

Abstract
Purpose
Mutations in the MFRP (membrane-type frizzled related protein) Gene leads to an entity characterized by retinitis pigmentosa (RP), nanophthalmos, optic disc drusen and macular changes, originally described as foveoschisis. Despite the association of MFRP gene mutation and increase in macular thickness, no treatment modality has been described for cystoid macular edema (CME) related to this particular entity so far.
Method
Case report: a 52 year-old woman presented with nanophthalmos, optic disc drusen, retinitis pigmentosa and increase in macular thickness. Genetic analysis revealed an MFRP gene mutation. The patient was treated with topical carbonic anydrase inhibitors (CAI).
Result
A progressive decrease in macular thickness and cystic changes was observed during the two month course of topical CAI treatment, and best corrected visual acuity improved from 20/100 to 20/50. Macular thickness remained stable after six months of follow up.
Conclusion
CME is part of the macular changes noted in the MFRP mutation-related Nanophthalmos-Retinitis Pigmentosa-Foveoschisis-Optic Disk Drusen Syndrome. Taking into account that resolution of CME in patients with RP may delay an irreversible decrease in visual acuity, treatment should be considered when cystic changes are suspected. Topical CAI was effective in decreasing macular thickness and cystic changes in the patient reported.
Keywords: Carbonic anydrase inhibitor, Cystoid macular edema, Electroretinogram, Foveoschisis, MFRP mutation, Nanophthalmos, Optic coherence tomography, Optic Disk Drusen, Posterior microphthalmos, Retinitis pigmentosa
INTRODUCTION
Mutations in the MFRP (membrane-type frizzled related protein) gene, located on chromosome 11q23, lead to a recently described entity characterized by autosomal recessively inherited retinitis pigmentosa (RP), nanophthalmos, optic disc drusen and macular changes, originally described as foveoschisis.1–3 MFRP mutations have long been related to isolated nanophthalmos without retinal changes. It is still not known why some mutations cause isolated nanophthalmos, while others originate the nanophthalmos-RP-foveoschisis-optic disc drusen complex. The various phenotypes observed with mutations at the same gene suggest that MFRP plays a key role in eye development, functioning as an axial length regulator and as a critical molecule for photoreceptor maintenance.4
Macular changes in patients with MFRP mutations differ among reported series of cases, as optic coherence tomography (OCT) shows localized or diffuse foveoschisis, as well as macular cystic changes, located at the inner or outer nuclear layers.1–3
CME is a well known cause of visual loss in patients with RP, with a prevalence ranging from 10 to 40%.5 Various treatment modalities have already been attempted for the treatment of CME in RP patients, including intravitreal triamcinolone acetonide, vitrectomy, systemic or topical carbonic anhydrase inhibitor (CAI).6–8
Despite of the well-known association of the MFRP gene mutation and increase in macular thickness, no treatment modality has been described for cystoid macular edema (CME) related to this particular entity so far. In the present study, we report the outcome of the pharmacological treatment of a patient with CME secondary to the nanophthalmos-RP-foveoschisis-optic disk drusen syndrome with a topical CAI.
CASE REPORT
A 52-year-old woman presented with a history of progressive nyctalopia and visual acuity (VA) loss over the last two decades. On ophthalmic examination, her best-corrected visual acuity (BCVA) was of 20/200 in both eyes (with a correction of +12,75 diopter sphere OU). Biomicoscopy showed symmetric shallow anterior chambers, with corneal diameters of 12,5mm, and moderate nuclear lens opacity.
Gonioscopy was performed and showed open angles, and IOP was 10mmHg OU. Fundoscopic examination disclosed large optic disc drusen, cystic macular changes, and retinal pigment epithelium (RPE) clumping and atrophy at the macular region and mid periphery in both eyes, as well as vascular sheathing (Figure 1).
Figure 1.
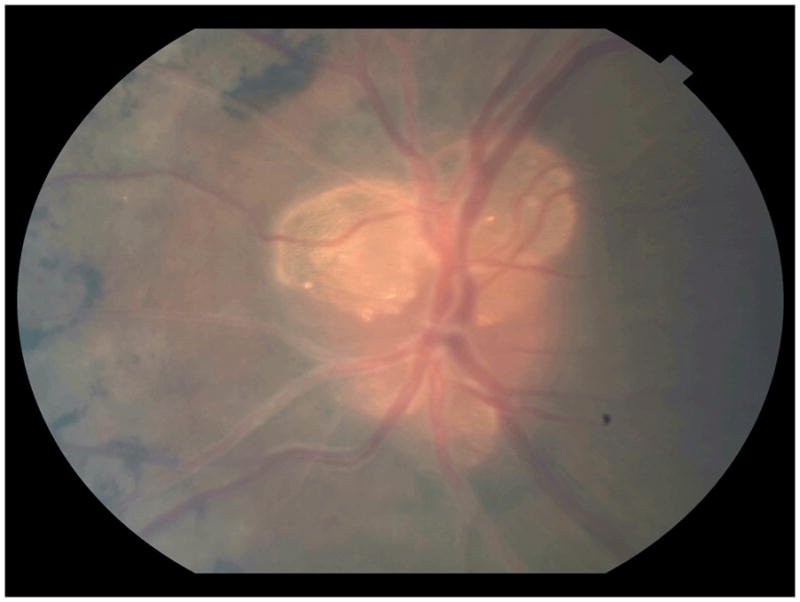
High magnification retinography of the left eye reveals several optic disc drusen, as well as bone spicule-shaped pigmentary deposits and sheathing of the retinal blood vessels.
Biometric scans showed short axial length (16.79 mm in the right eye and 15.40 mm in the left eye). Electroretinogram (ERG) showed abolished photopic and scotopic light response in both eyes, compatible with RP. OCT macular scans demonstrated a diffuse increase in retinal thickness and hyporeflective cystic changes at the inner and outer plexiform layers, as well as a small pocket of subfoveal fluid in the right eye. Central foveal thickness was of 694 ± 12 microns in the right eye and 483 ± 14 microns in the left eye. Central retinal volume was of 13.65mm3 in the right eye and 10.49mm3 in the left eye (Figures 2A and B).
Figure 2.
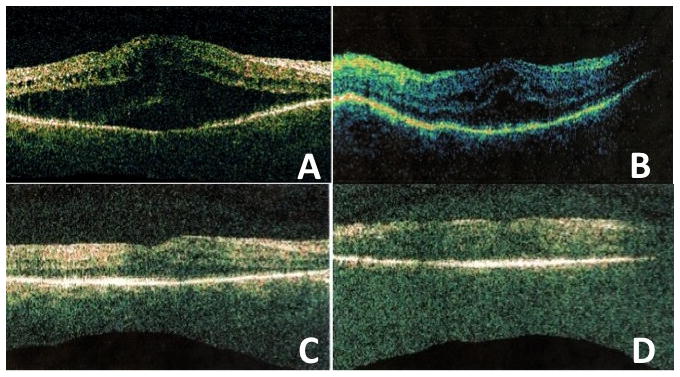
Optic coherence tomography (OCT) before and after topical carbonic anydrase treatment. A and B: Right and left eyes one week after phacoemulsification show diffuse increase in retinal thickness and hyporeflective cystic changes at the inner and outer plexiform layers, as well as a small pocket of subfoveal fluid. Aspect of the scans very similar before surgery (not presented due to very low resolution of the scans). C and D: right and left eye 2 months after topical carbonic anydrase inhibitor four times a day. There is marked decrease in retinal thickness and cystic changes. The anatomical outcome was maintained four months after discontinuation of the treatment.
The molecular analysis was performed and the patient was a compound heterozygote for two frameshift mutations in exons 5 (492delC) and 10 (1149dupC).
The patient was submitted to phacoemulsification in both eyes in a week interval, and a +40 dioptries intraocular lens (IOL) was implanted in both eyes. After cataract surgery and IOL implantation, vision was stable at 20/100. The patient was then treated with topical dorzolamide 2% 4 times a day as an attempt to reduce the cystoid macular edema.
Topical CAI was applied for two months. During that interval, the patient’s BCVA improved to 20/50 OU. A progressive decrease of macular thickness and of cystic changes, more pronounced at the right eye, was observed during the course of topical CAI treatment, and the central macular thickness improved to 355 ± 12 microns in the right eye (central volume of 10.31mm3) and 361 ± 22 microns in the left eye (central volume of 10.00mm3) (Figure 2C and D). Macular thickness remained stable after six months of follow up (four months after discontinuation of the treatment).
DISCUSSION
The MFRP gene was initially associated only with high hyperopia and reduced axial length, but no retinal degeneration. However, since 2006, a series of cases have reported the association of MFPR mutation and posterior microphthalmos, RP, macular abnormalities and optic disc drusen.1–3
Fundus examination of patients with the MFRP-related syndrome usually shows multiple, irregular, round yellow-white flecks located at the mid periphery, with2 or without1,3 “bone spicule” hyperpigmentation. ERG studies show a rod-cone pattern of dysfunction, with severe rod system involvement.2
The patient reported presented with mottled RPE changes and abolished photopic or scotopic ERG waves, compatible with RP and to previous MFRP-related reports.
Evidence suggest that the MFRP gene mutation causes a disturbance in the complex early retinal cell migrations that lead to normal foveal architecture, as macular OCTs show lack of foveal pit and thickening of the inner retina.1
Macular OCT description differs among case series.1,2 Previous literature agree that cystic changes may coexist with foveoschisis in cases of MFRP gene mutation. However, none of those previous reports attempted to pharmacologically treat the increase in macular thickness. It is possible that the combination of nanophthalmos (with a disturbance in retinal cell migration) and chronic macular cysts may lead to foveal schisis.1 The patient in this report clearly presented with CME, which regressed after a 40-day treatment of CAI. Considering that CME may be addressed in different ways, one should consider treating individuals with MFRP gene mutation and cystic changes seen on OCT, aiming for visual stabilization in this type of RP-related syndrome.
Summary statement.
Topical therapy with carbonic anydrase inhibitors is an Effective treatment against cystoid macular edema in a patient with MFRP related Nanophthalmos-Retinitis Pigmentosa- Foveoschisis- Optic Disk Drusen Syndrome
Acknowledgments
FUNDING: NIH grant EY013610
Footnotes
DISCLOSURES: NONE
References
- 1.Zenteno JC, Buentello-Volante B, Quiroz-Gonzáles M, Quiroz-Reyes MA. Compound heterozygosity for a novel and recurrent MFRP gene mutation in a family with the nanophthalmos-retinitis pigmentosa complex. Mol Vis. 2009;15:1794–8. [PMC free article] [PubMed] [Google Scholar]
- 2.Ayala-Ramirez R, Graue-Wiechers F, Robredo V, et al. A new autosomal recessive syndrome consisting of posterior microphthalmos, retinitis pigmentosa, foveoschisis, and optic disc drusen is caused by a MFRP gene mutation. Mol Vis. 2006;12:1483–9. [PubMed] [Google Scholar]
- 3.Sonmez K, Ozcan P. Angle-closure glaucoma in a patient with the nanophthalmos-ocular cystinosis-foveoschisis-pigmentary retinal dystrophy complex. BMC Ophthalmology. 2012;12:23. doi: 10.1186/1471-2415-12-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Won J, Smith RS, Peachey NS, Wu J, et al. Membrane frizzled-related protein is necessary for the normal development and maintenance of photoreceptor outer segments. Vis Neurosci. 2008;25:563–74. doi: 10.1017/S0952523808080723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hajali M, Fishman GA, Anderson RJ. The prevalence of cystoid macular oedema in retinitis pigmentosa patients determined by optical coherence tomography. Br J Ophthalmol. 2008;92:1065–8. doi: 10.1136/bjo.2008.138560. [DOI] [PubMed] [Google Scholar]
- 6.Fishman GA, Gilbert LD, Fiscella RG, et al. Acetazolamide for treatment of chronic macular edema in retinitis pigmentosa. Arch Ophthalmol. 1989;107:1445–52. doi: 10.1001/archopht.1989.01070020519031. [DOI] [PubMed] [Google Scholar]
- 7.Ikeda Y, Hisatomi T, Yoshida N, et al. The clinical efficacy of a topical dorzolamide in the management of cystoid macular edema in patients with retinitis pigmentosa. Graefes Arch Clin Exp Ophthalmol. 2012;250:809–14. doi: 10.1007/s00417-011-1904-5. [DOI] [PubMed] [Google Scholar]
- 8.Genead MA, Fishman GA. Efficacy of sustained topical dorzolamide therapy for cystic macular lesions in patients with retinitis pigmentosa and Usher Syndrome. Arch Ophthalmol. 2010;128:1146–50. doi: 10.1001/archophthalmol.2010.172. [DOI] [PMC free article] [PubMed] [Google Scholar]