

Abstract
Detection of Shiga toxin-producing Escherichia coli (STEC) by culture methods is advisable to identify the pathogen, but recovery of the strain responsible for the disease is not always possible. The use of DNA-based methods (PCR, quantitative PCR [qPCR], or genomics) targeting virulence genes offers fast and robust alternatives. However, detection of stx is not always indicative of STEC because stx can be located in the genome of temperate phages found in the samples as free particles; this could explain the numerous reports of positive stx detection without successful STEC isolation. An approach based on filtration through low-protein-binding membranes and additional washing steps was applied to reduce free Stx phages without reducing detection of STEC bacteria. River water, food, and stool samples were spiked with suspensions of phage 933W and, as a STEC surrogate, a lysogen harboring a recombinant Stx phage in which stx was replaced by gfp. Bacteria were tested either by culture or by qPCR for gfp while phages were tested using qPCR targeting stx in phage DNA. The procedure reduces phage particles by 3.3 log10 units without affecting the recovery of the STEC population (culturable or assessed by qPCR). The method is applicable regardless of phage and bacteria densities and is useful in different matrices (liquid or solid). This approach eliminates or considerably reduces the interference of Stx phages in the detection of STEC by molecular methods. The reduction of possible interference would increase the efficiency and reliability of genomics for STEC detection when the method is applied routinely in diagnosis and food analysis.
INTRODUCTION
Shiga toxin-producing Escherichia coli (STEC) bacteria are a class of enteric pathogens capable of causing severe gastrointestinal disease (hemorrhagic colitis) that can develop undesirable complications, such as acute kidney failure (hemolytic-uremic syndrome [HUS]) that could require lifelong treatment (1, 2). STEC strains belonging to different serotypes, notably strains of serotype O157:H7, have been the causative agents of large outbreaks of food-borne disease. However, the emergence of non-O157 STEC strains with various combinations of virulence genes also represents a serious challenge for the protection of consumers from food-borne disease (3, 4).
The identification of STEC strains, which requires culture enrichment on selective medium, is advisable to confirm and characterize the pathogen. However, recovery of the strain responsible for the disease is not always possible because it could be present in low concentrations, the cells might not be in a culturable state, or there could be interference from commensal E. coli within the microbiota in the sample. The need for early identification of STEC demands the use of faster and more robust methods.
STEC strains present genomic plasticity that complicates discrimination of the pathogenic strains among other E. coli strains present in a sample. For this reason, DNA and protein detection methods have been developed to target the genes related to virulence (5–7) and genes that help to identify a specific serotype (8). Reliable methods focused on the detection of virulent serotypes (for instance, O26, O45, O111, O103, O121, and O145, accounting for the non-O157 serotypes, plus O157:H7) (9) are very useful for known pathotypes or for epidemiological purposes. However, the recent outbreak caused by E. coli O104:H4 in Germany (10) highlighted the limitations of methods based on the detection of only well-known serotypes when an unusual one is present. In order to better assess the presence of an unknown STEC serotype in a sample, it appears to be necessary to establish protocols that target specific virulence genes, alone or preferentially in combination, instead of serotypes.
Many DNA-based methods for STEC detection include PCR techniques (5–7, 11–13), including standardized methods approved by legislation of the European Union (14, 15). Moreover, next-generation sequencing (NGS) protocols are emerging, and their implementation is advised to improve detection (10, 16).
The main virulence determinant of STEC, responsible for the most undesirable complications of infection such as HUS, is the Shiga toxin (Stx). stx genes are encoded in the genome of temperate bacteriophages (Stx phages) inserted as prophages into the STEC chromosome (17) or in enteric bacteria other than STEC, such as Citrobacter (18). Moreover, Stx phages can be found as free phages in environments that include urban sewage and animal wastewater (19, 20), human feces (21, 22), and even food (23), and they are extremely persistent in the natural environment (24–26).
When PCR-based and NGS methods are applied to detect STEC in a sample, the presence of free Stx phages in the sample (or other phages that could harbor virulence genes) represents a challenge since the phages can generate amplimers (PCR) or reads (NGS) that may be interpreted as belonging to STEC while they in fact belong to the phages. Moreover, when the protocols include a previous step of selective enrichment of the target bacteria, the step may also maintain or even propagate bacteriophages. The present study attempts (i) to confirm this possibility and (ii) to develop a method to eliminate or reduce the presence of free Stx phages in different samples, with the goal of minimizing their interference in DNA-based STEC detection methods.
MATERIALS AND METHODS
Bacteria, bacteriophage plasmids, and media.
Bacteriophage 933W was used in the experiments as a positive control. Phage 933W was induced from lysogenic E. coli strain C600 (933W) (27) and purified as described below. E. coli DH5α was used as a control of culturable bacteria. An E. coli C600 lysogen containing a modified 933W phage in which a fragment of the stx gene was replaced by the gene encoding the green fluorescent protein (gfp) and the gene encoding chloramphenicol resistance (cat), constructed as described below, was used to enumerate culturable bacteria and to distinguish bacteria from free Stx phages using quantitative PCR (qPCR). A pGEM-T-Easy cloning vector (Promega Co., Madison, WI) was used for the construction of standard curves. E. coli C600 (pGEM::stx2) (19) or plasmid pGreen TIR (28) containing the gfp gene was used to prepare the standards for the qPCR assays. Luria-Bertani (LB) broth or LB agar was used for bacteria cultivation and phage assays. When necessary, medium was supplemented with chloramphenicol (Cm) (20 μg/ml) (Sigma-Aldrich, Steinheim, Germany).
Preparation of bacteriophage 933W suspension.
Lysogen E. coli C600 (933W) was incubated under agitation (180 rpm) at 37°C to an optical density at 600 nm (OD600) of 0.3, as measured with a spectrophotometer (Spectronic 501; Milton Roy, Belgium). To induce 933W bacteriophages, mitomycin C (0.5 μg/ml) was added to the culture and incubated overnight at 37°C in the dark in a shaker. Phage lysates were prepared by filtration of the bacterial cultures through low-protein-binding 0.22-μm-pore-size membrane filters (Millex-GP; Millipore, Bedford, MA, USA). The supernatant was treated with DNase (100 units/ml of the phage lysate) to eliminate free DNA outside the phage particles. Phage DNA was extracted using different methods, as described below.
Standard and commercial DNA extraction methods.
A phage DNA purification method was used to extract DNA from the 933W suspension as described previously (29). The concentration and purity of the phage DNA extracted were determined using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies/Thermo Scientific Instruments, Wilmington, DE, USA).
In addition, phage 933W DNA was extracted using three commercial DNA extraction methods applied for bacterial or total DNA extraction, according to the manufacturers' instructions, as follows: QIAamp DNA Blood minikit (Qiagen GmbH, Hilden, Germany), QIAamp DNA Stool minikit (Qiagen), and NucliSENS miniMAG (bioMérieux España, Madrid, Spain). The phage count results from the commercial methods were compared with the specific protocol applied for bacteriophage DNA extraction.
Construction of the E. coli C600 (933W Δstx::gfp::cat) recombinant lysogen.
E. coli C600 lysogenic for phage 933W was used to prepare a recombinant strain in which stx was replaced by the gfp gene amplified from plasmid pGreen TIR (28) and a cat (chloramphenicol resistance) gene. A 224-bp fragment including the 5′ region of the stx2A subunit was amplified with tRNAup/Stx2a-gfpTir5 primers (Table 1), where the second primer overlaps gfp. The 753-bp gfp gene was amplified from plasmid pGreen TIR with primers GfpTir up/GfpTir lp-cm5, with the second primer overlapping cat. The 1,015-bp cat gene was amplified with primers cm5/cm3, and, finally, a 281-bp fragment of the stx2B subunit was amplified with primers Cm3-stx/Stx2Blp, with the first primer overlapping cat. A single stx2A-gfp-cat-stx2B amplimer was generated by overlap extension PCR and then purified and electroporated into the laboratory lysogen E. coli C600 (933W) containing pkD46 (30) with the Red recombinase system. The recombinant phage 933W Δstx2::gfp::cat (hereafter, 933Wgfp) was constructed using previously described methods (31) by recombination of the fragment into the late gene region of the Stx phage, replacing the sequence of the stx2 target gene (Fig. 1). The cat gene was introduced in the construct to prevent spontaneous induction of phage 933Wgfp from the E. coli C600 (933Wgfp) lysogen by growing the strain in the presence of Cm.
TABLE 1.
Oligonucleotides used in this study
| Name | Sequence (5′–3′)a | Amplimer size (bp) | Function or description | Reference or source |
|---|---|---|---|---|
| tRNAup | TCTGCATTATGCGTTGTT | 224 | Upstream stx2A | This study |
| Stx2a-gfpTir5 | TAAAGTTAATCAGAATTCAAAACCCAGTAACAGGCAC | Overlapping of gfp gene and stx2A | This study | |
| GfpTir up | GAATTCTGATTAACTTTA | 753 | Entire gfp gene from pGreen TIR vector | This study |
| GfpTir lp | TTATTTGTAGAGCTCATCC | |||
| GfpTir lp-cm5 | GAAGCAGCTCCAGCCTACACATTATTTGTAGAGCTCATCC | Overlapping of gfp gene and cat | ||
| Gfp short-up | TCCATCTTCAATGTTGTGTCT | 211 | gfp fragment for qPCR standard | This study |
| Gfp short-lp | GAACTATAAGACACGTGCTGA | |||
| Cm 5 | TGTGTAGGCTGGAGCTGCTTC | 1015 | cat gene | 31 |
| Cm-3 | CATATGAATATCCTCCTTAG | |||
| Cm3-stx | CTAAGGAGGATATTCATATGAGGAGTTAAGTATGAAGAAG | 281 | Overlapping cat-stx | 31 |
| Stx2Blp | TCAGTCATTATTAAACTG | Final codon of stx2B | This study | |
| UP378 | GCGTTTTGACCATCTTCGT | 378 | 378-bp stx2A fragment | 32 |
| LP378 | ACAGGAGCAGTTTCAGACAG | |||
| RR46 LP | GAGCTCTAAGGAGGTTAT | 457 | Red recombinase in pKD46 | 31 |
| RR46-UP | GTGCAGTACTCATTCGTT | |||
| GFPTir-F | GCTTCCATCTTCAATGTTGTGTCT | 121 | Real-time PCR for gfp gene | This study |
| GFPTir-R | CATTCTTGGACACAAATTGGAATACAACT | |||
| GFPTir- probe | FAM-CATGGCAGACAAACAA-NFQ | |||
| STX-Any f | ACGGACAGCAGTTATACCACTCT | 65 | Real-time PCR for stx2 gene | 29 |
| STX-Any r | ACGTTCCGGAATGCAAATCAG | |||
| STX-Any probe | FAM- CCAGCGCTGCGACACG-NFQ |
Overlap region is underlined.
FIG 1.
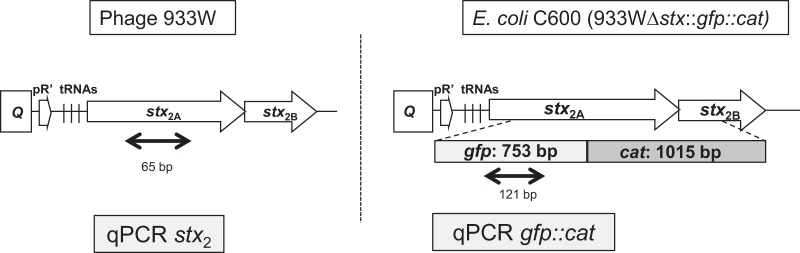
Schematic representation of the genetic organization of the stx operon in phage 933W in the STEC strain and the replacement of the stx2 gene by the gfp-cat fragment in E. coli C600 (933Wgfp). The schemes indicate where the fragments amplified by the qPCR assays for stx and gfp are located. Q, antiterminator gene; pR, late phage promoter.
Phage reduction procedure.
Phosphate-buffered saline (PBS) was spiked with various volumes of the 933W phage suspension. The spiked samples were evaluated in parallel with and without a previous filtration step. Volumes of 10 ml of each sample were filtered through 0.45-μm-pore-size mixed cellulose ester filters, commonly used for filtration in microbiological analyses (EZ-Pak membrane filter; Millipore, Bedford, MA, USA), or through 0.45-μm-pore-size low-protein-binding polyvinylidene fluoride (PVDF) membranes (Durapore membrane filter; Millipore, Bedford, MA, USA), which allowed bacteriophages to pass through but retained bacteria. To further remove phages retained in the filters, one or three washing steps were additionally applied. For each wash, 10 ml of PBS was added to the surface of the filter, gently agitated, and removed by filtration.
In addition, the phage reduction procedure with PVDF membranes was tested in PBS spiked with various volumes of E. coli DH5α or E. coli C600 (933Wgfp) bacterial cultures, and bacteria were recovered after three washing steps. Results of culturable bacteria were compared with and without a previous filtration step.
Phages and/or bacteria retained in the filter after the filtration procedure and the washing steps were recovered by suspending the membrane in the same volume of PBS as the original sample, shaking the membrane for 10 min by gentle rotation (horizontal shaker, Magna-AS15; Magna-Equipaments, Barcelona, Spain) at room temperature, and vortexing for 1 min. The filtrate was recovered in sterile 50-ml bottles and monitored for the presence of phages or bacteria as described below. Phages were monitored by qPCR as described below. Culturable E. coli was enumerated by serial decimal dilutions in PBS, and 100 μl was spread on LB agar plates or LB medium supplemented with Cm using a surface spread plate technique. The plates were incubated at 37°C for 18 h. To confirm that the reductions observed were due to the filtration procedure, in some experiments the filtrate was also analyzed for the presence of phages and bacteria.
Samples.
The procedure was tested in different matrices in addition to PBS. A mixture of stools obtained from five healthy adult volunteers (aged 25 to 30 years) was used in this study. Stool samples were collected, coded, and manipulated anonymously and were used only for this study. As food matrices, five salad and five minced-beef samples were purchased at different dates from local retailers. The samples of beef were freshly minced on request at local supermarkets, and salad samples were a commercially prepared mixture. Five river water samples were collected at different dates in the lower course of the River Llobregat, located in the Barcelona (Spain) area.
Samples were spiked with phages and bacteria as described above. Liquid samples were homogenized by magnetic stirring for 2 min and directly processed. Solid samples were mixed 1:5 (wt/vol) in phosphate-buffered saline (PBS), placed in stomacher bags with filters (Afora, Barcelona, Spain), and homogenized using a Masticator (IUL Instruments GmbH, Königswinter, Germany) for 2 min. The samples were then allowed to settle to precipitate the larger particles, and the homogenate was processed. To avoid filter clogging in solid samples, an additional soft centrifugation step (300 × g for 10 min) was added. The phage reduction procedure (with PVDF filters and three washing steps) was then applied to the samples, and phages and/or bacteria were recovered from the filters as described above. Phages and bacteria were monitored by qPCR as described below, and the results obtained were compared with the values of the controls processed without filtration.
Standard and qPCR procedures.
Endpoint PCR amplifications were performed with a GeneAmp PCR 2400 system (Perkin-Elmer, PE Applied Biosystems, Barcelona, Spain) with the primers described in Table 1.
Phage 933W quantification.
A stx2 qPCR assay was used for phage 933W quantification, as previously described (19). Since Stx phages are known to carry only one stx copy, the stx gene copy (GC) values can be extrapolated to the number of Stx phages in each sample.
Recombinant E. coli C600 (933Wgfp) quantification.
In the samples spiked with bacteria and phages, and to discriminate bacteria from free Stx phages, recombinant E. coli C600 (933Wgfp) was used; to quantify these recombinant bacteria, a TaqMan qPCR assay for the detection of gfp was designed using the primers GFPTir-F/GFPTir-R and a minor groove binding probe, GFPTir, with a 5′ 6-carboxyfluorescein (FAM) reporter and a nonfluorescent quencher (NFQ). The qPCR assays were performed under standard conditions in a StepOne Real Time PCR System (Applied Biosystems) in a 20-μl reaction mixture with TaqMan Environmental Real-Time PCR master mix 2.0 (Applied Biosystems). All the samples and standards were run in triplicate, and the GC number was the average of the triplicate data obtained. For the generation of a gfp qPCR standard, plasmid pGreen TIR (28) was used and prepared as described previously (19). The assay showed an efficiency of 99.39%.
Statistical analyses.
Data and statistical tests were performed using the Statistical Package for Social Science software (SPSS). One-way analysis of variance (ANOVA) with a P value of 0.05 was used to evaluate the differences between the DNA extraction methods and between filtered and nonfiltered samples.
RESULTS
Comparison of DNA extraction methods.
As expected, phage DNA was extracted from the 933W suspension with similar efficiencies by all the methods used, and quantification of stx in the phage DNA did not show any significant (P > 0.05) differences between the method designed specifically for phage DNA extraction and the commercial methods intended for total (bacterial and phage) DNA extraction (Fig. 2). These results confirm that common laboratory protocols can indeed include phage DNA in the results when food, environmental, and clinical samples are processed. The experiments we detail below were performed with a QIAamp DNA Blood minikit, and a single DNA extraction was used to quantify either bacterial or phage DNA.
FIG 2.
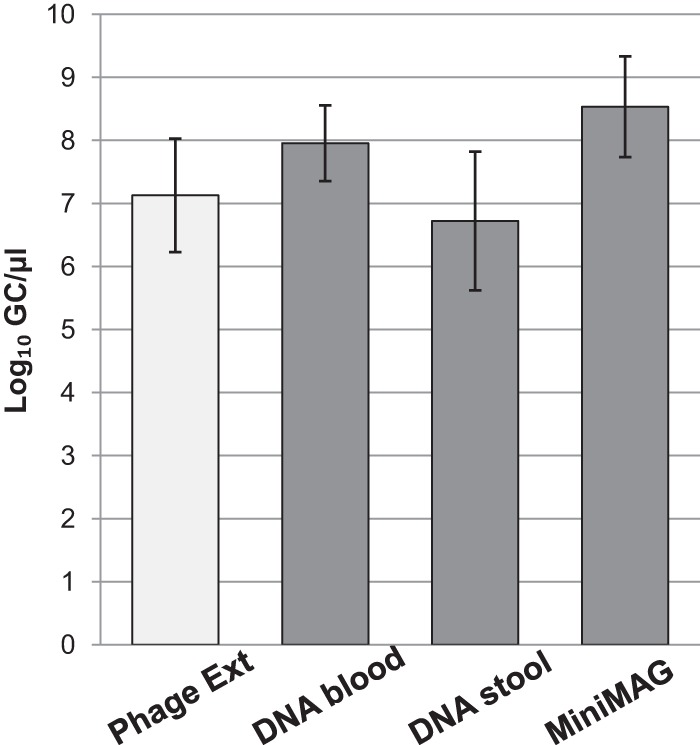
Concentrations of phage 933W evaluated by measurement of numbers of stx2 GC/μl from phage DNA extracted using conventional phage DNA extraction (Phage Ext) methods and commercial DNA extraction kits (QIAamp DNA Blood minikit, QIAamp DNA Stool minikit, and NucliSENS miniMAG).
Phage reduction procedure.
To determine whether the filtration step reduced the number of phage in a given sample, phage 933W was spiked in PBS. Samples were filtered through two different membrane filters: a 0.45-μm-pore-size mixed cellulose ester membrane and a 0.45-μm-pore-size PVDF membrane, described as a low-protein-binding membrane. To reduce phage content further, the filtration was followed by one washing step with PBS. After filtration, 933W was recovered from the filter and quantified by qPCR; results were then compared with those from the nonfiltered sample. All the filtration procedures significantly (P < 0.05) reduced the number of phage particles in the samples, but the use of low-protein-binding membranes resulted in a better reduction (Fig. 3A) (2.4 log10 GCs/μl) than filtration through mixed cellulose esters (0.8 log10 GCs/μl). Additional washing steps (three) were applied to the PVDF membrane to achieve a phage reduction of 3.3 log10 GCs/μl after filtration, compared with the initial sample (Fig. 3A). The experiments we describe below were therefore performed with filtration through PVDF membranes and three consecutive washing steps.
FIG 3.
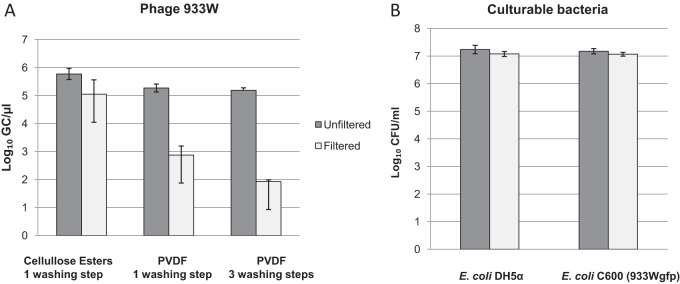
Concentrations of phage 933W (GC/μl) (A) and culturable E. coli strains DH5α and lysogen C600 (933Wgfp) (CFU/ml) (B) with or without application of the filtration procedure used to remove bacteriophage.
Analysis of the filtrate recovered after three washing steps allowed detection of 3.6 (0.13 standard deviation) log10 GCs/μl phage 933W, which is equivalent to the reduction observed by the filtration procedure.
Influence of the procedure on culturable bacteria.
The phage reduction procedure did not negatively affect recovery of culturable bacteria, as observed in experiments with PBS samples that were spiked with E. coli DH5α or with E. coli C600 laboratory lysogen containing phage 933Wgfp (Fig. 3B). This phage-containing lysogen was used to avoid manipulation of a pathogenic STEC strain and also as an STEC surrogate to evaluate an E. coli lysogen carrying an Stx phage under conditions as similar to those of an STEC strain as possible. No spontaneous release of the 933Wgfp phage during the cultivation of the strain before spiking the samples was expected due to the antibiotic selection, which allows growth of only the cells containing the phage. The numbers of culturable cells evaluated in LB agar for E. coli DH5α or in LB plus Cm for E. coli C600 (933Wgfp) at 37°C were not significantly different (P > 0.05) before and after the procedure (Fig. 3). Moreover, culturable E. coli strains DH5α and lysogen C600 (933Wgfp) (CFU/ml) were absent in the filtrate.
Evaluation of the procedure at different bacterial and phage concentrations.
To assess the influence of various densities of Stx phages or culturable bacterial cells in PBS, we evaluated the phage removal protocol at decreasing concentrations (from 105 to 102 CFU/ml) of culturable E. coli C600 (933Wgfp) in combination with decreasing concentrations (from 104 to 101 GCs/μl) of phage 933W as evaluated by qPCR. DNA extraction was performed for each mixture with and without application of the phage removal procedure (Fig. 4).
FIG 4.
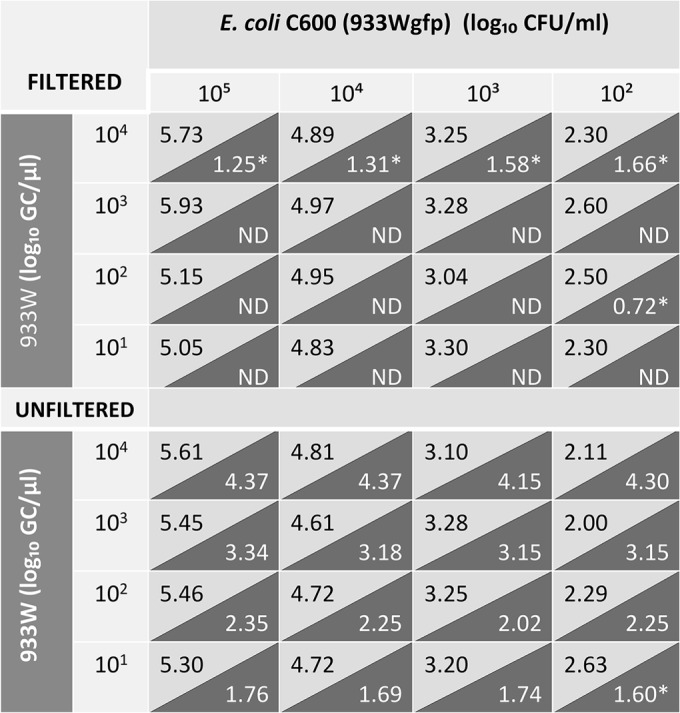
Concentrations of phage 933W (GC/μl) (dark gray) and culturable E. coli lysogenic strain C600 (933Wgfp) (CFU/ml) (light gray) in mixtures containing different phage and bacteria densities with or without the application of the filtration procedure used to remove bacteriophage. *, CT below the detection limit; ND, not detected.
As before, differences in numbers of culturable bacteria between the filtered and unfiltered samples were not significant (P > 0.05), indicating that the procedure did not interfere with bacterial determination. In contrast, the concentration of 933W fell below the limit of detection in the filtered samples. At the highest phage densities (105 GCs/μl), the phage was still detectable in the filtered samples although the values obtained were below the limit of detection required for positive samples in our qPCR assay, as determined by a threshold cycle (CT) of <32.0 (19). These results correspond to samples marked with an asterisk in Fig. 4. For the other dilutions, Stx phages were undetectable by the qPCR assay (CT of >33.5 or undetermined) (Fig. 4).
Application of the procedure for reduction of Stx phages in food and stool samples.
The phage reduction method was applied in different matrices: PBS (control), river water, salad, minced beef, and human feces. The samples were spiked with E. coli C600 (933Wgfp) and phage 933W at known densities (approximately 105 GCs/μl of bacteria or phage). To reproduce the real procedure, bacteria and phages were evaluated from a single DNA extraction and by qPCR (stx assay for phages and gfp assay for bacteria) (Fig. 1), and values were compared between filtered and unfiltered samples.
Significant (P < 0.05) reductions in the number of Stx phages were obtained by the procedure in all the matrices analyzed without any significant (P > 0.05) reduction in the densities of E. coli C600 (933Wgfp) (Fig. 5). The reduction efficiency varied between the different matrices. Liquid samples showed the best reductions; in solid samples, the procedure required sedimentation of the sample to avoid collapse of the filter membranes, which would reduce the efficacy of the procedure. Sedimentation was conducted by including soft centrifugation (300 × g for 10 min) that was confirmed not to reduce bacterial counts (Fig. 5).
FIG 5.

Simultaneous evaluation by qPCR of the densities of 933W and E. coli C600 (933Wgfp) in different matrices with and without the application of the filtration procedure.
Salad, meat, and stool samples were also used to evaluate the presence of Stx phages by endpoint PCR using primers UP378/LP378 (32). The samples were directly analyzed or spiked with 104 GCs/μl of phage 933W suspension, with or without filtration. Nonspiked samples were negatives for stx2. In the spiked samples, only unfiltered samples showed positive stx2 detection, while the filtered ones were negative by endpoint PCR.
DISCUSSION
An important volume of work reports positive detection of stx and therefore STEC by PCR or qPCR techniques but then fails to isolate an STEC strain (33, 34). This could be attributed to several causes, such as previous antibiotic treatment of the patient, a disease diagnosed late in its course, the presence of commensal E. coli and other commensal flora in the sample, even to the presence of viable but nonculturable microorganisms, or just because the target bacteria is present in very low numbers. However, here we present a new consideration: the presence of Stx phages in the samples and their interference in positive PCR detection or their inclusion in sequences generated by NGS. This possibility is very real since it has previously been shown that Stx phages are abundant in many environments, including the human gut (20, 21, 23, 29).
Our study confirms that the methods used for bacterial DNA extraction in these complex matrices (14, 35, 36) also extract phage DNA. While this result was expected, the efficiency of recovery of phage DNA had not been previously evaluated. Therefore, any virulence genes present in phage DNA, particularly stx but also other virulence genes described in prophages (e.g., nle and cdt), could be detected in the sample. When NGS is used, there is no easy way to establish whether the target detected is from bacteria or from a prophage if the contig is not a sequence long enough to contain bacterial DNA. Although it could be argued that detection of stx could be considered indicative of the presence of STEC, if it is from a phage, the threat for virulence in humans is much less important, and this should be considered. In terms of diagnosis or food safety analyses, false-positive results could interfere with determination of the real causative agent. Many PCR assays include multiple target genes that could help rule out false STEC detections (5, 7, 36), but, again, there is no confirmation that all genes detected are located in the same bacterial chromosome.
Moreover, from a more practical point of view, enrichment cultures used to selectively isolate bacteria can cause the propagation of any sort of phages present in a sample. Many natural samples carry virulent phages that will propagate during the culture by causing the lysis of the bacteria that is the intended target of the detection; this would cause a bias in the population of enriched bacteria (37). Phage reduction procedures offer the possibility of reducing the numbers of any sort of phages in food or stool samples, enhancing detection and plausibly isolation of the target pathogen.
Complete elimination of all phages present in the samples cannot be accomplished by the procedure presented here, and the fact that phages were still present in the samples after the procedure, even at low concentrations, could represent a problem because enrichment cultures will likely be performed with these samples, allowing phages to multiply. Nevertheless, a compromise can be found between an easy and feasible approach that could be applied in routine laboratories and that does not affect bacterial determination while causing a reasonable reduction on phage populations. In addition, the efficiency of the filtration would always be higher when aqueous samples or clear homogenates of solid samples are treated than when complex solid samples that require additional sedimentation steps are treated. In general, the additional time required to apply the procedure is around 15 min while the cost is negligible (limited only to the membranes), and the procedure requires basic laboratory equipment; the benefits should therefore be evaluated. Although the method has been checked on a laboratory basis using spiked samples, more studies using real STEC-positive samples, analyzed on a routine basis in parallel with or without phage reduction, should be conducted in future studies to validate and optimize this method.
New perspectives for pathogen detection are offered by NGS techniques (22, 38, 39). Regarding the implementation of these techniques for routine diagnosis of pathogens, the interference that phages could cause in the assembly of a genome sequence should be considered when DNA is obtained directly from the samples and not from isolates, particularly from the less abundant members of a microbial community (40). For example, virulence genes (stx or others) or antibiotic resistance genes, both reported in phages or phage-like elements (41–43), will certainly be detected, but the strain or species to which they belong, if any, will be unknown. The reduction of possible interferences would improve the efficiency and reliability of genomics when routinely applied.
ACKNOWLEDGMENTS
We thank S. Merino from the University of Barcelona for providing us with the plasmid pGreen TIR containing the green fluorescence protein.
This work was supported by the Spanish Ministry of Education and Science (AGL2012-30880) and the Generalitat de Catalunya (2009SGR1043) and by the Spanish Reference Network of Biotechnology (XeRBa). P.Q. and A.M.-C. received grants from the Spanish Ministry of Education and Science.
REFERENCES
- 1.Nataro JP, Kaper JB. 1998. Diarrheagenic Escherichia coli. Clin Microbiol Rev 11:142–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tarr PI, Gordon CA, Chandler WL. 2005. Shiga toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 365:1073–1086. doi: 10.1016/S0140-6736(05)71144-2. [DOI] [PubMed] [Google Scholar]
- 3.Paton JC, Paton AW. 1998. Pathogenesis and diagnosis of Shiga toxin-producing Escherichia coli infections. Clin Microbiol Rev 11:450–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson KE, Thorpe CM, Sears CL. 2006. The emerging clinical importance of non-O157 Shiga toxin-producing Escherichia coli. Clin Infect Dis 43:1587–1595. doi: 10.1086/509573. [DOI] [PubMed] [Google Scholar]
- 5.Paton JC, Paton AW. 2003. Methods for detection of STEC in humans. An overview. Methods Mol Med 73:9–26. [DOI] [PubMed] [Google Scholar]
- 6.Bibbal D, Loukiadis E, Kérourédan M, Peytavin de Garam C, Ferré F, Cartier P, Gay E, Oswald E, Auvray F, Brugère H. 2014. Intimin gene (eae) subtype-based real-time PCR strategy for specific detection of Shiga toxin-producing Escherichia coli serotypes O157:H7, O26:H11, O103:H2, O111:H8, and O145:H28 in cattle feces. Appl Environ Microbiol 80:1177–1184. doi: 10.1128/AEM.03161-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kagkli D-M, Weber TP, Van den Bulcke M, Folloni S, Tozzoli R, Morabito S, Ermolli M, Gribaldo L, Van den Eede G. 2011. Application of the modular approach to an in-house validation study of real-time PCR methods for the detection and serogroup determination of verocytotoxigenic Escherichia coli. Appl Environ Microbiol 77:6954–6963. doi: 10.1128/AEM.05357-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maurer JJ, Schmidt D, Petrosko P, Sanchez S, Bolton L, Lee MD. 1999. Development of primers to O-antigen biosynthesis genes for specific detection of Escherichia coli O157 by PCR. Appl Environ Microbiol 65:2954–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feng P, Weagant SD, Jinneman K. 2011. Diarrheagenic Escherichia coli, chapter 4A. In Bacteriological analytical manual online U. S. Food and Drug Administration, Silver Spring, MD: http://www.fda.gov/Food/FoodScienceResearch/LaboratoryMethods/ucm070080.htm. [Google Scholar]
- 10.Baquero F, Tobes R. Bloody coli: a gene cocktail in Escherichia coli O104:H4. mBio 4(1):e00066-13. doi: 10.1128/mBio.00066-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perelle S, Dilasser F, Grout J, Fach P. 2004. Detection by 5′-nuclease PCR of Shiga-toxin producing Escherichia coli O26, O55, O91, O103, O111, O113, O145 and O157:H7, associated with the world's most frequent clinical cases. Mol Cell Probes 18:185–192. doi: 10.1016/j.mcp.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 12.Gould LH, Bopp C, Strockbine N, Atkinson R, Baselski V, Body B, Carey R, Crandall C, Hurd S, Kaplan R, Neill M, Shea S, Somsel P, Tobin-D'Angelo M, Griffin PM, Gerner-Smidt P, Centers for Disease Control and Prevention (CDC) . 2009. Recommendations for diagnosis of Shiga toxin-producing Escherichia coli infections by clinical laboratories. MMWR Recomm Rep 58(RR-12):1–14. [PubMed] [Google Scholar]
- 13.Tseng M, Fratamico PM, Bagi L, Delannoy S, Fach P, Manning SD, Funk JA. 2014. Molecular characterization of Shiga toxin-producing E. coli (STEC) from finishing swine in a longitudinal study. Appl Environ Microbiol 80:6395–6402. doi: 10.1128/AEM.01761-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.International Organization for Standardization. 2012. Microbiology of food and animal feed. Real-time polymerase chain reaction (PCR)-based method for the detection of food-borne pathogens: horizontal method for the detection of Shiga toxin-producing Escherichia coli (STEC) and the determination of O157, O111, O26, O103 and O145 serogroups. ISO/TS 13136:2012. International Organization for Standardization, Geneva, Switzerland. [Google Scholar]
- 15.Commission of the European Communities. 2005. Commission regulation (EC) no. 2073/2005 on microbiological criteria for foodstuffs. Official Journal of the European Communities, L 388. Commission of the European Communities, Brussels, Belgium: http://faolex.fao.org/docs/pdf/eur61603.pdf. [Google Scholar]
- 16.Underwood AP, Dallman T, Thomson NR, Williams M, Harker K, Perry N, Adak B, Willshaw G, Cheasty T, Green J, Dougan G, Parkhill J, Wain J. 2013. Public health value of next-generation DNA sequencing of enterohemorrhagic Escherichia coli isolates from an outbreak. J Clin Microbiol 51:232–237. doi: 10.1128/JCM.01696-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muniesa M, Schmidt H. 2014. Shiga Toxin-encoding phages: multifunctional gene ferries, p 57–77. In Morabito S. (ed), Pathogenic Escherichia coli: molecular and cellular microbiology. Horizon Press, Norfolk, United Kingdom. [Google Scholar]
- 18.Schmidt H, Montag M, Bockemühl J, Heesemann J, Karch H. 1993. Shiga-like toxin II-related cytotoxins in Citrobacter freundii strains from humans and beef samples. Infect Immun 61:534–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Imamovic L, Serra-Moreno R, Jofre J, Muniesa M. 2010. Quantification of Shiga toxin 2-encoding bacteriophages, by real-time PCR and correlation with phage infectivity. J Appl Microbiol 108:1105–1114. doi: 10.1111/j.1365-2672.2010.04664.x. [DOI] [PubMed] [Google Scholar]
- 20.Rooks DJ, Yan Y, McDonald JE, Woodward MJ, McCarthy AJ, Allison HE. 2010. Development and validation of a qPCR-based method for quantifying Shiga toxin-encoding and other lambdoid bacteriophages. Environ Microbiol 12:1194–1204. doi: 10.1111/j.1462-2920.2010.02162.x. [DOI] [PubMed] [Google Scholar]
- 21.Martinez-Castillo A, Quirós P, Navarro F, Miró E, Muniesa M. 2013. Shiga toxin 2-encoding bacteriophages in human fecal samples from healthy individuals. Appl Environ Microbiol 79:4862–4868. doi: 10.1128/AEM.01158-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loman NJ, Constantinidou C, Christner M, Rohde H, Chan JZ-M, Quick J, Weir JC, Quince C, Smith GP, Betley JR, Aepfelbacher M, Pallen MJ. 2013. A culture-independent sequence-based metagenomics approach to the investigation of an outbreak of Shiga-toxigenic Escherichia coli O104:H4. JAMA 309:1502–1510. doi: 10.1001/jama.2013.3231. [DOI] [PubMed] [Google Scholar]
- 23.Imamovic L, Muniesa M. 2011. Quantification and evaluation of infectivity of Shiga toxin-encoding bacteriophages in beef and salad. Appl Environ Microbiol 77:3536–3540. doi: 10.1128/AEM.02703-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muniesa M, Lucena F, Jofre J. 1999. Comparative survival of free Shiga toxin 2-encoding phages and Escherichia coli strains outside the gut. Appl Environ Microbiol 65:5615–5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Allué-Guardia A, Martínez-Castillo A, Muniesa M. 2014. Persistence of infectious Shiga toxin-encoding bacteriophages after disinfection treatments. Appl Environ Microbiol 80:2142–2149. doi: 10.1128/AEM.04006-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rode TM, Axelsson L, Granum PE, Heir E, Holck A, L'abée-Lund TM. 2011. High stability of Stx2 phage in food and under food-processing conditions. Appl Environ Microbiol 77:5336–5341. doi: 10.1128/AEM.00180-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O'Brien AD, Newland JW, Miller SF, Holmes RK, Smith HW, Formal SB. 1984. Shiga-like toxin-converting phages from Escherichia coli strains that cause hemorrhagic colitis or infantile diarrhea. Science 226:694–696. doi: 10.1126/science.6387911. [DOI] [PubMed] [Google Scholar]
- 28.Miller WG, Lindow SE. 1997. An improved GFP cloning cassette designed for prokaryotic transcriptional fusions. Gene 191:149–153. doi: 10.1016/S0378-1119(97)00051-6. [DOI] [PubMed] [Google Scholar]
- 29.Imamovic L, Ballesté E, Jofre J, Muniesa M. 2010. Quantification of Shiga toxin-converting bacteriophages in wastewater and in fecal samples by real-time quantitative PCR. Appl Environ Microbiol 76:5693–5701. doi: 10.1128/AEM.00107-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Serra-Moreno R, Acosta S, Hernalsteens JP, Jofre J, Muniesa M. 2006. Use of the lambda Red recombinase system to produce recombinant prophages carrying antibiotic resistance genes. BMC Mol Biol 7:31. doi: 10.1186/1471-2199-7-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muniesa M, Jofre J. 1998. Abundance in sewage of bacteriophages that infect Escherichia coli O157:H7 and that carry the Shiga toxin 2 gene. Appl Environ Microbiol 64:2443–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martínez-Castillo A, Muniesa M. 2014. Implications of free Shiga toxin-converting bacteriophages occurring outside bacteria for the evolution and the detection of Shiga toxin-producing Escherichia coli. Front Cell Infect Microbiol 4:46. doi: 10.3389/fcimb.2014.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Urdahl AM, Solheim HT, Vold L, Hasseltvedt V, Wasteson Y. 2013. Shiga toxin-encoding genes (stx genes) in human faecal samples. APMIS 121:202–210. doi: 10.1111/j.1600-0463.2012.02957.x. [DOI] [PubMed] [Google Scholar]
- 35.Grys TE, Sloan LM, Rosenblatt JE, Patel R. 2009. Rapid and sensitive detection of Shiga toxin-producing Escherichia coli from nonenriched stool specimens by real-time PCR in comparison to enzyme immunoassay and culture. J Clin Microbiol 47:2008–2012. doi: 10.1128/JCM.02013-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Monday SR, Beisaw A, Feng PCH. 2007. Identification of Shiga toxigenic Escherichia coli seropathotypes A and B by multiplex PCR. Mol Cell Probes 21:308–311. doi: 10.1016/j.mcp.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 37.Muniesa M, Blanch AR, Lucena F, Jofre J. 2005. Bacteriophages may bias outcome of bacterial enrichment cultures. Appl Environ Microbiol 71:4269–4275. doi: 10.1128/AEM.71.8.4269-4275.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fricke WF, Rasko DA. 2014. Bacterial genome sequencing in the clinic: bioinformatic challenges and solutions. Nat Rev Genet 15:49–55. doi: 10.1038/nrg3624. [DOI] [PubMed] [Google Scholar]
- 39.Bertelli C, Greub G. 2013. Rapid bacterial genome sequencing: methods and applications in clinical microbiology. Clin Microbiol Infect 19:803–813. doi: 10.1111/1469-0691.12217. [DOI] [PubMed] [Google Scholar]
- 40.Relman DA. 2013. Metagenomics, infectious disease diagnostics, and outbreak investigations: sequence first, ask questions later? JAMA 309:1531–1532. doi: 10.1001/jama.2013.3678. [DOI] [PubMed] [Google Scholar]
- 41.Brüssow H, Canchaya C, Hardt W-D. 2004. Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol Mol Biol Rev 68:560–602. doi: 10.1128/MMBR.68.3.560-602.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Novick RP, Christie GE, Penadés JR. 2010. The phage-related chromosomal islands of Gram-positive bacteria. Nat Rev Microbiol 8:541–551. doi: 10.1038/nrmicro2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quirós P, Colomer-Lluch M, Martínez-Castillo A, Miró E, Argente M, Jofre J, Navarro F, Muniesa M. 2014. Antibiotic resistance genes in the bacteriophage DNA fraction of human fecal samples. Antimicrob Agents Chemother 58:606–609. doi: 10.1128/AAC.01684-13. [DOI] [PMC free article] [PubMed] [Google Scholar]