

Abstract
Background
The optimal laboratory assay for detecting KRAS mutations in different biospecimens from patients with metastatic colorectal cancer (mCRC), and the clinical relevance of these gene alterations is still in question. We analyzed the prognostic–predictive relevance of KRAS status, determined in tumor and plasma DNA by two different assays, in a large mono-institutional series of mCRC patients.
Methods
DNA sequencing and peptide-nucleic-acid-mediated-polymerase chain reaction clamping (PNA-PCR) were used to determine KRAS status in 416 tumor and 242 matched plasma DNA samples from mCRC patients who received chemotherapy only. Relationships with outcomes were analyzed with respect to the different assays and tissue types.
Results
PNA-PCR was significantly more sensitive in detecting KRAS mutations than sequencing (41% vs. 30%, p < 0.001). KRAS mutations were more frequent in tumor tissue than in plasma (sequencing, 38% vs. 17%, p < 0.001; PNA-PCR, 47% vs. 31%, p < 0.001). Median OS was consistently shorter in KRAS-mutated patients than KRAS wild-type patients, independent from the assay and tissue tested; the largest difference was in plasma samples analyzed by PNA-PCR (KRAS mutated vs. wild-type: 15.7 vs. 19.1 months, p = 0.009). No association was observed between KRAS status and other outcomes. When tumor and plasma results were considered together, median OS in patients categorized as tissue/plasma KRAS negative/negative, tissue/plasma KRAS discordant, and tissue/plasma KRAS positive/positive were 21.0, 16.9 and 15.4 months, respectively (p = 0.008).
Conclusions
KRAS mutation status is of prognostic relevance in patients with mCRC. KRAS mutations in both tumor tissue and plasma are a strong prognostic marker for poor outcomes.
Electronic supplementary material
The online version of this article (doi:10.1186/s13046-014-0104-7) contains supplementary material, which is available to authorized users.
Keywords: Kras, Colorectal cancer, Prognosis
Background
K-RAS is a key oncogene member of the mitogen-activated protein kinase signaling pathway [1,2]. Research primarily in colorectal cancer shows that KRAS mutations occur most commonly in codons 12 and 13, usually precede the development of malignancy [3,4], and are maintained in secondary disease sites [5].
Several studies have addressed the prognostic-predictive value of KRAS mutational status in colorectal cancer patients [6]. It is now well established that KRAS mutations are the main reason for resistance to anti-epidermal growth factor receptor (EGFR) antibodies [7,8], and account for nearly two-thirds of EGFR downstream effector alterations in colorectal cancer [9]. KRAS mutations are also a predictive factor for response to tyrosine kinase inhibitors [10].
Clinical data regarding the prognostic value of KRAS mutations in patients with metastatic colorectal cancer (mCRC) treated with chemotherapy remain inconclusive [1]. Most studies addressing this question were retrospective or included limited patient numbers [6]. Some prospective studies also reached discordant conclusions [11] because the data were derived from the control arm of randomized trials of anti-EGFR antibodies as first-line treatment in combination with chemotherapy. In these studies, cross-over to anti-EGFR therapy was permitted or patients received anti-EGFR therapy after study treatment, making prognostic interpretation difficult. The prognostic-predictive relevance of KRAS alterations to chemotherapy alone in mCRC has still to be determined in a large patient sample.
Although direct sequencing is widely accepted as the gold standard for mutation screening [12], this method requires that ≥25% of DNA alleles in the sample are mutated [6,13]. Over the past five years, several new techniques, including peptide-nucleic-acid-mediated polymerase chain reaction clamping (PNA-PCR), have emerged. These methods have higher sensitivity than direct sequencing, and permit the detection of lower mutation frequencies (1%-5%) [14-16].
Routine assessment of KRAS mutational status is generally performed in tumor samples and used to personalize treatment in patients with mCRC [14,17]. Oh et al. combined PNA-Mediated Asymmetric PCR with Melting Curve Analysis to detect several types of low-level KRAS mutations in colorectal cancer tissues [18]. However, a recent review of 11 clinical studies, reported that 29%-100% of patients presented with the same KRAS mutation in both blood and tumor samples suggesting that blood samples may also be suitable for determining KRAS status [19-21]. One study performed by Yu et al. has showed that PNA-PCR powered by pyrosequencing had the potenial to screen plasma KRAS mutations with high sensitivity and accuracy in pancreatic cancer patients [22].
We hypothesized that KRAS mutation status determined using PNA-PCR in tumor tissue and/or blood could be a powerful and easy-to-perform approach for planning treatment in patients with advanced colorectal cancer. In the present study, we analyzed the impact of KRAS status, determined by both direct sequencing and PNA-PCR methods in tumor and matched plasma samples, on clinical outcome in a large consecutive mono-institutional series of advanced colorectal cancer patients. All patients received oxaliplatin-based or irinotecan-based chemotherapy as first-line and second-line treatment, but never received biologic therapy.
Methods
Study design
Between January 2007 and June 2011, 566 consecutive patients with mCRC were admitted to the Affiliated Hospital Cancer Center of the Academy of Military Medical Sciences in Beijing and were treated with systemic chemotherapy. 416 patients meeting the inclusion criteria were enrolled into this study retrospectively. The study was approved by the ethical committee of Affiliated Hospital, Academy of Military Medical Sciences (ID-2011-91). All patients provide their written informed consent to participate in this study.
The inclusion criteria were: completion of ≥2 cycles of oxaliplatin-based or irinotecan-based chemotherapy as first-line treatment; no anti-EGFR or anti-vascular endothelial growth factor (VEGF) treatment; measurable disease; adequate follow-up for disease and survival assessment; adequate tumor tissue and paired plasma samples (if available) taken before chemotherapy; tumor specimens with ≥50% tumor cells confirmed by certified pathologists; and tumor specimens obtained from surgical resection, colonoscopy biopsy, or metastatic site biopsy. Written informed consent was obtained from each patient.
Reasons for excluding patients (n = 150) from the consecutive series were: subsequent treatment with anti-EGFR and/or anti-VEGF antibodies (n = 85); non-measurable disease (n = 23); received only 1 chemotherapy cycle (n = 11); inadequate follow-up (n = 9); tumor tissue unavailable (n = 15); <50% tumor cells confirmed by pathologists (n = 4); and inadequate tumor specimen (n = 3).
Tissue samples
Formalin-fixed, paraffin-embedded (FFPE) tumor tissues were retrieved at room temperature, assessed by sectioning, and hematoxylin-eosin stained by certified pathologists. The pathologist was responsible for tissue block selection and evaluation of neoplastic cellularity. Each evaluation was confirmed by two pathologists.
Whole blood samples were collected for DNA extraction from each participant before any invasive procedures or therapy. Five milliliters of peripheral blood were collected from each participant in a vacutainer system with lithium-heparin. Plasma was immediately separated from the cellular fraction by centrifugation at 1,500 × g for 10 min and, after aliquotation, frozen at −80°C.
Direct sequencing and PNA-PCR were performed with tumor and plasma samples to determine KRAS status.
DNA extraction and KRAS mutation analysis
Genomic DNA of tumor tissue was extracted using EZNA™ FFPE DNA Kit D3399 (Omega Bio-Tek, Inc, Norcross, GA, USA). Free plasma was purified by NucleoSpin® Plasma, N.740900 (Macherey-Nagel GmBH & Co, Düren, Germany). All DNA samples were stored at −80°C. Since most KRAS mutations occur at codons 12 or 13 of exon 2, PCR primers were designed to amplify the corresponding region. PCR products were purified by EZNA® Cycle Pure Kit (Omega Bio-Tek, Inc, Norcross, GA, USA).
Direct sequencing of KRAS in plasma or FFPE was performed with Applied Biosystems® 3100 Genetic Analyzer (Life Technologies, Carlsbad, CA, USA) according to manufacturer’s protocol.
All available tumor and plasma samples were tested by PNA-PCR. Peptide-nucleic-acid (PNA) oligomers are non-extendable oligonucleotides. In PNA-mediated PCR clamping, PNA oligomers suppress the amplification of the complementary sequence because PNA are not substrates for DNA polymerase. The PNA oligomers (with sequence of CTACGCCACCAGCTC) covered wild-type codons 12 and 13 of KRAS. The nested PCR system included: 50 ng genomic DNA; 25 μL MightyAmp Buffer; 1 μL MightyAmp DNA Polymerase (DR071, TaKaRa Bio Inc, Shiga, Japan); 0.01 μM KRAS primers; and 0.2 μM PNA. After initial denaturation at 94°C for 5 min, PCR amplification consisted of 35 cycles: 94°C for 30 s; 70°C for 10 s; 56°C for 30 s; and 68°C for 30 s. This was followed by elongation at 68°C for 5 min with final cooling at 4°C. The sequences of primer pairs in the first run were GTGTGACATGTTCTAATATAGTCA and GAATGGTCCTGCACCAGTAA, while the inner forward primer ATGTTCTAATATAGTCACATTTTC and reverse primer GGTCCTGCACCAGTAATATGCA were used in the second run. The KRAS mutations in the nested PCR products were detected by subsequent Sanger sequencing.
Personnel responsible for mutation analysis were blinded to clinical outcomes. All experiments were performed at the Affiliated Hospital Pharmacology Laboratory for Cancer Research, and all the samples were genotyped twice for quality control purposes.
Statistical analysis
The chi-squared test was used to determine the association between baseline characteristics, clinical response and KRAS status. Kaplan-Meier methods were used to estimate progression-free survival (PFS, defined as time between first day of first-line chemotherapy and disease progression or death from any cause) and overall survival (OS, defined as time between first day of first-line chemotherapy and death from any cause or date of last follow-up). Response was evaluated by the Response Evaluation Criteria in Solid Tumors. Cox proportional hazards models were used for multivariate analyses of baseline characteristics and KRAS mutations related to OS. In the multivariate Cox proportional hazard regression model, gender, age, Eastern Cooperative Oncology Group (ECOG) performance status, metastatic site, and KRAS status were used as covariates. The association between KRAS status in tumor tissue and plasma free DNA was examined by the Kappa test. All statistical tests were performed using SAS 9·2 software (SAS Institute, Cary NC, USA).
Results
Patient characteristics
A total of 416 patients met the enrollment criteria and had primary or metastatic tumor samples; 242 patients had paired plasma DNA samples. The characteristics of the patients were: 218 men and 198 women; median age of 56 (range, 26–87) years; and 365 patients (87.7%) had an ECOG performance status of 0–1. The characteristics of the study population are shown in Additional file 1: Table S1.
KRAS mutation analysis
In order to verify the accuracy of the assays in detecting KRAS mutation status, 658 samples (416 primary tumors; 242 plasma samples) were analyzed by both direct sequencing and PNA-PCR (Table 1). Overall, PNA-PCR identified a higher percentage of KRAS mutated cases than direct sequencing (41% and 30%, respectively; p < 0.001). This difference was evident in tumor tissue (47% and 38%, respectively; p < 10−8) and plasma samples (31% and 17%, respectively; p < 10−8). PNA-PCR was able to detect KRAS mutations in an additional 39 tumor and 35 plasma samples where direct sequencing failed (Table 1). There were no cases in which KRAS mutations were detected by direct sequencing but not PNA-PCR. When KRAS mutations were further characterized by location (i.e. codons 12 or 13), the sensitivity of PNA-PCR was consistently higher (Table 1).
Table 1.
KRAS status analyzed in tumor tissue and plasma samples by direct sequencing and PNA-PCR assays
| Tumor tissue | Plasma | Total | ||
|---|---|---|---|---|
| Assay | KRAS status | (n = 416) | (n = 242) | (n = 658) |
| Wild type | 260 (63) | 201 (83) | 461 (70) | |
| Mutated | 156 (38) | 41 (17) | 197* (30) | |
| Codon 12 | 118 (28) | 30 (12) | 148 (22) | |
| Direct sequencing | 12 D | 50 | 13 | 63 |
| 12 V | 39 | 10 | 49 | |
| Other | 29 | 7 | 36 | |
| Codon 13 | 38 (9) | 11 (5) | 49 (8) | |
| Wild type | 221 (53) | 166 (69) | 387 (59) | |
| Mutated | 195 (47) | 76 (31) | 271* (41) | |
| Codon 12 | 133 (32) | 56 (23) | 189 (29) | |
| PNA-PCR | 12 D | 57 | 26 | 83 |
| 12 V | 46 | 17 | 63 | |
| Other | 30 | 13 | 43 | |
| Codon 13 | 62 (15) | 20 (8) | 82 (12) |
Data expressed as n (%).
*p < 0.001.PNA-PCR, peptide-nucleic-acid-mediated polymerase chain reaction clamping.
KRAS mutations in tumor tissue and plasma
In order to ascertain whether plasma KRAS analysis is predictive of tumor KRAS mutation status, we assessed DNA samples from both tumor tissue and pre-surgery plasma of 242 patients (Table 2). Overall, KRAS mutations were more frequent in tumor than in plasma samples, and this was independent of the assay; KRAS mutation rates in tissue versus plasma were 38% vs. 17% with direct sequencing (p < 0.001) and 47% vs. 31.4% with PNA-PCR (p < 0.001) (Table 1). Only a small percentage of patients showed KRAS mutations in plasma but not in tumor tissue (5% by either method) (Table 2). The higher frequency of KRAS mutations in tumor tissue was also evident when different KRAS mutation subgroups were considered (Table 1).
Table 2.
Comparison of KRAS status in tumor tissue and pre-surgery plasma in 242 patients with available paired samples*
| Tumor tissue | |||||
|---|---|---|---|---|---|
| Plasma | Direct sequencing | ||||
| Wild type | Mutated | Total | Kappa | p value † | |
| Direct sequencing | |||||
| Wild-type (%) | 138 (57) | 63 (26) | 201 | ·278 | <0 · 001 |
| Mutated (%) | 11 (5) | 30 (12) | 41 | ||
| Total | 149 | 93 | 242 | ||
| PNA-PCR | PNA-PCR | ||||
| Wild-Type (%) | 113 (47) | 53 (22) | 166 | ·456 | <0 · 001 |
| Mutated (%) | 12 (5) | 64 (26) | 76 | ||
| Total | 125 | 117 | 242 | ||
*Kappa test was used to estimate the concordance of KRAS status between tumor tissue and plasma samples.
†All Wald statistical tests were two-sided.
PNA-PCR, peptide-nucleic-acid-mediated polymerase chain reaction clamping.
Clinical outcomes and prognostic analysis
The median follow-up duration (calculated from the first day of first-line chemotherapy) was 26.9 (range, 10.0-62.5) months. Overall, 125 patients (30.0%) achieved a partial response. The median PFS of all patients was 6.1 (95% confidence intervals [CI], 4.8-7.5) months and the median OS was 17.4 (95% CI, 16.0-18.8) months.
We retrospectively analyzed the association between KRAS status and patient characteristics. There were no significant differences between gender, ECOG, performance status (0–1 vs. 2), metastatic site, and second-line treatment in patients with KRAS wild-type vs. KRAS mutant samples. We also analyzed the association between KRAS status and clinical outcomes following first-line chemotherapy (Table 3).
Table 3.
Prognostic value of KRAS status analyzed in tumor tissue and plasma by different laboratory assays in colorectal cancer patients
| Prognostic value | ||||||||
|---|---|---|---|---|---|---|---|---|
| Tissue and testing method | PFS | OS | ||||||
| KRAS status | Median PFS (mo) | 95% Cl | P value * | Median OS (mo) | 95% Cl | P value * | ||
| Tumor | ||||||||
| Sequencing | Wild-type | 260 | 6·1 | 4·5-7·7 | ·473 | 18·3 | 16·2-20·4 | ·064 |
| Mutated | 156 | 5·8 | 3·4-8·2 | 15·9 | 14·4-17·4 | |||
| PNA-PCR | Wild-type | 221 | 6·2 | 4·5-7·9 | ·360 | 19·5 | 17·2-21·8 | ·025 |
| Mutated | 195 | 5·9 | 4·0-7·8 | 16·9 | 15·6-18·2 | |||
| Plasma | ||||||||
| Sequencing | Wild-type | 201 | 6·1 | 5·5-6·6 | ·489 | 18·3 | 15·9-20·7 | ·037 |
| Mutated | 41 | 5·4 | 4·9-5·8 | 15·7 | 9·3-22·1 | |||
| PNA-PCR | Wild-type | 166 | 6·1 | 5·5-6·7 | ·274 | 19·1 | 16·8-21·4 | ·009 |
| Mutated | 76 | 5·7 | 5·3-6·1 | 15·7 | 13·0-18·4 | |||
*Log-rank test. All statistical tests were two-sided.
CI, confidence interval; mo, month; OS, overall survival; PFS, progression-free survival; PNA-PCR, peptide-nucleic-acid-mediated polymerase chain reaction clamping.
Tumor DNA analysis
There was no significant relationship between clinical response in patients with and without KRAS mutations; this was also true when subsets of patients carrying specific alterations in codons 12 or 13 were analyzed separately (data not shown). No significant difference between PFS in patients with KRAS wild-type vs. KRAS mutant samples was evident, regardless of whether mutation status was determined by direct sequencing (6.1 vs. 5.8 months; p = 0.473) or PNA-PCR (6.2 vs. 5.9 months; p = 0.360) (Table 3). However, OS was prolonged in patients with KRAS wild-type vs. KRAS mutant tumor samples determined by DNA sequencing (18.3 vs. 15.9 months; p = 0.064), and was statistically significant when mutation status was determined by PNA-PCR (19.5 vs. 16.9 months; p = 0.025). Figure 1A shows OS curves according to KRAS mutation status and by codon in tumor; the worst prognosis was evident in the subgroup of patients carrying mutations different from V and D in codon 12.
Figure 1.
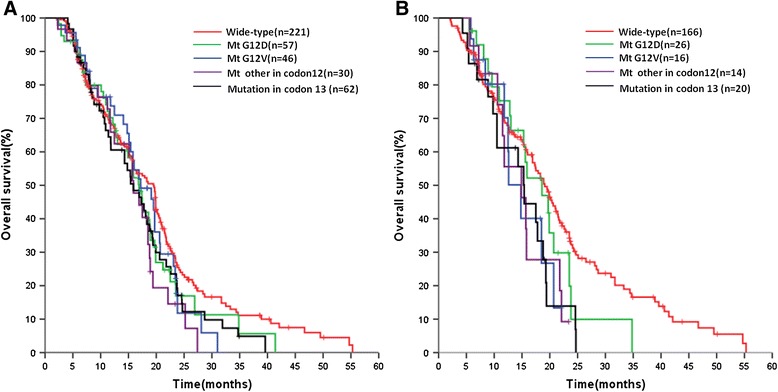
Overall survival by KRAS mutation status in tumor tissue (A) and plasma (B) samples detected by PNA-PCR (see text for further details).
The multivariate analysis showed that frequency and type of KRAS mutations was the only covariate, other than ECOG performance status, that were significantly associated with OS (ECOG performance status: odds ratio [OR] 3.03, 95% CI 2.21-3.41, p < 0.001; KRAS wild-type vs. mutated: OR 1.37, 95% CI 1.06-1.70, p < 0.008). This association was independent of the assay but stronger with PNA-PCR.
Plasma DNA analysis
No relationship between KRAS status in plasma DNA samples and clinical response (data not shown) or PFS was observed (Table 3). Median OS was significantly longer in patients with KRAS wild-type vs. KRAS mutant samples, in particular when mutation status was determined by PNA-PCR (19.1 vs. 15.7 months; p = 0.009). Figure 1B shows OS curves according to KRAS mutation status and by codon in plasma.
The multivariate analysis demonstrated that the frequency and type of KRAS mutations was the only covariate, other than ECOG performance status, significantly associated with OS (ECOG performance status: OR 3.06, 95% CI 2.22-3.46, p < 0.001; KRAS wild-type vs. mutated: OR 1.35, 95% CI 1.05-1.65, p < 0.008). This association was independent of the laboratory assay but was stronger for PNA-PCR.
Combined analysis of DNA from tumor and plasma
For the analysis of the prognostic impact of KRAS mutation status, we used data from patients for which DNA from both tumor tissue and plasma were available (n = 242). Each patient was classified as KRAS positive (i.e. KRAS mutations in tumor and plasma samples), KRAS negative (i.e. KRAS wild-type in tumor and plasma samples), or with discordant KRAS status in tumor and plasma samples. This analysis was performed using the results obtained with PNA-PCR only.
While there was no relationship between KRAS status and clinical response or PFS, the analysis of OS showed a statistically significant association between the three cohorts (Table 4 and Figure 2). This association was confirmed in a multivariate analysis (Additional file 2: Table S2).
Table 4.
Prognostic value of combined tumor/plasma KRAS status analyzed by PNA-PCR in a series of 242 metastatic colorectal cancer patients with matched samples
| KRAS status* | Median OS | |||
|---|---|---|---|---|
| Tumor/Plasma | No. | (mo) | 95% Cl | P value |
| Negative/Negative | 113 | 21.0 | 19.226-22.774 | 0.008 |
| Discordant | 65 | 16.9 | 14.184-19.616 | |
| Positive/Positive | 64 | 15.4 | 14.270-16.530 |
*KRAS status was categorized as: negative, mutation-negative tumor and matched plasma samples; discordant, discordant KRAS status in tumor and matched plasma samples; positive, mutation-positive tumor tissue and matched plasma samples.
CI, confidence interval; mo, month; OS, overall survival; PNA-PCR, peptide-nucleic-acid-mediated polymerase chain reaction clamping.
Figure 2.
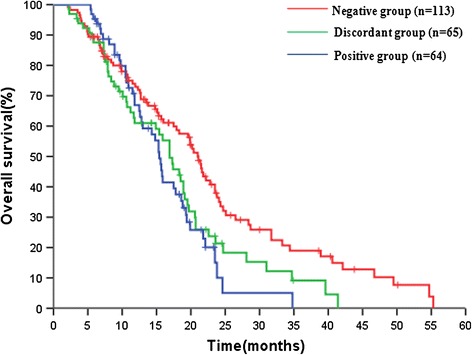
Prognostic value of KRAS status analysed either in tumor tissue or plasma samples by PNA-PCR. According to analyses of tumor tissue and plasma, KRAS status of each patient was categorized as: negative (KRAS wild type in tumor and matched plasma samples), discordant, (KRAS status discordant in tumor and matched plasma samples); positive (KRAS mutated either in tumor tissue and matched plasma samples.
Discussion
In spite of the clear role that KRAS alterations play in the pathogenesis and progression of colorectal cancer, several aspects concerning their clinical validation remain inconclusive or are not yet fully explored [1]. Among them, the role of more sensitive laboratory assays for KRAS mutation analysis, the possibility of using circulating DNA to acquire information on KRAS status, and the possible prognostic-predictive value of these assays. The present study addresses all of these questions in a large mono-institutional series of patients for whom colorectal cancer tissue and plasma samples were available.
PNA-PCR has been reported to have a higher sensitivity than direct sequencing, permitting the detection of mutations at frequencies as low as 1%-5% [23]. We performed 658 assays by PNA-PCR and direct sequencing and found a significantly higher percentage of KRAS gene alterations with PNA-PCR (41% vs. 30%; p < 0.001), which was independent of the sample source (FFPE tumor samples or frozen plasma). A similar result has been reported in other studies [24,25] . Further, there were no cases in our study in which mutations were detected by direct sequencing but not PNA-PCR. We suggest that PNA-PCR is more accurate than direct sequencing and may be a clinically useful assay.
It has been suggested that circulating DNA is a suitable and reliable source of DNA for tumor somatic gene alterations [26] , but is this the case for the KRAS gene? In our study, we observed KRAS mutation detection rates in plasma samples of 17% and 31% by direct sequencing and PNA-PCR, respectively. Recent research indicates that the mutation detection rates in tumor tissue and plasma are influenced by disease stage [27,28], metastatic sites and performance status, since the major source of plasma DNA is from necrotic or apoptotic tumor cells [26]. Therefore, there would be a greater likelihood of detecting KRAS mutations in patients with a larger tumor burden or after multiple lines of therapy. Our study population was chemotherapy-naïve, which would be expected to result in a lower detection rate in plasma DNA.
A recent literature review [15] confirmed that KRAS alterations in ctDNA from plasma are significantly predictive of KRAS tumor status with a concordance between tumor and plasma status ranging from 29% to 100%. We analyzed 242 paired tumor and plasma samples, the largest series reported to date, and showed that determining KRAS status in plasma is feasible and provides information on gene status concordant with tumor DNA analysis by PNA-PCR in 73% of cases (Table 2). Although we showed that PNA-PCR can detect more KRAS mutated cases and that the discrepancy might be due to cases showing a mutation in the primary tumor and not in plasma, and this fact is in agreement with other recent studies which addressed the same question [29,30], further optimization of this methods also might increase the concordance between tumor and plasma as the investigation of KRAS and BRAF mutations consistence in tumor tissue and cfDNA [31]. We also found that 5% of cases that are mutant in plasma but wide-type in the tissue with both methods. The reasonable explanation for this is the heterogenity of cancer tissue, which means that the sections used in the detection only represented part of the tumor and did not contain the DNA carrying KRAS mutations that released into blood from other parts of tumor. Our study indicates that, if a tumor sample is unavailable or insufficient, using a more sensitive method than PNA-PCR assay such as ultra deep sequencing technology on a plasma DNA sample might be an alternative way to determine KRAS status in metastatic colorectal cancer.
The prognostic-predictive value of KRAS mutations in mCRC remains controversial. Recently, Ren et al. [6] did a systematic review and meta-analysis on this topic and identified 13 studies which showed that KRAS mutations are significantly associated with OS, but no separate analysis for studies in metastatic disease was performed. Yokota [32] also did not consider mCRC separately. However, Loriot [1] reviewed 6 clinical studies specifically looking at KRAS status in stage IV colorectal cancer and reported that KRAS mutations have no predictive value with conventional chemotherapy. Our results confirm these observations. KRAS status, and KRAS mutations in codons 12 or 13, had no predictive relevance for clinical response to chemotherapy or PFS. This finding was independent of assay utilized and DNA source. The conclusions were different when OS was considered. OS was influenced by KRAS status analyzed by PNA-PCR in plasma free DNA (univariate analysis, 15.7 months for KRAS mutations vs. 19.1 months for KRAS wild-type; p < 0.009; multivariate analysis, OR 1.35, 95% CI 1.05-1.65, p < 0.008). Interestingly, patients with KRAS mutations, regardless of their location, had a poorer prognosis than KRAS wild-type patients (Figure 1), although the differences between them were not significantly different. Overall, our results seem to confirm that KRAS mutations are associated with a more aggressive form of colorectal cancer.
It is difficult to compare our results with previous studies, as few studies have differentiated between KRAS mutations at codon 12 and 13. The largest study on this topic, RASCAL II which involved 3439 colorectal cancer patients, concluded that different gene mutations have different impacts on outcome, and in particular the codon 12 glycine to valine mutation, but it did not look specifically at stage IV disease [33]. Only one study [34] has considered advanced disease treated with systemic chemotherapy and showed that only the OS of patients carrying KRAS codon 13 mutations was significantly worse than for patients with wild-type KRAS. We further demonstrated that the impact of KRAS status on OS is independent from assay and DNA source, thus reinforcing the reliability of the evidence.
When patients were divided according to their combined plasma/tissue KRAS status, the best prognosis was observed in patients without mutations in either tumor or plasma, while the worst was evident in patients with mutations in both tissues. The subgroup with a discordant KRAS status showed an intermediate long-term prognosis. The prognostic relevance of this classification was confirmed in the multivariate analysis. The biological implications of this observation are unknown, but it seems possible that DNA from both primary tumor and plasma (which includes DNA from different sites) may provide additional clinically relevant information.
Conclusion
This study showed that KRAS mutation status is able to predict long-term prognosis of patients with mCRC treated with conventional chemotherapy. This association was independent of the type of biospecimen and assay utilized for KRAS analysis, indicating that plasma DNA samples may provide an alternative biological source for KRAS mutation analysis. Further studies are required to corroborate this last hypothesis.
Acknowledgement
We thank Ming Lei Shi, MD, and Yan Zhang, MD, for their excellent technical work; Hui Gao, PhD, for his contribution in statistical analysis.
Supported by grant from National Natural Science Foundation Project (No. 81172280).
Additional files
Baseline characteristics.
Association of KRAS status with overall survival. Multivariate analysis with overall survival as dependent variable in a series of 242 advanced colorectal cancer patients; combined plasma/tumor KRAS status analyzed by PNA-PCR*.
Footnotes
Jian-Ming Xu and Xiao-Jing Liu contributed equally to this work.
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
J-MX and X-JL contributed to design the present study, to perform clinical activities, to revise and elaborate data, to write the paper. F-JG, LL, YW contributed to clinical management of patients, to revise and elaborate clinical and laboratory information. MRS, Z-YL to produce and review laboratory data, analysis and elaboration. ST participated to the elaboration of laboratory data analysis and contributed to manuscript writing. AP contributed to writing and analysis of the data, to write the manuscript, to critically review the content. All authors read and approved the final manuscript.
Contributor Information
Jian-Ming Xu, Email: jmxu2003@yahoo.com.
Xiao-Jing Liu, Email: lxj1982@163.com.
Fei-Jiao Ge, Email: gefj1981@163.com.
Li Lin, Email: lin100@sina.com.
Yan Wang, Email: pretty.wang@hotmail.com.
Manish R Sharma, Email: msharma@bsd.uchicago.edu.
Ze-Yuan Liu, Email: zyliu1961@sina.com.
Stefania Tommasi, Email: s.tommasi@oncologico.bari.it.
Angelo Paradiso, Email: paradiso@oncologico.bari.it.
References
- 1.Loriot Y, Mordant P, Deutsch E, Olaussen KA, Soria JC. Are RAS mutations predictive markers of resistance to standard chemotherapy? Nat Rev Clin Oncol. 2009;6:528–534. doi: 10.1038/nrclinonc.2009.106. [DOI] [PubMed] [Google Scholar]
- 2.Bos JL, Fearon ER, Hamilton SR, Verlaan-de Vries M, van Boom JH, van der Eb AJ, Vogelstein B. Prevalence of ras gene mutations in human colorectal cancers. Nature. 1987;327:293–297. doi: 10.1038/327293a0. [DOI] [PubMed] [Google Scholar]
- 3.Voorham QJ, Rondagh EJ, Knol DL, van Engeland M, Carvalho B, Meijer GA, Sanduleanu S. Tracking the molecular features of nonpolypoid colorectal neoplasms: a systematic review and meta-analysis. Am J Gastroenterol. 2013;108:1042–1056. doi: 10.1038/ajg.2013.126. [DOI] [PubMed] [Google Scholar]
- 4.Santini D, Loupakis F, Vincenzi B, Floriani I, Stasi I, Canestrari E, Rulli E, Maltese PE, Andreoni F, Masi G, Graziano F, Bald GG, Salvatore L, Russo A, Perrone G, Tommasino MR, Magnani M, Falcone A, Tonini G, Ruzzo A. High concordance of KRAS status between primary colorectal tumors and related metastatic sites: implications for clinical practice. Oncologist. 2008;13:1270–1275. doi: 10.1634/theoncologist.2008-0181. [DOI] [PubMed] [Google Scholar]
- 5.Tommasi S, Pinto R, Petriella D, Pilato B, Lacalamita R, Santini D, Zito F, Colucci G, Paradiso A, Silvestris N. Oncosuppressor methylation: a possible key role in colon metastatic progression. J Cell Physiol. 2011;226:1934–1939. doi: 10.1002/jcp.22524. [DOI] [PubMed] [Google Scholar]
- 6.Ren J, Li G, Ge J, Li X, Zhao Y. Is K-ras gene mutation a prognostic factor for colorectal cancer: a systematic review and meta-analysis. Dis Colon Rectum. 2012;55:913–923. doi: 10.1097/DCR.0b013e318251d8d9. [DOI] [PubMed] [Google Scholar]
- 7.Allegra CJ, Jessup JM, Somerfield MR, Hamilton SR, Hammond EH, Hayes DF, McAllister PK, Morton RF, Schilsky RL. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol. 2009;27:2091–2096. doi: 10.1200/JCO.2009.21.9170. [DOI] [PubMed] [Google Scholar]
- 8.Bardelli A, Siena S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J Clin Oncol. 2010;28:1254–1261. doi: 10.1200/JCO.2009.24.6116. [DOI] [PubMed] [Google Scholar]
- 9.De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, Penault-Llorca F, Rougier P, Vincenzi B, Santini D, Tonini G, Cappuzzo F, Frattini M, Molinari F, Saletti P, De Dosso S, Martini M, Bardelli A, Siena S, Sartore-Bianchi A, Tabernero J, Macarulla T, Di Fiore F, Gangloff AO, Ciardiello F, Pfeiffer P, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11:753–762. doi: 10.1016/S1470-2045(10)70130-3. [DOI] [PubMed] [Google Scholar]
- 10.Linardou H, Dahabreh IJ, Kanaloupiti D, Siannis F, Bafaloukos D, Kosmidis P, Papadimitriou CA, Murray S. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol. 2008;9:962–972. doi: 10.1016/S1470-2045(08)70206-7. [DOI] [PubMed] [Google Scholar]
- 11.Roth AD, Tejpar S, Delorenzi M, Yan P, Fiocca R, Klingbiel D, Dietrich D, Biesmans B, Bodoky G, Barone C, Aranda E, Nordlinger B, Cisar L, Labianca R, Cunningham D, Van Cutsem E, Bosman F. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60–00 trial. J Clin Oncol. 2010;28:466–474. doi: 10.1200/JCO.2009.23.3452. [DOI] [PubMed] [Google Scholar]
- 12.Eklof V, Wikberg ML, Edin S, Dahlin AM, Jonsson BA, Oberg A, Rutegard J, Palmqvist R. The prognostic role of KRAS, BRAF, PIK3CA and PTEN in colorectal cancer. Br J Cancer. 2013;108:2153–2163. doi: 10.1038/bjc.2013.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Phipps AI, Buchanan DD, Makar KW, Burnett-Hartman AN, Coghill AE, Passarelli MN, Baron JA, Ahnen DJ, Win AK, Potter JD, Newcomb PA. BRAF mutation status and survival after colorectal cancer diagnosis according to patient and tumor characteristics. Cancer Epidemiol Biomarkers Prev. 2012;21:1792–1798. doi: 10.1158/1055-9965.EPI-12-0674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jancik S, Drabek J, Berkovcova J, Xu YZ, Stankova M, Klein J, Kolek V, Skarda J, Tichy T, Grygarkova I, Radzioch D, Hajduch M. A comparison of direct sequencing, pyrosequencing, high resolution melting analysis, TheraScreen DxS, and the K-ras StripAssay for detecting KRAS mutations in non small cell lung carcinomas. J Exp Clin Cancer Res. 2012;31:79. doi: 10.1186/1756-9966-31-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tierling S, Sers C, Lehmann A, Walter J. A fast, cost-efficient and sensitive approach for KRAS mutation detection using multiplexed primer extension with IP/RP-HPLC separation. Int J Cancer. 2012;130:567–574. doi: 10.1002/ijc.26040. [DOI] [PubMed] [Google Scholar]
- 16.Tougeron D, Lecomte T, Pages JC, Villalva C, Collin C, Ferru A, Tourani JM, Silvain C, Levillain P, Karayan-Tapon L. Effect of low-frequency KRAS mutations on the response to anti-EGFR therapy in metastatic colorectal cancer. Ann Oncol. 2013;24:1267–1273. doi: 10.1093/annonc/mds620. [DOI] [PubMed] [Google Scholar]
- 17.Ellison G, Donald E, McWalter G, Knight L, Fletcher L, Sherwood J, Cantarini M, Orr M, Speake G. A comparison of ARMS and DNA sequencing for mutation analysis in clinical biopsy samples. J Exp Clin Cancer Res. 2010;29:132. doi: 10.1186/1756-9966-29-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oh JE, Lim HS, An CH, Jeong EG, Han JY, Lee SH, Yoo NJ. Detection of low-level KRAS mutations using PNA-mediated asymmetric PCR clamping and melting curve analysis with unlabeled probes. J Mol Diagn. 2010;12:418–424. doi: 10.2353/jmoldx.2010.090146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rogosnitzky M, Danks R. Validation of blood testing for K-ras mutations in colorectal and pancreatic cancer. Anticancer Res. 2010;30:2943–2947. [PubMed] [Google Scholar]
- 20.Kim HR, Lee SY, Hyun DS, Lee MK, Lee HK, Choi CM, Yang SH, Kim YC, Lee YC, Kim SY, Jang SH, Lee JC, Lee KY. Detection of EGFR mutations in circulating free DNA by PNA-mediated PCR clamping. J Exp Clin Cancer Res. 2013;32:50. doi: 10.1186/1756-9966-32-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Y, Liu B, Li XY, Li JJ, Qin HF, Tang CH, Guo WF, Hu HX, Li S, Chen CJ, Liu B, Gao HJ, Liu XQ. A comparison of ARMS and direct sequencing for EGFR mutation analysis and tyrosine kinase inhibitors treatment prediction in body fluid samples of non-small-cell lung cancer patients. J Exp Clin Cancer Res. 2011;30:111. doi: 10.1186/1756-9966-30-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu S, Wu J, Xu S, Tan G, Liu B, Feng J. Modified PNA-PCR method: a convenient and accurate method to screen plasma KRAS mutations of cancer patients. Cancer Biol Ther. 2012;13:314–320. doi: 10.4161/cbt.19075. [DOI] [PubMed] [Google Scholar]
- 23.Gilje B, Heikkila R, Oltedal S, Tjensvoll K, Nordgard O. High-fidelity DNA polymerase enhances the sensitivity of a peptide nucleic acid clamp PCR assay for K-ras mutations. J Mol Diagn. 2008;10:325–331. doi: 10.2353/jmoldx.2008.070183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kwon MJ, Lee SE, Kang SY, Choi YL. Frequency of KRAS, BRAF, and PIK3CA mutations in advanced colorectal cancers: comparison of peptide nucleic acid-mediated PCR clamping and direct sequencing in formalin-fixed, paraffin-embedded tissue. Pathol Res Pract. 2011;207:762–768. doi: 10.1016/j.prp.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 25.Kobunai T, Watanabe T, Yamamoto Y, Eshima K. The frequency of KRAS mutation detection in human colon carcinoma is influenced by the sensitivity of assay methodology: a comparison between direct sequencing and real-time PCR. Biochem Biophys Res Commun. 2010;395:158–162. doi: 10.1016/j.bbrc.2010.03.167. [DOI] [PubMed] [Google Scholar]
- 26.Gonzalez-Masia JA, Garcia-Olmo D, Garcia-Olmo DC. Circulating nucleic acids in plasma and serum (CNAPS): applications in oncology. Onco Targets Ther. 2013;6:819–832. doi: 10.2147/OTT.S44668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sorenson GD. Detection of mutated KRAS2 sequences as tumor markers in plasma/serum of patients with gastrointestinal cancer. Clin Cancer Res. 2000;6:2129–2137. [PubMed] [Google Scholar]
- 28.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, Antonarakis ES, Azad NS, Bardelli A, Brem H, Cameron JL, Lee CC, Fecher LA, Gallia GL, Gibbs P, Le D, Giuntoli RL, Goggins M, Hogarty MD, Holdhoff M, Hong SM, Jiao Y, Juhl HH, Kim JJ, Siravegna G, Laheru DA, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:224ra224. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morgan SR, Whiteley J, Donald E, Smith J, Eisenberg MT, Kallam E, Kam-Morgan L. Comparison of KRAS mutation assessment in tumor DNA and circulating free DNA in plasma and serum samples. Clin Med Insights Pathol. 2012;5:15–22. doi: 10.4137/CPath.S8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miyano S, Hanazawa K, Kitabatake T, Fujisawa M, Kojima K. Detecting KRAS mutations in peripheral blood of colorectal cancer patients by peptide nucleic acid clamp PCR. Exp Ther Med. 2012;4:790–794. doi: 10.3892/etm.2012.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thierry AR, Mouliere F, El Messaoudi S, Mollevi C, Lopez-Crapez E, Rolet F, Gillet B, Gongora C, Dechelotte P, Robert B, Del Rio M, Lamy PJ, Bibeau F, Nouaille M, Loriot V, Jarrousse AS, Molina F, Mathonnet M, Pezet D, Ychou M. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat Med. 2014;20:430–435. doi: 10.1038/nm.3511. [DOI] [PubMed] [Google Scholar]
- 32.Yokota T. Are KRAS/BRAF mutations potent prognostic and/or predictive biomarkers in colorectal cancers? Anti Cancer Agents Med Chem. 2012;12:163–171. doi: 10.2174/187152012799014968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andreyev HJ, Norman AR, Cunningham D, Oates J, Dix BR, Iacopetta BJ, Young J, Walsh T, Ward R, Hawkins N, Beranek M, Jandik P, Benamouzig R, Jullian E, Laurent-Puig P, Olschwang S, Muller O, Hoffmann I, Rabes HM, Zietz C, Troungos C, Valavanis C, Yuen ST, Ho JW, Croke CT, O'Donoghue DP, Giaretti W, Rapallo A, Russo A, Bazan V, et al. Kirsten ras mutations in patients with colorectal cancer: the ‘RASCAL II’ study. Br J Cancer. 2001;85:692–696. doi: 10.1054/bjoc.2001.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yokota T, Ura T, Shibata N, Takahari D, Shitara K, Nomura M, Kondo C, Mizota A, Utsunomiya S, Muro K, Yatabe Y. BRAF mutation is a powerful prognostic factor in advanced and recurrent colorectal cancer. Br J Cancer. 2011;104:856–862. doi: 10.1038/bjc.2011.19. [DOI] [PMC free article] [PubMed] [Google Scholar]