

Summary
Inducible Hsp70 (Hsp70i) is overexpressed in a wide spectrum of human tumors and its expression correlates with metastasis, poor outcomes, and resistance to chemotherapy in patients. Identification of small molecule inhibitors selective for Hsp70i could provide new therapeutic tools for cancer treatment. In this work, we used fluorescence-linked enzyme chemoproteomic strategy (FLECS) to identify HS-72, an allosteric inhibitor selective for Hsp70i. HS-72 displays the hallmarks of Hsp70 inhibition in cells, promoting substrate protein degradation and growth inhibition. Importantly, HS-72 is selective for Hsp70i over the closely related constitutively active Hsc70. Studies with purified protein show HS-72 acts as an allosteric inhibitor, reducing ATP affinity. In vivo HS-72 is well-tolerated, showing bioavailability and efficacy, inhibiting tumor growth and promoting survival in a HER2+ model of breast cancer. The HS-72 scaffold is amenable to resynthesis and iteration, suggesting an ideal starting point for a new generation of anticancer therapeutics targeting Hsp70i.
Introduction
The Heat shock protein 70 (Hsp70) family members have broad chaperone functions in cells that include folding of nascent proteins, refolding of misfolded proteins, removal of protein complexes, and control of regulatory proteins (Evans et al., 2010). These functions are driven by ATP hydrolysis in the N-terminal nucleotide-binding domain (NBD) of the Hsp70s. The Hsp70s are evolutionary conserved across species and there are 8 mammalian family members (Hunt and Morimoto, 1985). The inducible form of Hsp70 (Hsp70i, also called Hsp72, Hsp70-1, HspA1A/HspA1B) is present in low or undetectable levels in unstressed normal cells, however, expression levels rapidly increase in response to cellular stresses such as heat shock, viral infection or transformation. Deletion of its immediate paralog, the constitutive heat shock protein cognate 70 (Hsc70) is developmentally lethal, whereas deletion of Hsp70i results in sterility of male mice, but no other overt phenotype (Dix et al., 1996). Hsp70i and Hsc70 are highly related, sharing 90% sequence identity, with most of the amino acid variability confined to the NBD. There is greater sequence divergence with respect to other Hsp70 family members (50–80% identity), especially within the NBD (Daugaard et al., 2007). The close sequence similarity between Hsp70i and Hsc70 has contributed to past difficulties in separating the two proteins’ functions, both pharmacologically and with RNA interference approaches.
Overexpression of Hsp70i has been observed in several cancers, including breast, prostate, and colon, and this is thought to aid in resistance to apoptosis as well as to anti-cancer treatments (Shu and Huang, 2008). Hsp70i inhibits both intrinsic and extrinsic apoptosis pathways. This occurs by preventing TNF-related apoptosis-inducing ligand formation of the death-induced signaling complex through inhibition of death receptors 4 and 5, as well as by inhibiting events in mitochondrial-mediated apoptosis (Guo et al., 2005). In the later case, Hsp70i prevents Bax translocation to themitochondria, preventing release of cytochrome c, an apoptosis inducing factor (Yang et al., 2012). Additionally, Hsp70i mediates both caspase dependent and independent apoptotic pathways by binding Apaf-1, blocking recruitment of procaspase-9 to the apoptosome, and by inhibition of JNK, respectively (Beere et al., 2000; Massey et al., 2010). Hsp70i also protects cancer cells from oncogenic stress induced by up-regulation of specific oncogenes such as HER2. Increased expression of Hsp70i correlates with resistance to chemotherapy and radiation and therefore poor clinical outcomes by providing cancer cells a route to survive and proliferate in the presence of noxious stimuli such as hypoxia or denatured protein aggregates. These data have led to the proposal that cancer cells are dependent on Hsp70i for survival (Goloudina et al., 2012). This hypothesis is supported by Hsp70i depletion studies in which tumor cell death and sensitivity to chemotherapeutic drugs were evident, while non-tumorigenic cell lines were unaffected by Hsp70i depletion (Nylandsted et al., 2002).
From a drug discovery perspective, Hsp70i presents a number of challenges, not the least of which being its close sequence identity with Hsc70. Specific, physiological substrates of Hsp70i are poorly defined and high throughput assays based on chaperone or trafficking activities are limited (Kang et al., 2008). The crystal structure of the E. coli Hsp70, Dnak, shows the protein in either a closed nucleotide bound state or open unbound state (Qi et al., 2013). In the closed conformation, the bound nucleotide shows little solvent accessibility to the surface, limiting access to diffusible small molecule inhibitors. In cells, Hsp70s may be reminiscent of small G proteins in which the nucleotide-binding pocket is always occupied, undergoing GTP/GDP exchange upon activation, again limiting small molecule accessibility. In the case of Hsp70i, the protein has high affinity for ADP, which is likely exchanged with ATP through allosteric regulation (Powers et al., 2010). The chaperone activities of Hsp70i are also regulated by the C-terminus in cooperation with co-chaperones, such as Hsp40, Hip, Hop, CHIP and Bag1 (Tavaria et al., 1996). Crystallographic and NMR studies have shown that these co-chaperones induce altered conformational states (Evans et al., 2010). Because of these many complications, most Hsp70 inhibitors have either failed to discriminate between various Hsp70 family members or perform poorly in vivo (Massey, 2010).
To identify novel Hsp70 inhibitors that discriminate Hsp70i from Hsc70 and other family members, we used a fluorescence linked enzyme chemoproteomic strategy (FLECS) to screen an in house library of 3379 purine like molecules (Carlson et al., 2013). The screen identified HS-72, a highly selective and novel allosteric inhibitor of Hsp70i. HS-72 bears all the hallmarks of an Hsp70 inhibitor in cell models of breast cancer and in a cell culture model of Huntington’s disease. The molecule is well tolerated and is bioavailable in mice, and shows efficacy in the MMTV-neu mouse model of breast cancer. These data indicate that the HS-72 scaffold is an excellent starting point for development of highly selective inhibitors of Hsp70i.
Results
FLECS Screening Yields Highly Selective Hsp70i Interactors
For screening of Hsp70i inhibitors by FLECS, GFP-Hsp70i was expressed in HEK293 cells, extracts prepared, incubated directly with γ-phosphate-linked ATP-Sepharose and then eluted with ATP or ADP (Figure1A). These studies demonstrated that the GFP-fusion protein has a fully functional nucleotide binding pocket and that binding occurs in a reversible manner. Based on the crystal structure of the Hsp70 homologue DnaK with bound ATP, showing limited solvent accessibility, recovery of the fusion protein on γ-linked ATP resin was at first surprising (Figure 1B). This is because the γ-phosphate oxygen on the immobilized ATP is tethered to PEG, which is expected to sterically hinder Hsp70i binding. We therefore propose that the fusion protein is recovered through binding Hsp70i in the apo or ADP-bound form, which is subsequently driven into the ATP-bound conformation when exposed to the γ-linked ATP resin (Figure1C). Once bound, the protein is retained because of an inability to hydrolyze the PEG linked phosphate. The dramatic differences in elution between ATP compared to ADP shown in figure 1A are consistent with this hypothesis. Once bound, exposure to low µM [Mg2+ ATP] enables the protein to turn over and be released. In contrast, mM [ADP] were required to compete the bound fusion protein from the immobilized nucleotide. These findings suggested an opportunity to use the FLECS approach to identify selective inhibitors of Hsp70i that either act competitively at the ATP binding site or allosterically to regulate nucleotide binding.
Figure 1. Recombinant GFP-Hsp70i is readily captured on γ-phosphate linked ATP resin and is competitively released with ATP/ADP, consistent with nucleotide induced conformational changes.

(A) GFP-Hsp70i was bound to γ-phosphate-linked ATP-Sepharose then eluted with the indicted [ATP] or [ADP] (mean ± SEM). The solution structures shown in (B and C) suggest a mode of binding in which the GFP-fusion protein first recognizes the immobilized nucleotide in its open apo state. The protein closes around the immobilized ATP but cannot hydrolyze the γ phosphate. The presence of free Mg2+ATP, in contrast to ADP, however, enables rapid turnover and release of the bound protein. (B) Structure of Hsp70 (E. coli, DnaK) in the ATP-bound conformation (PDB 4B9Q) and (C) in the ADP-bound apo conformation (PDB 2KHO). NBD highlighted in blue, linker domain highlighted in green, and SBD highlighted in red. ATP represented in (B) as ball-and-stick model.
Assembly of our focused library, consisting of 3379 purine like compounds, was described previously (Carlson et al., 2013). Figure S1 describes FLECS, illustrating how individual compounds are screened in parallel against the ATP medium charged with GFP-Hsp70i. Briefly, GFP-Hsp70i is expressed in HEK 293T cells and crude cell lysate is then added to the ATP resin. Following several wash steps the bound GFP-Hsp70i is plated along with the compounds from the library in 96-well filter plates with ATP serving as a positive control, or with buffer to serve as a negative control. The lysates were eluted from the filter plate onto a catch plate by centrifugation and the fluorescence of the eluates was measured. Those compounds that disrupted the Hsp70i-ATP association resulted in an increased fluorescence signal over the buffer only samples. The primary screen identified 197 hits from the library, which were then sorted by their specificity towards GFP-Hsp70i over other purinome members that had also been screened against the same chemical library by FLECS (Figure 2A–B). The compounds that were active in multiple assays were removed from consideration. Next, the presence of GFP-Hsp70i in the eluates from the 197 primary hits was determined by Western blot. This reduced the collection to 60 compounds and also eliminated auto fluorescent falsepositive molecules (Figure S2A). Next we tested the ability of the 60 compounds for elution of native Hsp70 from the ATP resin using pig bladder extracts, a rich source of native Hsp70i (Figure S2B). This reduced the final collection to 22 diverse structures (0.65% of the library), showing selectivity towards both recombinant human and native mammalian Hsp70i (Figure S2C).
Figure 2. Heat maps showing summary of results of screening of Hsp70i by FLECS and the relative selectivity of hits against other purinome members.

In (A) FLECS identified 197 primary hits from a collection 3379 purine like molecules (color scale dark red = strong signal to light yellow = background). In (B), the relative selectivity of all hits against Hsp70i is shown compared with other distinct members of the broader purinome screened against the same library by FLECS including ACC1, DAPK3, PIMK 1, 2, 3, Hsp90, TRAP 1, FASN, IRAK 2, PfPK9, NEK9, DENV NS5, AMPK γ and α subunit (Color scale light blue = selective, Dark red = non-selective). (C) Structure of lead molecule HS-72.
See also Figure S1 and S2.
Identification of a caspase-activating cell-permeable compound that targets Hsp70i
To further narrow the number of compounds, we first tested the 22 hits for their ability to activate caspase-3/7 in various cancer cells, which is a hallmark of Hsp70i inhibition (Beere, 2001). Of the compounds tested, HS-72 ((S)-N-(1-propyl-1H-benzo[d]imidazol-2-yl)-1-(pyrazin-2-yl)piperidine-3-carboxamide) was most robust, inducing caspase activation in a dose dependent manner (Figure S2D). Other compounds were either less potent in this assay, or were cell impermeable and were not pursued herein. Furthermore, caspase activation by HS-72 was reproducible across several cancer cell lines at 6 hours and 24 hours in a dose dependent manner (Figure S2E–F). As a second test we examined the effect of HS-72 on the expression of Akt and HER2, two known client proteins of Hsp70i, in BT474 breast cancer cells. Figure S2G shows dose dependent reduction in Akt and HER2 with HS-72.
HS-72 specifically targets Hsp70i over other members of the Hsp70 superfamily
To test the selectivity of the HS-72 scaffold against the broader purinome, HEK 293T cell extracts or pig bladder tissue lysates were applied to the ATP resin and eluted with HS-72, as described for FLECS. The eluates were characterized by SDS-PAGE, silver stain, and MS analysis. Silver stain analysis for both HEK 293T cells and pig bladder lysates, confirmed elution of native Hsp70i and showed only a few non-specifically eluted proteins, indicating that HS-72 has a high degree of specificity within the wider purinome (Figure S3A–B). To more thoroughly determine the specificity of HS-72 for Hsp70i, the HEK 293T eluates were also analyzed for other Hsp70 family members Hsc70, Grp78, and Grp75, as well as Hsp90 by Western blot. This showed selective elution of Hsp70i by HS-72, with ATP serving as a positive control, showing elution of Hsp70 family members and Hsp90 (Figure S3C). Next, we synthesized an affinity resin using the HS- 72 scaffold. HEK 293T cell lysate was applied to the resin and subjected to several washes. To confirm selectivity of the HS-72 affinity resin for Hsp70i, free HS-72 was incubated with lysate to inhibit Hsp70i binding to the affinity resin. The linker on the resin was cleaved using sodium dithionite and the samples were subjected to SDS-PAGE and analyzed by Western blot, silver stain, and mass spectrometry. When probing for Hsp70i by Western blot, the HS-72 affinity resin binds Hsp70i and free HS-72 is able to block this interaction (Figure 3A). Importantly, when probing for the closely related Hsp70 family member, Hsc70, the HS-72 affinity resin does not pull down Hsc70 (Figure 3B). This indicates that the HS-72 scaffold is highly selective for the inducible Hsp70 over the constitutively active Hsc70. Furthermore, silver stain and MS analysis reveals the HS-72 affinity resin pulls down Hsp70i, and this association is blocked with free HS-72, with very few non-specific interactions (Figure 3C). While Hsp90 is also pulled down, as shown in Figure 3C, this is a non-specific interaction with the media itself because the association is not blocked by free HS-72. These studies identified HS-72 as the first example of a small molecule that can selectively discriminate Hsp70i from other members of the Hsp70 superfamily.
Figure 3. The HS-72 scaffold is highly selective for Hsp70i over the constitutively active Hsc70 and the wider purinome.
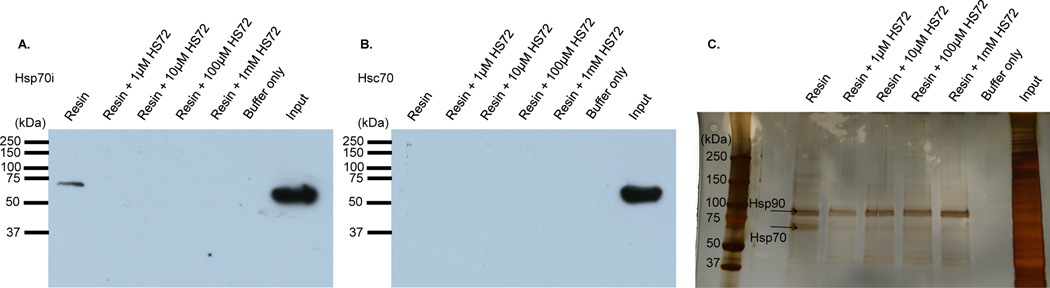
(A–C) Cleavable HS-72 affinity resin was synthesized and mixed with HEK293T crude cell lysate ± free HS-72. Following washing, sodium dithionite (25 mM) was used to cleave the ligand and the eluted proteins subjected to SDS-PAGE. (A) Western blot analysis for Hsp70i reveals Hsp70i binding to the affinity resin and this interaction is blocked by free HS-72. (B) Western blot against Hsc70 shows no binding to the resin. (C) Silver staining also demonstrates selectivity of the immobilized ligand for Hsp70i. Hsp90 recovery was non-competitive with respect to HS-72 and therefore nonspecifically bound to the media.
See also Figure S3.
HS-72 is an allosteric inhibitor of Hsp70
Because the initial isolate of HS-72 was a racemic mixture, the molecule was resynthesized in its R and S enantiomeric forms. Figure S4A shows the S enantiomer more effectively elutes GFP-Hsp70i from ATP resin than the R enantiomer (referred to herein as HS-71). To characterize the S and R enantiomers in more detail we tested their effects on the thermal stability of purified Hsp70i and Hsc70 (Figure 4 and Figure S4B–C). The Thermoflour assay is used to show direct binding of a small molecule as measured by a change in melting temperature (Tm), (Cummings et al., 2006). In general, ATP competitive inhibitors impart a large degree of thermal stability to purine binding proteins because of the number of potential contacts within the nucleotide-binding pocket (Cummings et al., 2006). For example, figure 4A shows that HS-10, an inhibitor of Hsp90, increases the Tm of this chaperone from 50° C to 60.5° C. Similarly, incubation of purified Hsp70i with ATP or ADP increased the Tm by 4–5° C (Figure 4B). However, when we repeated the study with HS-72, the Tm of Hsp70i decreased in a dose dependent manner (Figure 4C). Conversely, HS-71 had no effect on Tm of Hsp70i, indicating that any effects observed with HS-72 cannot be explained by artifacts in the Thermofluor assay, such as non-specific ionic interactions or hydrophobic binding or fluorophore quenching (Figure 4D). The effect of HS-72 on thermal stability was more obvious when the experiment was repeated in the presence of ATP, while HS-71 had no effect on Tm (Figure 4E–F). Significantly, HS-72 had no destabilizing effect on the ADP bound form (Figure 4G). Moreover, detergents had no effect on the ability of HS-72 to destabilize Hsp70i, eliminating the possibility of non-specific protein aggregation (Figure S4D). Furthermore, when HS-72 was tested with purified Hsc70 in the Thermofluor assay, the compound failed to trigger a significant shift in Hsc70 Tm in the presence or absence of ATP (Figure 4H–I). This further supports the selective nature of HS-72 for Hsp70i. Taken together these data suggest that although HS-72 is directly binding and selective for Hsp70i, its site(s) of interaction are unlikely to be in the ATP binding pocket. This hypothesis is consistent with data showing that HS-72 does not directly inhibit ATP hydrolysis in single turnover assays with Hsp70i (Figure 4J). Based on our Thermofluor data the most likely mechanism of HS-72 destabilization is via allosteric binding, which reduces the protein’s affinity for ATP. To explain HS-72’s action in this context, we hypothesize that upon binding to the ATP bound state the molecule induces large conformational changes, breaking a number of internal stabilizing contacts between the NBD and C terminal domain. This mechanism of action is also reminiscent of a small molecule allosteric inhibitor that decreases the Tm of RGS4 (Blazer et al., 2010).
Figure 4. Thermal stability and single turnover assays reveal HS-72 is an allosteric inhibitor of Hsp70i.

(A) Control study with HS-10, (Hsp90 inhibitor), increases the Tm of Hsp90 by 10.5° C. (B) ATP and ADP increase the Tm of Hsp70i in a dose dependent manner. (C) HS-72 decreases the Tm of Hsp70i by 0.1°C and 0.5°C at 10µM and 100µM respectively, indicating an allosteric effect (p<0.001 versus control). (D) HS-71 does not change Hsp70i Tm. (E) ATP increases the Tm of Hsp70i by 3°C, while HS-72 decreases ATP bound Hsp70i Tm 0.5°C and 1°C at 10µM and 100µM respectively (p<0.001 versus ATP). (F) HS-71 does not change the Tm of the ATP bound Hsp70i. (G) ADP increases the Tm of Hsp70i by 2.5°C, while HS-72 does not change the Tm of ADP Hsp70i. (H and I) HS-72 does not change Tm of Hsc70 ± ATP. (J) HS-72 does not reduce Hsp70 ATPase activity in the presence or absence of co-chaperone, Hlj1. Hsp70 + DMSO and Hsp70 + Hlj1 + DMSO indicated by dashed lines. Hsp70 + HS-72 is in green, and Hsp70 + Hlj1 + HS-72 is in red. Hlj1 + DMSO is indicated by dashed lines with squares. (K and L) HS-72 does not change Tm of the Hsp70i C306D mutant. ± ATP. Mean ± SEM. RFU, Relative Fluorescence Units. See also Figure S4.
Hsp70i C306D mutation perturbs HS-72 binding
Miyata et al., using site directed mutagenesis, showed that Hsp70i C306 is a potential allosteric regulatory site within the NBD (Miyata et al., 2012). Interestingly, C306 is not conserved amongst other Hsp70 family members, including Hsc70. Consistent with this earlier work, figure 4K–L show that the Tm of the Hsp70i C306D mutant was insensitive to HS-72, either in the presence or absence of ATP. The lack of effect of HS-72 on the thermal stability of Hsp70i C306D suggests that the molecule interacts either directly with C306 or that mutation of this residue results in a conformational change that renders the HS-72 binding site inaccessible.
HS-72 allosteric interaction induces a conformational change in Hsp70i
In an attempt to gain some insight how HS-72 might be interacting with Hsp70i, we conducted a docking study of HS-72 with the crystal structure of the human NBD of Hsp70i containing AMP-pnp using the SwissDock program (Grosdidier et al., 2011). Docking revealed 37 clusters, which were distributed between two binding sites on either side of the bound ATP analogue, further supporting an allosteric mechanism of action (Figure 5A–B). Along with the docking studies we used partial proteolysis to identify potential sites of interactions. Partial proteolysis, visualized through silver stain, reveals a profound difference in the proteolytic pattern of Hsp70i in the presence of HS-72, which indicates that HS-72 induces a conformational change in Hsp70i over several time points (Figure 5C–D and Figure 5SA). Furthermore, MS analysis of the proteolytic pattern revealed specific residues that are protected from trypsin digestion upon inhibitor binding. Specifically, after 24 hours, peptides 141–155, 326–342, and 518–533, were all present in the samples treated with HS-72, but absent in the samples lacking HS-72 (Figure 5D and Figure S5B–D). We propose that the conformational change induced byHS-72 results in sequences 141–155, 326–342, and 518–533 to be protected from digestion.
Figure 5. Molecular modeling and native protein digestion studies supports HS-72 as an allosteric inhibitor of Hsp70i.

(A and B) showing two 180° views of Hsp70i NBD (PDB: 2E8A) highlighting two potential binding sites of HS-72 (sticks) from docking studies. Views also show residues identified that are protected from trypsin digestion (highlighted in red). AMP-pnp is shown as a space-filling molecule in the center. (A-B) (Visualized using Chimera). (C and D) SDS-PAGE and silver stain showing results of native digests of Hsp70i ± HS-72 and ATP or ADP at 2 hours (C) and (D) 24 hours, revealing differences in digestion patterns. Full length Hsp70 (black arrow) and tryptic fragments (blue arrows).
See also Figure S5.
Collectively, these studies yield some insight as to the potential mode of HS-72 interaction with Hsp70i. The molecular docking studies reveal two putative binding sites that are distinct from the sequences that were protected from trypsin digestion (Figure 5A–B). Therefore it is likely that HS-72 is inducing a conformational change in Hsp70i that alters surface exposure to trypsin and therefore protects the identified sequences from trypsin digestion.
To further investigate HS-72 sites of interaction, HS-72 was tested in combination with VER-15508 (VER) or pifithrin-µ (PES) by Thermofluor. There was an increase in Hsp70i Tm with VER, consistent with previous work by Massey et al. showing binding of this compound in the active site of the NBD (Massey, 2010). When testing HS-72 and VER in combination there was no observed synergistic or additive effect on Hsp70i Tm in the absence or presence of ATP, indicating that these molecules do not target Hsp70i at the same sites (Figure S5E–F). This further supports an allosteric binding site of HS-72, since VER is known to bind the active site in the NBD. When testing PES alone there was no dose dependent effect on Hsp70i Tm (Figure S5G). Furthermore there was no synergistic or additive effect when testing HS-72 and PES in combination, indicating different binding sites on Hsp70i (Figure S5G–H). This indicates that HS-72 is not targeting the Hsp70i SBD, the primary site of PES binding (Leu et al., 2009).
HS-72 induces cellular protein aggregation
A hallmark of Hsp70i inhibition in cells is induction of protein aggregation, which was assayed using a cell culture model of Huntington’s disease. In this model, the PC12 rat neuronal cell line contains 74-glutamine repeats from exon 1 of human Huntington, fused to GFP (httQ74-GFP) (Wyttenbach et al., 2001). The htt Q74-GFP is expressed stably and is inducible through a doxycycline-regulated promoter. We found an induction in protein aggregates in the presence of HS-72 compared with untreated controls, shown by an increase in the insoluble associated pellet fraction (Figure 6A). Quantification of these bands shows a 50% increase in the insoluble associated pellet fraction in the HS-72 treated samples compared to untreated controls (Figure S6A).
Figure 6. HS-72 inhibits Hsp70i activity in a Huntington’s cell model and across multiple tumorigenic cell lines.

(A) HS-72 induces protein aggregation in a cell culture model of Huntington’s disease, as shown by an increase in httQ-GFP protein associated with the insoluble, pellet (P), fraction compared to the soluble (S) fraction. (B-G) HS-72 inhibits proliferation of (B) BT474, (C) MCF7, (D) SkBr3 breast cancer cell lines and (E) LnCaP prostate cancer cell lines. In contrast, non tumorigenic (F) MCF10A and (G) RWPE1 cells are insensitive to HS-72. (H and I) HS72 and the Hsp90 inhibitor HS10 show synergism in inhibiting cancer cell proliferation in (H) SkBr3 and (I) MCF7 cells. (Mean ± SEM. *, p<0.05. ***, p<0.001). See also Figure S6
HS-72 inhibits cancer cell proliferation
Upregulation of Hsp70i has been implicated in tumorigenicity in breast and prostate cancers (Goloudina et al., 2012; Shu and Huang, 2008). To determine if HS-72 discriminates between various cell lines we carried out proliferation assays. Figure 6B–G shows that the inhibitor has potent anti-proliferative activity against the tumorigenic breast and prostate lines while the non-tumorigenic lines continued to proliferate in the presence of HS-72. There was a significant inhibition (p<0.001) of proliferation in all tumorigenic cell lines tested (Figure 6B–E). In contrast, the non-tumorigenic MCF10A cells continue to grow at all concentrations, while the RWPE1 cells were only inhibited at the highest concentration tested (Figure 6F–G). HS-71 treatment results in a less potent effect on proliferation compared to HS-72, consistent with biochemical studies (Figure S6B–C).
To test if HS-72 acts synergistically with Hsp90 inhibitors we tested the effect of the Hsp90 inhibitor HS-10 in combination with HS-72 on the degradation of HER2 and Akt, which are classified as substrates or clients of Hsp70i and Hsp90, respectively. In the presence of HS-72, there is degradation of HER2 and Akt that is consistent with previous results (Figure S2G). HS-10 alone also induced degradation of HER2 and Akt, as well as increased Hsp70 protein levels as expected, due to the negative regulatory role that Hsp90 has on Heat Shock Transcription Factor 1. In combination, the levels of HER2 were completely abolished and there was significant Akt degradation (Figure S6D). Next we determined the effect of inhibitor combination on SkBr3 and MCF7 cell proliferation. Increasing amounts of HS-72 in addition to the HS-10 treatment resulted in an additive effect, potently inhibiting the proliferation of both cell lines more so than HS-72 or HS-10 alone (Figure 6H–I). These results add to the growing evidence that Hsp90 and Hsp70i inhibitor combinations are likely to have great therapeutic utility in the clinic (Guo et al., 2005; Powers et al., 2009).
HS-72 is bioavailable in vivo and reduces tumor growth in a spontaneous mouse mammary tumor model
Prior to testing the efficacy of HS-72 in vivo, we performed a preliminary experiment to assess dose dependent effects in wild type mice. Healthy wild-type mice were dose escalated biweekly for 35 days and no adverse events, reduction in body weight, or altered behavior were observed up to 30 mg/kg (mpk) (Figure S7A). Additionally, a complete blood workup was done following HS-72 treatment in wild-type mice. Mice that were treated with HS-72 showed no effect on complete blood count, no effect on kidney function, and no effect on liver function compared to control mice (Figure S7B–D). These data indicate that HS-72 is not toxic to wild-type mice. A limited PK study was also performed using wild-type mice, analyzing and quantifying HS-72 in the plasma, liver, and kidney. Each sample was spiked with an internal standard, HS-156, which is a close structural analogue to HS-72, and analyzed by LC-MS (Figure S7E). The ratio of the extracted ion chromatogram (EIC) comparing HS-72 to HS-156 was plotted on a standard curve to quantify [HS-72] present in each sample (Figure S7F–H and Supplemental Information 2). Figure 7A indicates that HS-72 is exponentially cleared from the plasma (T1/2 elimination = 0.4 ± 0.1 hours n=3, SEM) reaching 0.07 ± 0.03 mmol/ml (n=3, SEM) by 5 minutes post IP injection, clearing to 0.002 ± 0.0002 mmol/ml (n=3, SEM) by 8 hours, with only trace amounts detectable (<10 nmol/ml) by 24 hours. In kidney, HS-72 reached 0.43 ± 0.08 mmol/gram tissue (w.w.) (n=3, SEM) at 5 minutes and by 24 hours was retained at 0.02 ± 0.004 mmol/g (w.w.) (n=3, SEM) (Figure 7B). In contrast, in liver HS-72 uptake peaked by 8 hours at 2.26 ± 0.50 mmol/g (w.w.) (n=3, SEM) and was slowly cleared to 0.51 ± 0.11 mmol/g (w.w.) (n=3, SEM) by 24hours (Figure 7C). These findings show plasma HS-72 present at significant levels for at least 8 hours post I.P. and that HS-72 has a high degree of tissue bioavailability. In all 3 compartments, the parent MS ion of 365.2 Da was detected intact, with no evidence of rapid metabolism. The finding that HS-72 is absorbed to high [µM] levels following IP injection at 20mpk, particularly in liver, as well as kidney without adverse event, suggests the molecule is well tolerated in vivo.
Figure 7. Pharmacokinetic, distribution and efficacy studies with HS-72 in healthy mice and MMTV-neu mice.

(A–C) LC-MS analysis of plasma, kidney and liver following I.P. injection, 20mg/Kg (mpk), at the indicated time points over 24 hours show HS-72 has high degree of bioavailability in health mice. In (D) HS-72 promotes reduction in tumor volume in MMTV-neu mice treated I.P. with HS72 BiW (biweekly) at 20mpk (n = 7), compared to animals receiving no treatment (n = 26) with a significant decrease in tumor volume (p<0.05) at 21 days. Linear regression analysis comparing the slopes of the HS-72 tumor volume vs. no treatment tumor volume is trending towards significance (p=0.08). (E) The median survival of animals treated I.P. with HS-72 20mpk BiW or HS- 72 20mpk qd increased by 6 days and 13 days, respectively, than animals receiving no treatment. One set of control animals was used for multiple treatments, yielding more control animals than treated animals.
(A–D) Mean ± SEM. See also Figure S7.
The apparent safety and bioavailability of HS-72 allowed us to test the efficacy of HS-72 to reduce tumor growth in the MMTV-neu breast cancer model. In this model, HER2 is overexpressed under the transcriptional control of the mouse mammary tumor virus promoter/enhancer, leading to spontaneous development of mammary tumors (Taneja et al., 2009). To confirm that HS-72 would have efficacy in the MMTV-neu mouse model, HS-72 was tested in the NF639 cell line, which is derived from the mammary tumor of a MMTV-neu mouse. HS-72 was shown to potently inhibit proliferation in a manner similar to previously tested cell lines (Figure S7I). Furthermore, a synergistic effect on cell proliferation was observed when testing HS-72 and HS-10 in combination, thus highlighting the potential for combination therapy in the MMTV-neu mouse model (Figure S7J). Tumor bearing MMTV-neu mice were treated I.P. with HS-72 at 20mpk on a biweekly (BiW) schedule for 21 days. At 21 days there is a significant reduction (p<0.05) in tumor volume in the HS-72 treated mice compared to untreated mice (Figure 7D). A linear regression analysis comparing the slopes of the HS-72 tumor volume vs. no treatment tumor volume is trending towards significance (p=0.08) (Figure 7D). Furthermore, median survival of animals increased by 6 days in mice treated with HS-72 20mpk BiW, and by 13 days in animals treated with HS-72 20mpk qd (daily dosing) (Figure 7E). Collectively, these studies show that the HS-72 scaffold has no overt toxicities, exhibits tissue and tumor bioavailability, and demonstrates efficacy in a spontaneous mouse mammary tumor model even under conservative biweekly dosing conditions. Our findings indicate that further development of the HS-72 scaffold is warranted to improve its potency and drug like properties.
Discussion
This work identifies a new chemical scaffold, HS-72 ((S)-N-(1-propyl-1H-benzo[d]imidazol-2-yl)-1-(pyrazin-2-yl)piperidine-3-carboxamide) that exhibits specificity towards the inducible form of Hsp70, Hsp70i. Various biochemical approaches demonstrated the selectivity of HS-72 towards Hsp70i over other Hsp70 family members, in particular Hsc70. This includes selective elution from γ-phosphate linked ATP resin, creation of a highly selective HS-72 affinity resin, a selective thermo destabilizing effect in the presence of ATP compared with Hsc70 and Hsp90, altered protease digestion patterns in the presence and absence of the inhibitor, and sensitivity to the S enantiomer over the R form HS-71. Our studies also show that HS-72 has a unique mechanism of action in vitro by acting as an allosteric inhibitor of ATP binding, a feature that certainly underlies its ability to discriminate Hsp70i from other Hsp70 family members. This mode of action may also explain HS-72 selectivity against the broader purinome, since the molecule is not directly targeting the ATP binding pocket, which is also suggested by our molecular docking studies.
In cells, HS-72 bears all the hallmarks associated with inhibition of Hsp70i. At the molecular level this includes loss of HER2 and Akt expression in breast tumor cells and formation of protein aggregates in a cellular based model of Huntington’s disease. The inhibitor is also synergistic in combination with Hsp90 inhibitors, as determined by monitoring HER2 and Akt expression. In proliferation assays, HS-72 shows specificity towards more aggressive breast and prostate tumor cell lines, consistent with the specific role of Hsp70i in mediating metastatic progression in vivo. Importantly, HS-72 is well tolerated and bioavailable in mice with no evidence of overt toxicity at high doses. Collectively these data suggest that HS-72 is an attractive starting point for a medicinal chemistry campaign to improve the potency and pharmacological properties of the molecule. With this longer-term aim in mind we tested the efficacy of HS-72 in the MMTV model, a murine model of spontaneous breast cancer in humans (Taneja et al., 2009). On a conservative biweekly administration cycle, HS-72 demonstrated significant inhibition of tumor growth with evidence of improved survival. Further, our PK study showed that plasma [HS-72] are maintained at >20 µM for at least 8 hours, levels that reflect its potency against various tumor cell lines in vitro.
It is important to highlight that the HS-72 molecule was a raw hit from the FLECS screen with no SAR done to improve the compound. From a medicinal chemistry perspective the HS-72 scaffold is attractive and we have shown that it is highly amenable to resynthesis; we have synthesized a HS-72 affinity resin and analogs can readily be created to drive future SAR studies. The central piperidine carboxamide scaffold that makes up the core of HS-72 is structurally unique from all other described Hsp70i inhibitors. Prior inhibitors identified to target Hsp70s include NSC 630668-R/1, VER, MAL3-101, MKT-077, PES, Apoptozole, and YK5 (Powers et al., 2010; Rodina et al., 2013). There is considerable structural diversity amongst these inhibitors and generally the NBD domain has been favored for inhibitor development (Powers et al., 2010). However, the polar interactions present in the nucleotide binding pocket and its affinity for ATP have contributed to difficulties in selective inhibitor discovery (Massey, 2010). The full-length crystal structure of the nucleotide bound form shows that the nucleotide is completely enclosed, making the accessibility of small inhibitors difficult (Figure 1B). Importantly, none of the approaches adopted thus far have been able to target specific Hsp70 family members, especially Hsp70i from Hsc70. NSC 630668-R/1, inhibits ATPase activity but does not discriminate Hsp70i from Hsc70 (Fewell et al., 2001). VER shows broad specificity with other Heat shock protein family members, largely because it is a nucleotide derivative. It also contains two potentially labile, perhaps by design, benzyl groups (Massey, 2010). MAL3-101 has been shown to compromise co-chaperone-stimulated Hsp70 ATPase activity, suggesting it is an allosteric regulator, although the exact binding site of this molecule remains unknown (Braunstein et al., 2011). Like NSC 630668-R/1, MAL3-101 is quite large and has a number of labile ester groups. MKT-077 targets the NBD and inhibits proliferation in tumor cell lines, however, severe renal dysfunction in patients was observed in phase I clinical trials (Britten et al., 2000). PES has been shown to interact with the SBD of both Hsc70 and Hsp70i and disrupt client protein interaction in vitro (Leu et al., 2009). However, recent evidence suggests that the PES interaction with Hsp70 is through non-specific interactions (Schlecht et al., 2013). The molecule promotes caspase-dependent cell death only in tumor cells, suggesting some specificity to Hsp70i in vitro, although p53 binding has also been shown, which may explain its antitumor actions (Leu et al., 2009). Moreover, MKT-077 and PES have potential reactive groups that render them covalent modifiers, which may contribute to side effects in vivo. YK5 is an allosteric inhibitor of Hsp70, recently identified using modeling techniques, but this molecule does not discriminate between Hsp70i and Hsc70 (Rodina et al., 2013).
The FLECS assay employed in defining HS-72 is a simple quantitative binding assay that offers an alternate approach to defining inhibitors to enzymes/proteins that often seem intractable to conventional HT screens. Because of its chaperoning and trafficking functions in vivo, Hsp70i is a good example of such intractability. To date few HT screens have been designed that enable Hsp70i to be screened based on its chaperone function or ATPase activity (Kang et al., 2008; Rowlands et al., 2010; Wisen and Gestwicki, 2008). Although Hsp70i possess ATPase activity, following turnover, ADP remains bound to the enzyme (Mayer and Bukau, 2005). This means that competitive inhibitors acting at the ATP binding site must bind with a very high affinity to displace the bound nucleotide. In this respect, the Hsp70s are reminiscent of small G proteins, which have nM affinity for both GTP and GDP and are very difficult to drug directly. We propose that Hsp70i is recovered on our ATP media as a result of the ADP-bound or apo form being forced into and retained in the active state when exposed to the immobilized nucleotide. This enabled discovery of HS-72, which reduces the affinity of Hsp70i for the immobilized ligand. We are currently exploring the possibility of screening libraries containing molecules that selectively block interactions with this media, rather than displacing the bound protein through competitive or allosteric means. Finally, the FLECS approach enabled our ability to establish HS-72’s selectivity, both against the Hsp70 family and the wider purinome. In our view, when targeting purine-utilizing enzymes, the intrinsic selectivity of one’s lead scaffold towards the enzyme of interest is a greater priority than initial potency. This limits off-target liabilities that can be very difficult to remove during lead optimization studies. For this reason, our prioritization of HS-72 over other Hsp70i hits in our screen was driven by the intrinsic nature of the scaffold towards the protein over several other classes of purinome members screened against the same library (Figure 2). The finding that HS-72 is well tolerated in mice, reaching 2.26 mmol/g (w.w.) in liver without modification, is perhaps a testament to this prioritization strategy.
Significance
Inducible Hsp70 (Hsp70i) is generally recognized as a cutting edge drug target for the development of a new generation of anticancer drugs. However, drugging Hsp70i has proven extremely challenging. Hsp70i undergoes ATP/ADP exchange, leaving the nucleotide binding pocket occupied at all times. This presents a barrier to drug-like molecules that might act to inhibit Hsp70i through direct competition with bound nucleotide. Additionally, Hsp70i shares close sequence identity with other Hsp70 family members, in particular Hsc70, a constitutively active Hsp70 family member, whose activities are essential for normal cellular function. No groups to date have defined inhibitors that can discriminate Hsp70i from Hsc70. Screening by FLECS (fluorescence linked enzyme chemoproteomic strategy) has identified a new chemical scaffold, called HS-72 ((S)-N-(1-propyl-1H-benzo[d]imidazol-2-yl)-1-(pyrazin-2-yl)piperidine-3-carboxamide) that shows selectivity for Hsp70i over the closely related Hsc70 as well as the broader purinome. We show that HS-72 acts as an allosteric inhibitor of ATP binding, a feature that underlies its ability to discriminate Hsp70i from other Hsp70 family members. This discriminates HS-72 from all previous inhibitors targeting purine-utilizing proteins such as heat shock proteins or protein kinases, which are generally ATP competitive. In mice HS-72 is well tolerated and bioavailable and shows no evidence of overt toxicity at high doses. Furthermore, HS-72 shows signs of efficacy in the MMTV model of HER2+ breast cancer. While HS-72 is a raw hit from the FLECS screen, our data suggest the HS-72 scaffold is an appealing starting point for a medicinal chemistry campaign to improve its potency and pharmacological properties in vivo.
Experimental Procedures
FLECS Screen
A pEGFP-tagged Hsp70i was (plasmid 15215, Addgene, Cambridge, MA) used in the FLECS assay and was originally cloned by Evan Eisenburg (Zeng et al., 2004). ATP used in the assay was purchased from Sigma (St. Louis, MO) and a 200 mM stock was prepared with low salt buffer (150 mM NaCl, 25 mM Tris, pH 7.5, 60 mM MgCl2). The γ-phosphate ATP sepharose was synthesized as previously described and stored in low salt buffer (Carlson et al., 2013). FuGENE 6 transfection reagent (Roche, Mannheim, Germany) was used for transfection of GFP-Hsp70i into HEK 293T cells, following the manufacturer protocol. The transfection ensued for 48 hours, upon which time the cells were harvested and lysed in cell lysis buffer (150 mM NaCl, 50 mM Tris, pH 7.5, 1% Triton X-100, 1 mM EDTA, 1 mM DTT, and 1 tablet Complete Mini protease inhibitor(Roche)). Cell lysates were stored at −80°C until further use. Upon binding the resin lysates were washed 3× with high stringency wash buffer (1 M NaCl, 25 mM Tris, pH 7.5, 60 mM MgCl2, 1 mM DTT) and 3× with low stringency wash buffer (150 mM NaCl, 25 mM Tris, pH 7.5, 60 mM MgCl2, 1 mM DTT). Next the lysates were transferred to 0.2 µm PVDF filter 96-well plate (Corning, Corning, NY) sitting on top of a black flat-bottomed 96-well catch plate (Corning). The plates were spun down using an Eppendorf Centrifuge 5810 (Hamburg, Germany) at 2000 rpm for 2 min.
Caspase 3/7 Assay
The Amplite™ Fluorimetric Caspase3/7 assay kit (AAT Bioquest, Sunnyvale, CA) was used per the manufacturers instructions. Briefly, a fluorometric indicator, Ac-DEVD-AMC, was used to determine caspase activity. Cleavage of AMC by caspases resulted in a fluorescent signal that can be assessed at 440–460nm with an excitation of 340–350nm. Cells were seeded at 60,000 cells/well in a 96 well plate and treated with compound for the indicated period of time. Diluted caspase 3/7 assay solution was added to each well and incubated at room temperature for 2 hours protected from light, upon which time fluorescence was measured on the Victor X2 plate reader (Perkin Elmer, Waltham, MA).
Aggregation Assay
The PC12 rat neuronal cell line, which expresses Huntingtin exon 1 containing 74 glutamine repeats is fused to GFP and under the control of a doxycycline promoter was used. Cells were treated with HS-72 for 18 hours prior to a doxycycline addition for 48 hours. The soluble and pellet fraction were then separated by centrifugation at 14,000 rpm for 15 minutes and both fractions were assayed for httQ-GFP by solubilizing with SDS followed by western blotting with antibodies against GFP.
Thermofluor Assay
SYPRO orange (Molecular Probes, Eugene, OR) was diluted 1:1000 in 25mM HEPES, 5mM MgCl2, 10mM KCl (pH 7.5) and purified Hsp70i, Hsc70, Hsp70i C306, or Hsp90, was then added to a final dilution of 0.04 mg/ml. Where indicated 0.001% or 0.01% Triton X-100 (Sigma) was also added. The indicated compound or DMSO was then added at the specified concentration and each sample was added as 5 replicates to a 384 well plate (BioRad, Hercules, CA). A melt curve protocol (25°C to 90°C, increasing 0.5°C and a plate reading every 30 seconds) was run on a CFX384 Touch™ Real-Time PCR Detection System (BioRad). To determine the midpoint of the protein unfolding transition or Tm, GraphPad Prisim4 (La Jolla, CA) was used to normalize the melt curve and to calculate the first derivate of the melt curve, with the steepest point of the slope being the Tm.
Docking Studies
HS-72 was docked into the crystal structure of the Hsp70i NDB bound to AMP-pnp (PDB: 2E8A) using the SwissDock program. The returned clusters were distributed between two binding sites. Chimera was used to visualize the putative binding sites of HS-72 on the Hsp70i NBD.
Partial Proteolysis
Hsp70i (8µg for SDS-PAGE analysis, 2µg for mass spec analysis) was incubated with 1mM ATP, 1mM ADP, 100uM HS72, 100uM HS72 + 1mM ATP, or 100uM HS72 + 1mM ADP for 30 min. at room temperature. Hsp70i was digested by adding 0.1µg of trypsin (Promega, Madison, WI) per 2µg of protein and was quenched by addition of 25% TFA for mass spec analysis or addition of 5× SDS loading buffer and boiling for SDS-PAGE analysis at 2 hours, or 24 hours. Gels were visualized by silver stain.
HS-72 in vivo Efficacy Studies
MMTV-neu mice, a HER2 overexpression breast cancer mouse model in which HER2 is under the transcriptional control of the mouse mammary tumor virus promoter/enhancer, were treated with the indicated doses and dosing schedule (Taneja et al., 2009). All doses were delivered through IP injections using DMSO and their tumors were caliperedonce weekly. The mice were culled upon reaching tumor burden or if they expressed signs of toxicity as per MP1U standard protocol.
For further details regarding the experimental procedures used in this work, see the Supplemental Experimental Procedures section in the Supplemental Information. PK study raw data files from LC-MS can be found in Supplemental Information 2, located on figshare and can be found here: http://dx.doi.org/10.6084/m9.figshare.1209606
Supplementary Material
Acknowledgements
This work was funded by NIH grants R01-AI089526-04 to T.A.J.H. and R01-NS065890 to D.J.T, a Department of Defense Transformative Vision Award to T.A.J.H. and N.L.BS and by grant GM75061 to J.L.B. Thanks to Dr. Jason Gestwicki (UCSF) for clones expressing recombinant forms of Hsp70i, Hsc70, and Hsp70i mutant C306D. The structure and use of the HS-72 scaffold and its resynthesis have been disclosed to Duke University in accordance with Duke University guidelines concerning potential intellectual property.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
KB conducted the FLECS assay; MKH and KB conducted cell permeability experiments, substrate degradation, protein aggregation assays and proliferation assays; PFH synthesized HS-72, the HS-72 affinity resin, and HS-71; MKH performed selectivity studies; MKH, ETM, and AMJ conducted Thermofluor studies; PFH performed docking studies; MKH and DRL conducted partial proteolysis studies; MKH performed HS-72 and HS-10 combination studies; MKH and DAC performed PK studies; DBD, JLJ, and LMH conducted in vivo studies; TAG conducted single turnover ATPase assays and JLB analyzed the data; MKH and TAJH wrote the manuscript; all authors reviewed and edited the manuscript.
The authors have no competing financial interests to declare.
References
- Beere HM. Stressed to death: regulation of apoptotic signaling pathways by the heat shock proteins. Science's STKE : signal transduction knowledge environment 2001. 2001:re1. doi: 10.1126/stke.2001.93.re1. [DOI] [PubMed] [Google Scholar]
- Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, Tailor P, Morimoto RI, Cohen GM, Green DR. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nature cell biology. 2000;2:469–475. doi: 10.1038/35019501. [DOI] [PubMed] [Google Scholar]
- Blazer LL, Roman DL, Chung A, Larsen MJ, Greedy BM, Husbands SM, Neubig RR. Reversible, allosteric small-molecule inhibitors of regulator of G protein signaling proteins. Molecular pharmacology. 2010;78:524–533. doi: 10.1124/mol.110.065128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunstein MJ, Scott SS, Scott CM, Behrman S, Walter P, Wipf P, Coplan JD, Chrico W, Joseph D, Brodsky JL, et al. Antimyeloma Effects of the Heat Shock Protein 70 Molecular Chaperone Inhibitor MAL3-101. Journal of oncology. 2011;2011:232037. doi: 10.1155/2011/232037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britten CD, Rowinsky EK, Baker SD, Weiss GR, Smith L, Stephenson J, Rothenberg M, Smetzer L, Cramer J, Collins W, et al. A phase I and pharmacokinetic study of the mitochondrial-specific rhodacyanine dye analog MKT 077. Clinical cancer research : an official journal of the American Association for Cancer Research. 2000;6:42–49. [PubMed] [Google Scholar]
- Carlson DA, Franke AS, Weitzel D, Speer BL, Hughes PF, Hagerty L, Fortner CN, Veal JM, Barta TE, Zieba BJ, et al. Fluorescence Linked Enzyme Chemoproteomic Strategy for Discovery of a Potent and Selective DAPK1 and ZIPK Inhibitor. ACS chemical biology. 2013 doi: 10.1021/cb400407c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings MD, Farnum MA, Nelen MI. Universal screening methods and applications of ThermoFluor. Journal of biomolecular screening. 2006;11:854–863. doi: 10.1177/1087057106292746. [DOI] [PubMed] [Google Scholar]
- Daugaard M, Rohde M, Jaattela M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS letters. 2007;581:3702–3710. doi: 10.1016/j.febslet.2007.05.039. [DOI] [PubMed] [Google Scholar]
- Dix DJ, Allen JW, Collins BW, Mori C, Nakamura N, Poorman-Allen P, Goulding EH, Eddy EM. Targeted gene disruption of Hsp70-2 results in failed meiosis, germ cell apoptosis, and male infertility. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:3264–3268. doi: 10.1073/pnas.93.8.3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans CG, Chang L, Gestwicki JE. Heat shock protein 70 (hsp70) as an emerging drug target. Journal of medicinal chemistry. 2010;53:4585–4602. doi: 10.1021/jm100054f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fewell SW, Day BW, Brodsky JL. Identification of an inhibitor of hsc70-mediated protein translocation and ATP hydrolysis. The Journal of biological chemistry. 2001;276:910–914. doi: 10.1074/jbc.M008535200. [DOI] [PubMed] [Google Scholar]
- Goloudina AR, Demidov ON, Garrido C. Inhibition of HSP70: a challenging anti-cancer strategy. Cancer letters. 2012;325:117–124. doi: 10.1016/j.canlet.2012.06.003. [DOI] [PubMed] [Google Scholar]
- Grosdidier A, Zoete V, Michielin O. Fast docking using the CHARMM force field with EADock DSS. Journal of computational chemistry. 2011 doi: 10.1002/jcc.21797. [DOI] [PubMed] [Google Scholar]
- Guo F, Rocha K, Bali P, Pranpat M, Fiskus W, Boyapalle S, Kumaraswamy S, Balasis M, Greedy B, Armitage ES, et al. Abrogation of heat shock protein 70 induction as a strategy to increase antileukemia activity of heat shock protein 90 inhibitor 17-allylamino-demethoxy geldanamycin. Cancer research. 2005;65:10536–10544. doi: 10.1158/0008-5472.CAN-05-1799. [DOI] [PubMed] [Google Scholar]
- Hunt C, Morimoto RI. Conserved features of eukaryotic hsp70 genes revealed by comparison with the nucleotide sequence of human hsp70. Proceedings of the National Academy of Sciences of the United States of America. 1985;82:6455–6459. doi: 10.1073/pnas.82.19.6455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Taldone T, Clement CC, Fewell SW, Aguirre J, Brodsky JL, Chiosis G. Design of a fluorescence polarization assay platform for the study of human Hsp70. Bioorganic & medicinal chemistry letters. 2008;18:3749–3751. doi: 10.1016/j.bmcl.2008.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu JI, Pimkina J, Frank A, Murphy ME, George DL. A small molecule inhibitor of inducible heat shock protein 70. Molecular cell. 2009;36:15–27. doi: 10.1016/j.molcel.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey AJ. ATPases as drug targets: insights from heat shock proteins 70 and 90. Journal of medicinal chemistry. 2010;53:7280–7286. doi: 10.1021/jm100342z. [DOI] [PubMed] [Google Scholar]
- Massey AJ, Williamson DS, Browne H, Murray JB, Dokurno P, Shaw T, Macias AT, Daniels Z, Geoffroy S, Dopson M, et al. A novel, small molecule inhibitor of Hsc70/Hsp70 potentiates Hsp90 inhibitor induced apoptosis in HCT116 colon carcinoma cells. Cancer chemotherapy and pharmacology. 2010;66:535–545. doi: 10.1007/s00280-009-1194-3. [DOI] [PubMed] [Google Scholar]
- Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cellular and molecular life sciences : CMLS. 2005;62:670–684. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata Y, Rauch JN, Jinwal UK, Thompson AD, Srinivasan S, Dickey CA, Gestwicki JE. Cysteine reactivity distinguishes redox sensing by the heat621 inducible and constitutive forms of heat shock protein 70. Chemistry & biology. 2012;19:1391–1399. doi: 10.1016/j.chembiol.2012.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nylandsted J, Wick W, Hirt UA, Brand K, Rohde M, Leist M, Weller M, Jaattela M. Eradication of glioblastoma, and breast and colon carcinoma xenografts by Hsp70 depletion. Cancer research. 2002;62:7139–7142. [PubMed] [Google Scholar]
- Powers MV, Clarke PA, Workman P. Death by chaperone: HSP90, HSP70 or both? Cell Cycle. 2009;8:518–526. doi: 10.4161/cc.8.4.7583. [DOI] [PubMed] [Google Scholar]
- Powers MV, Jones K, Barillari C, Westwood I, van Montfort RL, Workman P. Targeting HSP70: the second potentially druggable heat shock protein and molecular chaperone? Cell Cycle. 2010;9:1542–1550. doi: 10.4161/cc.9.8.11204. [DOI] [PubMed] [Google Scholar]
- Qi R, Sarbeng EB, Liu Q, Le KQ, Xu X, Xu H, Yang J, Wong JL, Vorvis C, Hendrickson WA, et al. Allosteric opening of the polypeptide-binding site when an Hsp70 binds ATP. Nature structural & molecular biology. 2013;20:900–907. doi: 10.1038/nsmb.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodina A, Patel PD, Kang Y, Patel Y, Baaklini I, Wong MJ, Taldone T, Yan P, Yang C, Maharaj R, et al. Identification of an allosteric pocket on human hsp70 reveals a mode of inhibition of this therapeutically important protein. Chemistry & biology. 2013;20:1469–1480. doi: 10.1016/j.chembiol.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowlands M, McAndrew C, Prodromou C, Pearl L, Kalusa A, Jones K, Workman P, Aherne W. Detection of the ATPase activity of the molecular chaperones Hsp90 and Hsp72 using the TranscreenerTM ADP assay kit. Journal of biomolecular screening. 2010;15:279–286. doi: 10.1177/1087057109360253. [DOI] [PubMed] [Google Scholar]
- Schlecht R, Scholz SR, Dahmen H, Wegener A, Sirrenberg C, Musil D, Bomke J, Eggenweiler HM, Mayer MP, Bukau B. Functional analysis of Hsp70 inhibitors. PloS one. 2013;8:e78443. doi: 10.1371/journal.pone.0078443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu CW, Huang CM. HSP70s: From Tumor Transformation to Cancer Therapy. Clinical medicine Oncology. 2008;2:335–345. doi: 10.4137/cmo.s475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneja P, Frazier DP, Kendig RD, Maglic D, Sugiyama T, Kai F, Taneja NK, Inoue K. MMTV mouse models and the diagnostic values of MMTV-like sequences in human breast cancer. Expert review of molecular diagnostics. 2009;9:423–440. doi: 10.1586/ERM.09.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavaria M, Gabriele T, Kola I, Anderson RL. A hitchhiker's guide to the human Hsp70 family. Cell stress & chaperones. 1996;1:23–28. doi: 10.1379/1466-1268(1996)001<0023:ahsgtt>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisen S, Gestwicki JE. Identification of small molecules that modify the protein folding activity of heat shock protein 70. Analytical biochemistry. 2008;374:371–377. doi: 10.1016/j.ab.2007.12.009. [DOI] [PubMed] [Google Scholar]
- Wyttenbach A, Swartz J, Kita H, Thykjaer T, Carmichael J, Bradley J, Brown R, Maxwell M, Schapira A, Orntoft TF, et al. Polyglutamine expansions cause decreased CRE-mediated transcription and early gene expression changes prior to cell death in an inducible cell model of Huntington's disease. Human molecular genetics. 2001;10:1829–1845. doi: 10.1093/hmg/10.17.1829. [DOI] [PubMed] [Google Scholar]
- Yang X, Wang J, Zhou Y, Wang Y, Wang S, Zhang W. Hsp70 promotes chemoresistance by blocking Bax mitochondrial translocation in ovarian cancer cells. Cancer letters. 2012;321:137–143. doi: 10.1016/j.canlet.2012.01.030. [DOI] [PubMed] [Google Scholar]
- Zeng XC, Bhasin S, Wu X, Lee JG, Maffi S, Nichols CJ, Lee KJ, Taylor JP, Greene LE, Eisenberg E. Hsp70 dynamics in vivo: effect of heat shock and protein aggregation. Journal of cell science. 2004;117:4991–5000. doi: 10.1242/jcs.01373. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.