

Abstract
Thermostability is an important property of enzymes utilized for practical applications because it allows long-term storage and use as catalysts. In this study, we constructed an error-prone strain of the thermophile Geobacillus kaustophilus HTA426 and investigated thermoadaptation-directed enzyme evolution using the strain. A mutation frequency assay using the antibiotics rifampin and streptomycin revealed that G. kaustophilus had substantially higher mutability than Escherichia coli and Bacillus subtilis. The predominant mutations in G. kaustophilus were A · T→G · C and C · G→T · A transitions, implying that the high mutability of G. kaustophilus was attributable in part to high-temperature-associated DNA damage during growth. Among the genes that may be involved in DNA repair in G. kaustophilus, deletions of the mutSL, mutY, ung, and mfd genes markedly enhanced mutability. These genes were subsequently deleted to construct an error-prone thermophile that showed much higher (700- to 9,000-fold) mutability than the parent strain. The error-prone strain was auxotrophic for uracil owing to the fact that the strain was deficient in the intrinsic pyrF gene. Although the strain harboring Bacillus subtilis pyrF was also essentially auxotrophic, cells became prototrophic after 2 days of culture under uracil starvation, generating B. subtilis PyrF variants with an enhanced half-denaturation temperature of >10°C. These data suggest that this error-prone strain is a promising host for thermoadaptation-directed evolution to generate thermostable variants from thermolabile enzymes.
INTRODUCTION
Enzymes catalyze numerous reactions that are difficult to perform using chemical catalysts and are commercially utilized in industry (1). The starch industry is a good example; it has a great demand for α-amylase, β-amylase, glucoamylase, pullulanase, and glucose isomerase for producing glucose and its related sugars. In addition, biosensors for monitoring of human blood glucose levels use glucose oxidase or glucose dehydrogenase. Recently developed detergents also contain protease, α-amylase, β-amylase, lipase, and/or cellulase. Thus, enzymes have potential for commercial use, but many are not practical despite possessing catalytic activities suitable for use. One reason is enzyme thermolability, which hinders the prolonged use of enzymes as catalysts and their long-term storage at around room temperature. Although enzymes identified in extreme thermophiles show excellent thermostability, they generally show low activity at moderate temperatures and accordingly possess low utility in applications that require efficient enzymatic activity at moderate temperatures (e.g., as biosensors and in chemical production and medical fields). Therefore, thermostability enhancement of thermolabile enzymes while maintaining catalytic activities at high levels at moderate temperatures is an important approach to expanding the commercial use of enzymes.
Thermostability enhancement has been achieved by two main approaches or their combination (2–4). One approach uses an in silico rational design to predict enzyme variants with enhanced thermostability (3). This approach requires three-dimensional structure information for target enzymes but allows the rapid prediction of candidates. The other approach uses in vitro random mutations to construct an enzyme variant library from which desired variants are screened. This approach is proven but may require in vitro enzymatic characterization of numerous enzyme variants (4). To facilitate the screening process, thermophiles have been used as library hosts because they allow in vivo enzymatic characterization at high temperatures without cell death. Examples are identification of thermoadaptive variants of Escherichia coli hygromycin B phosphotransferase (5), Streptoalloteichus hindustanus bleomycin-binding protein (6), Geobacillus stearothermophilus α-galactosidase (7), Saccharomyces cerevisiae 3-isopropylmalate dehydrogenase (8), and Staphylococcus aureus kanamycin nucleotidyltransferase (9–11) in Thermus thermophilus and/or G. stearothermophilus. Intriguingly, some of these variants have been generated, not via in vitro random mutations, but via spontaneous mutations in thermophiles, termed thermoadaptation-directed enzyme evolution (5, 7–9).
Spontaneous mutations primarily occur through formation of base-base mismatches caused by replication errors and DNA damage, such as deamination, 7,8-dihydro-8-oxoguanine (8-oxoG), and depurination lesions (12, 13). Deamination of adenine produces hypoxanthine and causes A · T→G · C transitions (14, 15), whereas deamination of cytosine and 5-methylcytosine produces U · G and T · G mismatches, respectively, leading to C · G→T · A transitions (12, 16–18). Oxidative damage to guanine produces the aberrant base 8-oxoG, which causes the C · G→A · T transversion because of the ability to form base pairs with adenine and cytosine (19, 20). The 8-oxoG lesion is also observed in the nucleotide pool as 8-oxo-dGTP, from which 8-oxoG can be misincorporated in opposing template adenines, causing the A · T→C · G transversion (20). Replication errors and depurination events can potentially cause any transversion or transition.
Because of the deleterious effects of mutations, base-base mismatches are corrected by several DNA repair systems. In E. coli, mismatches arising from replication errors are corrected by methyl-dependent mismatch repair that involves MutS, MutL, and MutH proteins (21). Although most organisms lack MutH homologs, MutL from species that lack MutH harbors a latent endonuclease activity similar to that of MutH (22), suggesting a MutH-independent mechanism (23). Mismatches arising from DNA damage are commonly corrected through base excision repair pathways, which are initiated by DNA glycosylases that excise damaged bases, such as Ung (uracil-DNA glycosylase) for uracil excision (17, 24), alkyladenine DNA glycosylase for hypoxanthine excision (16), MutM for 8-oxoG excision from C · 8-oxoG mismatches (20, 25, 26), and MutY for adenine excision from A · 8-oxoG mismatches (20, 25, 26). In addition, MutT homologs and transcription-coupled repair systems have been identified in several organisms. MutT degrades 8-oxo-dGTP to prevent 8-oxoG incorporation during DNA replication (20), and transcription-coupled repair corrects damage on the transcribed DNA strand, with preference over nontranscribed strands and nontranscribed double-stranded DNA (27). In E. coli, transcription-coupled repair is mediated by a transcription repair coupling factor encoded by mfd, which facilitates the removal of RNA polymerases stalled at DNA lesions that block transcriptional elongation and the recruitment of DNA repair proteins to lesions on transcribed strands (27, 28).
Recently, we have studied genetic tools for the aerobic, Gram-positive, Bacillus-related thermophile Geobacillus kaustophilus HTA426 with the aim of using the strain for biotechnological applications and biological studies of the genus Geobacillus (29–32). In these studies, HTA426 cells efficiently produced descendants constitutively resistant to the antibiotics rifampin (Rif) and streptomycin (Str). Because Rif and Str resistance (Rifr and Strr) often arise from spontaneous mutations in certain genes (33–38), this observation suggests that the HTA426 strain has considerable mutability. Thus, in the present study, we evaluated G. kaustophilus mutability in detail and constructed an error-prone strain from the thermophile. We further investigated the generation of enzyme variants that are more thermostable than the parent enzyme using the error-prone strain.
MATERIALS AND METHODS
Bacterial strains and media.
Table 1 summarizes G. kaustophilus strains. G. kaustophilus MK242 was constructed from strain MK93 (29) using the same procedure as for construction of MK244 from MK72 (31). If not otherwise specified, G. kaustophilus strains were grown at 60°C in Luria-Bertani (LB) medium and minimal medium (MM). MM consisted of K2SO4 (0.3 g liter−1), Na2HPO4 · 12H2O (2.5 g liter−1), NH4Cl (1 g liter−1), MgSO4 (0.4 g liter−1), MnCl2 · 4H2O (3 mg liter−1), CaCl2 · 2H2O (5 mg liter−1), FeCl3 · ·6H2O (7 mg liter−1), 0.1% trace element solution (39), 10 mM Tris-HCl (pH 7.0), d-glucose (10 g liter−1), and Casamino Acids (1 g liter−1; Becton, Dickinson and Company, Franklin Lakes, NJ). Kanamycin (5 mg liter−1) and uracil (10 mg liter−1) were added when necessary. E. coli DH5α (TaKaRa Bio, Otsu, Japan) and pCR4Blunto-TOPO (Life Technologies, Rockville, MD) were used for DNA manipulations. E. coli BL21(DE3) and pET-16b (Merck KGaA, Darmstadt, Germany) were used for recombinant-protein production. E. coli cells were grown at 37°C in LB medium. Ampicillin (50 mg liter−1) was added when necessary. Bacillus subtilis 168 was obtained from the Bacillus Genetic Stock Center (Columbus, OH) and grown in LB medium at 37°C.
TABLE 1.
G. kaustophilus strains
| Strain | Relevant descriptiona | Doubling time (min)b |
|---|---|---|
| MK242 | Derivative of the wild-type strain HTA426; ΔpyrF ΔpyrR ΔhsdM1S1R1 Δ(mcrB1-mcrB2-hsdM2S2R2-mrr) GK0707::Pgk704-bgaB | ND |
| MK480 | Derivative of the strain MK242; ΔpyrF ΔpyrR ΔhsdM1S1R1 Δ(mcrB1-mcrB2-hsdM2S2R2-mrr) GK0707::Pgk704-bgaB Δ(mutS-mutL) ΔmutY Δung Δmfd | ND |
| MK242p70 | MK242 derivative; trpE::pGKE70 | 20.2 ± 0.5 |
| MK480p70 | MK480 derivative; trpE::pGKE70 | 21.8 ± 0.5 |
| ΔmutSLp70 | MK242 derivative; Δ(mutS-mutL) trpE::pGKE70 | 20.4 ± 0.8 |
| ΔmutMp70 | MK242 derivative; ΔmutM trpE::pGKE70 | 21.6 ± 0.8 |
| ΔmutYp70 | MK242 derivative; ΔmutY trpE::pGKE70 | 20.0 ± 1.0 |
| ΔmutTp70 | MK242 derivative; ΔmutT trpE::pGKE70 | 21.9 ± 1.8 |
| Δungp70 | MK242 derivative; Δung trpE::pGKE70 | 20.4 ± 0.7 |
| Δmfdp70 | MK242 derivative; Δmfd trpE::pGKE70 | 22.7 ± 1.6 |
| ΔmutSLcm | MK242 derivative; Δ(mutS-mutL) trpE::pGKE70-mutSL | 21.9 ± 0.3 |
| ΔmutMcm | MK242 derivative; ΔmutM trpE::pGKE70-mutM | 22.6 ± 1.5 |
| ΔmutYcm | MK242 derivative; ΔmutY trpE::pGKE70-mutY | 22.9 ± 1.0 |
| ΔmutTcm | MK242 derivative; ΔmutT trpE::pGKE70-mutT | 22.9 ± 0.7 |
| Δungcm | MK242 derivative; Δung trpE::pGKE70-ung | 20.5 ± 0.9 |
| Δmfdcm | MK242 derivative; Δmfd trpE::pGKE70-mfd | 20.7 ± 0.7 |
| MK242BSpyrF | MK242 derivative; trpE::pGKE70-BSpyrFwt | ND |
| MK480BSpyrF | MK480 derivative; trpE::pGKE70-BSpyrFwt | ND |
Δ(mutS-mutL) encodes codons mutS1–106 fused with mutL534–630. ΔmutM, ΔmutY, ΔmutT, Δung, and Δmfd are deleted in codons mutM11–255, mutY14–347, mutT13–150, ung16–218, and mfd33–1060, respectively. trpE is an internal gene for anthranilate synthase component I. The strains MK242 and MK480 lack the native pyrF gene encoding orotidine-5′-phosphate decarboxylase, but MK242BSpyrF and MK480BSpyrF harbor BSpyrFwt, which encodes orotidine-5′-phosphate decarboxylase in B. subtilis.
G. kaustophilus strains were cultured at 60°C in LB medium with monitoring of OD600. The binary logarithm of OD600 data during the logarithmic growth phase was plotted against incubation time to calculate doubling times. The data are presented as means ± standard deviations (SD) (n = 3 to 16). ND, not determined.
Construction of pGKE25 derivatives.
Primer sequences are listed in Table 2. The plasmid constructs pΔmutSL, pΔmutM, pΔmutY, pΔmutT, pΔung, and pΔmfd were used for in-frame deletions of the mutS-mutL, mutM, mutY, mutT, ung, and mfd genes, respectively, in G. kaustophilus MK242. For pΔmutSL construction, the upstream region (1.5 kb) of mutS (GK1306) was amplified using primers 1306upF and 1306upR, and the mutL (GK1307) downstream region (1.5 kb) was amplified using primers 1307dwF and 1307dwR. The two amplified fragments were combined using overlap extension PCR and were cloned into the BamHI site of pGKE25 (31) to yield pΔmutSL. In addition, ΔmutM (GK2728), ΔmutY (GK0463), ΔmutT (GK3067), Δung (GK3421), and Δmfd (GK0048) fragments were generated using appropriate primers and cloned into pGKE25 to yield pΔmutM, pΔmutY, pΔmutT, pΔung, and pΔmfd, respectively.
TABLE 2.
Primers used in this study
| Primer | Sequence (5′-3′) | Target region | Underlined site |
|---|---|---|---|
| 0048F | GCCGCATGCTTTCGTTGCATCGCTATTTAG | mfd full sequence | SphI |
| 0048R | GCCGGATCCTTATGCCGTCACCGACTTCTC | mfd full sequence | BamHI |
| 0048upF | GCCGGATCCCTTCGTGAAGGCGGGCGCAC | mfd upstream | BamHI |
| 0048upR | GACCATTTCCTCACGCAGCCCGGCGACAAGCTG | mfd upstream | |
| 0048dwF | CAGCTTGTCGCCGGGCTGCGTGAGGAAATGGTC | mfd downstream | |
| 0048dwR | GGCGGATCCCGACGATGCCATAAATCGGG | mfd downstream | BamHI |
| 0463F | GCCCCTGCAGGCACAAGAGAAACAGAGCG | mutY full sequence | Sse8387I |
| 0463R | GCCGGATCCTTAATCGGGGCGGCGTAC | mutY full sequence | BamHI |
| 0463upF | GCCGGATCCCTTGGATTTGGCGGATGAAC | mutY upstream | BamHI |
| 0463upR | CCAAACACGTTGATGAAACTCGCGCGCTGG | mutY upstream | |
| 0463dwF | CCAGCGCGCGAGTTTCATCAACGTGTTTGG | mutY downstream | |
| 0463dwR | GCCGGATCCACATTCGTCCATGCGTGCTC | mutY downstream | BamHI |
| 1306F | GGTGTGTACGATGGCATAATCCCCCTCCTGTC | mutSL full sequence | |
| 1306R | GACAGGAGGGGGATTATGCCATCGTACACACC | Pgk704 fragment | |
| 1306upF | GCCGGATCCTCCACCAAAACGAAAAAGTG | mutS upstream | BamHI |
| 1306upR | CCACGTCGGATGGGAGAGCTGCACAACTTC | mutS upstream | |
| 1307R | GCCAGATCTCTACATCACCCGTTTAAATAG | mutSL full sequence | BglII |
| 1307dwF | GAAGTTGTGCAGCTCTCCCATCCGACGTGG | mutS downstream | |
| 1307dwR | GCCGGATCCATACCGCGTTTTCCAATAGG | mutS downstream | BamHI |
| 2204F | GCCGAATTCTGACGAATCGCCGTGGGCGC | trpE internal sequence | EcoRI |
| 2204R | GGCGAATTCCTGTCCGGATGGCGATGCAC | trpE internal sequence | EcoRI |
| 2728F | GCCGCATGCCGGAATTGCCGGAGGTG | mutM full sequence | SphI |
| 2728R | GCCGGATCCCTAGCGCTGGCAGCGCGGGC | mutM full sequence | BamHI |
| 2728upF | GCCGGATCCGCCGGAGTGAAAGTGGATAC | mutM upstream | BamHI |
| 2728upR | GACCGTTTTTTCAATGATCGTTTCCACCTC | mutM upstream | |
| 2728dwF | GAGGTGGAAACGATCATTGAAAAAACGGTC | mutM downstream | |
| 2728dwR | GCCGGATCCTAATCGCAGAATGCGGATTC | mutM downstream | BamHI |
| 3067F | GCCCCTGCAGGCGTGAACGAATTGCAACGGG | mutT full sequence | Sse8387I |
| 3067R | GCCGGATCCTCAGCTCGGATCGAGCCG | mutT full sequence | BamHI |
| 3067upF | GCCGGATCCAATACATGTGGCGCCGACAG | mutT upstream | BamHI |
| 3067upR | GAGCCGATATGAAAGGTTTGTCACCCGTTG | mutT upstream | |
| 3067dwF | CAACGGGTGACAAACCTTTCATATCGGCTC | mutT downstream | |
| 3067dwR | GCCGGATCCTTCGCCTTGCATACTTCCTG | mutT downstream | BamHI |
| 3421F | GCCGCATGCCGATTCTCAAAAACGAC | ung full sequence | SphI |
| 3421R | GCCGGATCCTCATTCAGCGCGGGCGCCGATG | ung full sequence | BamHI |
| 3421upF | GCCGGATCCCTTGGATGTACCATGCTTTG | ung upstream | BamHI |
| 3421upR | GTTCTCGATTTGCCACTCCTCTTCAAGCAG | ung upstream | |
| 3421dwF | CTGCTTGAAGAGGAGTGGCAAATCGAGAAC | ung downstream | |
| 3421dwR | GCCGGATCCTTTCATCCGATGCGACGTCC | ung downstream | BamHI |
| P704F | GCCAAGCTTTTTCTTTTTCCTCCTTTGTTATC | Pgk704 fragment | HindIII |
| pyrFF | GTACATATGCGAAACAACCTGCCCATC | BSpyrFwt sequence | NdeI |
| pyrFR | GCCGGATCCTTAAGATTTGATTCCCTCCC | BSpyrFwt sequence | BamHI |
Construction of pGKE70 derivatives.
The plasmid pGKE70 (Fig. 1) was constructed to integrate genes into the trpE locus (GK2204) and to force gene expression under the control of the Pgk704 promoter (29). To construct pGKE70, the Pgk704 fragment was excised from pGAM48 (29) and the trpE internal sequence (positions 157 to 1360) was amplified using primers 2204F and 2204R. The Pgk704 fragment was cloned between the HindIII and SphI sites of the plasmid pTK19 (30), and the trpE fragment was then cloned in the MunI site to obtain pGKE70. To construct pGKE70-mutSL, the mutS-mutL and Pgk704 fragments were amplified using primers 1306F and 1307R and P704F and 1306R, respectively. These two fragments were combined to create the Pgk704-mutSL fragment, which was trimmed using HindIII and BglII and cloned between the HindIII and BamHI sites of pGKE70 to generate pGKE70-mutSL. In addition, mutM, ung, and mfd were amplified and cloned between SphI and BamHI sites of pGKE70 to generate pGKE70-mutM, pGKE70-ung, and pGKE70-mfd, respectively. mutY and mutT were amplified and trimmed using Sse8387I and BamHI and subsequently cloned between the PstI and BamHI sites of pGKE70 to generate pGKE70-mutY and pGKE70-mutT, respectively. The gene encoding orotidine-5′-phosphate decarboxylase of B. subtilis (BSpyrFwt) was amplified from the strain 168 chromosome and subcloned between the SphI and BamHI sites of pGKE70 to generate pGKE70-BSpyrFwt.
FIG 1.
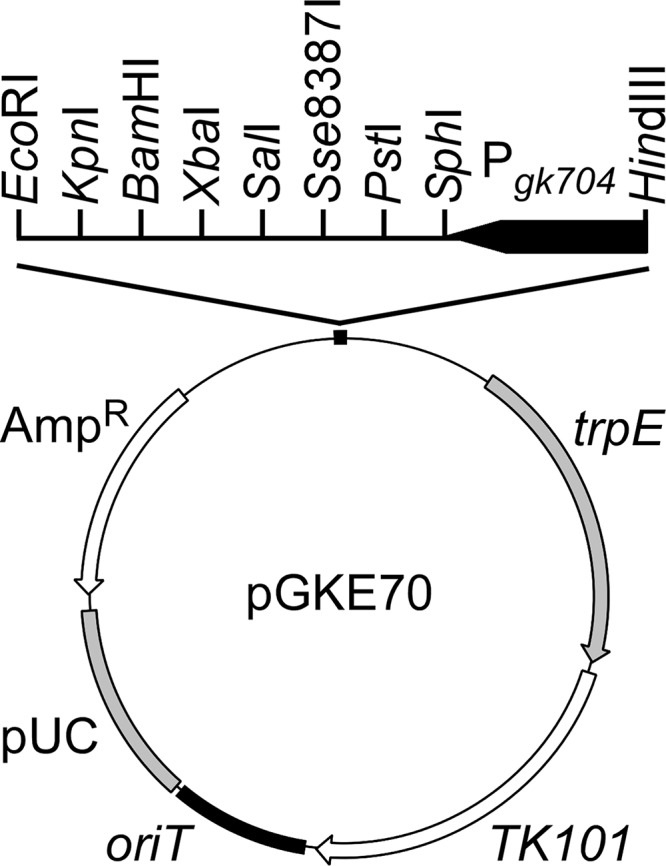
pGKE70 structure. pGKE70 was constructed to integrate genes into the trpE locus and to force gene expression under the control of the Pgk704 promoter. AmpR, ampicillin resistance gene; pUC, pUC replicon; oriT, conjugative-transfer origin; TK101, thermostable kanamycin nucleotidyltransferase gene; Pgk704, the Pgk704 promoter functional in G. kaustophilus (29); and trpE, an internal and defective gene for anthranilate synthase component I of strain HTA426. Restriction enzyme sites unique to multiple cloning sites are also indicated.
Plasmid introduction into G. kaustophilus.
Derivatives of pGKE25 and pGKE70 were introduced into G. kaustophilus cells using ternary conjugative transfer from E. coli DH5α (31). Transconjugants integrating pGKE25 derivatives in the chromosome were selected using uracil prototrophy. Transconjugants integrating pGKE70 derivatives at the trpE locus were selected using kanamycin resistance and tryptophan auxotrophy.
Construction of G. kaustophilus mutants deficient in DNA repair genes.
DNA repair genes in G. kaustophilus MK242 were deleted by two-step reciprocal crossovers using pyrF-based counterselection (30) with plasmids pΔmutSL, pΔmutM, pΔmutY, pΔmutT, pΔung, and pΔmfd. The mutants were transformed with pGKE70, yielding the ΔmutSLp70, ΔmutMp70, ΔmutYp70, ΔmutTp70, Δungp70, and Δmfdp70 mutants. Strain MK480 was constructed by simultaneous deletions of mutSL, mutY, ung, and mfd genes in MK242. Strains MK242 and MK480 were also transformed with pGKE70 to generate strains MK242p70 and MK480p70, respectively. For gene complementation, ΔmutSL, ΔmutM, ΔmutY, ΔmutT, Δung, and Δmfd mutants were transformed with plasmids pGKE70-mutSL, pGKE70-mutM, pGKE70-mutY, pGKE70-mutT, pGKE70-ung, and pGKE70-mfd to generate the ΔmutSLcm, ΔmutMcm, ΔmutYcm, ΔmutTcm, Δungcm, and Δmfdcm mutants, respectively.
Southern blotting.
Total DNA was digested using restriction endonucleases and separated on a 0.8% (wt/vol) agarose gel by electrophoresis. DNA was transferred to a nylon membrane and hybridized with digoxigenin (DIG)-labeled DNA probes that were synthesized using a PCR DIG Probe synthesis kit (Roche, Basel, Switzerland). Hybridized DNA was detected by the chromogenic method using a DIG Nucleic Acid Detection Kit (Roche).
Mutation frequency assay.
G. kaustophilus, E. coli, and B. subtilis strains were cultured overnight in liquid LB medium and mixed with glycerol to a final concentration of 20%. The mixture was divided into aliquots and stored at −80°C until use. Glycerol stocks were thawed, and an aliquot (106 cells containing <1 Rifr and Strr cell stochastically) was inoculated into liquid LB medium (20 ml) in an Erlenmeyer flask (100 ml) with a silicon plug. The flask was incubated with shaking at 180 rpm, and the optical density at 600 nm (OD600) was monitored using an OD-MonitorA instrument (Taitec, Saitama, Japan). On reaching early stationary phase (2 h after the OD600 reached 1.0), cells were spread on LB plates containing efficacious Rif (10 and 50 mg liter−1 for G. kaustophilus and B. subtilis; 50 and 250 mg liter−1 for E. coli) and Str (10 and 50 mg liter−1 for G. kaustophilus and E. coli; 500 and 3,000 mg liter−1 for B. subtilis). Cells were also spread on LB plates without Rif or Str to determine viable-cell concentrations. After subsequent incubation for 24 h, grown colonies were counted, and the numbers of Rifr or Strr cells per 109 viable cells were determined.
Generation of BSpyrFe1 and BSpyrFe2.
G. kaustophilus MK242 and MK480 were transformed with pGKE70-BSpyrFwt to generate MK242BSpyrF and MK480BSpyrF, respectively. The strains were cultivated in liquid MM (without uracil) at 60°C for 24 h, and then the grown cells were cultivated at 65°C for 24 h. Cells were spread on MM plates and incubated at 65°C to isolate single clones. From the clones, BSpyrFwt genes were amplified and sequenced to identify the mutant genes BSpyrFe1 and BSpyrFe2.
Preparation of BSpyrFwt, BSpyrFe1, and BSpyrFe2 proteins.
The BSpyrFwt, BSpyrFe1, and BSpyrFe2 genes were amplified using primers pyrFF and pyrFR and cloned in pCR4Blunt-TOPO. After sequence checking, the genes were subcloned between the NdeI and BamHI sites of pET-16b. Using the resulting plasmids, E. coli BL21(DE3) was transformed and cultured at 37°C in liquid LB medium containing 1% lactose. After 6 h of incubation, the cells were harvested and stored at −80°C until use. All of the subsequent operations were performed at 4°C. Cells were suspended in buffer (20 mM sodium phosphate, 0.5 M NaCl, pH 7.4), followed by sonication. The cell lysates were clarified by centrifugation (18,000 × g for 10 min at 4°C) and applied to a Talon column (0.2 ml; TaKaRa Bio) equipped with the same buffer. The column was washed with buffer containing 20 mM imidazole (2 ml) followed by 50 mM imidazole (0.5 ml). Recombinant proteins were subsequently eluted with buffer containing 200 mM imidazole and dialyzed against 10 mM sodium phosphate (pH 7.0) with 0.5 M NaCl. The protein purity was evaluated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
Thermostability assay of BSpyrFwt, BSpyrFe1, and BSpyrFe2 proteins.
For analysis of thermal denaturation curves, protein solutions (20 μl; 0.4 g liter−1 protein) were incubated at 30.0 to 55.6°C for 1 h using a gradient temperature heater (Thermal Cycler Dice Gradient; TaKaRa Bio) and then centrifuged (18,000 × g for 10 min at 4°C) to remove aggregated proteins. Proteins retained in the supernatant were analyzed by the Bradford method using bovine serum albumin as a standard. T1/2, the temperature at 50% residual protein, was calculated by linear regression using thermal denaturation data (BSpyrFwt, 35.6 to 45.6°C; BSpyrFe1, 48.0 to 53.2°C; BSpyrFe2, 48.0 to 54.4°C). For time course analysis, protein solutions were incubated at 37°C for 7 days. After centrifugation, proteins retained in the supernatant were analyzed by the Bradford method.
RESULTS
G. kaustophilus mutability.
G. kaustophilus MK242 was constructed as a laboratory strain that lacks genes related to pyrimidine biosynthesis (pyrF and pyrR) and DNA restriction modification in the HTA426 strain. It was transformed with pGKE70 for coordinating analytical conditions with other G. kaustophilus mutants (see below). The resulting mutant, MK242p70, was used to assess G. kaustophilus mutability (Fig. 2). The mutability assay was based on generation frequencies of Rifr and Strr cells, because Rifr and Strr often arise from mutations in rpoB (encoding RNA polymerase β subunit) and rpsL (encoding ribosomal subunit protein S12) genes, respectively (33–38). The generation frequencies of Rifr cells (per 109 viable cells) from MK242p70 cells were 930 ± 350 (for 10 mg liter−1 Rif) and 520 ± 270 (for 50 mg liter−1 Rif), and those of Strr cells were 120 ± 60 (for 10 mg liter−1 Str) and 70 ± 40 (for 50 mg liter−1 Str). These frequencies were higher than those of E. coli and B. subtilis, confirming substantial mutability of G. kaustophilus.
FIG 2.

Mutability of G. kaustophilus, E. coli, and B. subtilis. Generation frequencies of Rifr or Strr cells (per 109 viable cells) from G. kaustophilus MK242p70, E. coli DH5α, and B. subtilis 168 were analyzed using Rif and Str at low (solid bars) and high (open bars) concentrations. Analysis was performed using four independent culture experiments (n = 4), and the data are presented as means and standard errors (SE).
Mutability of G. kaustophilus mutants.
The G. kaustophilus HTA426 genome (40) contains genes that may be involved in DNA repair (mutS, mutL, mutM, mutY, mutT, ung, and mfd). To evaluate gene functions, six deletion mutants (ΔmutSL, ΔmutM, ΔmutY, ΔmutT, Δung, and Δmfd) were constructed. After correct deletions were verified by Southern blotting (Fig. 3), these mutants were further transformed with pGKE70 or its derivatives to construct six mutants (ΔmutSLp70, ΔmutMp70, ΔmutYp70, ΔmutTp70, Δungp70, and Δmfdp70) and their complementary mutants (ΔmutSLcm, ΔmutMcm, ΔmutYcm, ΔmutTcm, Δungcm, and Δmfdcm) and analyzed for generation frequencies of Rifr and Strr cells (Fig. 4). All mutants showed growth rates comparable with that of strain MK242P70 (Table 1), but mutation frequencies were markedly enhanced in the ΔmutSLp70 (30- to 400-fold), ΔmutYp70 (400- to 2,000-fold), Δungp70 (100- to 400-fold), and Δmfdp70 (100- to 400-fold) mutants. Their complementary mutants showed substantially suppressed frequencies, confirming the absence of polar effects accompanying the deletions. For the mutM and mutT genes, both deletion and complementation had relatively small effects on the frequencies. These data suggest that the mutSL, mutY, ung, and mfd genes have important roles in DNA repair but mutM and mutT have small contributions.
FIG 3.

In-frame deletions in DNA repair genes in G. kaustophilus MK242. Correct deletions to generate the ΔmutSL (A), ΔmutM (B), ΔmutY (C), ΔmutT (D), Δung (E), and Δmfd (F) mutants were verified by Southern blotting. The open boxes represent DNA probes. The dotted lines indicate the deletion regions. The numbers indicate DNA fragment lengths (kb).
FIG 4.

Effects of deletion and complementation of mutSL, mutM, mutY, mutT, ung, and mfd genes on G. kaustophilus mutability. The generation frequencies of Rifr or Strr cells were evaluated using Rif and Str at 10 mg liter−1 (solid bars) and 50 mg liter−1 (open bars). Analyses were performed in three to five independent experiments for each condition (n = 3 to 5). The data are presented as means and SE of fold changes relative to the mean generation frequency of the MK242p70 strain.
Mutations generated in G. kaustophilus.
The rpoB and rpsL genes in Rifr and Strr cells that were derived from strain MK242p70 were sequenced to analyze mutations generated in cells (Table 3). Rifr cells contained any of the 12 single mutations in the rpoB gene, and Strr cells contained any of the six single mutations in the rpsL gene. All mutations occurred independently and are probably responsible for the antibiotic resistance. Overall, A · T→G · C and C · G→T · A transitions were predominant, whereas A · T→C · G, A · T→T · A, C · G→A · T, and C · G→G · C transversions were less frequent. We also analyzed Rifr and Strr clones derived from ΔmutSLp70, Δungp70, ΔmutYp70, and Δmfdp70 mutants (Table 3). In addition to single mutations observed in MK242p70 cells, three single mutations in rpoB and one single mutation in rpsL were identified. Some Strr cells showed no mutation in rpsL. Cells may contain mutations in rrn for 16S rRNA because it is known that certain rrn mutations result in Strr in E. coli (37, 38). The ΔmutSLp70 and Δmfdp70 mutants showed markedly increased frequencies of A · T→G · C and A · T→C · G mutations, respectively. The ΔmutYp70 mutant showed high frequencies of the C · G→A · T transversion, suggesting that MutY suppresses the transversion by repairing A · 8-oxoG mispairs, similar to E. coli (20, 25) and B. subtilis homologs (26).
TABLE 3.
Mutations generated in G. kaustophilus mutants
| Mutation | Mutation frequencya |
|||||
|---|---|---|---|---|---|---|
| MK242p70 | ΔmutSLp70 | ΔmutYp70 | Δungp70 | Δmfdp70 | MK480p70 | |
| rpoB mutations in Rifr | ||||||
| T1393C | 3/48 | 2/19 | ||||
| C1405A | 2/48 | 1/22 | 3/21 | |||
| A1406G | 5/20 | |||||
| A1406T | 3/48 | |||||
| G1414T | 2/48 | |||||
| A1415T | 2/48 | |||||
| A1416T | 1/48 | |||||
| C1433A | 3/22 | |||||
| C1433T | 2/48 | 1/19 | 1/20 | |||
| C1444G | 1/21 | |||||
| C1444T | 14/48 | 2/19 | 3/22 | 3/21 | 1/20 | |
| A1445G | 4/48 | 11/19 | 1/21 | |||
| A1445T | 1/48 | |||||
| C1460A | 1/48 | 1/19 | 15/22 | 24/32 | ||
| C1460T | 13/48 | 2/19 | 13/21 | 13/20 | 8/32 | |
| rpsL mutations in Strr | ||||||
| A167C | 2/48 | 9/21 | ||||
| A167G | 18/48 | 1/22 | 2/22 | 10/21 | ||
| A168T | 1/48 | |||||
| A301G | 13/48 | 4/22 | 6/22 | 7/22 | 1/21 | 20/32 |
| A302G | 11/48 | 10/22 | 4/22 | 10/22 | 1/21 | 3/32 |
| C311A | 3/48 | 1/22 | 9/22 | 1/22 | ||
| C311T | 2/22 | 1/22 | 7/32 | |||
| Not identified | 6/22 | 1/22 | 1/22 | 2/32 | ||
| Mutation spectra | ||||||
| A · T→C · G | 2/96 (2.1) | 9/41 (22) | ||||
| A · T→G · C | 49/96 (51) | 28/35 (80) | 10/43 (23) | 20/42 (48) | 17/41 (41) | 23/62 (37) |
| A · T→T · A | 8/96 (8.3) | |||||
| C · G→A · T | 8/96 (8.3) | 2/35 (5.7) | 28/43 (65) | 4/42 (9.5) | 24/62 (39) | |
| C · G→G · C | 1/42 (2.4) | |||||
| C · G→T · A | 29/96 (30) | 5/35 (14) | 5/43 (12) | 17/42 (40) | 15/41 (37) | 15/62 (24) |
G. kaustophilus mutants that were grown on LB plates containing 50 mg liter−1 Rif or Str were analyzed for rpoB and rpsL sequences, respectively. The fraction represents sequences carrying the mutation per total number of sequences determined. Mutation spectra summarize sequences that arise from the mutation per total sequences carrying mutations (excluding those not identified). Values in parentheses indicate percentages (%). Empty cells indicate that the mutation was not observed.
The error-prone thermophile G. kaustophilus MK480.
G. kaustophilus MK480 was constructed from strain MK242 by simultaneous deletions of the mutSL, mutY, ung, and mfd genes, which were confirmed to be genes involved in functional DNA repair systems in G. kaustophilus, as described above. The properties were evaluated using the mutant MK480p70, which showed 700- to 9,000-fold-increased generation frequencies of Rifr and Strr cells (Fig. 5A). The growth curves of MK242p70 and MK480p70 were almost identical (Fig. 5B), with comparable doubling times (Table 1). The predominant mutations were A · T→G · C, C · G→T · A, and C · G→A · T (Table 3).
FIG 5.
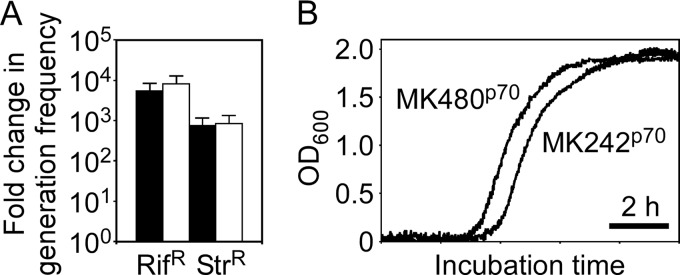
Comparison of mutability (A) and growth curves (B) between the G. kaustophilus strains MK242p70 and MK480p70. (A) Generation frequencies of Rifr or Strr cells from MK480p70 were evaluated using Rif and Str at 10 mg liter−1 (solid bars) and 50 mg liter−1 (open bars). The data are presented as means and SE (n = 5) of fold changes relative to the mean generation frequency of the MK242p70 strain. (B) The strains MK242p70 and MK480p70 were cultured at 60°C in LB medium with monitoring of the OD600.
Generation of BSpyrFe1 and BSpyrFe2 genes in G. kaustophilus MK480.
Because G. kaustophilus MK242 and MK480 are auxotrophic for uracil owing to a deficiency in the native pyrF gene, we investigated thermostability-directed evolution of the BSpyrFwt protein (encoded by B. subtilis pyrF) using the strains MK242BSpyrF and MK480BSpyrF. The MK242BSpyrF strain grew at 60°C in liquid MM (without uracil) but not at 65°C, probably because of BSpyrFwt thermal denaturation (Fig. 6A). Similar observations were made for MK480BSpyrF. However, after cultivation at 60°C, MK480BSpyrF cells grew even at 65°C, whereas MK242BSpyrF cells did not. MK480BSpyrF cells efficiently formed colonies on MM plates. Among the colonies grown, eight were analyzed for BSpyrFwt sequences to identify the mutant genes BSpyrFe1 (6/8 clones) and BSpyrFe2 (2/8 clones). BSpyrFe1 carried a C497T mutation responsible for A166V substitutions in the BSpyrFwt amino acid sequence (Fig. 6B). In addition to the C497T mutation, BSpyrFe2 carried a C169A mutation responsible for the L57I substitution.
FIG 6.

Generation of thermostable variants BSpyrFe1 and BSpyrFe2 from BSpyrFwt. (A) Phenotype transition from prototrophy to auxotrophy for uracil in G. kaustophilus MK480BSpyrF. G. kaustophilus MK480p70, MK480BSpyrF, and MK242BSpyrF were incubated in MM at 60°C (left) and 65°C (middle). MK480BSpyrF and MK242BSpyrF cells that had been grown at 60°C were incubated in MM at 65°C (right). (B) Mutations in BSpyrFe1 and BSpyrFe2 genes. BSpyrFe1 carried C497T, and BSpyrFe2 carried C169A and C497T mutations. The C169A and C497T mutations are responsible for L57I and A166V amino acid substitutions, respectively.
Thermostability of BSpyrFwt, BSpyrFe1, and BSpyrFe2 proteins.
Recombinant BSpyrFwt, BSpyrFe1, and BSpyrFe2 proteins were produced as proteins fused with an octahistidine tag at the N terminus and purified to homogeneity using immobilized metal affinity chromatography (Fig. 7A). To assess their thermostability, the protein solution was incubated at high temperatures and analyzed for the amounts of protein free of thermal aggregation (Fig. 7B). The BSpyrFwt protein was completely aggregated when incubated at 45°C, whereas BSpyrFe1 and BSpyrFe2 were almost completely retained in the supernatant. Complete aggregation of BSpyrFe1 was observed at >53°C and that of BSpyrFe2 at >54°C. The T1/2 values for BSpyrFwt, BSpyrFe1, and BSpyrFe2 were 40.3, 50.0, and 51.0°C, respectively, showing that these variants were much more thermostable than the BSpyrFwt protein. This finding was further supported by time course analysis of protein aggregation (Fig. 7C). After incubation at 30°C for 1 day, BSpyrFwt was completely aggregated, whereas BSpyrFe1 retained 77% of its total amount. BSpyrFe2, which retained 84% at 30°C and 38% at 37°C even after 7 days, was notably more stable. These results suggest that this approach has potential for markedly improving the storage stability of thermolabile enzymes at ordinary temperatures.
FIG 7.

Thermostability assay of BSpyrFwt, BSpyrFe1, and BSpyrFe2 proteins. (A) SDS-PAGE analysis of purified recombinant BSpyrFwt (lane 1), BSpyrFe1(lane 2), and BSpyrFe2 (lane 3) proteins with molecular markers (lane M; kDa). (B) Thermal denaturation curves of BSpyrFwt (solid circles), BSpyrFe1 (open circles), and BSpyrFe2 (solid squares) proteins. The protein solution was incubated for 1 h at the indicated temperatures and centrifuged. Proteins retained in the supernatant were analyzed by the Bradford method. The data are presented as means and SE (n = 4 to 8). The amount of protein remaining after 30°C incubation was taken to be 100%. (C) Time course analysis of BSpyrFwt (solid circles), BSpyrFe1 (open circles), and BSpyrFe2 (solid squares) aggregations. The protein solution was incubated at 30°C (left) and 37°C (right) for the indicated periods. After centrifugation, proteins retained in the supernatant were analyzed by the Bradford method. The data are presented as means (n = 4; SE are less than 3% [not shown]).
DISCUSSION
Among the types of DNA damage, deamination, depurination, and destruction of deoxyribose residues increase at high temperatures (13, 15, 41, 42). Because of such high-temperature-associated DNA damage, thermophiles may undergo more frequent DNA damage during growth, but the hyperthermophile Sulfolobus acidocaldarius exhibits mutability comparable with that of mesophiles, reflecting efficient functioning of DNA repair genes in the hyperthermophile (43). However, our preliminary observation suggests that G. kaustophilus HTA426 may have relatively high mutability, leading us to evaluate its mutability in detail. As expected, a mutation assay using Rif and Str revealed the substantial mutability of G. kaustophilus. This high mutability may partially be due to high-temperature-associated DNA damage and insufficient DNA repair genes, because mutation analysis of G. kaustophilus showed frequent A · T→G · C and C · G→T · A transitions, which can increase at high temperature through deamination of adenine and cytosine, respectively (12, 14–18). Although spontaneous adenine deamination is much less frequent than cytosine deamination in general (15, 16), A · T→G · C transition was more frequent than C · G→T · A transition in G. kaustophilus. In the ΔmutSLp70 mutant, the frequency of A · T→G · C was as high as 80%. These observations imply a defect in deaminated-adenine (i.e., hypoxanthine) repair in G. kaustophilus. In addition, gene deletion analysis indicated small contributions of mutM and mutT to DNA repair. In E. coli (20) and B. subtilis (26), MutM suppresses the C · G→A · T transversion cooperatively with MutY, and MutT suppresses A · T→C · G transversion. Although MutY and Mfd contribute to suppressing these transversions in G. kaustophilus (Table 3), it is likely that G. kaustophilus is exposed to relatively high risks of the transversions.
We constructed G. kaustophilus MK480 as an error-prone thermophile by deleting functional DNA repair genes that we had identified and investigated thermoadaptive gene mutations in the error-prone strain using BSpyrFwt as a model. G. kaustophilus MK480BSpyrF was originally auxotrophic for uracil at 65°C but became prototrophic after culture in MM at 60°C, followed by 65°C, along with BSpyrFe1 and BSpyrFe2 generation. This phenotype transition can be explained by generation and selection, during incubation at 60°C, of mutant genes that encode enzyme variants that are more thermostable than the BSpyrFwt protein. The thermostability assay confirmed that the variants BSpyrFe1 and BSpyrFe2 are actually more thermostable than BSpyrFwt and that BSpyrFe2 is more thermostable than BSpyrFe1. These observations indicate that both A166T and L57I substitutions contribute to BSpyrFwt thermostabilization. In the (β/α)8 barrel fold of the BSpyrFwt protein (44), the residues L57 (replaced in BSpyrFe2) and A166 (replaced in BSpyrFe1 and BSpyrFe2) constitute β-strand β3 and α-helix α2, respectively (Fig. 8). The side chain of L57 hydrophobically interacts with the side chains of the F39, V47, and L59 residues to occupy the void between β-strand β3 and α-helix α2 (Fig. 8A). Similar hydrophobic interactions were observed for the A166 residue, the side chain of which interacts with Y170 and T179 to occupy the void between α-helix α6 and β-strand β7 (Fig. 8B). Therefore, it is possible that thermostability enhancement by L57I and A166V substitutions involves optimizing hydrophobic interactions between α-helices and β-strands in the barrel fold, thereby making the structure robust.
FIG 8.

Three-dimensional structures surrounding the L57 (A) and A166 (B) residues of the BSpyrFwt protein.
Thermoadaptation-directed evolution using strain MK480 requires no construction of random mutant libraries because mutations are generated in cells during successive culture. Although the mutations may be less frequent than mutations obtained using in vitro mutagenesis approaches, one amino acid alteration potentially confers sufficient enhancement of enzyme thermostability, as shown by BSpyrFe1 and other examples (45). Moreover, strain MK480 can employ in vivo enzymatic characterization at high temperatures to select thermoadaptive enzymes, as described below: (i) when cell growth is accelerated by catalytic activity of target enzymes, candidate genes encoding thermoadaptive enzymes can be concentrated, because thermolabile enzymes undergo thermal denaturation during cell growth at high temperatures, as in BSpyrFe1 and BSpyrFe2 generation, and (ii) when catalytic activities of target enzymes can be detected by chromogenic or fluorogenic assay in vivo, candidate genes can be selected by a plate assay without cell death. Even in other cases, candidate genes may be efficiently selected by the activity-independent method described by Chautard et al. (46). The availability of high-temperature in vivo assays is a notable advantage of strain MK480 over error-prone mesophiles for thermoadaptation-directed evolution. In addition, strain MK480 showed 700- to 9,000-fold-increased mutability. This enhancement is comparable to that achieved by abolishing (5,000-fold) or silencing (2,000-fold) DNA repair genes in E. coli (47, 48), while G. kaustophilus had higher intrinsic mutability than E. coli (Fig. 2). Therefore, G. kaustophilus MK480 has higher mutability than E. coli strains engineered for increased mutability, an additional advantage over error-prone mesophiles.
There are some reports of thermoadaptation-directed enzyme evolution in thermophiles (5, 7–9), although not using an error-prone thermophile. A 3-isopropylmalate dehydrogenase variant having five substitutions with a 12°C-higher T1/2 than the parent enzyme was generated by iterative incubation for approximately 10 days in T. thermophilus (8). T. thermophilus cells were also used to generate an α-galactosidase variant possessing one substitution with an enhanced T1/2 of 3°C by 11 days of incubation (7). Compared with these examples, strain MK480 generated thermoadaptive variants with excellent thermostability enhancement (>10°C) in a short time (2 days), probably due to high mutability. These data suggest that strain MK480 has an advantage over not only error-prone mesophiles, but also thermophiles used for thermoadaptation-directed evolution to date. It is also noteworthy that in strain MK480, every mutation can theoretically occur, given that although the A · T→C · G, A · T→T · A, and C · G→G · C mutations were not observed in the strain, they were found in the parent strain (MK242p70) or the Δung mutant (Δungp70). Moreover, G. kaustophilus allows a wide range of selection temperatures (42 to 74°C), which is especially favorable for thermoadaptation-directed evolution of very thermolabile enzymes. Thus, strain MK480 is a practical resource for generating thermostable enzyme variants by thermoadaptation-directed evolution and provides new opportunities for the facile generation of thermostable enzymes.
ACKNOWLEDGMENTS
This work was supported by the Programme for Promotion of Basic and Applied Researches for Innovations in Bio-Oriented Industry (BRAIN), Japan, and in part by the Institute for Fermentation, Osaka, Japan.
REFERENCES
- 1.Haki GD, Rakshit SK. 2003. Developments in industrially important thermostable enzymes: a review. Bioresour Technol 89:17–34. doi: 10.1016/S0960-8524(03)00033-6. [DOI] [PubMed] [Google Scholar]
- 2.Bommarius AS, Broering JM, Chaparro-Riggers JF, Polizzi KM. 2006. High-throughput screening for enhanced protein stability. Curr Opin Biotechnol 17:606–610. doi: 10.1016/j.copbio.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Chen J, Yu H, Liu C, Liu J, Shen Z. 2012. Improving stability of nitrile hydratase by bridging the salt-bridges in specific thermal-sensitive regions. J Biotechnol 164:354–362. doi: 10.1016/j.jbiotec.2013.01.021. [DOI] [PubMed] [Google Scholar]
- 4.Steffler F, Guterl J-K, Sieber V. 2013. Improvement of thermostable aldehyde dehydrogenase by directed evolution for application in synthetic cascade biomanufacturing. Enzyme Microb Technol 53:307–314. doi: 10.1016/j.enzmictec.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 5.Nakamura A, Takakura Y, Kobayashi H, Hoshino T. 2005. In vivo directed evolution for thermostabilization of Escherichia coli hygromycin B phosphotransferase and the use of the gene as a selection marker in the host-vector system of Thermus thermophilus. J Biosci Bioeng 100:158–163. doi: 10.1263/jbb.100.158. [DOI] [PubMed] [Google Scholar]
- 6.Brouns SJJ, Wu H, Akerboom J, Turnbull AP, de Vos WM, van der Oost J. 2005. Engineering a selectable marker for hyperthermophiles. J Biol Chem 280:11422–11431. doi: 10.1074/jbc.M413623200. [DOI] [PubMed] [Google Scholar]
- 7.Fridjonsson O, Watzlawick H, Mattes R. 2002. Thermoadaptation of α-galactosidase AgaB1 in Thermus thermophilus. J Bacteriol 184:3385–3391. doi: 10.1128/JB.184.12.3385-3391.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tamakoshi M, Nakano Y, Kakizawa S, Yamagishi A, Oshima T. 2001. Selection of stabilized 3-isopropylmalate dehydrogenase of Saccharomyces cerevisiae using the host-vector system of an extreme thermophile, Thermus thermophilus. Extremophiles 5:17–22. doi: 10.1007/s007920000168. [DOI] [PubMed] [Google Scholar]
- 9.Liao H, McKenzie T, Hageman R. 1986. Isolation of a thermostable enzyme variant by cloning and selection in a thermophile. Proc Natl Acad Sci U S A 83:576–580. doi: 10.1073/pnas.83.3.576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoseki J, Yano T, Koyama Y, Kuramitsu S, Kagamiyama H. 1999. Directed evolution of thermostable kanamycin-resistance gene: a convenient selection marker for Thermus thermophilus. J Biochem 126:951–956. doi: 10.1093/oxfordjournals.jbchem.a022539. [DOI] [PubMed] [Google Scholar]
- 11.Matsumura M, Aiba S. 1985. Screening for thermostable mutant of kanamycin nucleotidyltransferase by the use of a transformation system for a thermophile, Bacillus stearothermophilus. J Biol Chem 260:15298–15303. [PubMed] [Google Scholar]
- 12.Smith KC. 1992. Spontaneous mutagenesis: experimental, genetic and other factors. Mutat Res 277:139–162. doi: 10.1016/0165-1110(92)90002-Q. [DOI] [PubMed] [Google Scholar]
- 13.Lindahl T. 1993. Instability and decay of the primary structure of DNA. Nature 362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 14.Parry TE. 2007. On the mutagenic action of adenine. Leuk Res 31:1621–1624. doi: 10.1016/j.leukres.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 15.Karran P, Lindahl T. 1980. Hypoxanthine in deoxyribonucleic acid: generation by heat-induced hydrolysis of adenine residues and release in free form by a deoxyribonucleic acid glycosylase from calf thymus. Biochemistry 19:6005–6011. doi: 10.1021/bi00567a010. [DOI] [PubMed] [Google Scholar]
- 16.Wanner RM, Castor D, Güthlein C, Böttger EC, Springer B, Jiricny J. 2009. The uracil DNA glycosylase UdgB of Mycobacterium smegmatis protects the organism from the mutagenic effects of cytosine and adenine deamination. J Bacteriol 191:6312–6319. doi: 10.1128/JB.00613-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.López-Olmos K, Hernández MP, Contreras-Garduño JA, Robleto EA, Setlow P, Yasbin RE, Pedraza-Reyes M. 2012. Roles of endonuclease V, uracil-DNA glycosylase, and mismatch repair in Bacillus subtilis DNA base-deamination-induced mutagenesis. J Bacteriol 194:243–252. doi: 10.1128/JB.06082-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krokan HE, Drabløs F, Slupphaug G. 2002. Uracil in DNA—occurrence, consequences and repair. Oncogene 21:8935–8948. doi: 10.1038/sj.onc.1205996. [DOI] [PubMed] [Google Scholar]
- 19.Shibutani S, Takeshita M, Grollman AP. 1991. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature 349:431–434. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- 20.Tajiri T, Maki H, Sekiguchi M. 1995. Functional cooperation of MutT, MutM and MutY proteins in preventing mutations caused by spontaneous oxidation of guanine nucleotide in Escherichia coli. Mutat Res 336:257–267. doi: 10.1016/0921-8777(94)00062-B. [DOI] [PubMed] [Google Scholar]
- 21.Kunkel TA, Erie DA. 2005. DNA mismatch repair. Annu Rev Biochem 74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 22.Pillon MC, Lorenowicz JJ, Uckelmann M, Klocko AD, Mitchell RR, Chung YS, Modrich P, Walker GC, Simmons LA, Friedhoff P, Guarné A. 2010. Structure of the endonuclease domain of MutL: unlicensed to cut. Mol Cell 39:145–151. doi: 10.1016/j.molcel.2010.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simmons LA, Davies BW, Grossman AD, Walker GC. 2008. Beta clamp directs localization of mismatch repair in Bacillus subtilis. Mol Cell 29:291–301. doi: 10.1016/j.molcel.2007.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lindahl T. 1974. An N-glycosidase from Escherichia coli that releases free uracil from DNA containing deaminated cytosine residues. Proc Natl Acad Sci U S A 71:3649–3653. doi: 10.1073/pnas.71.9.3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Michaels ML, Cruz C, Grollman AP, Miller JH. 1992. Evidence that MutY and MutM combine to prevent mutations by an oxidatively damaged form of guanine in DNA. Proc Natl Acad Sci U S A 89:7022–7025. doi: 10.1073/pnas.89.15.7022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sasaki M, Kurusu Y. 2004. Analysis of spontaneous base substitutions generated in mutator strains of Bacillus subtilis. FEMS Microbiol Lett 234:37–42. doi: 10.1111/j.1574-6968.2004.tb09510.x. [DOI] [PubMed] [Google Scholar]
- 27.Selby CP, Witkin EM, Sancar A. 1991. Escherichia coli mfd mutant deficient in “mutation frequency decline” lacks strand-specific repair: in vitro complementation with purified coupling factor. Proc Natl Acad Sci U S A 88:11574–11578. doi: 10.1073/pnas.88.24.11574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park J-S, Marr MT, Roberts JW. 2002. E. coli transcription repair coupling factor (Mfd protein) rescues arrested complexes by promoting forward translocation. Cell 109:757–767. doi: 10.1016/S0092-8674(02)00769-9. [DOI] [PubMed] [Google Scholar]
- 29.Suzuki H, Yoshida K, Ohshima T. 2013. Polysaccharide-degrading thermophiles generated by heterologous gene expression in Geobacillus kaustophilus HTA426. Appl Environ Microbiol 79:5151–5158. doi: 10.1128/AEM.01506-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suzuki H, Murakami A, Yoshida K. 2012. Counterselection system for Geobacillus kaustophilus HTA426 through disruption of pyrF and pyrR. Appl Environ Microbiol 78:7376–7383. doi: 10.1128/AEM.01669-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suzuki H, Wada K, Furukawa M, Doi K, Ohshima T. 2013. A ternary conjugation system for the construction of DNA libraries for Geobacillus kaustophilus HTA426. Biosci Biotechnol Biochem 77:2316–2318. doi: 10.1271/bbb.130492. [DOI] [PubMed] [Google Scholar]
- 32.Suzuki H, Yoshida K. 2012. Genetic transformation of Geobacillus kaustophilus HTA426 by conjugative transfer of host-mimicking plasmids. J Microbiol Biotechnol 22:1279–1287. doi: 10.4014/jmb.1203.03023. [DOI] [PubMed] [Google Scholar]
- 33.Jin DJ, Gross CA. 1988. Mapping and sequencing of mutations in the Escherichia coli rpoB gene that lead to rifampicin resistance. J Mol Biol 202:45–58. doi: 10.1016/0022-2836(88)90517-7. [DOI] [PubMed] [Google Scholar]
- 34.Nicholson WL, Maughan H. 2002. The spectrum of spontaneous rifampin resistance mutations in the rpoB gene of Bacillus subtilis 168 spores differs from that of vegetative cells and resembles that of Mycobacterium tuberculosis. J Bacteriol 184:4936–4940. doi: 10.1128/JB.184.17.4936-4940.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Noguchi N, Sasatsu M, Kono M. 1993. Genetic mapping in Bacillus subtilis 168 of the aadK gene which encodes aminoglycoside 6-adenylyltransferase. FEMS Microbiol Lett 114:47–52. doi: 10.1111/j.1574-6968.1993.tb06549.x. [DOI] [PubMed] [Google Scholar]
- 36.Timms AR, Steingrimsdottir H, Lehmann AR, Bridges BA. 1992. Mutant sequences in the rpsL gene of Escherichia coli B/r: mechanistic implications for spontaneous and ultraviolet light mutagenesis. Mol Gen Genet 232:89–96. doi: 10.1007/BF00299141. [DOI] [PubMed] [Google Scholar]
- 37.Montandon PE, Wagner R, Stutz E. 1986. Escherichia coli ribosomes with a C912 to U base change in the 16S rRNA are streptomycin resistant. EMBO J 5:3705–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Melançon P, Lemieux C, Brakier-Gingras L. 1988. A mutation in the 530 loop of Escherichia coli 16S ribosomal RNA causes resistance to streptomycin. Nucleic Acids Res 16:9631–9639. doi: 10.1093/nar/16.20.9631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amartey SA, Leak DJ, Hartley BS. 1991. Development and optimization of a defined medium for aerobic growth of Bacillus stearothermophilus LLD-15. Biotechnol Lett 13:621–626. doi: 10.1007/BF01086315. [DOI] [Google Scholar]
- 40.Takami H, Takaki Y, Chee G-J, Nishi S, Shimamura S, Suzuki H, Matsui S, Uchiyama I. 2004. Thermoadaptation trait revealed by the genome sequence of thermophilic Geobacillus kaustophilus. Nucleic Acids Res 32:6292–6303. doi: 10.1093/nar/gkh970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lindahl T, Nyberg B. 1972. Rate of depurination of native deoxyribonucleic acid. Biochemistry 11:3610–3618. doi: 10.1021/bi00769a018. [DOI] [PubMed] [Google Scholar]
- 42.Lindahl T, Nyberg B. 1974. Heat-induced deamination of cytosine residues in deoxyribonucleic acid. Biochemistry 13:3405–3410. doi: 10.1021/bi00713a035. [DOI] [PubMed] [Google Scholar]
- 43.Jacobs KL, Grogan DW. 1997. Rates of spontaneous mutation in an archaeon from geothermal environments. J Bacteriol 179:3298–3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Appleby TC, Kinsland C, Begley TP, Ealick SE. 2000. The crystal structure and mechanism of orotidine 5′-monophosphate decarboxylase. Proc Natl Acad Sci U S A 97:2005–2010. doi: 10.1073/pnas.259441296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Imanaka T. 1990. Enhancement of thermostability of neutral proteases. Ann N Y Acad Sci 613:347–351. doi: 10.1111/j.1749-6632.1990.tb18176.x. [DOI] [PubMed] [Google Scholar]
- 46.Chautard H, Blas-Galindo E, Menguy T, Grand'Moursel L, Cava F, Berenguer J, Delcourt M. 2007. An activity-independent selection system of thermostable protein variants. Nat Methods 4:919–921. doi: 10.1038/nmeth1090. [DOI] [PubMed] [Google Scholar]
- 47.Greener A, Callahan M, Jerpseth B. 1997. An efficient random mutagenesis technique using an E. coli mutator strain. Mol Biotechnol 7:189–195. doi: 10.1007/BF02761755. [DOI] [PubMed] [Google Scholar]
- 48.Nakashima N, Tamura T. 2009. Conditional gene silencing of multiple genes with antisense RNAs and generation of a mutator strain of Escherichia coli. Nucleic Acids Res 37:e103. doi: 10.1093/nar/gkp498. [DOI] [PMC free article] [PubMed] [Google Scholar]