

Abstract
IGSF1 is a membrane glycoprotein highly expressed in the anterior pituitary. Pathogenic mutations in the IGSF1 gene (on Xq26.2) are associated with X-linked central hypothyroidism and testicular enlargement in males. In this study we tested the hypothesis that IGSF1 is involved in the development of pituitary tumors, especially those that produce growth hormone (GH). IGSF1 was sequenced in 21 patients with gigantism or acromegaly and 92 healthy individuals. Expression studies with a candidate pathogenic IGSF1 variant were carried out in transfected cells and immunohistochemistry for IGSF1 was performed in sections from GH-producing adenomas, familial somatomammotroph hyperplasia and in normal pituitary. In two male patients, and in one female, with somatomammotroph hyperplasia from the same family, we identified the sequence variant p.N604T, which in silico analysis suggested could affect IGSF1 function. Of 60 female controls, two carried the same variant, and seven were heterozygous for other variants. Immunohistochemistry showed increase IGSF1 staining in the GH-producing tumor from the patient with the IGSF1 p.N604T variant compared to a GH-producing adenoma from a patient negative for any IGSF1 variants and to normal control pituitary tissue. The IGSF1 gene appears polymorphic in the general population. A potentially pathogenic variant identified in the germline of three patients with gigantism from the same family (segregating with the disease) was also detected in two healthy female controls. Variations in IGSF1 expression in pituitary tissue in patients with or without IGSF1 germline mutations point to the need for further studies of IGSF1 action in pituitary adenoma formation.
Keywords: pituitary tumor, IGSF1, growth hormone, overgrowth, acromegaly, gigantism
Introduction
Gigantism, a rare condition that causes abnormal growth in children, often has a genetic etiology. Indeed, cases running in a family were first reported in the late 19th century (Herder 2012). Growth hormone (GH)-secreting pituitary adenomas and/or hyperplasia are the main causes of gigantism in childhood. These lesions, much more common in adults, have an annual incidence of approximately 3 per 1,000,000 and a prevalence of about 60 per 1,000,000 (Ezzat, et al. 2004).
IGSF1 is a plasma membrane glycoprotein encoded by the “Ig superfamily, member 1” (IGSF1) gene, located on Xq26.2. This gene is conserved in mammals and is expressed as transcripts of different lengths in many tissues, including muscle, heart, brain, testis, and pancreas (Frattini, et al. 1998). A previous study by Sun, et al. (Sun, et al. 2012) demonstrated that IGSF1 is highly expressed in the Rathke’s pouch and the adult anterior pituitary in humans. IGSF1 deficiency has been linked to congenital hypothyroidism of central origin (CeH), hypoprolactinemia, delayed puberty, testicular enlargement, increased body weight, and GH deficiency (Joustra, et al. 2013; Nakamura, et al. 2013; Sun, et al. 2012; Tajima, et al. 2013), which is mainly observed in males, as expected from an X-linked genetic defect. In Igsf1 knockout (KO) mice, a decrease in pituitary and circulating thyroid stimulating hormone (TSH) was observed, most probably secondary to impaired thyrotropin-releasing hormone (TRH) receptor expression and signaling (Sun, et al. 2012).
Based on this recent work from Sun, et al. (Sun, et al. 2012), we investigated IGSF1 germline variations in patients with gigantism and/or familial acromegaly from the NIH data registry and in healthy controls. We also test the expression of IGSF1 in GH-producing adenomas. Although our data do not prove a definitive link between pituitary tumor formation and IGSF1, the variation in its pituitary expression and its polymorphic content suggest that IGSF1 should be studied further as a possible modifier of somatomammotropinomas formation and/or their clinical expression.
Materials and Methods
Subjects & Protocol
The IGSF1 gene was screened for germline mutations in 21 patients (7 females and 14 males; one female and two males from the same family) with gigantism or acromegaly and in 92 previously described controls (100% white Americans, 60 females and 32 males) with a negative family history of endocrine disorders (Horvath, et al. 2009). All patients were previously reported (Glasker, et al. 2011; Stratakis, et al. 2010). Gigantism or acromegaly were diagnosed based on established criteria (Cook, et al. 2004): high IGF-1 levels according to age and sex, and serum GH concentration >1 ng/ml after a 2-hour 75 g oral glucose tolerance test (OGTT) in an appropriate clinical context, and of pituitary macro- (>10 mm) or micro- (<10 mm) adenomas or pituitary hyperplasia in magnetic resonance imaging (MRI) imaging. Leukocyte DNA was obtained from each patient. Written informed consent was isolated from all participants and the study was approved by the Institutional Review Boards of the participating institutions.
IGSF1 sequencing analysis
DNA was extracted from peripheral blood leucocytes according to manufacturer’s protocols (Qiagen, Valencia, CA, USA). For all patients and controls, the complete IGSF1-coding and flanking intronic sequence was analyzed, as previously described (Faucz, et al. 2011) using the primers and conditions described in Supplementary Table 1.
In silico analyses
The SIFT (Sorting Intolerant from Tolerant) (http://sift.bii.a-star.edu.sg/), the Align-GVGD (Grantham Variation Grantham Deviation) (http://agvgd.iarc.fr/) and PolyPhen (Polymorphism Phenotyping v2) (http://genetics.bwh.harvard.edu/pph2/) software packages were used to predict the pathogenic potential of the identified missense variants in IGSF1.
Cell cultures
GH3 cells (rat somatomammotroph pituitary cell line) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with high glucose (4500 mg/liter), 10% fetal bovine serum (FBS), penicillin (100 IU/ml), and streptomycin (100 mg/ml) in a humidified atmosphere at 37°C with 5% CO2. HEK293 cells were cultured as previously described (Sun, et al. 2012).
Transfection and ELISA
The p.N604T (c.1811A>C, rs146462069, NM_001170961.1) IGSF1 mutation was introduced by overlapping PCR in a pCMV6 IGSF1 gene open reading frame plasmid (ORIGENE – Rockville, MD, USA - cat#209621) or by site-directed mutagenesis as described in Sun, et al (Sun, et al. 2012). Approximately 1 × 105 GH3 cells were plated per well in 12-well cluster dishes overnight (37°C), washed and replenished with Opti-MEM. GH3 cell lines were transiently transfected by electroporation using Basic Nucleofector™ Kit for Primary Mammalian Endothelial Cells (Lanza, Basel, Switzerland, cat#VCA-1001) following manufacturer’s protocol. Cells were transfected with plasmid DNA (6 μg) expressing either the wild type (WT) or the variant form of IGSF1 and harvested 48 h after the transfection. The supernatant from GH3 transfected cells was analyzed for GH expression levels using the Rat/Mouse Growth Hormone ELISA Kit (catalog # EZRMGH-45K, Millipore, St. Charles, Missouri, USA) following manufacturer’s instructions.
Pulse-Chase Analysis (Metabolic Labeling)
Metabolic labeling studies were performed as described by Rejon, et al. (Rejon, et al. 2013). Briefly, 106 HEK293 cells were seeded in 35 mm dishes and transiently transfected the following day with 2 μg HA-tagged WT or p.N604T variant IGSF1 expression vector using Lipofectamine 2000 (Invitrogen), following the manufacturer’s instructions. Cells were cultured in methionine and cysteine-free DMEM (supplemented with 4 mM glutamine) for 3 h at 37°C. The culture medium was then additionally supplemented with 198 μCi [35S] methionine/[35S] cysteine (per mL) for 15 min. Cells were washed twice with warm PBS, then incubated in DMEM/10% FBS supplemented with 2 mM methionine, 2 mM cysteine, and 4 mM glutamine. Following 2, 4, 8, or 24 h at 37°C cells were lysed for 15 min, on ice, in 200 μl lysis buffer [PBS with 0.5% deoxycholate, 1% Triton X-100, 10 mM EDTA (pH 8.0), 1 mM phenylmethylsulfonyl fluoride and Complete™ Mini Protease Inhibitor Cocktail tablets, Roche, Nutley, NJ, USA]. Cells at the various time points were lysed after 15 min incubation with [35S] methionine/[35S] cysteine. Protein extracts were centrifuged for 15 min at 16,000 ×g to remove insoluble material. Ten microliter of supernatant was saved to assess the effectiveness of the labeling (‘total’) and the remaining immunoprecipitate (IP) with EZ-view Red anti-HA affinity gel, according to the manufacturer’s (Sigma, St. Louis, MO, USA) instructions. Gel-bound proteins were eluted by adding 16 μl of 2× loading buffer containing β-mercaptoethanol and boiling for 5 min at 95°C min. Proteins were resolved by SDS-PAGE (8% Tris-Glycine) and visualized by autoradiography.
Membrane trafficking analyses
Membrane transport of WT and p.N604T variant forms of IGSF1 was assessed by immunofluorescence or cell-surface biotinylation of transiently transfected HEK293 cells, as previously described (Sun, et al. 2012; Nakamura, et al. 2012).
Immunohistochemistry
Human pituitary tissue used for immunostaining was obtained during surgery and was fixed in formalin, paraffin embedded and sections (5μm) mounted onto 3-aminopropyl-triethoxylasine coated slides (Sigma Chemical Co., St Louis, MO). Routine staining with haematoxylin and eosin (H&E, Histoserv Inc, Germantown, MD, USA) was performed on several sections across each sample. Unstained slides were used for immunostaining, the procedure is outlined as follows: Sections were deparaffinized with Histoclear (National Diagnostics, Atlanta, GA) and rehydrated with a graded series of ethanol (absolute, 95%, 70% and 50% ethanol and distilled water), followed by antigen retrieval, which was performed by boiling the tissue sections in Antigen Unmasking Solution (pH 6; Vector Laboratories, Burlingame, CA) for 20 min in a pressure cooker; sections were then allowed to cool to room temperature (20 min). Sections were immunostained using the antibody against IGSF1 (rabbit anti-human diluted 1:1000, Abcam ab66509, Cambridge, MA, USA). Immunostaining was identified by colorimetric staining using the ImmPRESS Polymer Detection Kit (Rabbit; Vector Laboratories, Burlingame, CA) and counterstained with haematoxylin. Briefly, once sections had cooled to room temperature following antigen retrieval, sections were blocked in 0.3% (v/v) H2O2 (Sigma, CA) made in methanol for 30 minutes. After a wash with Tris-buffered Saline (TBS, pH 7.4; diluted to 1X from a 10X stock, Quality Biologicals, Inc., Gaithesburg, MD) containing 0.01% Tween-20 (Life Technologies, Carlsbad, CA) (TBS-T; 1 × 15 min) sections were blocked for 1 h at room temperature using 2.5% normal horse serum (provided with the ImmPRESS Polymer Detection Kit). Tissues sections were then incubated with primary antibody made in blocking serum, in a humidified chamber overnight at 4°C. Sections were subsequently washed with TBS-T (3 × 5 min) and incubated for 30 minutes at room temperature with the ImmPRESS reagent, followed by another washing step in TBS-T (3 × 5 min). Sections were incubated with counterstain ImmPACT DAB peroxidase substrate (Vector Laboratories, Burlingame, CA), as per manufacturer’s instructions. Sections were then counterstained with Mayer’s hematoxylin, mounted and coverslips applied with VectaMount AQ Aqueous Mounting Medium (Vector Laboratories, Burlingame, CA), or mounted without counterstaining.
Homology model
Homology models of the 6th Ig loop in IGSF1 with Asn or Thr at position 604 were generated using Phyre2 (http://www.sbg.bio.ic.ac.uk/phyre2/) and Quark (http://zhanglab.ccmb.med.umich.edu/QUARK/). Figures of the resulting models were generated using PyMol (http://pymol.org/).
Statistical analysis
Data are described as frequencies and percentages, mean ± standard deviation or median (inter-quartile range), as appropriate, and were analyzed using SAS v9.1 (SAS Inc, Cary, NC). All experiments were repeated at least three times. Categorical data were compared using the Fisher’s exact test. A p-value ≤ 0.05 was considered statistically significant.
Results
We identified a nonsynonymous genetic IGSF1 variant (p.N604T, c.1811A>C, rs146462069) in two males and one female (from the same family – a mother and two sons). All three patients showed evidence of central hypothyroidism, however they had either pituitary somatomammotroph tumors and/or hyperplasia or were seen in our hospital after they had their first operation. Eighteen additional unrelated patients were negative for the variant. All patients have been described previously (Glasker, et al. 2011; Stratakis et al. 2010). The minor allele frequency of this variant is 0.01 (http://www.1000genomes.org/). In silico analysis predicted this variant as possibly damaging. Of the 60 healthy females controls, 2 carried this variant, and 7 carried various other nonsynonimous variants (1 nonsense, 1 frameshift and 6 missense, Table 1). The variant was not seen in any of the 32 healthy males studied.
Table 1.
Variants found in 92 healthy controls
| Chrom X Position (in hg19) | DNA change# | protein change | exon | SNP ID | Domains | in silico modeling prediction
|
||
|---|---|---|---|---|---|---|---|---|
| SIFT | Polyphen | Align-GVGD | ||||||
| 130,420,579 | c.70C>T | p.R24W | 2 | COSM1465661 | signal peptide | tolerated | benign | probably damaging |
| 130,419,882 | c.238A>G | p.I80V | 4 | Novel | - x - | tolerated | benign | benign |
| 130,419,793 | c.327G>T | p.W109C | 4 | Novel | - x - | tolerated | probably damaging | probably damaging |
| 130,417,192 | c.714G>A | p.M238I | 6 | Novel | - x - | tolerated | benign | benign |
| 130,412,680 | c.1811A>C | p.N604T | 12 | rs146462069 | CTC | tolerated | probably damaging | possibly damaging |
| 130,412,680 | c.1811A>C | p.N604T | 12 | rs146462069 | CTC | tolerated | probably damaging | possibly damaging |
| 130,412,078 | c.2091dupC | p.Thr698Hisfs*24 | 13 | Novel | CTC | |||
| 130,412,018 | c.2147G>A | p.G716E | 13 | Novel | CTC | tolerated | benign | probably damaging |
| 130,408,107 | c.3844delC | p.V1282X | 19 | Novel | CTC | |||
CTD: C-terminal domain;
All DNA changes were found in females in heterozygosity; GVGD Grantham variation / Grantham deviation
To investigate whether the p.N604T variant could be related to gigantism in our patients, we examined its impact on IGSF1 protein function. Transfection of GH3 cells with the p.N604T IGSF1 variant did not significantly affect GH production compared to cells transfected with WT IGSF1 (P=0.1561, data not shown). Metabolic studies showed that both the WT and mutant proteins exhibit the same pattern of maturation and stability when expressed in heterologous cells (Supplemental Figure 1). Both proteins were detected in the plasma membrane by immunofluorescence (Figure 1A) or cell surface biotinylation (Figure 1B). By western blot, the WT and p.N604T proteins were indistinguishable (Fig. 1B, lower panels, lanes 2 and 3).
Figure 1.

IGSF1 p.N604T traffics to the plasma membrane. A – HEK293 cells were transfected with pcDNA3, IGSF1 WT or IGSF1 p.N604T constructs and subjected to immunofluorescence under permeabilizing (bottom panels) and non-permeabilizing conditions (top panels). The proteins were detected with an antibody that recognizes the N-terminus of the IGSF1 C-terminal domain. B – HEK293 cells were transfected with the indicated constructs and subjected to cell surface biotinylation followed by immunoprecipitation. Streptavidin-HRP signals of equal intensity was detected in cell lysates from WT and p.N604T transfected cells (top panel). The middle panel shows equivalent IP of the two proteins whereas the bottom panel shows equivalent expression of the two proteins. Note that IGSF1 migrates as a double on SDS-PAGE. The lower band corresponds to the core (immature) glycoform whereas the upper band is the mature glycoform.
Asn604 maps to Ig loop 6 (of a total of 12) in the IGSF1 protein. IGSF1 is co-translationally cleaved into N- and C-terminal domains (NTD and CTD, respectivaly). Ig loop 6 is at the N-terminus of the CTD. We generated a homology model of Ig loop 6 and mapped Asn604 to a solvent exposed surface (Figure 2). Though no IGSF1 interacting partners have yet been identified, it is possible that the Asn 604 Thr substitution could affect a protein-protein interaction surface.
Figure 2.

3D homology model of the IGSF1 Ig loop 6 in which the variant p.N604T resides. The arrows show the position of Asn 604 and Thr 604.
Finally, immunohistochemical analysis of our patient’s pituitary tumors carrying the IGSF1 variant showed increased IGSF1 staining compared to normal pituitary control or a GH-producing adenoma from a patient without the IGSF1 variant (Figure 3). Additional 3 GH-producing adenomas from patients with acromegaly and no IGSF1 variants showed a similar pattern to that of the tumor shown in Figure 3 (data not shown).
Figure 3.
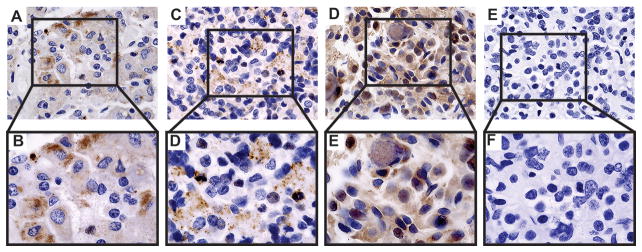
Immunohistochemistry of IGSF1 in normal pituitary and in GH-producing pituitary adenomas with and without p.N604T IGSF1 sequencing variant. A/B – Normal Pituitary; C/D – Patient with a GH producing adenoma without IGSF1 sequencing variants; E/F – Patient with Pituitary hyperplasia with p.N604T IGSF1 variant; G/H – negative control.
Discussion
We identified a hemizygous/heterozygous IGSF1 variant (p.N604T) segregating with gigantism in three patients in a family. Mutations in this gene were recently linked with congenital central hypothyroidism (CCH) both in humans and mice (Nakamura et al. 2013; Sun et al. 2012; Tajima et al. 2013), perhaps secondary to loss of TRH receptor expression and signaling in the pituitary (Sun et al. 2012). Considering its involvement in pituitary function and particularly in abnormal TSH secretion, as well as its demonstrated expression in murine somatotrope cells, we examined whether IGSF1 plays a causative role in our cohort of patients with gigantism or early onset acromegaly.
Sequencing analysis of our 21 patients revealed an IGSF1 sequencing variant that we also observed in 2 female heterozygous controls. To date, 14 pathogenic mutations have been described in the IGSF1 gene (Nakamura et al. 2013; Sun et al. 2012; Tajima et al. 2013), all impair either protein maturation or membrane trafficking. Among these mutations, 2 are complete gene deletions and the remaining 12 are located in the extracellular portion of the C-terminal domain (CTD), an indication that this may represent an important functional domain of the protein.
Eight different variants (1 nonsense, 1 frameshift and 6 missense) were identified in our control group; all in females. We would expect that if the nonsense and frameshift mutations were carried by males, they might lead to some IGSF1 deficiency phenotypes, as CCH and several of the other clinical features of IGSF1 deficiency syndrome. Among the missense variants, two (p.W109C and p.N604T) were predicted as damaging by two programs and the remaining four (p.R24W, p.I80V, p.M238I and p.G716E) were predicted as benign by at least two programs. At a minimum, these observations reveal considerable polymorphism in this gene, including variants that could impair protein function.
A difference in the expression of IGSF1 in human pituitary was also noted; IGSF1 was intensely expressed in the tumor from one of the hemizygous (male) patients carrying the IGSF1 variant compared to patients with GH-producing adenomas without an IGSF1 variant or to a normal pituitary tissues. However, we did not observe differences in GH secretion from GH3 cells engineered to express the p.N604T variant versus WT IGSF1, indicating that the variant is not sufficient to directly affect GH production and secretion.
Previously described pathogenic IGSF1 mutations affect the maturation and plasma membrane trafficking of the protein’s CTD. However we failed to detect any effect of the p.N604T IGSF1 variant on protein expression, maturation, stability, or membrane trafficking in heterologous cells. Therefore, if this variant alters IGSF1 function, it is likely through its extracellular activities. Given the prediction that the modified residue is solvent exposed and modifies the surface charge in the 6th Ig loop (N-terminus of the CTD), it is tempting to speculate that the variant might alter IGSF1’s interaction with an extracellular partner. However, this possibility must await the identification of extracellular ligands for the protein.
Although the sequence variant detected in our patients does not appear to affect GH levels, it may be acting as a modifier in GH and prolactin production and secretion. This is supported by the observation that the same variant was also found in a Finnish family with apparent X-linked delayed puberty, the index male case showing tall stature (197 cm) and hyperprolactinemia, with normal brain MRI (Joustra, et al. 2014).
In conclusion, the IGSF1 gene is polymorphic. The variant identified in three of our patients with gigantism was also detected in apparently healthy female controls. It is also possible that the variant-bearing allele is in linkage disequilibrium with the actual genetic cause of the phenotype in our family. Indeed, the apparent increase in pituitary IGSF1 expression in patients with an IGSF1 germline variant may indicate that IGSF1, while not be playing a causative role in pituitary tumor development, could act synergistically in tumor development.
Supplementary Material
HEK293 cells were transfected with pcDNA3 (p3) or expression vectors for wild type or the N604T variant of IGSF1. Cells were cultured in the presence of 35S-labeled methionine and cysteine for 15 min. Cells were then harvested immediately (time 0 h) or cultured with unlabeled amino acids for an additional 2, 4, 8, or 24 hours. Protein lysates were immunoprecipitated (IP) with an HA antibody and run on SDS-PAGE. Gels were then dried and exposed to film. IGSF1 migrates in two predominant forms: core glycosylated (closed arrow) or as a mature glycoform (open arrow). The bottom blot shows protein labeling prior to IP. Molecular weight markers (in kDa) are presented at the left.
Acknowledgments
Funding
This study was supported by the Intramural Research Program, Eunice Kennedy Shriver National Institute of Child Health & Human Development (NICHD), National Institutes of Health (NIH) (to Constantine A. Stratakis). Additional support was provided from a post-doctoral fellowship grant from CNPq and from the University of Brasilia, Brazil (to Monalisa F. Azevedo; mentor: Dr. Stratakis), in part, by a grant from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Process: 311166/2011-3 - PQ-2 (to Fabio Rueda Faucz), and in part by a grant from Government of Canada-Canadian Institutes of Health Research, grant number CIHR MOP-133557 (to Daniel J. Bernard).
The authors thank Dr. Sjoerd Joustra for sharing unpublished data on a family with the same gene variant, Dr. Dmitry Rodionov for his assistance with the homology modeling, Dr. Tanya Silander for her help with preparing some of the figures and Dr. Stephen Butler for assistance in specimen collection.
Footnotes
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Author contributions
The contribution of each author is as follows: Fabio R. Faucz performed the in silico analyses, some of the molecular experiments described in the manuscript and prepared the manuscript. Anelia D. Horvath and Monalisa F. Azevedo, Allison D. Manning, Rodrigo Bertollo de Alexandre and Giampaolo Trivellin performed most of the molecular experiments. Isaac Levy and Emmanouil Saloustros were involved in immunohistochemical studies. Beata Bak and Ying Wang performed in vitro analyses of the IGSF1 variant. Paraskevi Xekouki and Eva Szarek participated in manuscript preparation and editing Evgenia Gourgari, Maya Lodish, Paul Hofman, Yvonne C. Anderson, Ian Holdaway and Nienke R. Biermasz were the clinicians involved in clinical analysis, patients’ caring and samples colection. Edward Oldfield was the physician who took care of the described kindred at the NIH. Prashant Chitiboina was involved in pathology evaluation. Maria Nesterova prepared the clinical specimens for genetic analysis. Jan M. Wit was involved in the interpretation of the data and in revising the various draft versions of the manuscript. Daniel J. Bernard directed the in vitro investigations of the IGSF1 variant, generated the homology model, and contributed to the writing of the manuscript. Constantine A. Stratakis was the senior investigator at NICHD, which provided most of the funding for this project under the NIH Intramural Research Program, and overall supervised the experiments, presentation of results, design of figures, and writing the manuscript.
References
- Cook DM, Ezzat S, Katznelson L, Kleinberg DL, Laws ER, Jr, Nippoldt TB, Swearingen B, Vance ML, Force AAGT. AACE Medical Guidelines for Clinical Practice for the diagnosis and treatment of acromegaly. Endocrine practice : official journal of the American College of Endocrinology and the American Association of Clinical Endocrinologists. 2004;10:213–225. doi: 10.4158/EP.10.3.213. [DOI] [PubMed] [Google Scholar]
- Ezzat S, Asa SL, Couldwell WT, Barr CE, Dodge WE, Vance ML, McCutcheon IE. The prevalence of pituitary adenomas: a systematic review. Cancer. 2004;101:613–619. doi: 10.1002/cncr.20412. [DOI] [PubMed] [Google Scholar]
- Faucz FR, Horvath A, Rothenbuhler A, Almeida MQ, Libe R, Raffin-Sanson M-L, Bertherat J, Carraro DM, Soares FA, Molina GdC, et al. Phosphodiesterase 11A (PDE11A) genetic variants may increase susceptibility to prostatic cancer. The Journal of clinical endocrinology and metabolism. 2011;96:E135–140. doi: 10.1210/jc.2010-1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frattini A, Faranda S, Redolfi E, Allavena P, Vezzoni P. Identification and genomic organization of a gene coding for a new member of the cell adhesion molecule family mapping to Xq25. Gene. 1998;214:1–6. doi: 10.1016/s0378-1119(98)00253-4. [DOI] [PubMed] [Google Scholar]
- Glasker S, Vortmeyer AO, Lafferty AR, Hofman PL, Li J, Weil RJ, Zhuang Z, Oldfield EH. Hereditary pituitary hyperplasia with infantile gigantism. The Journal of clinical endocrinology and metabolism. 2011;96:E2078–2087. doi: 10.1210/jc.2011-1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herder WWd. Familial gigantism. Clinics (Sao Paulo, Brazil) 2012;67(Suppl 1):29–32. doi: 10.6061/clinics/2012(Sup01)06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath A, Korde L, Greene MH, Libe R, Osorio P, Faucz FR, Raffin-Sanson ML, Tsang KM, Drori-Herishanu L, Patronas Y, et al. Functional phosphodiesterase 11A mutations may modify the risk of familial and bilateral testicular germ cell tumors. Cancer Res. 2009;69:5301–5306. doi: 10.1158/0008-5472.CAN-09-0884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joustra SD, Schoenmakers N, Persani L, Campi I, Bonomi M, Radetti G, Beck-Peccoz P, Zhu H, Davis TM, Sun Y, et al. The IGSF1 deficiency syndrome: characteristics of male and female patients. The Journal of clinical endocrinology and metabolism. 2013;98:4942–4952. doi: 10.1210/jc.2013-2743. [DOI] [PubMed] [Google Scholar]
- Joustra SD, Wehkalampi K, Oostdijk W, Biermasz NR, Howard S, Silander T, Bernard DJ, Wit JM, Dunkel L, Losekoot M. IGSF1 variants in boys with familial delayed puberty. European Journal of Pediatrics. 2014 doi: 10.1007/s00431-014-2445-9. In press. [DOI] [PubMed] [Google Scholar]
- Nakamura A, Bak B, Silander TLR, Lam J, Hotsubo T, Yorifuji T, Ishizu K, Bernard DJ, Tajima T. Three Novel IGSF1 Mutations in Four Japanese Patients With X-Linked Congenital Central Hypothyroidism. The Journal of clinical endocrinology and metabolism. 2013;98:E1682–1691. doi: 10.1210/jc.2013-1224. [DOI] [PubMed] [Google Scholar]
- Rejon CA, Ho CC, Wang Y, Zhou X, Bernard DJ, Hebert TE. Cycloheximide inhibits follicle-stimulating hormone beta subunit transcription by blocking de novo synthesis of the labile activin type II receptor in gonadotrope cells. Cellular signalling. 2013;25:1403–1412. doi: 10.1016/j.cellsig.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Stratakis CA, Tichomirowa MA, Boikos S, Azevedo MF, Lodish M, Martari M, Verma S, Daly AF, Raygada M, Keil MF, et al. The role of germline AIP, MEN1, PRKAR1A, CDKN1B and CDKN2C mutations in causing pituitary adenomas in a large cohort of children, adolescents, and patients with genetic syndromes. Clin Genet. 2010;78:457–463. doi: 10.1111/j.1399-0004.2010.01406.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Bak B, Schoenmakers N, van Trotsenburg ASP, Oostdijk W, Voshol P, Cambridge E, White JK, le Tissier P, Gharavy SNM, et al. Loss-of-function mutations in IGSF1 cause an X-linked syndrome of central hypothyroidism and testicular enlargement. Nature genetics. 2012;44:1375–1381. doi: 10.1038/ng.2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima T, Nakamura A, Ishizu K. A novel mutation of IGSF1 in a Japanese patient of congenital central hypothyroidism without macroorchidism. Endocrine journal. 2013;60:245–249. doi: 10.1507/endocrj.ej13-0009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
HEK293 cells were transfected with pcDNA3 (p3) or expression vectors for wild type or the N604T variant of IGSF1. Cells were cultured in the presence of 35S-labeled methionine and cysteine for 15 min. Cells were then harvested immediately (time 0 h) or cultured with unlabeled amino acids for an additional 2, 4, 8, or 24 hours. Protein lysates were immunoprecipitated (IP) with an HA antibody and run on SDS-PAGE. Gels were then dried and exposed to film. IGSF1 migrates in two predominant forms: core glycosylated (closed arrow) or as a mature glycoform (open arrow). The bottom blot shows protein labeling prior to IP. Molecular weight markers (in kDa) are presented at the left.