

Abstract
The novel anti-epileptic drug lacosamide (LCM; SPM927, Vimpat®) has been heralded as having a dual-mode of action through interactions with both the voltage-gated sodium channel and the neurite outgrowth-promoting collapsin response mediator protein 2 (CRMP2). Lacosamide’s ability to dampen neuronal excitability through the voltage-gated sodium channel likely underlies its efficacy in attenuating the symptoms of epilepsy (i.e. seizures). While the role of CRMP2 in epilepsy has not been well-studied, given the proposed involvement of circuit reorganization in epileptogenesis, the ability of lacosamide to alter CRMP2 function may prove disease-modifying. Recently, however, the validity of lacosamide’s interaction with CRMP2 has come under scrutiny. In this review, we address the contradictory reports concerning the binding of lacosamide to CRMP2, as well as, the ability of lacosamide to directly impact CRMP2 function. Additionally, we address similarly contradicting reports regarding the potential disease-modifying effect of lacosamide on the development and progression of epilepsy. As the vast majority of anti-epileptic drugs influence only the symptoms of epilepsy, the ability to hinder disease progression would be a major breakthrough in efforts to cure or prevent this debilitating syndrome.
Keywords: Lacosamide, CRMP2, Sodium channels, Slow inactivation, Epileptogenesis
Introduction: Lacosamide as a novel anti-epileptic
Lacosamide (LCM; SPM927, Vimpat®) (R-N-benzyl 2-acetamido-3-methoxypropionamide) is a first in class antiepileptic drug (AED) approved by the United States Food and Drug Administration and the European Medicines Agency for adjunctive treatment of partial-onset epilepsy with or without secondary generalization in adults (for review see [1]). Lacosamide was first identified as the lead compound in a class of functionalized amino acids demonstrating antiepileptic properties in the maximal electroshock model [2,3]. Lacosamide reduced seizure activity in the genetically susceptible Frings mouse as well as the hippocampal kindled rat [4]. Additionally lacosamide demonstrated antiepileptic potential in the 6 Hz psychomotor seizure model of partial epilepsy [5,6]. The efficacy of lacosamide in these diverse models separated it from currently available anti-epileptic drugs. However, lacosamide was unable to ameliorate tonic-clonic seizures associated with subcutaneous administration of picrotoxin, bicuculline, or pentylenetetrazole [6]. Phase III clinical trials evaluated the ability of lacosamide to control partial-onset seizures when used in conjunction with 1–3 other AEDs (for review see [7]). The proportion of subjects who reported a ≥50% seizure reduction rate increased from 23% with placebo to 40% with 400 mg lacosamide treatment. Unfortunately, seizure freedom remained out of reach, with only 5 of 107 subjects reporting total loss of seizure activity within the 28 day trial period [8]. While improvement was observed with the addition of lacosamide treatment, results were modest. However, the advantage of lacosamide lies within its low potential for drug-drug interactions, allowing it to be combined with almost every class of AED as a concomitant treatment option [7]. While pharmacokinetic studies report a limited impact of age and gender on lacosamide serum levels [9], no influence on efficacy was identified in clinical trials [10]. Lacosamide treatment was also associated with low incidence of severe adverse advents, with the most commonly reported side effects being dizziness, headache, nausea, and diplopia [11] (For an in-depth review of the clinical value and limitations of lacosamide please see [1]).
Mechanism of action
Despite its success in both pre-clinical and clinical studies, the mechanism by which lacosamide alleviated seizure activity remained unknown [5]. Radioligand displacement studies attempted to identify a target for lacosamide within the central nervous system. While the ability of lacosamide to weakly displace H3-Batrachotoxin indicated some level of binding to voltage-gated sodium channels (VGSCs), lacosamide treatment did not affect steady-state gating kinetics or current density [12]. It was ultimately determined that lacosamide reduces excitability through selective enhancement of VGSC slow inactivation [13]. It was hypothesized that this unique method of action would allow for discrimination between steady-state and aberrantly increased levels of firing. Lacosamide derivatives were employed as affinity baits in an attempt to identify any additional targets within the central nervous system [14–16]. The results of these studies suggested that, aside from the VGSC, lacosamide may also target collapsin response mediator protein 2 (CRMP2). CRMP2, also known as DPYSL2/DRP2, Unc-33, Ulip, or TUC2, is an intracellular phosphoprotein first identified for its role in regulating growth cone extension/retraction [17,18]. The canonical roles of CRMP2 include its involvement in neurite outgrowth, axon/dendrite specification, neuronal polarity, progenitor proliferation, and radial migration (for review see [19,20,18]). Central to many of these functions is the ability of CRMP2 to regulate microtubule dynamics. Through interactions with the anterograde motor protein kinesin, CRMP2 aids in the transport of tubulin dimers from the soma to distal projections [21]. Additionally, CRMP2 stabilizes the growing end of the microtubule and promotes the inherent GTPase activity of tubulin [22]. The aforementioned processes are governed by the phosphorylation state of CRMP2, whereby phosphorylation by GSK3β, Cdk5, ROCK, CaMKII, or FYN kinase renders CRMP2 inactive, promoting neurite retraction and growth cone collapse [23–31]. Recent work has uncovered a multitude of additional roles for CRMP2 in neurons including neurodegeneration, cell death, and trafficking control [32–39]. Indeed, the CRMP2 interactome has expanded to include proteins involved in microtubule dynamics, synaptic assembly, vesicle recycling and endocytosis, neurotransmitter release, and calcium channel regulation (for review see [40]). Given the number and wide variety of functions and interacting partners, it is not surprising that CRMP2 has been implicated in numerous disease states. Therefore, the potential impact of lacosamide interacting with CRMP2 is substantial.
Does lacosamide have a dual mode of action?
Despite the original evidence supporting CRMP2 as a target of lacosamide, the validity of the interaction remains controversial. The first evidence supporting the interaction can be found in the aforementioned preclinical report published by Beyreuther and colleagues [14]. Direct evidence supporting their claims that CRMP2 is a target of lacosamide can be found within a patent application filed by three of the authors: Beyreuther, Stohr, and Freitag [41]. The most convincing data within the application are the radioligand binding studies where they demonstrated competitive and specific binding of [14C]-lacosamide to crude fractions isolated from Xenopus oocytes transfected with CRMP2, as well as rat brain membranes. These studies reported a Kd-value lower than 5 μM. Importantly, radioligand binding could be competed off with an excess of cold, unlabeled lacosamide. Additionally, no specific binding was reported from control Oocyte fractions not containing CRMP2. Based on these results, along with others supporting the interaction of CRMP2 and lacosamide, the application states the following: “CRMP2 is therefore regarded as a target of lacosamide in the nervous system in epilepsy, pain, essential tremor, dyskinesias, amyotrophic lateral sclerosis, schizophrenia, and other disease conditions” [41]. Soon after, Park and colleagues demonstrated affinity-bait capture of CRMP2 by lacosamide in two separate reports [15,16]. Affinity-bait methods aid in the identification of unknown protein binding partners (Figure 1). In these experiments, two additional moieties are added to the ligand in question (i.e. lacosamide), an “affinity bait moiety” (AB) and a “chemical reporter moiety” (CR). Upon binding of the ligand to a potential target, the AB moiety covalently modifies the target creating an irreversible interaction. Binding partners can be identified by in-gel fluorescence of the CR moiety. The addition of the AB and CR groups can potentially modify the ligand; therefore, it is imperative for the final product to mimic the original ligand in functionality. Lacosamide agents containing the AB and CR moieties (lacosamide AB-CR) were determined to provide antiepileptic relief in the MES model similar to that of the parent compound [15,16]. Soluble mouse brain lysates were incubated with 5 μM of the lacosamide AB-CR compound for 30 minutes at room temperature and separated using electrophoresis. Proteins captured by the lacosamide AB-CR compound were visualized by in-gel fluorescence of the CR moiety. Reactivity was observed around 62 kDa, which correlates to the size of CRMP2. CRMP2 immunoreactivity was also confirmed at 62 kDa from the samples. To confirm the identity of the 62 kDa protein captured by the affinity bait experiment, the experiment was repeated using a biotinylated lacosamide AB-CR compound. Lacosamide AB-CR/target protein complexes could then be isolated using streptavidin precipitation. Mass spectrometry of the gel-excised 62 kDa band captured by the AB-CR compound confirmed its identity to be CRMP2. Importantly, the capture of CRMP2 by lacosamide AB-CR could be prevented in a dose-dependent manner by excesses of the original lacosamide compound [16]. Other potential targets have since been identified using this method, such as 14-3-3 [42]. However, the impact of this interaction has not yet been determined.
Figure 1. Affinity bait capture of lacosamide-interacting proteins.
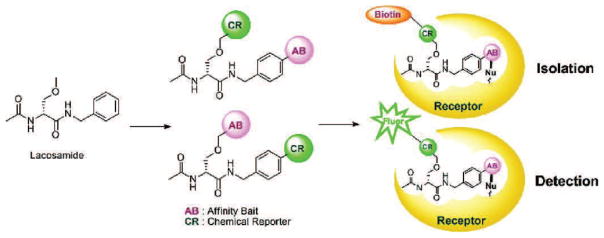
Lacosamide compounds modified to contain affinity bait (AB) and chemical reporter (CR) moieties. Capture and covalent modification of interaction targets occurs at the AB site. Addition of the CR site allows for the isolation (via biotin) or detection (via fluorescence) of the captured complex. Adapted with permission from Park and colleagues [88].
A 2010 report by our laboratory verified the ability to capture CRMP2 with lacosamide affinity bait compounds [43]. Furthermore, in silico docking was used to identify putative binding sites for lacosamide within the CRMP2 protein. The technique uses the known structure of the target protein (CRMP2) to predict the structure of the intermolecular complex when bound to a ligand (lacosamide) (for review see [44]). A total of 100 runs were carried out over the surface of the CRMP2 protein to yield five pockets capable of coordinating lacosamide binding. Interestingly, it was observed that CRMP2 expression levels could influence the ability of lacosamide to transition voltage-gated sodium channels to the slow-inactivated state in a neuronal cell line. Site-directed mutagenesis of key residues within the previously identified binding pockets on CRMP2 prevented the impact of CRMP2-overexpression on modulation of VGSC slow inactivation by lacosamide.
While evidence suggested that CRMP2 might be a target of lacosamide, it was unclear if this interaction would impact the function of CRMP2. Calcium dysregulation has been suggested to play a large role in the pathophysiology of various epilepsies [45]. As CRMP2 is a positive regulator of N-type calcium channels, our laboratory sought to determine if acute or chronic lacosamide treatment could impact calcium channel currents. Primary cultured hippocampal neurons were treated with 300 μM lacosamide for 0.5–24 hours. Whole cell patch clamp recordings revealed that neither acute nor chronic treatment altered current density or kinetics of activation or inactivation [46]. As the L-type calcium channel currents were inhibited by the presence of nifedipine, currents predominantly represented calcium carried through N-type channels, with a small percentage attributed to P/Q-type channels at this age in culture [47]. Consistent with previous findings, overexpression of CRMP2 led to an ~60% increase in current density, which was not altered by the presence of lacosamide. We then investigated if lacosamide could impact the canonical role of CRMP2 in neurite outgrowth. Sholl analysis was used to measure neurite length and complexity in primary cultured cortical neurons. This technique measures the number of neurites crossing concentric circles (denoted as intersections or branch points) at various radial distances from the cell soma [48]. This consecutive-circles (cumulative intersection) analysis identifies dendritic geometry, ramification abundance, and branching patterns. Overnight application of 300 μM lacosamide led to a ~30% decrease in neurite outgrowth, which could not be replicated with the application of other sodium channel inhibitors [49]. Specificity of this outcome was confirmed, as lacosamide was unable to further reduce outgrowth following siRNA knockdown of CRMP2. Concentration-response curves yielded an IC50 of ~25 μM, a concentration which was unable to alter sodium channel slow inactivation, suggesting that lacosamide may alter CRMP2 function at concentrations previously considered to be sub-therapeutic. However, the distinct mechanism by which lacosamide impaired CRMP2-mediated neurite outgrowth remained unclear. CRMP2 promotes outgrowth through two separate and distinct mechanisms: (1) linking tubulin dimers to the motor protein kinesin to aid in anterograde transport [21] and (2) enhancing the intrinsic GTPase activity of tubulin [22]. The ability of CRMP2 to co-immunoprecipitate tubulin was not affected by upwards of 300 μM lacosamide [49]. Tubulin polymerization, as measured by a turbidimetric assay, is greatly increased in the presence of purified CRMP2 protein. While high concentrations of lacosamide (300 μM) did not alter basal levels of tubulin polymerization, the addition of 3 μM lacosamide was sufficient to prevent increases in polymerization by CRMP2. Previously, our laboratory had created a mutant CRMP2 protein (CRMP25ALA) in which 5 residues, identified by in silico docking to be the most critical for lacosamide binding, had been mutated to alanines. Lacosamide was unable to block the enhancement of tubulin polymerization by CRMP25ALA. Based on these results, we concluded that lacosamide reduces CRMP2-mediated neurite outgrowth by selectively impairing the ability of CRMP2 to enhance tubulin polymerization.
The aforementioned findings supporting a direct interaction between lacosamide and CRMP2 are in contrast to a 2012 report by Wolff and colleagues [50]. Previous radioligand binding studies reported a Kd of ~5 μM; however these employed [14C] lacosamide which was reported to have low specific radioactivity, limiting further binding studies. Therefore, Wolff and colleagues used [3H] lacosamide, whose specific activity was 1000 fold higher than that of [14C] lacosamide. No specific binding was observed in fractions taken from rat cortex, hippocampus, striatum, or cerebellum that were incubated with 300 nM [3H] lacosamide. To maximize CRMP2 levels, fractions were taken from CRMP2-expressing oocytes and incubated with 969 nM [3H] lacosamide. Still, no specific binding was observed. CRMP2 expressed in Cos-7 cells was sequestered by coupling to SPA beads prior to incubation with 600 nM [3H] lacosamide. Again, no specific binding was reported. Ultimately, surface plasmon resonance (SPR) was employed to address binding between immobilized CRMP2 and lacosamide. SPR monitors changes in resonance angle of light off a surface as a direct result of changes in the refraction index of the surface. Ligands (lacosamide) interacting the proteins (CRMP2) immobilized onto the surface alters the refraction index of the surface which is detected through changes in the light’s resonance angle (for review see [51,52]). SPR studies revealed no detectable interaction between immobilized CRMP2 and lacosamide, ranging in concentration from 0.39–100 μM. However, due to the harsh conditions required for the immobilization process, it is necessary to demonstrate functionality of the protein before lack of binding can be verified. Also, availability of the binding site must also be considered. While the immobilized protein should exist in a variety of confirmations, there is a slight possibility that one orientation is energetically favored, resulting in the majority of the immobilized protein in one confirmation. For these reasons, the use of SPR to confirm lack of binding is considered controversial. Based on the lack of specific binding in this report, the authors infer that any effects of lacosamide on CRMP2, or vice versa, must be indirect in nature. The authors suggest that results gleaned from affinity bait studies do not provide direct evidence that CRMP2 is a target of lacosamide. As the structure of the ligand required modification (addition of AB and CR moieties), the observed interaction may be a result of an altered receptor interaction profile and may not be reflective of interactions between the parent compound and the proposed target.
Recently, our laboratory has employed the novel technique of microscale thermophoresis (MST) to investigate potential binding between lacosamide and CRMP2. MST employs the movement of proteins induced by microscopic temperature gradients to measure interactions between ligands and target proteins. This movement is determined by the entropy of the hydration shell surrounding the target protein. Binding of other proteins, peptides, or small molecules alters the hydration shell, resulting in a change in thermophoretic movement that is visualized as a change in fluorescence [53] (Figure 2A). Primary-amine labeled CRMP2 or CRMP25ALA were incubated with varying concentrations of lacosamide (0.009–150 μM and 0.03–1000 μM, respectively). Standard capillaries (Monolith NT Capillaries, NanoTemper) were filled with ~5 μl of sample mixture and thermophoresis analysis was performed on a NanoTemper Monolith NT.115 instrument. MST curves were fitted using GraphPad Prism software to obtain relative Kd values. Thermophoresis of labeled CRMP2 was altered by increasing concentrations of lacosamide, indicating an interaction with Kd value of 1 ± 0.04 μM (Figure 2B–C). Thermophoresis of labeled CRMP25ALA was not affected by lacosamide, even at concentrations as high as 1 mM (Figure 2D–F). The lack of association with CRMP25ALA suggests both that the ability of lacosamide to alter thermophoresis of wildtype CRMP2 is not due to non-specific binding and that the interaction between lacosamide and CRMP2 is coordinated by the binding pockets previously identified via in silico docking.
Figure 2. Binding of lacosamide ((R)-LCM) to wildtype but not mutant CRMP2 in solution.

(A) Representative microscale thermophoresis (MST) curve. A standard capillary containing NT647-labeled protein is locally heated by an IR laser (green arrow). Labeled protein diffuses away from the heated spot, causing a local depletion and drop in fluorescence. Fluorescence returns following cessation of the IR laser (red arrow). Dashed yellow lines indicate the point at which the degree of thermodiffusion is measured. (B). MST time traces of wildtype CRMP2 and (R)-LCM (0.009–150 μM). Thermodiffusion of CRMP2 was altered by increasing concentrations of (R)-LCM. (C) Logarithmic concentration-response curve used to determine the dissociation constant of (R)-LCM to fluorescently labeled CRMP2. Values represent mean ± SEM from 3 separate trials. (D) CRMP2 surface representation highlighting the location of the (R)-LCM binding pocket (green). The box represents an enlarged view of the binding pocket highlighting the helices and beta-strands (gold) involved in coordinating (R)-LCM binding. The mutated residues comprising CRMP25ALA are indicated in single amino acid letter code. (E) MST time traces of CRMP25ALA and (R)-LCM (0.030–1000 μM). Thermodiffusion of CRMP25ALA was not altered by increasing concentrations of (R)-LCM. (F) Logarithmic concentration-response curve further demonstrates that (R)-LCM does not interact with CRMP25ALA.
While the MST results provide strong evidence in support of a direct interaction between lacosamide and CRMP2, more research is needed in order to definitively conclude the nature of the interaction, such as irreversibility and dynamics. However, the ability of lacosamide to impair neurite outgrowth and, more specifically, tubulin polymerization induced by wildtype but not mutant CRMP2 provides very strong evidence that lacosamide directly impairs CRMP2 function, irrespective of the nature of the physical interaction. The question remains as to what therapeutic impact this interaction may have. The majority of approved anti-epileptic drugs are effective at treating the symptoms of epilepsy, yet cannot be considered “disease modifying” as they do not directly impede disease progression. Partial-onset seizures are associated with acquired epilepsy as they often develop as a direct result of CNS trauma, such as stroke, traumatic brain injury (TBI), infections, or tumors [54]. Unfortunately, the mechanisms underlying epileptogenesis, particularly in acquired epilepsies, are not well understood.
Therapeutic potential of an interaction between CRMP2 and lacosamide
One hallmark of acquired epilepsy is the presence of a potentially months-long asymptomatic latent period between the instigating injury and the development of spontaneous recurrent seizures. This latent period provides a unique opportunity for prophylactic intervention if therapeutic targets were to be identified. Animal models have revealed significant morphological changes occurring during this period, which may be attributed to the development of epilepsy. One of the more striking changes is aberrant sprouting of axons in response to injury, which has been observed both in the hippocampus and the neocortex in a variety of models [55–57]. Sprouting of injured axon fibers is associated with the formation of recurrent excitatory circuits [58], which directly contribute to the development of spontaneous seizure activity [59,60]. Robust axon sprouting and elongation has also been observed following in vitro lesion of axon collaterals and is sufficient to induce hyperexcitability and increased polysynaptic, epileptiform burst discharges [61]. A previous study of post-mortem tissue samples from patients with mesial temporal lobe epilepsy observed a decrease in a 55 kDa band associated with CRMP2, without a change in the higher molecular weight band [62]. It is now known that as the two splice variants of CRMP2 are expressed at 75- and 64 kDa [63], the 55 kDa band likely reflects a calpain-mediated breakdown product of CRMP2 [64]. What will be of particular interest are potential changes in phosphorylation state within the hippocampus of temporal lobe epilepsy patients, as the ability of CRMP2 to mediate neurite outgrowth is regulated via numerous phosphorylation events [65–67,30,25,27,68–70]. Indeed, changes in phosphorylation of CRMP2 have been observed following traumatic events often associated with the development of temporal lobe epilepsy, such as hypoxia-ischemia and TBI [71,72]. Interestingly, recent work has linked the sprouting of mossy fibers in the hippocampus following status epilepticus with CRMP2 [73]. The ability of lacosamide to target not only sodium channel activity, but CRMP2-mediated synaptic reorganization as well, may afford efficacy in the prevention of epileptogenesis, where so many classical antiepileptics have failed. However, the anti-epileptogenic potential of lacosamide relies heavily on the assumption that the relationship between aberrant sprouting and epileptogenesis is causal in nature.
Work by Brandt and colleagues demonstrated that lacosamide treatment could hinder the progression of kindling [74], a model often used to investigate mechanisms of epileptogenesis (for review see [75]). As the majority of AEDs are not disease-modifying, they do not alter kindling acquisition (for review see [76]). Rats were kindled by once-daily stimulation of the amygdala 0.5 hrs following intra-peritoneal injection of 3, 10, or 30 mg/kg lacosamide. Both the severity of seizures and duration of after-discharges elicited by each current injection were decreased in animals treated with 10 or 30 mg/kg lacosamide. Additionally, lacosamide treatment led to a ~90% increase in the number of stimulations required for kindling. In fact, while all of the vehicle-treated rats were successfully kindled, kindling in the 10 and 30 mg/kg treatment groups was unsuccessful in 2/10 and 3/10 rats, respectively. To determine if the effect of lacosamide was long-lasting, after 22–23 days of stimulation rats were allowed a 2.5 month washout period, during which they received no current injections. All rats could be rekindled within 4 stimulations, regardless of prior lacosamide treatment. This outcome can be explained in a variety of ways. Of the rats used for the washout experiments, 7/10 lacosamide treated rats and 10/10 vehicle treated rats had reached kindled status, which by definition insinuates that all rats underwent class V seizures in response to the same current injection. Therefore, as the seizure severity was relatively conserved across treatment groups in the presence of treatment, differences following a treatment-free washout period were unlikely. For this reason, previous work involving washout periods following amygdala stimulations excluded animals which had reached kindled status prior to washout [77]. In these studies, which identified potential anti-epileptogenic properties of the AED levetiracetam, the rate of kindling following washout was slower in previously exposed animals. Once kindling was obtained, however, focal seizure threshold was not different from controls. Therefore it is possible that while the kindling model provides a framework for anti-epileptogenic intervention, once kindling status has been reached the opportunity for prophylaxis may have passed. Alternatively, as lacosamide is now known to lower excitability by enhancing sodium channel slow-inactivation, treatment directly prior to stimulation may have simply dampened the response to the current injections, thereby impeding the kindling process.
Aside from kindling, models involving spontaneous recurring seizures following sustained status epilepticus (SE) in rodents have been extensively used to evaluate anti-epileptogenic potential of therapeutic agents. Animals were given a single dose of lacosamide, 10 minutes following initiation of perforant path stimulation, which lasted 30 minutes. Lacosamide treatment was associated with a dose-dependent decrease in the number of spontaneous recurrent seizures (SRS) 6 weeks following induction of status epilepticus [78]. As the development of SRS is dependent upon successful, sustained status epilepticus, it is possible that the reduction of seizures is a direct result of lacosamide interfering with the induction process. Therefore, a second group of animals received lacosamide treatment 40 minutes following perforant path stimulation. While all of the vehicle-treated animals demonstrated SRS, they were only observed in 3 of 9 lacosamide-treated animals. Interestingly, of the animals that did develop SRS, there was no difference in the number of seizures across groups, suggesting that the development of SRS may be an all or nothing process. That late lacosamide treatment was able to prevent the development of SRS in 6 of 9 animals suggests that lacosamide may have some anti-epileptogenic potential. Although its direct role in epileptogenesis has not been confirmed, extensive mossy fiber sprouting occurs following SE and has been linked to changes in CRMP2 function [73]. Our laboratory recently employed the undercut (neocortical isolation) model of post-traumatic epileptogenesis to explore the ability of lacosamide to alter CRMP2-mediated changes in excitatory connectivity [49]. In this model of posttraumatic epileptogenesis, axon sprouting has been demonstrated by morphological reconstruction of axons of the layer V pyramidal neurons [55] and by functional mapping of excitatory synaptic connectivity using laser scanning photostimulation of caged glutamate [58]. Animals received daily lacosamide treatments for 1 week following surgery, upon which time the animals were allowed to an additional week. While field potential recordings did not reveal a difference in the frequency of evoked epileptiform events, the mean amplitude of the recordings was significantly lower in lacosamide treated animals. Importantly, electrophysiological recordings from layer V pyramidal neurons revealed a decrease in the frequency of spontaneous excitatory post-synaptic currents. A decrease in frequency without a change in the amplitude or kinetics (rise time, decay time, constant, and ½ width of events) is indicative of a decrease in the number of synaptic connections. Indeed glutamate uncaging studies demonstrated a decrease in excitatory connectivity following lacosamide treatment. As animals had undergone a 1-week washout period prior to all recordings, these results suggest that lacosamide may have had a disease-modifying effect following neocortical isolation.
As previously mentioned, traumatic brain injury is a common instigator of acquired epilepsy. Mice receiving closed-head TBI were treated with lacosamide 30 minutes following injury and continuing twice daily for 3 days. Lacosamide treatment was associated with a decrease in neuronal injury that was correlated by a delay in the expression of pro-inflammatory mediator genes and reduced microglial activation [79]. Lacosamide-treated mice demonstrated improved function in both the rotorod and Morris water maze paradigms. Interestingly, contrary to previous reports that CRMP2 activity is important in mossy fiber sprouting and that lacosamide can impact CRMP2 function, the emergence of mossy fiber projections into the inner molecular layer was not altered by lacosamide treatment. However, mossy fiber sprouting is viewed as a progressive process and is typically measured in much later stages following injury. It would be interesting to extend treatment well into the latent period when mossy fiber sprouting is at its highest. In a recent study of rats exposed to the lateral fluid percussion model of TBI, however, lacosamide given at 30 min post-injury and continued at 8 hr intervals for 3 days had no effect on neuroscore, beam walking, or water maze performance [80]. Furthermore, histological analysis revealed no difference in the extent of neurodegeneration, axonal injury, or hilar neuron loss within the hippocampus of lacosamide-treated rats compared to vehicle treatment. Recently, Licko and colleagues sought to expand previous reports on the anti-epileptogenic potential of lacosamide following status epilepticus [81]. To avoid any possible interference with the induction of sustained SE, lacosamide treatment began following its cessation and was continued for 23 days. On the molecular level, many hallmarks associated with status epilepticus were altered by lacosamide treatment. Chronic lacosamide treatment prevented both neurogenesis of granule neurons as well as cell loss within the piriform cortex and CA1 region of the hippocampus yet did not impede the aberrant migration of neurons into the hilus. Unfortunately, the extent of mossy fiber sprouting was not determined in this report. However, lacosamide treatment was associated with a decrease in the number of SE-induced persistent basal dendrites present in the hilar region. Despite the impact on molecular changes induced by SE and contrary to previous reports, lacosamide treatment did not successfully prevent the development of SRS. Additionally, latency to first seizure and the frequency of spontaneous seizures were not attenuated by chronic lacosamide treatment [81]. While the treatment paradigm was somewhat unorthodox, combining oral administration and continuous infusion via osmotic minipump, it is unlikely that this would account for the lack of efficacy in preventing the development of spontaneous seizures. In light of all the aforementioned work in vivo, it is difficult to determine whether lacosamide is anti-epileptogenic in nature.
Combined in vivo results suggest that lacosamide may have potential as a disease-modifying AED; however, more work is needed in 2 critical areas: (1) It is necessary to determine whether lacosamide prevents aberrant sprouting, such as that of mossy fibers within the hippocampus, in multiple of models of acquired epilepsy. While it is likely that there are common mechanisms involved in aberrant sprouting induced by different insults, even slight disparities may account for incongruous reports using different models. It is important not to dismiss the therapeutic potential of a compound based on the results from one model alone, especially as it is unclear which model most directly mimics the human condition. (2) Is preventing aberrant sprouting sufficient to prevent epileptogenesis? The relationship between epileptogenesis and circuit reorganization is still highly debated. In theory, the formation of recurrent excitatory circuits should increase excitability and decrease the threshold for spontaneous epileptiform events [82]. However, recent work by the Buckmaster group has demonstrated the ability to attenuate mossy fiber sprouting without impacting seizure frequency following status epilepticus in mice [83]. While the results suggest a disconnect between mossy fiber sprouting and epileptogenesis, the authors advise that rapamycin, at the doses required to completely block mossy fiber sprouting, has many other effects. According to the authors, some of these effects may be pro-epileptogenic in nature, thereby canceling out any potential anti-epileptogenic effect that the prevention of mossy fiber sprouting may carry. Therefore, we still cannot definitively identify the exact role of mossy fiber sprouting without more selective means.
Additionally, as CRMP2 is also involved with important developmental processes such as neuronal migration and polarity, we must also consider the potential negative impact of targeting CRMP2 in pediatric patients. Despite these concerns, however, the safety profile of lacosamide in children is reminiscent of that in adults [84]. In fact, lacosamide has recently shown promising potential in the treatment of refractory focal epilepsy with high tolerability in two separate reports utilizing patient populations with a median age of 13.9 and 2.7 years [85,86]. Nevertheless, limited data is available for the long-terms effects of lacosamide treatment in pediatric patients or those exposed prenatally [87]. Given the role of neurite outgrowth in neuronal plasticity, of particular interest will be the long-term impact of lacosamide treatment on learning and memory.
Conclusion
Here we have addressed two controversial issues surrounding the novel anti-epileptic drug lacosamide. First, is CRMP2 a bona fide target of lacosamide? Second, does lacosamide’s proposed dual mode of action provide disease-modifying properties? Unfortunately, direct answers exist for neither of these issues. However, new data presented here of a specific association between lacosamide and CRMP2 in solution, in combination with previous reports of a direct impact on CRMP2 function, provide strong evidence in support of a direct interaction between lacosamide and CRMP2. Future work should seek to address the disease-modifying potential of lacosamide. Given the two distinct mechanisms through which lacosamide may impact epileptogenesis (hyperexcitability via the VGSC and circuit reorganization via CRMP2) it would be of great interest to dissect out the individual importance of each on the full mechanism of action of lacosamide. Additionally, understanding the molecular and mechanistic framework for lacosamide’s actions on CRMP2 will consolidate the involvement of CRMP2 as a potentially important player in neuropathology and as a novel drug target for epilepsy.
Figure 3. Mechanisms through which lacosamide (LCM) may provide disease-modifying effects.

Enhancement of sodium channel slow inactivation leads to a decrease in the fraction of available voltage-gated sodium channels. Through this mechanism, LCM is able to hinder epileptiform activity. Inhibition of CRMP2-mediated tubulin polymerization may attenuate the aberrant circuit reorganization commonly observed within the epileptic hippocampus and neocortex. While the exact contribution of this reorganization to epileptogenesis is not yet fully understood, it is possible that the two functions of lacosamide may provide the ability to affect disease progression.
References
- 1.Biton V. Lacosamide for the treatment of partial-onset seizures. Expert review of neurotherapeutics. 2012;12 (6):645–655. doi: 10.1586/ern.12.50. [DOI] [PubMed] [Google Scholar]
- 2.Cortes S, Liao ZK, Watson D, Kohn H. Effect of structural modification of the hydantoin ring on anticonvulsant activity. J Med Chem. 1985;28 (5):601–606. doi: 10.1021/jm50001a012. [DOI] [PubMed] [Google Scholar]
- 3.Choi D, Stables JP, Kohn H. The anticonvulsant activities of functionalized N-benzyl 2-acetamidoacetamides. The importance of the 2-acetamido substituent. Bioorganic and Medicinal Chemistry. 1996;4 (12):2105–2114. doi: 10.1016/s0968-0896(96)00225-8. [DOI] [PubMed] [Google Scholar]
- 4.Bialer M, Johannessen SI, Kupferberg HJ, Levy RH, Loiseau P, Perucca E. Progress report on new antiepileptic drugs: a summary of the Fifth Eilat Conference (EILAT V) Epilepsy research. 2001;43 (1):11–58. doi: 10.1016/s0920-1211(00)00171-6. [DOI] [PubMed] [Google Scholar]
- 5.Duncan GE, Kohn H. The novel antiepileptic drug lacosamide blocks behavioral and brain metabolic manifestations of seizure activity in the 6 Hz psychomotor seizure model. Epilepsy research. 2005;67 (1–2):81–87. doi: 10.1016/j.eplepsyres.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 6.Stöhr T, Kupferberg HJ, Stables JP, Choi D, Harris RH, Kohn H, Walton N, White HS. Lacosamide, a novel anti-convulsant drug, shows efficacy with a wide safety margin in rodent models for epilepsy. Epilepsy research. 2007;74 (2–3):147–154. doi: 10.1016/j.eplepsyres.2007.03.004. http://dx.doi.org/10.1016/j.eplepsyres.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 7.Perucca E, Yasothan U, Clincke G, Kirkpatrick P. Lacosamide. Nat Rev Drug Discov. 2008;7 (12):973–974. doi: 10.1038/nrd2764. [DOI] [PubMed] [Google Scholar]
- 8.EMA EMA. Vimpat. EPAR summary for the public. 2012 http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000863/human_med_001139.jsp&mid=WC0b01ac058001d124.
- 9.Markoula S, Teotonio R, Ratnaraj N, Duncan JS, Sander JW, Patsalos PN. Lacosamide Serum Concentrations in Adult Patients With Epilepsy: The Influence of Gender, Age, Dose, and Concomitant Antiepileptic Drugs. Therapeutic drug monitoring. 2014 doi: 10.1097/FTD.0000000000000051. [DOI] [PubMed] [Google Scholar]
- 10.Chung S, Ben-Menachem E, Sperling MR, Rosenfeld W, Fountain NB, Benbadis S, Hebert D, Isojarvi J, Doty P. Examining the clinical utility of lacosamide: pooled analyses of three phase II/III clinical trials. CNS drugs. 2010;24 (12):1041–1054. doi: 10.2165/11586830-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 11.Beydoun A, D’Souza J, Hebert D, Doty P. Lacosamide: pharmacology, mechanisms of action and pooled efficacy and safety data in partial-onset seizures. Expert review of neurotherapeutics. 2009;9 (1):33–42. doi: 10.1586/14737175.9.1.33. [DOI] [PubMed] [Google Scholar]
- 12.Errington AC, Coyne L, Stohr T, Selve N, Lees G. Seeking a mechanism of action for the novel anticonvulsant lacosamide. Neuropharmacology. 2006;50 (8):1016–1029. doi: 10.1016/j.neuropharm.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 13.Errington AC, Stohr T, Heers C, Lees G. The investigational anticonvulsant lacosamide selectively enhances slow inactivation of voltage-gated sodium channels. Molecular Pharmacology. 2008;73 (1):157–169. doi: 10.1124/mol.107.039867. [DOI] [PubMed] [Google Scholar]
- 14.Beyreuther BK, Freitag J, Heers C, Krebsfanger N, Scharfenecker U, Stohr T. Lacosamide: a review of preclinical properties. CNS Drug Rev. 2007;13 (1):21–42. doi: 10.1111/j.1527-3458.2007.00001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park KD, Morieux P, Salome C, Cotten SW, Reamtong O, Eyers C, Gaskell SJ, Stables JP, Liu R, Kohn H. Lacosamide isothiocyanate-based agents: novel agents to target and identify lacosamide receptors. Journal of Medicinal Chemistry. 2009;52 (21):6897–6911. doi: 10.1021/jm9012054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park KD, Stables JP, Liu R, Kohn H. Proteomic searches comparing two (R)-lacosamide affinity baits: An electrophilic arylisothiocyanate and a photoactivated arylazide group. Organic & Biomolecular Chemistry. 2010;8 (12):2803–2813. doi: 10.1039/c000987c. [DOI] [PubMed] [Google Scholar]
- 17.Wang LH, Strittmatter SM. A family of rat CRMP genes is differentially expressed in the nervous system. J Neurosci. 1996;16 (19):6197–6207. doi: 10.1523/JNEUROSCI.16-19-06197.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmidt EF, Strittmatter SM. The CRMP family of proteins and their role in Sema3A signaling. Advances in Experimental Medicine and Biology. 2007;600:1–11. doi: 10.1007/978-0-387-70956-7_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Charrier E, Reibel S, Rogemond V, Aguera M, Thomasset N, Honnorat J. Collapsin response mediator proteins (CRMPs): involvement in nervous system development and adult neurodegenerative disorders. Molecular Neurobiology. 2003;28 (1):51–64. doi: 10.1385/MN:28:1:51. [DOI] [PubMed] [Google Scholar]
- 20.Hensley K, Venkova K, Christov A, Gunning W, Park J. Collapsin Response Mediator Protein-2: An Emerging Pathologic Feature and Therapeutic Target for Neurodisease Indications. Molecular Neurobiology. 2011 doi: 10.1007/s12035-011-8166-4. [DOI] [PubMed] [Google Scholar]
- 21.Fukata Y, Itoh TJ, Kimura T, Menager C, Nishimura T, Shiromizu T, Watanabe H, Inagaki N, Iwamatsu A, Hotani H, Kaibuchi K. CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat Cell Biol. 2002;4 (8):583–591. doi: 10.1038/ncb825. [DOI] [PubMed] [Google Scholar]
- 22.Chae YC, Lee S, Heo K, Ha SH, Jung Y, Kim JH, Ihara Y, Suh PG, Ryu SH. Collapsin response mediator protein-2 regulates neurite formation by modulating tubulin GTPase activity. Cellular Signalling. 2009;21 (12):1818–1826. doi: 10.1016/j.cellsig.2009.07.017. [DOI] [PubMed] [Google Scholar]
- 23.Arimura N, Inagaki N, Chihara K, Menager C, Nakamura N, Amano M, Iwamatsu A, Goshima Y, Kaibuchi K. Phosphorylation of collapsin response mediator protein-2 by Rho-kinase. Evidence for two separate signaling pathways for growth cone collapse. Journal of Biological Chemistry. 2000;275 (31):23973–23980. doi: 10.1074/jbc.M001032200. [DOI] [PubMed] [Google Scholar]
- 24.Brown M, Jacobs T, Eickholt B, Ferrari G, Teo M, Monfries C, Qi RZ, Leung T, Lim L, Hall C. Alpha2-chimaerin, cyclin-dependent Kinase 5/p35, and its target collapsin response mediator protein-2 are essential components in semaphorin 3A-induced growth-cone collapse. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2004;24 (41):8994–9004. doi: 10.1523/jneurosci.3184-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cole AR, Knebel A, Morrice NA, Robertson LA, Irving AJ, Connolly CN, Sutherland C. GSK-3 phosphorylation of the Alzheimer epitope within collapsin response mediator proteins regulates axon elongation in primary neurons. The Journal of biological chemistry. 2004;279 (48):50176–50180. doi: 10.1074/jbc.C400412200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arimura N, Menager C, Kawano Y, Yoshimura T, Kawabata S, Hattori A, Fukata Y, Amano M, Goshima Y, Inagaki M, Morone N, Usukura J, Kaibuchi K. Phosphorylation by Rho kinase regulates CRMP-2 activity in growth cones. Mol Cell Biol. 2005;25 (22):9973–9984. doi: 10.1128/MCB.25.22.9973-9984.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uchida Y, Ohshima T, Yamashita N, Ogawara M, Sasaki Y, Nakamura F, Goshima Y. Semaphorin3A signaling mediated by Fyn-dependent tyrosine phosphorylation of collapsin response mediator protein 2 at tyrosine 32. The Journal of biological chemistry. 2009;284 (40):27393–27401. doi: 10.1074/jbc.M109.000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Uchida Y, Ohshima T, Sasaki Y, Suzuki H, Yanai S, Yamashita N, Nakamura F, Takei K, Ihara Y, Mikoshiba K, Kolattukudy P, Honnorat J, Goshima Y. Semaphorin3A signalling is mediated via sequential Cdk5 and GSK3beta phosphorylation of CRMP2: implication of common phosphorylating mechanism underlying axon guidance and Alzheimer’s disease. Genes to Cells. 2005;10 (2):165–179. doi: 10.1111/j.1365-2443.2005.00827.x. [DOI] [PubMed] [Google Scholar]
- 29.Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A, Kaibuchi K. GSK-3[beta] Regulates Phosphorylation of CRMP-2 and Neuronal Polarity. Cell. 2005;120 (1):137–149. doi: 10.1016/j.cell.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 30.Cole AR, Causeret F, Yadirgi G, Hastie CJ, McLauchlan H, McManus EJ, Hernandez F, Eickholt BJ, Nikolic M, Sutherland C. Distinct priming kinases contribute to differential regulation of collapsin response mediator proteins by glycogen synthase kinase-3 in vivo. The Journal of biological chemistry. 2006;281 (24):16591–16598. doi: 10.1074/jbc.M513344200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hou ST, Jiang SX, Aylsworth A, Ferguson G, Slinn J, Hu H, Leung T, Kappler J, Kaibuchi K. CaMKII phosphorylates collapsin response mediator protein 2 and modulates axonal damage during glutamate excitotoxicity. Journal of Neurochemistry. 2009;111 (3):870–881. doi: 10.1111/j.1471-4159.2009.06375.x. [DOI] [PubMed] [Google Scholar]
- 32.Brittain JM, Chen L, Wilson SM, Brustovetsky T, Gao X, Ashpole NM, Molosh AI, You H, Hudmon A, Shekhar A, White FA, Zamponi GW, Brustovetsky N, Chen J, Khanna R. Neuroprotection against traumatic brain injury by a peptide derived from the collapsin response mediator protein 2 (CRMP2) The Journal of biological chemistry. 2011;286 (43):37778–37792. doi: 10.1074/jbc.M111.255455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brittain JM, Pan R, You H, Brustovetsky T, Brustovetsky N, Zamponi GW, Lee WH, Khanna R. Disruption of NMDAR-CRMP-2 signaling protects against focal cerebral ischemic damage in the rat middle cerebral artery occlusion model. Channels (Austin) 2012;6(1) doi: 10.4161/chan.18919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nishimura T, Fukata Y, Kato K, Yamaguchi T, Matsuura Y, Kamiguchi H, Kaibuchi K. CRMP-2 regulates polarized Numb-mediated endocytosis for axon growth. Nat Cell Biol. 2003;5 (9):819–826. doi: 10.1038/ncb1039. [DOI] [PubMed] [Google Scholar]
- 35.Kawano Y, Yoshimura T, Tsuboi D, Kawabata S, Kaneko-Kawano T, Shirataki H, Takenawa T, Kaibuchi K. CRMP-2 is involved in kinesin-1-dependent transport of the Sra-1/WAVE1 complex and axon formation. Molecular and cellular biology. 2005;25 (22):9920–9935. doi: 10.1128/MCB.25.22.9920-9935.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kimura T, Watanabe H, Iwamatsu A, Kaibuchi K. Tubulin and CRMP-2 complex is transported via Kinesin-1. J Neurochem. 2005;93 (6):1371–1382. doi: 10.1111/j.1471-4159.2005.03063.x. [DOI] [PubMed] [Google Scholar]
- 37.Lykissas MG, Batistatou AK, Charalabopoulos KA, Beris AE. The role of neurotrophins in axonal growth, guidance, and regeneration. Current neurovascular research. 2007;4 (2):143–151. doi: 10.2174/156720207780637216. [DOI] [PubMed] [Google Scholar]
- 38.Arimura N, Kimura T, Nakamuta S, Taya S, Funahashi Y, Hattori A, Shimada A, Ménager C, Kawabata S, Fujii K, Iwamatsu A, Segal RA, Fukuda M, Kaibuchi K. Anterograde Transport of TrkB in Axons Is Mediated by Direct Interaction with Slp1 and Rab27. Developmental Cell. 2009;16 (5):675–686. doi: 10.1016/j.devcel.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 39.Rahajeng J, Giridharan SS, Naslavsky N, Caplan S. Collapsin response mediator protein-2 (Crmp2) regulates trafficking by linking endocytic regulatory proteins to dynein motors. Journal of Biological Chemistry. 2010 doi: 10.1074/jbc.C110.166066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khanna R, Wilson SM, Brittain JM, Weimer J, Sultana R, Butterfield A, Hensley K. Opening Pandora’s jar: a primer on the putative roles of CRMP2 in a panoply of neurodegenerative, sensory and motor neuron, and central disorders. Future Neurol. 2012;7 (6):749–771. doi: 10.2217/fnl.12.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beyreuther B, Stohr T, Freitag J. Google Patents. 2009. Method for identifying crmp modulators. [Google Scholar]
- 42.Park KD, Kim D, Reamtong O, Eyers C, Gaskell SJ, Liu R, Kohn H. Identification of a lacosamide binding protein using an affinity bait and chemical reporter strategy: 14-3-3 zeta. Journal of the American Chemical Society. 2011;133 (29):11320–11330. doi: 10.1021/ja2034156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Y, Brittain JM, Jarecki BW, Park KD, Wilson SM, Wang B, Hale R, Meroueh SO, Cummins TR, Khanna R. In silico docking and electrophysiological characterization of lacosamide binding sites on collapsin response mediator protein 2 (CRMP-2) identifies a pocket important in modulating sodium channel slow inactivation. Journal of Biological Chemistry. 2010 doi: 10.1074/jbc.M110.128801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sousa SF, Fernandes PA, Ramos MJ. Protein-ligand docking: current status and future challenges. Proteins. 2006;65 (1):15–26. doi: 10.1002/prot.21082. [DOI] [PubMed] [Google Scholar]
- 45.Delorenzo RJ, Sun DA, Deshpande LS. Cellular mechanisms underlying acquired epilepsy: the calcium hypothesis of the induction and maintainance of epilepsy. Pharmacology & therapeutics. 2005;105 (3):229–266. doi: 10.1016/j.pharmthera.2004.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Y, Khanna R. Calcium channels are not affected by the anti-epileptic drug lacosamide. Translational Neuroscience. 2011 doi: 10.2478/s13380-011-0002-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang JF, Randall AD, Ellinor PT, Horne WA, Sather WA, Tanabe T, Schwarz TL, Tsien RW. Distinctive pharmacology and kinetics of cloned neuronal Ca2+ channels and their possible counterparts in mammalian CNS neurons. Neuropharmacology. 1993;32 (11):1075–1088. doi: 10.1016/0028-3908(93)90003-l. [DOI] [PubMed] [Google Scholar]
- 48.Sholl DA. Dendritic organization in the neurons of the visual and motor cortices of the cat. Journal of Anatomy. 1953;87 (4):387–406. [PMC free article] [PubMed] [Google Scholar]
- 49.Wilson SM, Xiong W, Wang Y, Ping X, Head JD, Brittain JM, Gagare PD, Ramachandran PV, Jin X, Khanna R. Prevention of posttraumatic axon sprouting by blocking collapsin response mediator protein 2-mediated neurite outgrowth and tubulin polymerization. Neuroscience. 2012;210:451–466. doi: 10.1016/j.neuroscience.2012.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wolff C, Carrington B, Varrin-Doyer M, Vandendriessche A, Van der Perren C, Famelart M, Gillard M, Foerch P, Rogemond V, Honnorat J, Lawson A, Miller K. Drug Binding Assays do not Reveal Specific Binding of Lacosamide to Collapsin Response Mediator Protein 2 (CRMP-2) CNS Neuroscience & Therapeutics. 2012;18 (6):493–500. doi: 10.1111/j.1755-5949.2012.00313.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Johnsson B, Lofas S, Lindquist G. Immobilization of proteins to a carboxymethyldextran-modified gold surface for biospecific interaction analysis in surface plasmon resonance sensors. Anal Biochem. 1991;198 (2):268–277. doi: 10.1016/0003-2697(91)90424-r. [DOI] [PubMed] [Google Scholar]
- 52.Phizicky EM, Fields S. Protein-protein interactions: methods for detection and analysis. Microbiological reviews. 1995;59 (1):94–123. doi: 10.1128/mr.59.1.94-123.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wienken CJ, Baaske P, Rothbauer U, Braun D, Duhr S. Protein-binding assays in biological liquids using microscale thermophoresis. Nat Commun. 2010;1:100. doi: 10.1038/ncomms1093. [DOI] [PubMed] [Google Scholar]
- 54.Chang BS, Lowenstein DH. Epilepsy. The New England journal of medicine. 2003;349 (13):1257–1266. doi: 10.1056/NEJMra022308. [DOI] [PubMed] [Google Scholar]
- 55.Salin P, Tseng GF, Hoffman S, Parada I, Prince DA. Axonal sprouting in layer V pyramidal neurons of chronically injured cerebral cortex. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1995;15 (12):8234–8245. doi: 10.1523/JNEUROSCI.15-12-08234.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Golarai G, Greenwood AC, Feeney DM, Connor JA. Physiological and structural evidence for hippocampal involvement in persistent seizure susceptibility after traumatic brain injury. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2001;21 (21):8523–8537. doi: 10.1523/JNEUROSCI.21-21-08523.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kharatishvili I, Nissinen JP, McIntosh TK, Pitkanen A. A model of posttraumatic epilepsy induced by lateral fluid-percussion brain injury in rats. Neuroscience. 2006;140 (2):685–697. doi: 10.1016/j.neuroscience.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 58.Jin X, Prince DA, Huguenard JR. Enhanced excitatory synaptic connectivity in layer v pyramidal neurons of chronically injured epileptogenic neocortex in rats. Journal of Neuroscience. 2006;26 (18):4891–4900. doi: 10.1523/JNEUROSCI.4361-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Graber KD, Prince DA. A critical period for prevention of posttraumatic neocortical hyperexcitability in rats. Annals of neurology. 2004;55 (6):860–870. doi: 10.1002/ana.20124. [DOI] [PubMed] [Google Scholar]
- 60.Prince DA, Parada I, Scalise K, Graber K, Jin X, Shen F. Epilepsy following cortical injury: Cellular and molecular mechanisms as targets for potential prophylaxis. Epilepsia. 2009;50:30–40. doi: 10.1111/j.1528-1167.2008.02008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McKinney RA, Debanne D, Gahwiler BH, Thompson SM. Lesion-induced axonal sprouting and hyperexcitability in the hippocampus in vitro: implications for the genesis of posttraumatic epilepsy. Nat Med. 1997;3 (9):990–996. doi: 10.1038/nm0997-990. [DOI] [PubMed] [Google Scholar]
- 62.Czech T, Yang JW, Csaszar E, Kappler J, Baumgartner C, Lubec G. Reduction of hippocampal collapsin response mediated protein-2 in patients with mesial temporal lobe epilepsy. Neurochem Res. 2004;29 (12):2189–2196. doi: 10.1007/s11064-004-7025-3. [DOI] [PubMed] [Google Scholar]
- 63.Bretin S, Reibel S, Charrier E, Maus-Moatti M, Auvergnon N, Thevenoux A, Glowinski J, Rogemond V, Premont J, Honnorat J, Gauchy C. Differential expression of CRMP1, CRMP2A, CRMP2B, and CRMP5 in axons or dendrites of distinct neurons in the mouse brain. The Journal of comparative neurology. 2005;486 (1):1–17. doi: 10.1002/cne.20465. [DOI] [PubMed] [Google Scholar]
- 64.Zhang Z, Ottens AK, Sadasivan S, Kobeissy FH, Fang T, Hayes RL, Wang KK. Calpain-mediated collapsin response mediator protein-1, -2, and -4 proteolysis after neurotoxic and traumatic brain injury. Journal of neurotrauma. 2007;24 (3):460–472. doi: 10.1089/neu.2006.0078. [DOI] [PubMed] [Google Scholar]
- 65.Arimura N, Menager C, Kawano Y, Yoshimura T, Kawabata S, Hattori A, Fukata Y, Amano M, Goshima Y, Inagaki M, Morone N, Usukura J, Kaibuchi K. Phosphorylation by Rho kinase regulates CRMP-2 activity in growth cones. Molecular and cellular biology. 2005;25 (22):9973–9984. doi: 10.1128/MCB.25.22.9973-9984.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Arimura N, Inagaki N, Chihara K, Menager C, Nakamura N, Amano M, Iwamatsu A, Goshima Y, Kaibuchi K. Phosphorylation of collapsin response mediator protein-2 by Rho-kinase. Evidence for two separate signaling pathways for growth cone collapse. J Biol Chem. 2000;275 (31):23973–23980. doi: 10.1074/jbc.M001032200. [DOI] [PubMed] [Google Scholar]
- 67.Brown M, Jacobs T, Eickholt B, Ferrari G, Teo M, Monfries C, Qi RZ, Leung T, Lim L, Hall C. Alpha2-chimaerin, cyclin-dependent Kinase 5/p35, and its target collapsin response mediator protein-2 are essential components in semaphorin 3A-induced growth-cone collapse. J Neurosci. 2004;24 (41):8994–9004. doi: 10.1523/JNEUROSCI.3184-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Uchida Y, Ohshima T, Sasaki Y, Suzuki H, Yanai S, Yamashita N, Nakamura F, Takei K, Ihara Y, Mikoshiba K, Kolattukudy P, Honnorat J, Goshima Y. Semaphorin3A signalling is mediated via sequential Cdk5 and GSK3beta phosphorylation of CRMP2: implication of common phosphorylating mechanism underlying axon guidance and Alzheimer’s disease. Genes to cells: devoted to molecular & cellular mechanisms. 2005;10 (2):165–179. doi: 10.1111/j.1365-2443.2005.00827.x. [DOI] [PubMed] [Google Scholar]
- 69.Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A, Kaibuchi K. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell. 2005;120 (1):137–149. doi: 10.1016/j.cell.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 70.Hou ST, Jiang SX, Aylsworth A, Ferguson G, Slinn J, Hu H, Leung T, Kappler J, Kaibuchi K. CaMKII phosphorylates collapsin response mediator protein 2 and modulates axonal damage during glutamate excitotoxicity. Journal of neurochemistry. 2009;111 (3):870–881. doi: 10.1111/j.1471-4159.2009.06375.x. [DOI] [PubMed] [Google Scholar]
- 71.Sato Y, Ishida-Nakajima W, Kawamura M, Miura S, Oguma R, Arai H, Takahashi T. Hypoxia-ischemia induces hypo-phosphorylation of collapsin response mediator protein 2 in a neonatal rat model of periventricular leukomalacia. Brain research. 2011;1386:165–174. doi: 10.1016/j.brainres.2011.02.027. [DOI] [PubMed] [Google Scholar]
- 72.Wilson SM, Yeon SK, Yang XF, Park KD, Khanna R. Differential regulation of collapsin response mediator protein 2 (CRMP2) phosphorylation by GSK3β and CDK5 following traumatic brain injury. Frontiers in Cellular Neuroscience. 2014;8:14. doi: 10.3389/fncel.2014.00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lee C-Y, Jaw T, Tseng H-C, Chen IC, Liou H-H. Lovastatin Modulates Glycogen Synthase Kinase-3β Pathway and Inhibits Mossy Fiber Sprouting after Pilocarpine-Induced Status Epilepticus. PloS one. 2012;7 (6):e38789. doi: 10.1371/journal.pone.0038789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brandt C, Heile A, Potschka H, Stoehr T, Löscher W. Effects of the Novel Antiepileptic Drug Lacosamide on the Development of Amygdala Kindling in Rats. Epilepsia. 2006;47 (11):1803–1809. doi: 10.1111/j.1528-1167.2006.00818.x. [DOI] [PubMed] [Google Scholar]
- 75.Loscher W, Brandt C. Prevention or modification of epileptogenesis after brain insults: experimental approaches and translational research. Pharmacological reviews. 2010;62 (4):668–700. doi: 10.1124/pr.110.003046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Loscher W. Animal models of epilepsy for the development of antiepileptogenic and disease-modifying drugs. A comparison of the pharmacology of kindling and post-status epilepticus models of temporal lobe epilepsy. Epilepsy research. 2002;50 (1–2):105–123. doi: 10.1016/s0920-1211(02)00073-6. [DOI] [PubMed] [Google Scholar]
- 77.Loscher W, Honack D, Rundfeldt C. Antiepileptogenic effects of the novel anticonvulsant levetiracetam (ucb L059) in the kindling model of temporal lobe epilepsy. The Journal of pharmacology and experimental therapeutics. 1998;284 (2):474–479. [PubMed] [Google Scholar]
- 78.Wasterlain CG, Stohr T, Matagne A. The acute and chronic effects of the novel anticonvulsant lacosamide in an experimental model of status epilepticus. Epilepsy research. 2011;94 (1–2):10–17. doi: 10.1016/j.eplepsyres.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang B, Dawson H, Wang H, Kernagis D, Kolls BJ, Yao L, Laskowitz DT. Lacosamide Improves Outcome in a Murine Model of Traumatic Brain Injury. Neurocrit Care. 2012 doi: 10.1007/s12028-012-9808-8. [DOI] [PubMed] [Google Scholar]
- 80.Pitkanen A, Immonen R, Ndode-Ekane X, Grohn O, Stohr T, Nissinen J. Effect of lacosamide on structural damage and functional recovery after traumatic brain injury in rats. Epilepsy research. 2014;108 (4):653–665. doi: 10.1016/j.eplepsyres.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 81.Licko T, Seeger N, Zellinger C, Russmann V, Matagne A, Potschka H. Lacosamide treatment following status epilepticus attenuates neuronal cell loss and alterations in hippocampal neurogenesis in a rat electrical status epilepticus model. Epilepsia. 2013 doi: 10.1111/epi.12196. [DOI] [PubMed] [Google Scholar]
- 82.Santhakumar V, Aradi I, Soltesz I. Role of mossy fiber sprouting and mossy cell loss in hyperexcitability: a network model of the dentate gyrus incorporating cell types and axonal topography. Journal of neurophysiology. 2005;93 (1):437–453. doi: 10.1152/jn.00777.2004. [DOI] [PubMed] [Google Scholar]
- 83.Heng K, Haney MM, Buckmaster PS. High-dose rapamycin blocks mossy fiber sprouting but not seizures in a mouse model of temporal lobe epilepsy. Epilepsia. 2013;54 (9):1535–1541. doi: 10.1111/epi.12246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Verrotti A, Loiacono G, Pizzolorusso A, Parisi P, Bruni O, Luchetti A, Zamponi N, Cappanera S, Grosso S, Kluger G, Janello C, Franzoni E, Elia M, Spalice A, Coppola G, Striano P, Pavone P, Savasta S, Viri M, Romeo A, Aloisi P, Gobbi G, Ferretti A, Cusmai R, Curatolo P. Lacosamide in pediatric and adult patients: comparison of efficacy and safety. Seizure: the journal of the British Epilepsy Association. 2013;22 (3):210–216. doi: 10.1016/j.seizure.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 85.Kim JS, Kim H, Lim BC, Chae JH, Choi J, Kim KJ, Hwang YS, Hwang H. Lacosamide as an adjunctive therapy in pediatric patients with refractory focal epilepsy. Brain & development. 2013 doi: 10.1016/j.braindev.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 86.Grosso S, Parisi P, Spalice A, Verrotti A, Balestri P. Efficacy and safety of lacosamide in infants and young children with refractory focal epilepsy. European journal of paediatric neurology: EJPN: official journal of the European Paediatric Neurology Society. 2014;18 (1):55–59. doi: 10.1016/j.ejpn.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 87.Tomson T, Landmark CJ, Battino D. Antiepileptic drug treatment in pregnancy: changes in drug disposition and their clinical implications. Epilepsia. 2013;54 (3):405–414. doi: 10.1111/epi.12109. [DOI] [PubMed] [Google Scholar]
- 88.Park KD, Morieux P, Salome C, Cotten SW, Reamtong O, Eyers C, Gaskell SJ, Stables JP, Liu R, Kohn H. Lacosamide isothiocyanate-based agents: novel agents to target and identify lacosamide receptors. Journal of medicinal chemistry. 2009;52 (21):6897–6911. doi: 10.1021/jm9012054. [DOI] [PMC free article] [PubMed] [Google Scholar]