

Abstract
Background
Although Rapamycin (RPM) have been studied extensively in ischemia models, its functional mechanisms remains to be defined.
Methods
We determined how RPM impacted the pathogenesis of ischemia reperfusion injury (IRI) in a murine liver partial warm ischemia model, with emphasis on its regulation of hepatocyte death.
Results
RPM protected livers from IRI in the presence of fully developed liver inflammatory immune response. RPM enhanced liver autophagy induction at the reperfusion stage. Dual mTOR1/2 inhibitor Torin 1, despite its ability to induced autophagy, failed to protect livers from IRI. The treatment with RPM, but not Torin 1, resulted in the enhanced activation of the mTORC2-Akt signaling pathway activation in livers post reperfusion. Inactivation of Akt by Triciribine abolished liver protective effect of RPM. The differential cytoprotective effect of RPM and Torin 1 was confirmed in vitro in hepatocyte cultures. RPM, but not Trin 1, protected hepatocytes from stress and TNF-α induced cell death; and inhibition of either autophagy by chloroquine or Akt by Triciribine abolished RPM-mediated cytoprotection.
Conclusion
RPM protected livers from IRI via both autophagy and mTORC2-Akt activation mechanisms.
Keywords: Liver Injury, mTOR complex (mTORC), Autophagy, Cell Death, Inflammation
Introduction
Rapamycin (RPM) becomes a choice of immunosuppressive agent in liver transplantation, particularly in patients experiencing post-Tx nephrotoxicity and with hepatocellular carcinoma (1, 2). The mammalian (mechanic) target of rapamycin (mTOR) is an evolutionally conserved serine/threonine kinase, which regulates cell proliferation, metabolism, and aging by responding to growth factors, nutrients, cellular energy, as well as immune stimulations of Ag-specific receptors, Toll-like-receptors (TLRs), and cytokine receptors (3). RPM binds to an intracellular protein, FK506-binding protein 12, to form a gain-of-function complex to interact with and inhibit mTOR, which results in the suppression of protein synthesis, leading to the blockade of cell cycle progression at the G1 to S phase. Thus, RPM inhibits cell proliferation, and blocks T cell differentiation into effectors (Th1, Th2 and Th17), which constitute the basis of its anti-tumor and immunosuppressive property (1, 4). At the molecular level, mTOR interacts with multiple proteins to form two signaling complexes, mTORC1 and mTORC2. Majority of functions associated with mTOR responses to nutrient and energy conditions are mediated by mTORC1, whereas, the activation and function of mTORC2 are much less well characterized. Currently, the PI3 kinase is known to control mTORC2 activities and active mTORC2 phosphorylates Akt at Ser 473 position (3). Importantly, RPM inhibits only mTORC1, but not mTORC2, while new generation inhibitors inhibit both by targeting the catalytic subunit of mTOR (5).
In contrast to its immune suppressive effect in T cells, mTOR inhibition was found recently to promote pro-inflammatory IL-12, but inhibit anti-inflammatory IL-10 cytokine productions in myeloid phagocytes (e.g., macrophages) (6, 7), which has been accounted for the clinical finding that patients treated with RPM suffer higher frequencies of variety of inflammatory disorders (e.g., interstitial pneumonitis) (4). As liver IRI results from innate inflammatory immune activation and IL-10 is a critical regulatory cytokine to control liver inflammation and homeostasis, the pro-inflammatory property of RPM raises the question of whether its use would potentially be detrimental to liver grafts. Furthermore, as a nutrient sensing kinase, mTOR is a key regulator of autophagy via its direct phosphorylation of autophagy initiating proteins (8). The question of whether RPM regulates liver autophagy induction in vivo during IR has not been determined. Although autophagy has been established in recent years as an essential homeostatic mechanism in cells and its upregulation is a highly conserved adaptive mechanism to promote cell survival under conditions of starvation, energy deprivation and metabolic stress (9), its roles in the pathogenesis of IRI is controversial(10, 11).
In this study, we investigated whether and how mTOR inhibition regulated the development of liver IRI, by analyzing its impact on hepatocyte death and innate immune activation both in vivo and in vitro. Liver autophagy induction and autophagy flux during IR were measured. RPM and a dual mTORC 1/2 inhibitor, Torin 1 were studied comparatively. Our results indicated that RPM, but not Torin 1, attenuated liver IRI by protecting hepatocytes from inflammatory cell death via autophagy induction and mTORC2-Akt activation.
Results
Rapamycin protects livers from IRI without inhibiting inflammatory immune activation
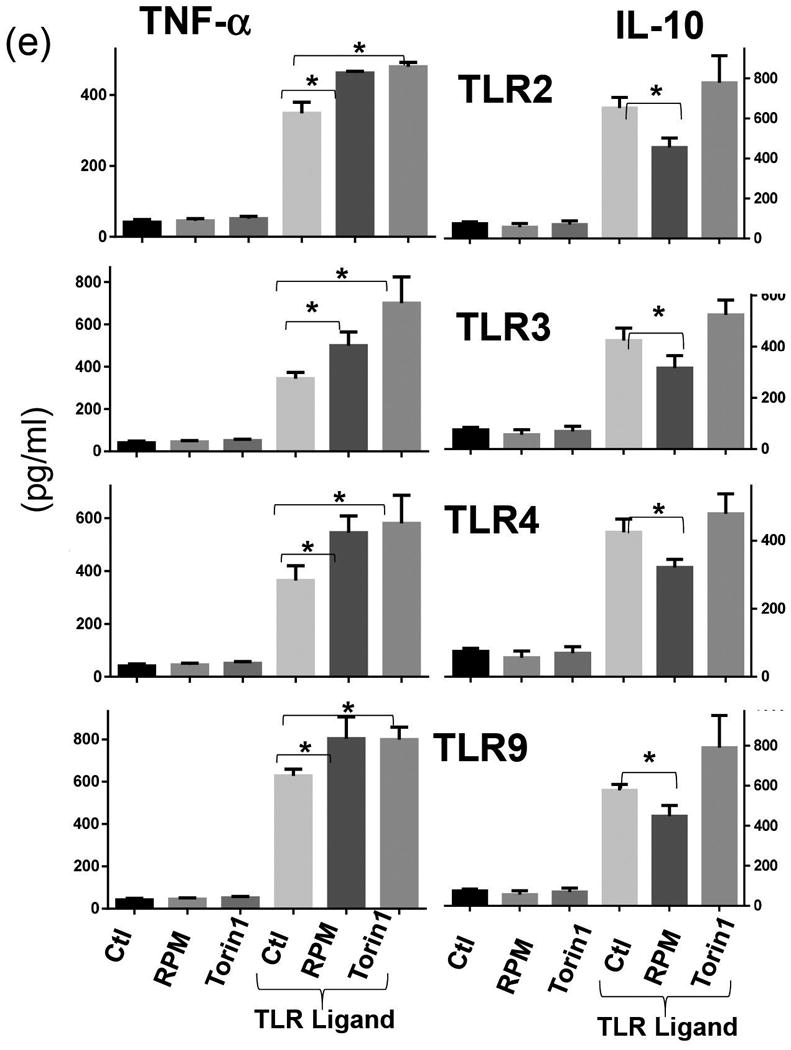
To determine the effect of RPM in liver IRI, we administered the drug peri-operationally in our murine model of partial liver warm ischemia. Liver injuries were evaluated at 6h post reperfusion. RPM at doses of both 1 and 5 mg/kg, i.p. 1h prior to the start of liver ischemia provided potent protection of livers from IRI. Serum ALT levels were significantly lower and liver architectures were preserved much better (low Suzuki scores) in treated mice, as compared with those in controls (Fig. 1a, b). Post ischemia administration of RPM also showed some protective effect, but not statistically significant. RPM did not inhibit liver inflammatory immune activation, as measured by both qRT-PCR of tissue cytokine/chemokine gene expression levels, including TNF-α, IL-1β, IL-6 and CXCL10 (Fig.1c), and ELISA of serum cytokines. In fact, the serum IL-10 level post IR was lower in RPM-treated mice, while TNF-α levels were comparable between controls and treated ones (Fig.1d). In vitro study of KCs in their responses to various TLR stimulation confirmed that RPM enhanced TNF-α, but decreased IL-10, production (Fig.1e).
Figure 1.
Rapamycin protects livers from IRI in the presence of inflammation, (a) Serum ALT levels in different groups of mice subjected to either sham operation or 90m ischemia/6h reperfusion treated with vehicle (DMSO) or RPM at 1 or 5mg/kg pre- or post-liver ischemia, as described in the material and methods, (b) Liver histology (H/E stain) and Suzuki scores. Representative tissue sections from either sham-operated or IR livers from DMSO or 1mg/kg RPM treated mice. (c) Quantitative RT-PCR analysis of inflammatory gene expressions in sham-operated and IR livers from DMSO or 1mg/kg RPM treated mice. The ratios of target gene vs. HPRT were plotted against different experimental groups. Note. There were trends of decrease in TNF-α, and CXCL-10 expression levels by RPM, without statistical differences, (d) Serum cytokine levels post liver IR (90m/6h) of different groups of mice. Representative results of at least 2 different experiments. n=3-4/group. *p<0.05, **p<0.01.
(e) Cytokine productions by Kupffer cells in response to TLR stimulation in vitro. KCs were isolated from B6 mouse livers by in situ collagenase digestion and enriched by selective adherence to petri dishes. After an overnight culture, cells were stimulated with TLR2, or 3, or 4, or 9 ligands for 24hrs in the presence or absence of RPM or Torin 1. TNF-α and IL-10 productions were measured by ELISA.
Representative results of at least 2 different experiments. 3 replicates/group, *p<0.05.
Rapamycin enhances liver autophagy induction during reperfusion
Western blot analysis of liver ribosomal p70S6 kinase (S6K) showed that mTORC1 signaling pathway was activated by IR, with the peak level of phosphorylated S6K at 1h post reperfusion. RPM treatment completely abolished S6K phosphorylation (Fig.2a). To measured liver autophagy induction and flux, chloroquine (CQ) was administered in vivo in combination with either vehicle control or RPM. LC3B II levels were measured at both 0 and 6h post reperfusion by Western blots (Fig.2b). Although ischemia increased LC3B II levels, autophagy flux was inhibited, as there were no further increases of LC3B II levels by CQ in ischemic livers, while CQ did increased LC3B II levels in sham livers. (Fig.2b). RPM did not further increase LC3B II levels in ischemic livers, nor did it restored autophagy flux inhibited by ischemia, at Oh post reperfusion. At 6h post-reperfusion, LC3B II levels in ischemic livers became similar to those in sham, which was further increased by the CQ treatment. These indicated that autophagy flux was recovered by reperfusion in ischemia livers. Importantly, RPM now enhanced liver autophagy induction, as shown by higher levels of LC3B II, as compared with those in sham and ischemic livers. Autophagy flux was not enhanced by RPM, as further increases of LC3B II by CQ were less pronounced in RPM-treated ischemic livers, as compared with those in sham or control ischemic livers (Fig.2b). These data indicate that RPM enhanced liver autophagy induction but not flux, during reperfusion. Functionally, we tested whether inhibition of autophagy induction by 3-MA would interfere with the liver protection by RPM. Chloroquine was not chosen due to its direct immune suppressive effect in liver IR models (12). Although 3-MA did not increase liver injuries in control mice, it restored full scale liver IRI in RPM treated mice (Fig.2c), supportive of a role of autophagy in RPM therapeutic effect in liver IRI.
Figure 2.
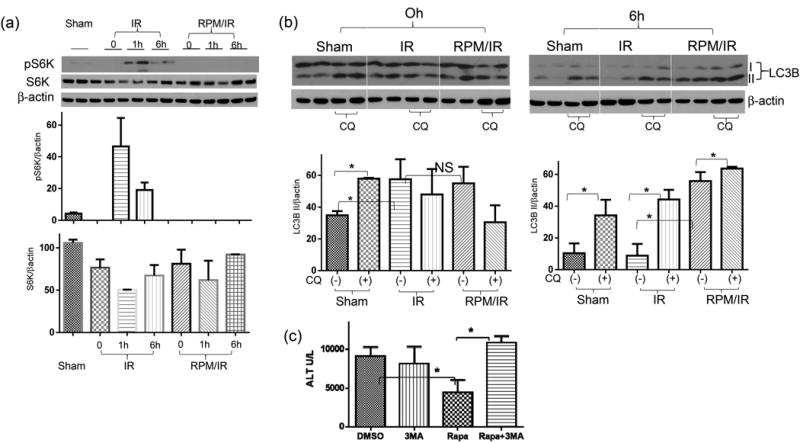
Rapamycin enhances liver autophagy during reperfusion. (a) Western blots of p70S6K in liver tissues post IR. Livers were harvested from sham or IR ones after 0, 1, 6 hrs of reperfusion (duplicate samples). Tissue protein lysates were prepared and separated by SDS-PAGE. S6K, phosphorylated S6K and β-actin levels were measured by Western blots, and protein bands were quantitated as ratios against β actin. (b) Western blots of LC3B in IR livers. Liver tissue proteins were prepared from mice after sham operation or ischemia and 0 or 6h reperfusion. To measure autophagy flux, groups of mice received CQ prior to liver ischemia, as described in the material and methods. Average LC3B II band intensities were quantitated as ratios against β actin. For tissue Western blot analysis, 2 samples/group, (c) Average serum ALT levels in mice subjected to 90m ischemia/6h reperfusion treated with vehicle (DMSO) or 3-MA, or RPM, or 3-MA/RPM prior to the start of liver ischemia, as described in the material and methods. n=4-6 mice/group.
Representative results of 2 different experiments. *p<0.05.
Torin 1 failed to protect livers from IRI
As the new generation of mTOR inhibitors target the kinase catalytic site and inhibit both mTORC1 and mTORC2, they are more potent than RPM in autophagy induction (5). Indeed, Torine-1 enhanced autophagy flux in vitro in Hepa1 cells more significantly than RPM, as documented by the Western blot result of LC3B (Fig.3a). When we tested its therapeutic effect in vivo, however, it failed to protect livers from IRI. Serum ALT levels and liver histological analysis showed similar degree of hepatocellular injuries between Torin 1 treated and control mice (Fig.3b, c). Similar to RPM, Torin 1 did not inhibit liver inflammatory immune response against IR, as measured by post-IR serum TNF-α and IL-10 levels (Fig.1d).
Figure 3.
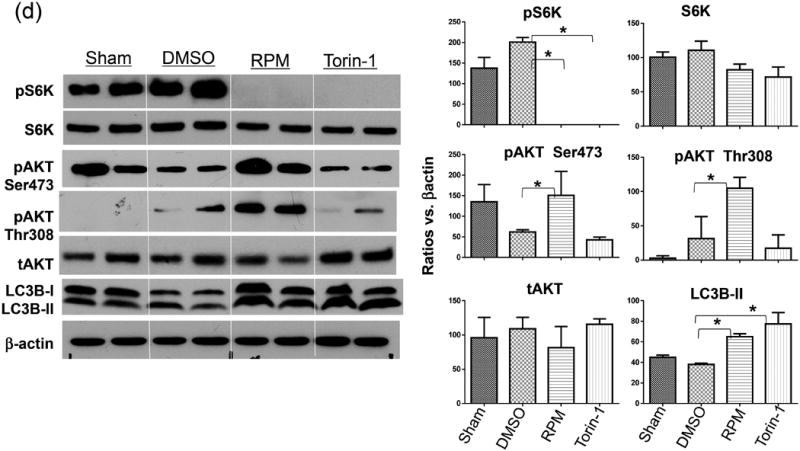
Dual mTOR inhibitor Torin 1 fails to protect livers from IRI. (a) Torin 1 enhances autophagy flux in vitro. Hepa1 cells were incubated with vehicle (Ctl) or Torin 1 or RPM in the absence or presence of CQ, as described in the material and methods. Lysates were collected at 6h, and analyzed by Western blots to measure LC3B levels. Average LC3B II band intensities were quantitated as ratios against β actin. 2 samples/group/experiment, (b) Average serum ALT levels in different groups of mice subjected to either sham operation or 90m ischemia/6h reperfusion treated with vehicle (DMSO) or RPM, or Torin 1, or TCN, or RPM/TCN prior to the start of liver ischemia, as described in the material and methods. n=4-6 mice/group, (c) Representative liver histology (H/E stain) after IR in different mouse groups as in (b). (d) mTOR inhibitors regulate liver autophagy and intracellular signaling pathways. Liver tissues were harvested after sham operation or 90m ischemia/6h reperfusion from different groups of mice treated with either DMSO, RPM or Torin 1, as described in the material and methods. Protein levels of total and phosphorylated S6K and Akt, as well as LC3B were measured by Western blots. Target protein band intensities were quantitated as rations against β actin. 2 samples/group.
Representative results of 2 different experiments. *p<0.05.
The key role of mTORC2-Akt activation in Rapamycin-mediated liver protection
As mTORC2 was differentially targeted by Torin 1 and RPM, we next determined roles of mTORC2 in the RPM liver protection mechanism by measuring its substrate Akt phosphorylation in livers post IR. The mTORC2-mediated Akt phosphorylation at Serine 473 was inhibited by IR. RPM, but not Torin 1, enhanced its activation significantly. The Akt phosphorylation at Thr308 (mediated by PI3K-PDK1) was also enhanced selectively by RPM (Fig.3d), indicating an overall increase of Akt activation by RPM. Both mTOR inhibitors inhibited the phosphorylation of S6K, the direct substrate of mTORC1, and increased LC3B II levels in IR livers.
To test the functional significance of the mTORC2-Akt signaling pathway activation, we administered Triciribine (TCN), which inhibited Akt phosphorylation, together with RPM in mice. Although TCN by itself did not increase liver IRI, it completely abrogated the liver protective effect of RPM, as documented by sALT measurements and liver histological analysis (Fig.3b, c). TCN did not affect liver inflammatory immune activation against IR, as measured by post-IR serum TNF-α and IL-10 levels (Fig.1d).
Rapamycin protected hepatocytes from stress and TNF-α induced cell death
As RPM protected livers in the presence of inflammation in vivo, we next determined in vitro whether mTORC1 inhibition decreased hepatocyte death. Hepa1 cell death was induced by either ER stress inducer Tg or a combined treatment of Tg and TNF-α. Indeed, RPM reduced cell death in both conditions that LDH levels were significantly lower in RPM-treated cells (Fig.4a, b). In contrast, Torin 1 had no cytoprotective effect against Tg (Fig.4c). The differential cytoprotective effect of RPM and Torin 1 was confirmed in primary murine hepatocytes (data not shown).
Figure 4.
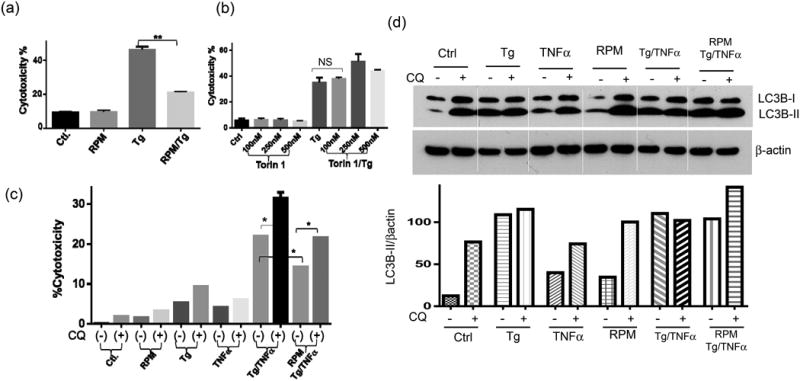
mTOR inhibitors regulate hepatocyte death. Hepa1 cells were incubated with Tg in the absence or presence of RPM (a), or Torin 1 at different concentrations (b); or incubated with Tg/TNF-α in the presence of RPM w/or w/o CQ (c), as described in the material and methods. Cell death was measured 24hrs later by LDH assays. Average cytotoxicities (% cell death) in different cell culture groups were plotted. 3 replicates/expt. group, (d) LC3B levels in different Hepa1 cell culture groups as in (c) were measured by Western blots. Cell lysates were harvested after 6 hrs of incubation with various stimuli. LC3B II band intensities were quantitated as ratios against β actin. 2 replicates/expt. group.
Representative results of 2 different experiments. *p<0.05, **p<0.01.
Rapamycin protected hepatocytes via both autophagy induction and mTORC2-Akt activation
To test whether autophagy induction played a role in the cytoprotective mechanism of RPM, we added chloroquine (CQ) together with RPM. Indeed, CQ increased Hepa1 cell death induced by Tg/TNF-α both in the absence and presence of RPM, which indicated that both basal and RPM-induced autophagy played cytoprotective roles (Fig.5c). Western blot results indicated that although Tg treatment increased cellular LC3B II levels, it inhibited autophagy flux (No further increases in LC3B II levels by CQ) in both Tg or Tg/TNF-αtreated cells. RPM partially restored autophagy flux in Tg/TNF-α-treated cells (Fig.4d). To test the role of the mTORC2-Akt signaling pathway activation in hepatocyte cytoprotection, we first measured S6K and Akt S473 phosphorylation in Hepa1 cells in response to Tg. Both kinases were activated by ER stress. RPM inhibited S6K, but spared Akt, activation, while Torin 1 inhibited both kinases (Fig.5a). Torin 1 increased LC3B II levels more consistently than RPM in these cells (Fig.5a). Inhibition of Akt phosphorylation by TCN increased hepatocyte death against Tg and abrogated RPM cytoprotective effect (Fig.5b). These results indicated that both autophagy and mTORC2-Akt activation played critical roles in the cytoprotection mechanism of RPM in vitro.
Figure 5.
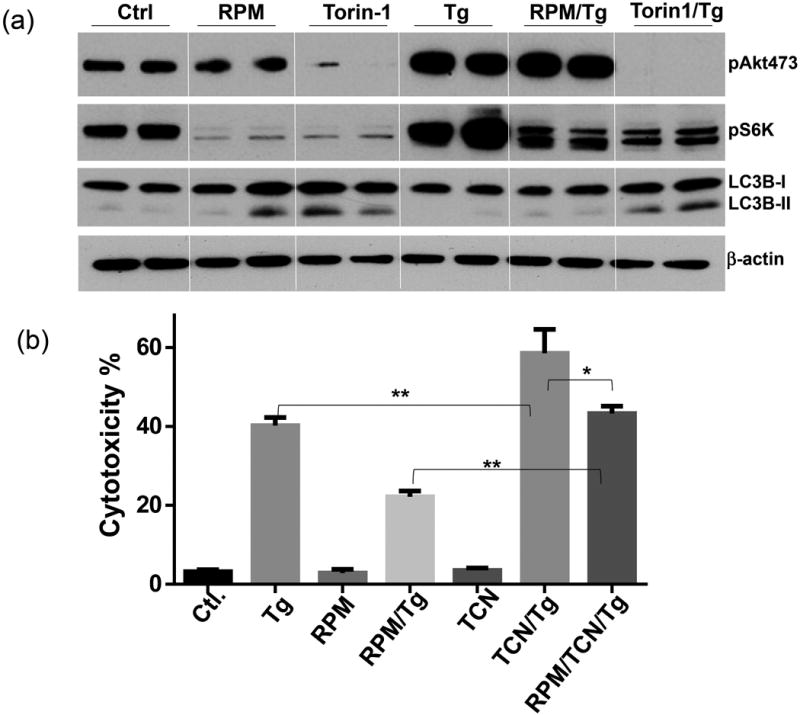
RPM protects hepatocytes via the mTORC2-Akt pathway, (a) mTOR inhibitors regulate hepatocyte mTORC2-Akt signaling pathway. Hepa1 cells were incubated with vehicle (Ctl) or RPM, or Torin 1 or Tg, or RPM/Tg, or Torin 1/Tg, as described in the material and methods. Cell lysates were collected at 6h, and analyzed by Western blots to measure levels of phosphorylated S6K, Akt (S473) and LC3B. 2 replicates/expt. group, (b) Akt inhibition abrogated cytoprotective effect of RPM. Hepa1 cells were incubated with vehicle (Ctl) or Tg, or RPM, or RPM/Tg, or TCN or RPM/TCN/Tg, as described in the material and methods. Cell death was measured at 24h by LDH assay. 3 replicates/expt. group.
Representative results of 2 different experiments. *p<0.05, **p<0.01
Discussion
As an immune-suppressive and anti-proliferative agent, RPM therapeutic effects and functional mechanisms in the pathogenesis of IRI remain controversial (1, 4, 13). Our results showed that RPM pre-treatment was able to protect livers from IRI in the presence of undiminished inflammatory immune activation by enhancing both hepatocyte autophagy, as well as the activation of mTORC2-Akt signaling pathway. Therefore, dual mTORC1 and 2 inhibitor Torin1 failed to protect livers. Inhibition of autophagy induction by 3-MA or Akt phosphorylation (downstream of mTORC2 activation) by TCN abrogated RPM liver protection effect. In vitro, RPM protected hepatocytes from both ER stress- or TNF-α-induced cell death, which was also dependent on autophagy induction and mTORC2-Akt activation. Although RPM directly targets mTORC1, it could interfere with mTORC2 assembly and activities as well when used at high concentrations or for long-term. (14). The key role of mTORC2 activation in the liver protective mechanism of RPM against IR may provide us an in vivo measurement to evaluate its therapeutic effect in IR models.
Both beneficial and detrimental effects of RPM have been shown in IRI models (15-20). Early literatures documented its T-cell-independent anti-inflammatory effects by downregulating TNF-α and decreasing neutrophil chemoattractant, in small bowl and liver models (15, 16). Its organ protective effects were also shown in kidneys and pancreases by improving microcirculation post IR (17, 21). Direct cytoprotective effects were demonstrated in cardiac infarct models and in vitro cell cultures that RPM protected cardiomyocytes against necrosis and apoptosis induced by simulated ischemia and reoxygenation (22, 23). The opening of the mitochondrial KATP channel and activation of JAK2-Stat3 signaling pathway seemed to play key roles. On the other hand, RPM treatment was shown to impair the recovery of renal function post IR, due to increased apoptosis and decreased proliferation of tubular cells (20) (24) mediated possibly by inhibitions of p70S6K (18) or Akt (19) or HO-1 repression (25). In a rat liver Tx model, recipients treated with RPM showed decreased cholangiocyte proliferation and delayed biliary recovery after liver transplantation, as a result of STAT3 inhibition (26). More recently, RPM regulation of autophagy became the focus of its role in IRI. The enhancement of autophagy by RPM treatment protected tissue parenchymal cells; while inhibitions of autophagy by either chemicals or gene KO exacerbated IRI and abrogated its protective effects (27-32).
Although autophagy is generally a cytoprotective mechanism against metabolic stress, its involvement and functions in the pathologic process of IRI were complicated with controversies (10, 33). In murine heart ischemia models, it was shown that autophagy was induced in myocardium by IR (34) via AMPK or Beclin-1-dependent mechanisms, depending on stages of the disease process (35). However, the opposite, i.e., diminished autophagy in ischemic heart tissues, has also been observed (36). An in vitro cell culture study showed that only mild ischemia up-regulated autophagy and severe ischemia trigged necrotic cell death (37), and that anoxia impaired autophagy due to mitochondrial dysfunction (38). Functionally, both protective and damaging roles of autophagy have been reported in the reperfusion cardiomyocyte death (35, 39-43). It has been hypothesized that induction of autophagy without increasing autophagy flux will causes a cytotoxic buildup of the lysosomal compartment, leading to more severe cellular damages. In such contexts, inhibitors that block autophagy induction may become cytoprotective. In livers, a role of autophagy in IRI was shown in aged mice which had decreased abilities of autophagy in hepatocytes due to loss of Atg4B (44). Activation of autophagy was also able to protect against acetaminophen-induced hepatotoxicity (45).
In our current study, we measured both LC3B II levels and autophagy flux at different stages of IR to define the impact of RPM. Thus, although LC3B II levels were increased in ischemic livers, autophagy flux was inhibited due possibly to the extended ischemia time. Autophagy flux was restored during reperfusion. RPM was able to enhanced autophagy induction at reperfusion but not ischemia stages of the disease. Our results further revealed that autophagy induction was not sufficient for RPM cytoprotective effect, as Torin1, a more potent autophagy inducer, was neither effective in protecting livers from IRI in vivo, nor hepatocytes from inflammatory cell death in vitro. We documented for the first time in livers that mTORC2 activation was a critical cytoprotective mechanism. Roles of mTORC2 in heart ischemic damages were demonstrated recently (46). Mice treated with Torin1 and Rictor siRNA adenovirus had increased myocardial damages after infarction. As the key downstream substrate of mTORC2, Akt activation is a pro-survival signal in cells against IR (47-49). Relevant to our finding that RPM treatment resulted in mTORC2 activation in IR livers, it has been documented in vitro that mTORC1 -activated S6K1 could phosphorylated Rictor to inhibit mTORC2 activities (50).
In summary, we performed a comprehensive analysis of RPM in the pathological mechanisms of liver IRI both in vivo and in vitro. Results indicate that a dual regulation of autophagy and the mTORC2-Akt signaling pathway is critical for its cytoprotective effect in liver inflammatory tissue injuries.
Materials and Methods
Mice
Male wide-type (WT) C57BL/6 mice (6-8 weeks old) were purchased from the Jackson Laboratory (Bar Harbor, ME). Animals were housed in the UCLA animal facility under specific pathogen-free conditions, and received humane care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals” prepared by the National Academy of Sciences and published by the National Institute of Health (NIH publication 86-23 revised 1985).
Model of warm liver IRI
A model of partial liver warm IRI was used (51). In brief, an atraumatic clip was used to interrupt the arterial and portal venous blood supply to the cephalad lobes of the liver for 90 min. Sham controls underwent the same procedure, but without vascular occlusion. Rapamycin (1-5mg/kg) or Torin 1(10mg/kg), or chloroquine (CQ, 60mg/kg) (Tocris Bioscience, Cambridge, UK) or 3-Methyladenine (3-MA, 30mg/kg), or Triciribine (1mg/kg) (Sigma, St. Luis, MO), were administered, i.p., 1 h prior to the onset of liver ischemia.
Mice were sacrificed after 6h of reperfusion. Serum alanine aminotransferase (sALT) levels were measured by IDEXX Laboratories. Part of liver specimens were fixed in 10% buffered formalin and embedded in paraffin. Liver sections (4μm) were stained with hematoxylin and eosin, and then analyzed blindly. The severity of liver IRI was graded blindly using Suzuki's criteria on a scale from 0 to 4 (52).
Cell cultures
Hepa 1 cells were plated in 48-well plate at 3×104/well. After o/n culture, Cells were first incubated with RPM (100nM) or Torin 1 (100-500nM) for 3 hrs. prior to the addition of Thapsigargin (Tg, 1μM). Tg-induced cell death was measured 24h later by LDH assay (Biochain Institute, Hayward, CA) with culture supernatants, according to the manufacturer's instruction. Alternatively, cells were incubated with Tg for 1h. These cells were then washed with warm culture media prior to the addition of recombinant mouse TNF-α (25ng/ml, R&D System, Minneapolis, MN). Inflammatory cell death was evaluated at 24h. For Western blot analysis, cells were harvested at 4h post either Tg or TNF-a stimulation. To measure autophagy flux, chloroquine (CQ, 50mM, Sigma Aldrich) was added 1h before the addition of RPM or Torin 1.
Quantitative RT-PCR
Total RNA (2μg) was reverse-transcribed into cDNA using SuperScriptTM III First-Strand Synthesis System (Invitrogen, Carlsbad, CA). Quantitative-PCR was performed using the DNA Engine with Chromo 4 Detector (MJ Research, Waltham, MA). In a final reaction volume of 20 μl, the following were added: 1xSuperMix (Platinum SYBR Green qPCR Kit, Invitrogen, Carlsbad, CA), cDNA and 0.5 mM of each primer. Amplification conditions were: 50°C (2 min), 95°C (5 min) followed by 50 cycles of 95 °C (15 s), 60 °C (30 s). Primers used to amplify a specific mouse gene fragments are the same as described previously (53).
Western blots
Protein was extracted from cultured cells or liver tissue with ice cold lysis buffer (1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 10% glycerol, 137mM sodium chloride, 20mM Tris, pH 7.4). Proteins (20 μg) were subjected to 12% SDS-PAGE electrophoresis and transferred to PVDF nitrocellulose membrane. Antibodies against phosphorylated or total S6K, Akt, and LC3B, β-actin (Cell Signaling Technology, Danvers, MA) were used. Membranes were probed with primary antibody (1:500-1000) in 10 ml blocking buffer overnight at 4°C. After washing, membranes were further probed with appropriate HRP-conjugated secondary antibody (1:1000) in 10 ml of blocking buffer for 1.5 h at room temperature. SuperSignal® West Pico Chemiluminescent Substrates (Thermo Fisher Scientific, Rockford, IL) were used for chemo-luminescence development. ImageJ 1.47v software was used to quantitate western blot bands.
ELISA
Cytokines (TNF-α and IL-10) in cell culture supernatants or serum were measured by ELISA, according to the manufacturer's protocols (eBioscience, San Diego, CA).
Statistical analysis
Results are shown as mean±SD. Unpaired Student t-test or one-way ANOVA test were used for comparison of two groups or more than two groups. A two-tailed P value less than 0.05 was considered to be statistically significant.
Acknowledgments
This work was supported NIH Grants R01 DK083408 (YZ) and The Dumont Research Foundation.
Abbreviations
- RPM
Rapamycin
- CQ
chloroquine
- 3-MA
3-Methyladenine
- Tx
Transplantation
- IRI
Ischemia reperfusion injury
- mTOR
mammalian (mechanical) target of Rapamycin
- mTORC1/2
mTOR complex1/2
- TLR
Toll-like-receptor
- KCs
Kupffer cells
- ALT
Alanine aminotransferase
- CQ
Chloroquine
- Tg
Thapsigargin
- LDH
Lactate dehydrogenase
- KO
Knock-out
- LPS
Lipopolysaccharide
- NPC
Non-parenchymal cell
- TNF-α
Tumor necrosis factor alpha
Footnotes
Footnote: JZ, TL and SY designed experiments and performed in vitro assays; XS and FG performed in vivo experiments; RWB and JWK revised manuscript; XQ, YZ designed, supervise the study and wrote manuscript.
All authors declare no conflict of interest.
References
- 1.Kawahara T, Asthana S, Kneteman NM. m-TOR inhibitors: what role in liver transplantation? J Hepatol. 2011;55(6):1441. doi: 10.1016/j.jhep.2011.06.015. [DOI] [PubMed] [Google Scholar]
- 2.Otto G. Liver transplantation: an appraisal of the present situation. Dig Dis. 2013;31(1):164. doi: 10.1159/000347213. [DOI] [PubMed] [Google Scholar]
- 3.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saemann MD, Haidinger M, Hecking M, Horl WH, Weichhart T. The multifunctional role of mTOR in innate immunity: implications for transplant immunity. Am J Transplant. 2009;9(12):2655. doi: 10.1111/j.1600-6143.2009.02832.x. [DOI] [PubMed] [Google Scholar]
- 5.Thoreen CC, Kang SA, Chang JW, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284(12):8023. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weichhart T, Costantino G, Poglitsch M, et al. The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity. 2008;29(4):565. doi: 10.1016/j.immuni.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 7.Schmitz F, Heit A, Dreher S, et al. Mammalian target of rapamycin (mTOR) orchestrates the defense program of innate immune cells. Eur J Immunol. 2008;38(11):2981. doi: 10.1002/eji.200838761. [DOI] [PubMed] [Google Scholar]
- 8.Inoki K, Kim J, Guan KL. AMPK and mTOR in cellular energy homeostasis and drug targets. Annu Rev Pharmacol Toxicol. 2012;52:381. doi: 10.1146/annurev-pharmtox-010611-134537. [DOI] [PubMed] [Google Scholar]
- 9.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40(2):280. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gottlieb RA, Mentzer RM. Autophagy during cardiac stress: joys and frustrations of autophagy. Annu Rev Physiol. 2010;72:45. doi: 10.1146/annurev-physiol-021909-135757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murrow L, Debnath J. Autophagy as a stress-response and quality-control mechanism: implications for cell injury and human disease. Annu Rev Pathol. 2013;8:105. doi: 10.1146/annurev-pathol-020712-163918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fang H, Liu A, Dahmen U, Dirsch O. Dual role of chloroquine in liver ischemia reperfusion injury: reduction of liver damage in early phase, but aggravation in late phase. Cell Death Dis. 2013;4:e694. doi: 10.1038/cddis.2013.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hwang SK, Kim HH. The functions of mTOR in ischemic diseases. BMB Rep. 2011;44(8):506. doi: 10.5483/bmbrep.2011.44.8.506. [DOI] [PubMed] [Google Scholar]
- 14.Sarbassov DD, Ali SM, Sengupta S, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22(2):159. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 15.Puglisi RN, Strande L, Santos M, Schulte G, Hewitt CW, Whalen TV. Beneficial effects of cyclosporine and rapamycin in small bowel ischemic injury. J Surg Res. 1996;65(2):115. doi: 10.1006/jsre.1996.0352. [DOI] [PubMed] [Google Scholar]
- 16.Matsuda T, Yamaguchi Y, Matsumura F, et al. Immunosuppressants decrease neutrophil chemoattractant and attenuate ischemia/reperfusion injury of the liver in rats. J Trauma. 1998;44(3):475. doi: 10.1097/00005373-199803000-00009. [DOI] [PubMed] [Google Scholar]
- 17.Inman SR, Davis NA, Olson KM, Lukaszek VA, McKinley MR, Seminerio JL. Rapamycin preserves renal function compared with cyclosporine A after ischemia/reperfusion injury. Urology. 2003;62(4):750. doi: 10.1016/s0090-4295(03)00475-8. [DOI] [PubMed] [Google Scholar]
- 18.Lieberthal W, Fuhro R, Andry CC, et al. Rapamycin impairs recovery from acute renal failure: role of cell-cycle arrest and apoptosis of tubular cells. Am J Physiol Renal Physiol. 2001;281(4):F693. doi: 10.1152/ajprenal.2001.281.4.F693. [DOI] [PubMed] [Google Scholar]
- 19.Loverre A, Ditonno P, Crovace A, et al. Ischemia-reperfusion induces glomerular and tubular activation of proinflammatory and antiapoptotic pathways: differential modulation by rapamycin. J Am Soc Nephrol. 2004;15(10):2675. doi: 10.1097/01.ASN.0000139932.00971.E4. [DOI] [PubMed] [Google Scholar]
- 20.Lui SL, Chan KW, Tsang R, Yung S, Lai KN, Chan TM. Effect of rapamycin on renal ischemia-reperfusion injury in mice. Transpl Int. 2006;19(10):834. doi: 10.1111/j.1432-2277.2006.00361.x. [DOI] [PubMed] [Google Scholar]
- 21.Serr F, Lauer H, Armann B, et al. Sirolimus improves early microcirculation, but impairs regeneration after pancreatic ischemia-reperfusion injury. Am J Transplant. 2007;7(1):48. doi: 10.1111/j.1600-6143.2006.01589.x. [DOI] [PubMed] [Google Scholar]
- 22.Khan S, Salloum F, Das A, Xi L, Vetrovec GW, Kukreja RC. Rapamycin confers preconditioning-like protection against ischemia-reperfusion injury in isolated mouse heart and cardiomyocytes. J Mol Cell Cardiol. 2006;41(2):256. doi: 10.1016/j.yjmcc.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 23.Das A, Salloum FN, Durrant D, Ockaili R, Kukreja RC. Rapamycin protects against myocardial ischemia-reperfusion injury through JAK2-STAT3 signaling pathway. J Mol Cell Cardiol. 2012;53(6):858. doi: 10.1016/j.yjmcc.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fuller TF, Freise CE, Serkova N, Niemann CU, Olson JL, Feng S. Sirolimus delays recovery of rat kidney transplants after ischemia-reperfusion injury. Transplantation. 2003;76(11):1594. doi: 10.1097/01.TP.0000095897.38634.30. [DOI] [PubMed] [Google Scholar]
- 25.Gonçalves GM, Cenedeze MA, Feitoza CQ, et al. The role of heme oxygenase 1 in rapamycin-induced renal dysfunction after ischemia and reperfusion injury. Kidney Int. 2006;70(10):1742. doi: 10.1038/sj.ki.5001893. [DOI] [PubMed] [Google Scholar]
- 26.Chen LP, Zhang QH, Chen G, Qian YY, Shi BY, Dong JH. Rapamycin inhibits cholangiocyte regeneration by blocking interleukin-6-induced activation of signal transducer and activator of transcription 3 after liver transplantation. Liver Transpl. 2010;16(2):204. doi: 10.1002/lt.21985. [DOI] [PubMed] [Google Scholar]
- 27.Carloni S, Buonocore G, Balduini W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol Dis. 2008;32(3):329. doi: 10.1016/j.nbd.2008.07.022. [DOI] [PubMed] [Google Scholar]
- 28.Carloni S, Buonocore G, Longini M, Proietti F, Balduini W. Inhibition of rapamycin-induced autophagy causes necrotic cell death associated with Bax/Bad mitochondrial translocation. Neuroscience. 2012;203:160. doi: 10.1016/j.neuroscience.2011.12.021. [DOI] [PubMed] [Google Scholar]
- 29.Jiang M, Wei Q, Dong G, Komatsu M, Su Y, Dong Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012;82(12):1271. doi: 10.1038/ki.2012.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakagawa S, Nishihara K, Inui KI, Masuda S. Involvement of autophagy in the pharmacological effects of the mTOR inhibitor everolimus in acute kidney injury. Eur J Pharmacol. 2012;696(1-3):143. doi: 10.1016/j.ejphar.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 31.Kang JW, Cho HI, Lee SM. Melatonin Inhibits mTOR-Dependent Autophagy during Liver Ischemia/Reperfusion. Cell Physiol Biochem. 2014;33(1):23. doi: 10.1159/000356647. [DOI] [PubMed] [Google Scholar]
- 32.Li Q, Zhang T, Wang J, et al. Rapamycin attenuates mitochondrial dysfunction via activation of mitophagy in experimental ischemic stroke. Biochem Biophys Res Commun. 2014;444(2):182. doi: 10.1016/j.bbrc.2014.01.032. [DOI] [PubMed] [Google Scholar]
- 33.Przyklenk K, Dong Y, Undyala VV, Whittaker P. Autophagy as a therapeutic target for ischaemia/reperfusion injury? Concepts, controversies, and challenges. Cardiovasc Res. 2012;94(2):197. doi: 10.1093/cvr/cvr358. [DOI] [PubMed] [Google Scholar]
- 34.Yan L, Vatner DE, Kim SJ, et al. Autophagy in chronically ischemic myocardium. Proc Natl Acad Sci U SA. 2005;102(39):13807. doi: 10.1073/pnas.0506843102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsui Y, Takagi H, Qu X, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100(6):914. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 36.French CJ, Taatjes DJ, Sobel BE. Autophagy in myocardium of murine hearts subjected to ischemia followed by reperfusion. Histochem Cell Biol. 2010;134(5):519. doi: 10.1007/s00418-010-0748-0. [DOI] [PubMed] [Google Scholar]
- 37.Loos B, Genade S, Ellis B, Lochner A, Engelbrecht AM. At the core of survival: autophagy delays the onset of both apoptotic and necrotic cell death in a model of ischemic cell injury. Exp Cell Res. 2011;317(10):1437. doi: 10.1016/j.yexcr.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 38.Kim JS, Nitta T, Mohuczy D, et al. Impaired autophagy: A mechanism of mitochondrial dysfunction in anoxic rat hepatocytes. Hepatology. 2008;47(5):1725. doi: 10.1002/hep.22187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hamacher-Brady A, Brady NR, Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem. 2006;281(40):29776. doi: 10.1074/jbc.M603783200. [DOI] [PubMed] [Google Scholar]
- 40.Gurusamy N, Lekli I, Gorbunov NV, Gherghiceanu M, Popescu LM, Das DK. Cardioprotection by adaptation to ischaemia augments autophagy in association with BAG-1 protein. J Cell Mol Med. 2009;13(2):373. doi: 10.1111/j.1582-4934.2008.00495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hariharan N, Zhai P, Sadoshima J. Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid Redox Signal. 2011;14(11):2179. doi: 10.1089/ars.2010.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhai P, Sciarretta S, Galeotti J, Volpe M, Sadoshima J. Differential roles of GSK-3beta during myocardial ischemia and ischemia/reperfusion. Circ Res. 2011;109(5):502. doi: 10.1161/CIRCRESAHA.111.249532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanemura M, Ohmura Y, Deguchi T, et al. Rapamycin causes upregulation of autophagy and impairs islets function both in vitro and in vivo. Am J Transplant. 2012;12(1):102. doi: 10.1111/j.1600-6143.2011.03771.x. [DOI] [PubMed] [Google Scholar]
- 44.Wang JH, Ahn IS, Fischer TD, et al. Autophagy suppresses age-dependent ischemia and reperfusion injury in livers of mice. Gastroenterology. 2011;141(6):2188. doi: 10.1053/j.gastro.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ni HM, Bockus A, Boggess N, Jaeschke H, Ding WX. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology. 2012;55(1):222. doi: 10.1002/hep.24690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Volkers M, Konstandin MH, Doroudgar S, et al. Mechanistic target of rapamycin complex 2 protects the heart from ischemic damage. Circulation. 2013;128(19):2132. doi: 10.1161/CIRCULATIONAHA.113.003638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vasudevan KM, Garraway LA. AKT signaling in physiology and disease. Curr Top Microbiol Immunol. 2010;347:105. doi: 10.1007/82_2010_66. [DOI] [PubMed] [Google Scholar]
- 48.Zhu M, Feng J, Lucchinetti E, et al. Ischemic postconditioning protects remodeled myocardium via the PI3K-PKB/Akt reperfusion injury salvage kinase pathway. Cardiovasc Res. 2006;72(1):152. doi: 10.1016/j.cardiores.2006.06.027. [DOI] [PubMed] [Google Scholar]
- 49.Kamo N, Ke B, Busuttil RW, Kupiec-Weglinski JW. PTEN-mediated Akt/beta-catenin/Foxo1 signaling regulates innate immune responses in mouse liver ischemia/reperfusion injury. Hepatology. 2013;57(1):289. doi: 10.1002/hep.25958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Julien LA, Carriere A, Moreau J, Roux PP. mTORC1-activated S6K1 phosphorylates Rictor on threonine 1135 and regulates mTORC2 signaling. Mol Cell Biol. 2010;30(4):908. doi: 10.1128/MCB.00601-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shen XD, Ke B, Zhai Y, et al. CD154-CD40 T-cell costimulation pathway is required in the mechanism of hepatic ischemia/reperfusion injury, and its blockade facilitates and depends on heme oxygenase-1 mediated cytoprotection. Transplantation. 2002;74(3):315. doi: 10.1097/00007890-200208150-00005. [DOI] [PubMed] [Google Scholar]
- 52.Suzuki S, Toledo-Pereyra LH, Rodriguez FJ, Cejalvo D. Neutrophil infiltration as an important factor in liver ischemia and reperfusion injury. Modulating effects of FK506 and cyclosporine. Transplantation. 1993;55(6):1265. doi: 10.1097/00007890-199306000-00011. [DOI] [PubMed] [Google Scholar]
- 53.Zhai Y, Shen XD, Gao F, et al. CXCL10 regulates liver innate immune response against ischemia and reperfusion injury. Hepatology. 2008;47(1):207. doi: 10.1002/hep.21986. [DOI] [PubMed] [Google Scholar]