrenton LS, Oh A, Pitts K, Anderson DJ, Skelton NJ, Fauber BP, Pan B, Malek S, Stokoe D, et al. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:5299–5304. doi: 10.1073/pnas.1116510109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen DP, Gee KR, Kang HC, Neubig RR. Fluorescent BODIPY-GTP analogs: real-time measurement of nucleotide binding to G proteins. Analytical biochemistry. 2001;291:109–117. doi: 10.1006/abio.2001.5011. [DOI] [PubMed] [Google Scholar]
- McEwen DP, Gee KR, Kang HC, Neubig RR. Fluorescence approaches to study G protein mechanisms. Methods in enzymology. 2002;344:403–420. doi: 10.1016/s0076-6879(02)44730-1. [DOI] [PubMed] [Google Scholar]
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of computational chemistry. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohba Y, Mochizuki N, Yamashita S, Chan AM, Schrader JW, Hattori S, Nagashima K, Matsuda M. Regulatory proteins of R-Ras, TC21/R-Ras2, and M-Ras/R-Ras3. The Journal of biological chemistry. 2000;275:20020–20026. doi: 10.1074/jbc.M000981200. [DOI] [PubMed] [Google Scholar]
- OpenEye Scientific Software, I.w.e.c.
- Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–551. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patgiri A, Yadav KK, Arora PS, Bar-Sagi D. An orthosteric inhibitor of the Ras-Sos interaction. Nature chemical biology. 2011;7:585–587. doi: 10.1038/nchembio.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierre S, Bats AS, Coumoul X. Understanding SOS (Son of Sevenless) Biochemical pharmacology. 2011;82:1049–1056. doi: 10.1016/j.bcp.2011.07.072. [DOI] [PubMed] [Google Scholar]
- Porollo A, Meller J. POLYVIEW-MM: web-based platform for animation and analysis of molecular simulations. Nucleic acids research. 2010;38:W662–666. doi: 10.1093/nar/gkq445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nature reviews Cancer. 2011;11:761–774. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AE, Araki T, Swanson KD, Montgomery KT, Schiripo TA, Joshi VA, Li L, Yassin Y, Tamburino AM, Neel BG, et al. Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nature genetics. 2007;39:70–74. doi: 10.1038/ng1926. [DOI] [PubMed] [Google Scholar]
- Rojas JM, Oliva JL, Santos E. Mammalian son of sevenless Guanine nucleotide exchange factors: old concepts and new perspectives. Genes & cancer. 2011;2:298–305. doi: 10.1177/1947601911408078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schopel M, Jockers KF, Duppe PM, Autzen J, Potheraveedu VN, Ince S, Yip KT, Heumann R, Herrmann C, Scherkenbeck J, et al. Bisphenol A Binds to Ras Proteins and Competes with Guanine Nucleotide Exchange: Implications for GTPase-Selective Antagonists. Journal of medicinal chemistry. 2013;56:9664–9672. doi: 10.1021/jm401291q. [DOI] [PubMed] [Google Scholar]
- Shang X, Marchioni F, Evelyn CR, Sipes N, Zhou X, Seibel W, Wortman M, Zheng Y. Small-molecule inhibitors targeting G-protein-coupled Rho guanine nucleotide exchange factors. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:3155–3160. doi: 10.1073/pnas.1212324110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang X, Marchioni F, Sipes N, Evelyn CR, Jerabek-Willemsen M, Duhr S, Seibel W, Wortman M, Zheng Y. Rational design of small molecule inhibitors targeting RhoA subfamily Rho GTPases. Chemistry & biology. 2012;19:699–710. doi: 10.1016/j.chembiol.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondermann H, Soisson SM, Boykevisch S, Yang SS, Bar-Sagi D, Kuriyan J. Structural analysis of autoinhibition in the Ras activator Son of sevenless. Cell. 2004;119:393–405. doi: 10.1016/j.cell.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Stengel KR, Zheng Y. Essential role of Cdc42 in Ras-induced transformation revealed by gene targeting. PloS one. 2012;7:e37317. doi: 10.1371/journal.pone.0037317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Burke JP, Phan J, Burns MC, Olejniczak ET, Waterson AG, Lee T, Rossanese OW, Fesik SW. Discovery of small molecules that bind to K-Ras and inhibit Sos-mediated activation. Angew Chem Int Ed Engl. 2012;51:6140–6143. doi: 10.1002/anie.201201358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia M, Pennacchio LA, Zhao C, Yadav KK, Fodale V, Sarkozy A, Pandit B, Oishi K, Martinelli S, Schackwitz W, et al. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nature genetics. 2007;39:75–79. doi: 10.1038/ng1939. [DOI] [PubMed] [Google Scholar]
- Tian X, Feig LA. Basis for signaling specificity difference between Sos and Ras-GRF guanine nucleotide exchange factors. The Journal of biological chemistry. 2001;276:47248–47256. doi: 10.1074/jbc.M107407200. [DOI] [PubMed] [Google Scholar]
- Timofeeva OA, Zhang X, Ressom HW, Varghese RS, Kallakury BV, Wang K, Ji Y, Cheema A, Jung M, Brown ML, et al. Enhanced expression of SOS1 is detected in prostate cancer epithelial cells from African-American men. International journal of oncology. 2009;35:751–760. [PMC free article] [PubMed] [Google Scholar]
- Vigil D, Cherfils J, Rossman KL, Der CJ. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nature reviews Cancer. 2010;10:842–857. doi: 10.1038/nrc2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienken CJ, Baaske P, Rothbauer U, Braun D, Duhr S. Protein-binding assays in biological liquids using microscale thermophoresis. Nature communications. 2010;1:100. doi: 10.1038/ncomms1093. [DOI] [PubMed] [Google Scholar]
- Zimmermann G, Papke B, Ismail S, Vartak N, Chandra A, Hoffmann M, Hahn SA, Triola G, Wittinghofer A, Bastiaens PI, et al. Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature. 2013;497:638–642. doi: 10.1038/nature12205. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.