

Summary
Wnt signaling is involved in T cell development, activation, and differentiation. However, the role for Wnt signaling in mature naïve T cells has not been investigated. Here, we report that activation of Wnt signaling in T cell lineages by deletion of the Apc gene causes spontaneous T cell activation and severe T lymphopenia. The lymphopenia is the result of rapid apoptosis of newly exported, mature T cells in the periphery and is not due to defects in thymocyte development or emigration. Using chimera mice consisting of both WT and Apc-deficient T cells, we found that loss of naïve T cells is due to T-cell intrinsic dysregulation of Wnt signaling. Since Apc deletion causes over-expression of the Wnt target gene cMyc, we generated mice with combined deletion of the cMyc gene. Since combined deletion of cMyc and Apc attenuated T cell loss, cMyc over-expression is partially responsible for spontaneous T cell apoptosis and lymphopenia. Cumulatively, our data reveals a missing link between Wnt signaling and survival of naïve T cells.
Introduction
Hematopoietic progenitors, from the bone marrow, migrate into the thymus and undergo a well-regulated developmental program to produce T lymphocytes (1, 2). Once functionally mature, T lymphocytes emigrate from the thymus to populate the spleen and lymph nodes, where they wait for stimulation by their cognate antigen to mount a protective immune response (3, 4). While the developmental and activation programs have been well characterized, the program that maintains peripheral naïve T cells in the resting stage remains poorly understood. Recent studies from our team and others have implicated critical roles in regulation by mTOR activity (5, 6) and by the FoxO and FoxP1 transcription factors (7–9).
The Wnt signaling pathway is an evolutionarily conserved pathway that regulates cell proliferation, differentiation, cell survival, migration, and polarity (10–13). Wnt stimulation releases β-catenin from a destruction complex scaffolded by Apc, thus allowing β-catenin to regulate its transcriptional targets by interacting with T cell factors, such as Tcf-1(14–16). Mice lacking different components of the Wnt signaling pathway reveal a broad dysfunction in various stages of T cell development, including the generation of CD4−CD8− (DN) thymocytes and differentiation/survival of multiple functional T cell subsets in the periphery. Tcf1 deficient mice show an age-dependent reduction in thymocyte production and a corresponding loss of early thymic progenitors (17). Deletion of Ctnnb1 (the gene that encodes β-catenin) results in a developmental blockage at the DN3 to DN4 stage(18). In activated T cells, ectopic expression of the β-catenin partner TCF1 stimulates differentiation to Th2 (19), while that of a Wnt signaling inhibitor, DKK-1, abrogates it (20). Ectopically expressing a β-catenin mutant that evades Apc-mediated destruction also enhances the survival of T regulatory cells (21). A recent study suggests that heterozygous mutation of the Apc gene in the Apcmin/+ mice partially attenuates regulatory T cell function (22). Perhaps because of the difficulties in deleting Apc in mature naive T cells, the role for Wnt signaling in mature naïve T cells in the periphery has not been investigated.
To address this gap, we used mice with exon 14-floxed Apc locus (23) and a CD4-Cre transgene to induce exon 14 deletion in the T cell lineage (24). Deleting exon 14 in APC generates a truncated polypeptide that lacks most of the functional domains of APC (25), including seven repeated sequences of 20 amino acids, each in the central region of the APC protein. Since these repeats are critical for APC binding to β-catenin, the key step in canonical Wnt signaling (23), the mutant cells will have constitutive activation of the Wnt pathway. The mutant also lacks the binding sites for EB1 and microtubules that are responsible for cell polarity and mitosis (26, 27). The truncated APC is still capable of encoding a polypeptide that contains the oligomerization domain (28) and some of the armadillo repeats, which have been shown to interact with the APC-stimulated guanine nucleotide exchange factor (Asef)(29). Thus, while the truncated APC may still have a role in stabilization and motility of the actin cytoskeleton network through its interaction with Asef and Rac and Rho GTP binding proteins (30), the essential role for Apc in canonical Wnt signaling is completely inactivated. This tool provided us with a unique opportunity to investigate the role of Wnt signaling in naïve T cell function. Surprisingly, we found that deletion of exon 14 of the Apc gene, using CD4-Cre, activated Wnt signaling without affecting T cell development. Our data revealed that inactivation of Apc resulted in a drastic loss of mature naïve T cells in the periphery and severe T cell lymphopenia. This loss is at least due, in part, to over-expression of cMyc, as it is attenuated by deletion of the cMyc gene. Our data unveils an unexpected impact of Wnt signaling on the survival of naïve T cells in the periphery.
Materials and Methods
Mice
CD45.1 C57BL/6 mice were obtained from Charles River Laboratories, through a contract with the National Cancer Institute. Mice with homozygous knockin of the floxed Apc (23) and transgenic mice expressing the Cre recombinase, under the control of either the proximal Lck promoter (31) or CD4 promoter (24), were obtained from Jackson Laboratories. Mice with floxed cMyc locus (32) were kindly provided by Dr. De Alboran (National Center for Biotechnology CNB/CSIC, Madrid, Spain). All mice used in this study have been backcrossed to C57BL/6 background for at least 10 generations. These strains were maintained in our animal facilities under pathogen-free conditions. All experiments were performed in accordance with the guidelines of the Institutional Animal Care and Use Committees of the University of Michigan and the Children’s National Medical Center.
Intrathymic injections
Five week old mice were anesthetized with isoflurane. A midline incision was used to expose the ribs. Ten μl of 1 mM CFSE (Life Technology) was injected into each lobe of the thymus. Mice were sacrificed at either 6 or 24 hours after CFSE injection. The thymus, spleen, and lymph nodes were collected and analyzed by flow cytometry.
Adoptive transfer of T cells
Mature CD24− thymocytes were isolated by Dynal negative selection using biotinylated anti-CD24 mAb M1/69 (Ebioscience). Cells were labeled with 10 μM CFSE and injected intravenously to CD45.1 recipients (1×106/mouse).
Western blot analysis
Tissues were lysed in a protein lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.5% NP-40) supplemented with a protease inhibitor cocktail (Pierce). Cell lysates were separated by SDS-PAGE and transferred to polyvinylidene fluoride membranes. The membranes were incubated with anti-c-Myc (Abcam, AU016757, Ab39688,1:2000) and anti-actin (Abcam ACTN05, Ab1801, 1:5000). Either anti-rabbit or anti-mouse IgG horseradish peroxidase-linked antibody (Santa Cruz) was used as a secondary antibody. A chemiluminescence kit (Life Technology) was used to visualize blots.
Flow cytometric analysis
Mice were sacrificed at 6–8 weeks of age. Thymus and spleen tissues were homogenized to generate a single-cell suspension. Cells were stained at 4°C for 20 minutes in phosphate-buffered saline with 2% FBS with the following antibodies from BD Bioscience (1:200): CD4 (RM4-5), CD8 (SK-1), B220 (HIS24), CD44 (IM7), CD62L (MEL-14), CD24 (30-F1), CD69 (H1.2F3), CCR7 (4B12), beta 7 integrin (FIB205), β-catenin (15B8), δγ TCR (UC7-13D5) and CD25 (PC61.5). Samples were analyzed by a BD LSR II Flow Cytometer.
For the co-localization experiment, splenocytes were stained for CD3 and permeabilized using the BD fix/perm kit™ followed by anti-β-catenin (15B8) staining overnight. DRAQ5 (Ebioscience) was added (1:10,000) before analysis. Samples were analyzed by an Amnis Imaging Flow Cytometry. Analysis was carried out using IDEAS® software. Calculations were made by following IDEAS® software’s wizard.
Irradiation chimera
CD45.1+ B6 mice were lethally irradiated (11 Gy delivered in 2 installments, 4 hours apart, as reported(33)) and reconstituted with of 2.5×106 CD45.1+ bone marrow cells in conjunction with equal numbers of either APC cKO or Ctrl bone marrow cells. Reconstitution was confirmed at 8 weeks from peripheral blood staining of CD45.1 and CD45.2. Bone marrow chimeras were analyzed for T cell numbers and phenotypes at 10 weeks post-transplantation.
Statistics
Data was analyzed using 2-tailed unpaired Student’s t-tests. All statistics were performed using GraphPad Prism, version 5 (GraphPad Software). *P<0.05, **P<0.01, ***P<0.001.
Results
Deletion of exon 14 floxed Apc by CD4-Cre does not grossly disturb normal thymocyte development
A previous study suggested that deletion of Apc using Lck-Cre disturbs thymocyte development (34). In order to study the impact of Wnt signaling in mature naïve T cells, we used CD4-Cre to delete Apc and used a more commonly known Apc conditional allele that has loxP sites inserted into introns 13 and 14 of the endogenous Apc gene (Apcfl)(23). These mice were labeled as cKO, while their Cre− littermates were used as a control and labeled as Ctrl. Based on the known specificity of the Cre promoter, Apcfl/fl;CD4-Cre+ mice should have a frameshift mutation at codon 580 and a truncated APC polypeptide beginning at the CD4−CD8−CD25+CD44− (DN3) stage. To confirm Apc deletion among thymocytes, we FACS-sorted CD4−CD8− (DN) thymocytes based on their expression of CD4, CD8, CD44, and CD25. Using polymerase chain reaction (PCR) that yielded differently sized products from floxed (fl) and deleted (del) Apc alleles, we determined the kinetics of Apc deletion in developing DN thymocytes. As expected, no deletion was observed in DN1-4 of the Ctrl mice. In the cKO thymocytes, nearly equal amounts of fl and del alleles were observed in DN3. By DN4, most products were derived from the del allele (Fig. 1a). Thus, the Apc deletion began at the DN3 stage and was largely completed at the CD4−CD8−CD25−CD44− DN4 stage in the cKO mice. Since the primary function of the Apc protein is to mediate the destruction of β-catenin, we used intracellular accumulation of the protein as the primary readout for functional Apc inactivation throughout thymocyte development. As shown in Fig. 1b, despite a nearly 50% deletion of the Apc gene, no increase of intracellular β-catenin was observed in the DN3 stage. Likewise, although the Apc deletion was nearly complete in the DN4 stage, only a small subset showed functional inactivation of the Apc protein. However, significant accumulation of β-catenin was observed from the bulk of CD4+CD8+ (DP), CD4+CD8− (CD4 SP), and CD4−CD8+ (CD8 SP) thymocytes. The delayed functional inactivation of the Apc protein in relation to the gene deletion is likely due to residual Apc protein and/or mRNA. To investigate whether β-catenin accumulation is adequate for its translocation into the nucleus, we stained for CD3− and β-catenin in the splenocytes from ctrl and cKO mice. Using an Aminis Imaging flow cytometry, we detected the presence of β-catenin in the nucleus, which was also stained with a nuclear stain, DRAQ5. As seen in Fig 1c, cKO, clearly results in an accumulation of β-catenin that in over 50% of the case co-localizes in the nucleus, thus demonstrating that without APC, β-catenin will stabilization and translocate into the nucleus thereby activating Wnt signaling. Regardless, the robust activation of Wnt signaling from the DP stage allowed us to assess the function of Wnt signaling in T cell development and function starting at the DP stage.
Fig. 1.

Activation of Wnt signaling by CD4-Cre-driven Apc deletion does not affect thymocyte development. a. Kinetics of Apc deletion by CD4-Cre. CD4−CD8− thymocytes from 6–8 week old Apcfl/fl; Cd4-Cre+(cKO) mice were sorted based on expression of CD4, CD8, CD44 and CD25 into DN1-4 (CD4−CD8−CD25−CD44+, CD4−CD8−CD25+CD44+, CD4−CD8−CD25+CD44−, and CD4−CD8−CD25−CD44−, respectively). The genomic DNA was amplified by PCR, yielding distinct sizes for floxed (fl, 314 bp) and deleted (del, 258 bp). Similar data were obtained in two experiments. b. Activation of Wnt signaling as indicated by levels of intracellular β-catenin in different thymocyte subsets from 6–8 week old Ctrl (Apcfl/fl;Cd4-Cre−) or cKO mice. Data shown are histograms depicting the distribution of β-catenin among the gated thymocyte subsets. DP indicates CD4+CD8+, CD4 SP indicates CD4+CD8−, and CD8 SP indicates CD4−CD8+. Similar results were obtained in 9 pairs of mice. c. Spleens of cntrl or cKO were stained with CD3, β-catenin, and DRAQ5. Samples were analyzed by an Amnis Imagining flow cytometry. % β-catenin was calculated from the total number of CD3+ that were in focus. % co-localization was calculated by determining the number of CD3+ that had co-localization of both β-catenin and DRAQ5. Experiment was repeated twice, n=3. d. Thymic cellularity of 6–8 week Ctrl and APC cKO (closed squares) mice (n=15). e. Representative profiles of thymocyte subsets based on expression of CD4 and CD8 in Ctrl or cKO thymocytes. f. Absolute numbers of DP, CD8 SP, and CD4 SP thymocytes (n=9). g. Representative profiles of the DN subset among cKO and Ctrl thymocytes. h. Absolute number of DN populations from 6–8 week old Ctrl or cKO mice (n=8). No statistically significant differences were observed in any of the parameters depicted in d, f, and h. Data from b–h are either representative or summary of those from three independent experiments.
As shown in Fig. 1d, Ctrl and cKO thymi had comparable cellularity. Distribution of CD4 and CD8 markers among thymocytes revealed no gross abnormality in thymocyte development as the % (Fig. 1e) and numbers (Fig. 1f) of DP, CD4 SP, and CD8 SP thymocytes were similar. As expected from the largely normal β–catenin levels in cKO DN thymocytes, the frequency (Fig. 1g) and number (Fig. 1h) of double negative cells (CD4−CD8−, DN) were also comparable throughout stages 1–4. Thus, the deletion of Apc that began at the DN3 stage did not affect thymocyte development. However, we did observe an increase in apoptosis of Apc- deficient thymocytes (Supplemental Fig. S1a), although apoptosis was more pronounced in peripheral T cells (Supplemental Fig. S1b).
The differences observed between our results and those in a previous study (34) was not entirely due to a subtle difference between the Lck-Cre used in the previous study and the CD4-Cre used here. In our hands, both CD4-Cre and Lck-cre had largely normal thymocyte development. (supplemental Fig. S2). However, the modest but statistically significant reduction in cellularity in Lck-Cre mice suggests that even minor differences in timing of gene deletion can affect the outcomes. One potential phenotypic difference between Lck-Cre and CD4-Cre is in the expression of CD44 a known Wnt target gene. In the periphery, both CD8 and CD4 T cells from Lck-Cre and CD4-Cre upregulate CD44 at much higher levels than in the ctrl mice. However, thymocytes show a different pattern, where SP CD4 and CD8 thymocytes in CD4-Cre mice express much higher levels of CD44, while Lck-Cre levels of CD44, though higher, appears much more like ctrl mice. In addition, we observed a loss of thymic cellularity and an apparent increase of DN thymocytes in older CD4-Cre-induced cKO mice that became moribund (supplemental Fig. S1c and S1d). This raised the intriguing possibility that abnormal thymocyte development may be secondary effect of overall immune activation in the mice, as reported by Martin et al. (35). Overall, our data demonstrates that Wnt signaling starting at DN4 did not directly disturb normal T cell development in healthy young mice. This normal T cell development allowed us to study the function of Wnt signaling after T cell maturation.
Apc deletion causes rapid loss of T cells after their emigration from the thymus
Despite normal T cell development, we observed a marked reduction of T cell numbers in the periphery. Thus, the frequencies of CD4+ or CD8+ T cells in the cKO mice were reduced by 10-fold or more in comparison to the Ctrl mice (Fig. 2a). In addition to the loss of CD3+CD4+ and CD3+CD8+ T cells, approximately 1/3 of the T cells in the cKO mice lost expression of CD4 and CD8 (Fig. 2b). This loss of CD4 and CD8, however, is not the result of a lineage switch to γδT cells, as cKO mice have similar numbers of γδ T cells in the thymus and the spleen (Supplemental Fig. S3a and S3b). The ontogeny of these cells remains to be determined. Moreover, in both CD4+ (Fig. 2c) and CD8+ (Fig. 2d) T cell compartments (supplemental Fig. S4a and S4b), a profound loss of naïve T cells was observed in the spleen. A similar reduction was observed in the lymph nodes (supplemental Fig. S4c, S4d and S4e). While more than 60% of the T cells in Ctrl mice exhibited the naïve phenotype, CD44loCD62Lhi, approximately 5% or less of the cKO T cells are naïve. In fact, most of the cKO T cells displayed markers associated with effector memory T cells (CD44hiCD62Llo), although significant increases in the central memory compartment were also observed. In addition, expression of CD69 was also elevated in the cKO T cells (Fig. 2e).
Fig. 2.

T-lineage-specific activation of Wnt signaling causes T cell lymphopenia and spontaneous activation. a. Wnt signaling causes massive reductions in both CD4 and CD8 T cells. Representative profile depicting the frequencies of CD4 and CD8 T cells in 6–8 week old WT and cKO spleens are shown on the left, while the summary is presented on the right (n=13). b. Wnt signaling causes loss of CD4 and CD8 markers in T cells. Representative profiles for CD4 and CD8 markers among CD3+ spleen cells are shown on the left, while the summary is shown on the right (n=11). c, d. Wnt signaling causes loss of naïve CD4 (c) and CD8 (d) T cells in the spleen. The left panels show representative profiles depicting the distribution of CD44 and CD62L markers, while the right panels show the summary of the frequencies of naïve (CD62Lhi CD44lo), central memory (CD62Lhi CD44hi), or effector memory (CD44hiCD62Llo) cells (n=13). e. Increased CD69 expression among cKO T cells. Data presented are % of CD69+ T cells among CD4 and CD8 T cells (n=9). Data are either representative or summary of those from at least three independent experiments.
The loss of T cells in the periphery could be due to defects in either T cell emigration or survival. To elucidate whether the rate of thymic output was reduced in the Apc- deficient thymocytes, we injected CFSE intrathymically and tracked recent thymic emigrants in the periphery. To avoid potential artifacts associated with peripheral labeling, we monitored the labeling of the thymus, excluding any samples that had less than 3% labeling of either CD4 SP or CD8 SP thymocytes. Moreover, since the efficacy of CFSE labeling of thymocytes is variable in different mice, we normalized the % of CFSE+ T cells in the lymph node by dividing them with the % CFSE+ of either CD4 or CD8 SP thymocytes. At 6 and 24 hours after CFSE injections, we detected comparable frequencies of CFSE+ T cells in the lymph nodes of cKO and Ctrl mice (Fig 3a, b). This data demonstrates that T cell emigration from the thymus was unaffected by the deletion of the Apc gene and suggests that the reduced accumulation of Apc-deficient T cells was due to loss of T cells after their emigration from the thymus.
Fig. 3.

Comparable emigration of T cells in mature Ctrl and cKO thymocytes. Thymocytes were labeled by intrathymic injections of CSFE. After 6 or 24 hours, Ctrl and cKO thymi and lymph nodes were harvested to analyze the frequency of CFSE+ CD4 or CD8 T cells. FACS profiles depicting the CFSE+ cells among gated CD4 or CD8 T cells in the thymus or lymph nodes are presented in a, while the summary from one experiment involving 6–14 mice per group from 2–3 independent experiments are shown in b. Rare mice with <3% labeling of either CD4 or CD8 SP thymocytes were excluded from the analyses.
To confirm this notion, we isolated CD24− mature thymocytes from CD45.2+ cKO and Ctrl mice, labeled them in vitro with CFSE and injected them intravenously into CD45.1+ congenic hosts and followed their persistence in peripheral blood and lymphoid organs. As shown in Fig. 4a upper panel, similar numbers of CFSE+ Ctrl and cKO T cells were observed in the spleen of the congenic host at 6 hours after transfer. By 24 hours, the numbers of cKO T cells in the spleen was less than 1/3 of their Ctrl counterparts. The frequencies of cKO T cells in the pooled lymph nodes were reduced by more than 10-fold (Fig. 4b). The rapid loss of T cells was attributable to cell death as there was a higher % of 7-AAD+ and/or Annexin V+ T cells in the cKO mice (Fig. 4c, d).
Fig. 4.

Rapid loss of mature cKO thymocytes in the periphery. CD24− mature T cells were isolated from Ctrl or cKO mice (CD45.2) and injected intravenously into congenic CD45.1 mice. The frequency of CD45.2+CFSE+ cells in the spleen (a) or lymph nodes (b) are summarized from three independent experiments. c. Increased apoptosis of mature cKO thymocytes at 6 hours after adoptive transfer. % apoptosis was detected via staining of 7-AAD and Annexin V. Representative FACS profiles are shown in the left, while summary data involving 3–4 mice per group were shown in the right. The data have been reproduced in two independent experiments.
To determine whether the loss of peripheral T cells in the cKO mice was cell intrinsic, we created bone marrow chimeras using cKO or Ctrl donor-type (CD45.2) cells mixed with an equal number of recipient-type (CD45.1) bone marrow cells. After 10 weeks, we harvested the thymi and spleens to investigate the reconstitution of CD45.2 donor cells. As shown in Fig. 5a, the % of CD45.2+ cells in the thymus was unaffected by the deletion of Apc. Based on the distribution of CD4 and CD8 markers, the development of both Ctrl and cKO thymocytes was grossly normal, as all major subsets were present among CD45.2+ thymocytes (Fig. 5b). However, the % of SP thymocytes were reduced by approximately 2-fold (Fig. 5b), perhaps due to a subtle competitive disadvantage of the cKO thymocytes. Among SP thymocytes, the % of Qa-2hi and CD24lo thymocytes were comparable between cKO and Ctrl groups (Fig. 5c).
Fig. 5.
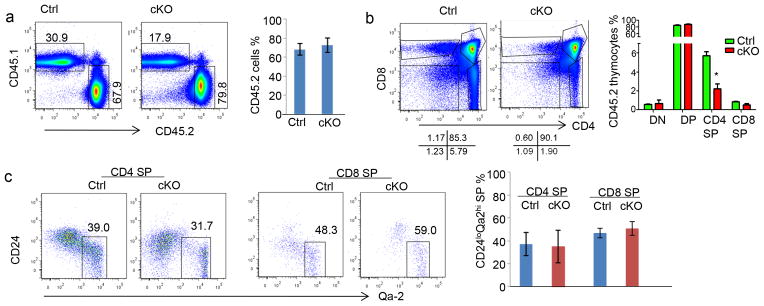
Grossly normal development of Apc-deficient T cells in bone marrow chimera mice. Bone marrow chimeras were generated by injecting 2.5×106 of either Ctrl or cKO bone marrow cells, in conjunction with an equal number of recipient CD45.1-type bone marrow cells into lethally irradiated mice. Mice were sacrificed 10 weeks after bone marrow transplantation. For all panels, representative profiles are shown on the left, while the summarized data from one experiment involving 4 mice per group are presented on the right. All data have been reproduced in two independent experiments. a. In the chimeric mouse thymus, Apc deletion does not affect thymocyte production. Data shown are frequencies of donor-derived T cells. b. Development of CD45.2+ thymocytes was assessed based on the distribution of CD4 and CD8 markers. c. Normal thymocyte maturation was assessed based on CD24 and Qa-2 expression on donor-type CD4 SP and CD8 SP thymocytes.
In sharp contrast to a roughly normal thymocyte development, severe cKO T-cell loss was observed in the spleens of chimera mice. Thus, while the frequency of CD45.2 donor leukocytes was unaffected by Apc deletion (Fig. 6a, b), more than a 20-fold reduction was observed in the frequency of T cells within the CD45.2 splenocytes (Fig. 6c, d). In addition to loss of T cell cellularity, the remaining T cells exhibited a marked difference in cell surface markers. Even though the majority of the Ctrl T cells displayed CD44loCD62Lhi markers of naïve T cells, most of the cKO T cells had either central or effector memory markers (Fig. 6e, f). Loss of naïve T cell markers and acquisition of memory/effector T cell markers in cKO T cells were reminiscent of what was observed in the Ctrl and cKO mice. On the other hand, cellularity of the recipient-type T cells was not affected in a trans-fashion by the genotype of the donor-type cells in the recipients (Supplemental Fig. S3c and S3d). Since the cKO T cells have developed and emigrated into a normal environment, both the loss of cellularity and the acquisition of activation markers following Apc deletion is cell-intrinsic.
Fig. 6.

T-cell intrinsic function of Wnt signaling and T cell survival. See Fig. 5 legends for the generation of chimera mice. a, b. Chimerism in the spleen was assessed based on the expression of congenic markers. c, d. Cell-intrinsic lymphopenia after lineage-specific Apc deletion was assessed based on 5-color flow cytometry using antibodies specific for CD45.1, CD45.2, CD3, CD4, and CD8. e, f. Spontaneous loss of naïve T cells is due to cell-intrinsic Apc deletion. The frequencies of naïve, central, and effector memory T cells were determined using CD45.2, CD44, CD62L, CD4 and CD8. Representative FACS profiles are shown in a, c and e, while summary data from two independent experiments involving 4 mice per group are shown in b, d and f.
cMyc over-expression contributes to apoptosis in mature T cells
cMyc is a β-catenin target gene that causes T-cell apoptosis through Fas/FasL interaction, death ligand tumor necrosis factor, and tumor necrosis factor related apoptosis-inducing ligand (36–38). As the first step to investigate its potential contribution to lymphopenia in cKO mice, we evaluated cMyc over-expression in CD24− thymocytes from Ctrl and cKO mice by quantitative PCR. As shown Fig 7a, the cMyc transcript was doubled in cKO mice. However, cMyc protein was not obviously over-expressed among the mature thymocytes. In contrast, peripheral cKO T cells had drastically higher levels of cMYC than Ctrl T cells (Fig 7b). We therefore generated the cMycfl/+Apcfl/fl;CD4-Cre+ (Myc+/−cKO) mice to diminish the impact of transcriptional activation of cMyc by Wnt signaling. Heterozygous deletion of cMyc resulted in a substantial reduction of the cMyc protein when compared to cKO T cells (Fig. 7b). Nevertheless, heterozygous deletion of cMyc partially restored the numbers of cKO T cells in the periphery (Fig. 7c). An approximately 3-fold increase of both CD4 and CD8 T cells was also observed in Myc+/− cKO mice in comparison to cMyc+/+cKO mice (Fig. 7d). To further demonstrate that over-expression of c-Myc caused apoptosis of Apc−/− T cells, we adoptively transferred either Apc−/− or Apc−/−cMyc+/− mature thymocytes into WT mice and followed the survival of the T cells in the host at 6 hours post-injection. As shown in Fig. 7e, Apc−/−cMyc+/− T cells exhibited reduced apoptosis and therefore increased survival in the WT hosts. This data demonstrates that poor survival of Apc−/− T cells can be rescued by down-regulation of cMyc expression. In addition to an overall increase in T cell numbers, the % of naïve CD4 and CD8 T cells increased by 3-fold or more, while that of naïve CD8 T cells increased by 50% (Fig. 7f, g). A corresponding reduction of central and effector memory T cells was also observed in CD4 T cells, although the accumulation of effector memory CD8 T cells was less affected by cMyc deletion (Fig. 7f,g).
Fig. 7.

Heterozygous deletion of cMyc partially attenuate the T cell defects caused by Apc deletion. a. Apc deletion increases cMyc mRNA in CD24− mature thymocytes as measured by quantitative PCR. Data shown are means+/− SEM (n=3). These data have been reproduced 3 times. b. Western blot analysis of c-Myc protein in pooled CD24− mature thymocytes from Ctrl (n=3), cMyc+/−cKO (n=6), and cMyc+/+cKO (n=9). These data have been reproduced twice. c. Representative profiles of CD4 and CD8 T cells in Ctrl, Myc+/−cKO, or cKO spleens. d. Summary of the frequencies of CD4 or CD8 T cells in in Ctrl, Myc+/− cKO, or cKO spleens (n=9). e. Heterozygous deletion of cMyc in T cells attenuates apoptosis of CD24− mature thymocytes at 6 hours after adoptive transfer via intravenous route. Aliquots of 1×106 CFSE-label thymocytes were injected into congenic CD45.1 recipients. Apoptosis was measured by staining with 7-AAD and Annexin V. Data shown are means and SEM (n=9). f. Representative profiles depicting the distribution of CD44 and CD62L markers on CD4 and CD8 T cells in Ctrl, Myc+/− cKO, or cKO spleens. g. Frequencies of naïve, central memory, or effector memory splendid T cells in 6–8 week old Ctrl, cMyc+/−cKO, or cKO mice (n=9). Data from c–g are either representative or summary of those from three independent experiments.
Our data in Fig. 7b demonstrated that heterozygous deletion of cMyc failed to reduce cMyc protein levels in cKO mice to WT levels. To determine whether the partial effect of a heterozygous cMyc deletion was due to an insufficient reduction in cMyc protein, we generated mice with a homozygously floxed cMyc locus. Deletion of cMyc had no effect on thymocyte development (Fig. 8a) and T cell cellularity in the periphery (Fig. 8b), although a reduction of CD44 levels was observed on both CD4 and CD8 T cells (Fig. 8c). Combinational deletion of both Apc and cMyc did not affect thymocyte development (Fig. 8d). However, a homozygous deletion of cMyc resulted in a further rescue of T cell survival defects in the cKO mice, although it did not completely restore T cell cellularity and survival defects (Fig. 8e, f).
Fig. 8.

Homozygous deletion of cMyc substantially rescues lymphopenia and spontaneous activation of Apc-deficient T cells. a–c. cMyc deletion has minimal impact on thymocyte development and T lymphocyte cellularity but reduces CD44 expression in spleen T cells. a. Representative profiles depicting thymocyte development based on distribution of CD4 and CD8 markers. c. Summary data showing the frequencies of CD4 and CD8 T cells, data shown are means and SEM (n=9). d. cMyc deletion reduces CD44 expression. Data shown are means and SEM (n=9) of mean fluorescence intensities, depicting CD44 expression among CD4 and CD8 T cells. d. Summary data on thymocytes subsets among 6–8 weeks old mice with or without deletion of Apc and genes. Data shown are means and SEM (n=9). e. Frequencies of CD4 or CD8 T cell from Ctrl (n=6), cKO (n=2), or, cMyc+/−cKO (n=5), or cMyc−/−cKO (n=3). f. Frequencies of naïve, central memory and effector memory T cells in the spleen from Ctrl (n=6), cKO (n=2), Myc+/− cKO (n=5), or cMyc−/−cKO (n=3) mice.
Discussion
Previous studies demonstrated that β-catenin overexpression produces multiple developmental defects in thymocyte development (39–43). The most dramatic phenotype was reported in mice with a deletion of the Apc locus, with largely depleted mature T cells and immature DP thymocytes (34). Surprisingly, with the use of a different Cre system (CD4-cre rather than Lck-cre) and floxed Apc allele, we observed no changes in either thymic cellularity nor percentages and numbers of DN, DP, and CD4/8 SP thymocytes. We took advantage of the largely normal T cell development in mice with CD4-Cre-driven deletion of the Apc exon 14 to show an unappreciated role for regulated Wnt signaling in survival of naïve T cells, including a massive loss of cellularity and a lack of the naïve T cell phenotype. Since CD44 is a target for Wnt signaling (44), it is of interest to consider whether loss of the naïve T cell phenotype is a reflection of increased Wnt signaling. It should be recognized that Wnt signaling may directly upregulates CD44 expression so the “true” naïve T cell phenotype may be masked. We consider it unlikely as the overwhelming majority of CD44+ T cells also lost CD62L expression. The concurrent loss of CD62L, massive loss of cellularity and high levels of apoptosis are more consistent with activation-induced cell death.
Since the phenotype of the Apc deletion is significantly attenuated by simultaneous deletion of the cMyc gene, at least part of the T cell loss is due to over-activation of cMyc. Since the homozygous deletion of cMyc in T cells was insufficient to restore T cell cellularity, additional signaling pathways down-stream of Wnt signaling maybe involved. In this context, it is of interest to note that Wnt signaling is upstream of the mTOR pathway (12), and studies by us and others have established a critical role for mTOR in survival of naïve T cells (5, 6). Additional studies are needed to establish whether mTOR and cMyc constitutes two parallel pathways for defective T cell survival caused by unregulated Wnt signaling.
It is of note that unlike the high rates and early onset of thymic lymphoma reported by another group (45, 46), we observed only rare cases of late-onset thymic lymphoma (Data not shown). However, our data are consistent with lack of lymphoma in the Apcmin/+ mice (47). Additional studies are needed to reconcile the differences. Since the previously reported lymphoma requires the Rag recombinase that peaks at the DN3 stage (46), and since the β-catenin activation was not detectable at DN3 in our model, we suspect that the lack of concurrent Rag and β-catenin activities explains the lower risk of thymic lymphoma in our model.
A previous report showed a dramatic arrest of thymocytes at the DN4 stage by an Apc deletion (34). The developmental block of thymocytes was also noted using retroviral vector over-expressing stabilized β-catenin (39, 40). However, This phenotype was not recapitulated when the interaction between β-catenin and Apc was inactivated by deletion of the Apc-binding domain on β-catenin (48). Our data clearly demonstrated that deletion of the Apc gene, starting at DN3 stage, had no discernible impact on thymocyte development, as revealed by the comparable cellularity and a normal distribution of the CD3, CD4, CD8, CD44, CD25, CD24, and Qa2 markers. One likely explanation is the kinetics of functional inactivation of Apc. The previous study found β-catenin increased in most of the cells from the DN3 stage and on, while our study observed an increase in only a small fraction of DN4 and all of the DP and SP thymocytes. The delayed functional inactivation of Apc in our model allowed normal thymocyte development. The reason for the delayed activation of Wnt signaling is unknown, as both the Cre-driver and the Apcfl differed in these studies. Since the CD4-Cre is activated later than the Lck-Cre in DN (49), we crossed the Apcfl allele with the Lck-Cre. We also found largely normal thymocyte development and severe lymphenia when the Lck-Cre was used. Although subtle differences exist between the two: Lck-Cre- and CD4-Cre-induced cKO, these differences are not sufficient to account for the dramatic discrepancy between our study and the earlier work (34). One explanation could be due to the use of different Apcfl alleles rather than different Cre-drivers. Finally, since activation of T-cells in the periphery can cause loss of thymocytes (35), the possibility that activation of T cells in the periphery may account for a part of the previously described phenotype (34) remains to be excluded.
Regardless of how the differences are reconciled, the lack of significant impact of T cell development allows us to reveal a critical role for regulated Wnt signaling in T cell survival. Given the significant role for lymphopenia in autoimmune diseases (50, 51), the missing link between T cell survival and the Wnt-cMyc pathway may be important for our understanding of the pathogenesis of autoimmune diseases.
Supplementary Material
Acknowledgments
We thank Drs. Eric Fearon, Yuan Zhu, Philip King and Beth Moore for helpful discussions throughout the study and Dr. Kaoru Sakabe for critical reading of the manuscript. Part of the study was performed at University of Michigan where the authors were previously employed.
This study is supported by grants from National Institute of Health (R01 AG036690-A1,PI: Zheng and R01 AI64350, PI: Liu). CW is supported by T32 AI007413 and Rackham Merit Fellowship.
Abbreviations used
- Apc
adenomatous polyposis coli
- cKO
Apcfl/flCD4-Cre+
- Ctrl
Apcfl/flCD4-Cre−
- DP
CD4+CD8+
- DN
CD4−CD8−
- SP
CD4+CD8− or CD4−CD8+
Footnotes
The authors have no financial conflict of interest.
References
- 1.Jameson SC, Conquest KA, Bean MJ. Positive selection of thymocytes. Annul Rev Immunol. 1995;13:93–126. doi: 10.1146/annurev.iy.13.040195.000521. [DOI] [PubMed] [Google Scholar]
- 2.von Boomer H. Developmental biology of T cells in T cell-receptor transgenic mice. Annul Rev Immunol. 1990;8:531–556. doi: 10.1146/annurev.iy.08.040190.002531. [DOI] [PubMed] [Google Scholar]
- 3.Love PE, Banderole A. Signal integration and crosswalk during thymocyte migration and emigration. Nat Rev Immunol. 2011;11:469–477. doi: 10.1038/nri2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Enrich MA, Conquest KA. Thymic emigration: when and how T cells leave home. J Immunol. 2008;181:2265–2270. doi: 10.4049/jimmunol.181.4.2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang K, Neal G, Green DR, et al. The tumor suppressor Tsc1 enforces quiescence of naive T cells to promote immune homeostasis and function. Nat Immunol. 2011;12:888–897. doi: 10.1038/ni.2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu Q, Liu Y, Chen C, et al. The tuberous sclerosis complex–mammalian target of repaying pathway maintains the quiescence and survival of naive T cells. J Immunol. 2011;187:1106–1112. doi: 10.4049/jimmunol.1003968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feng X, Impolite GC, Tina L, et al. Foxp1 is an essential transcriptional regulator for the generation of quiescent naive T cells during thymocyte development. Blood. 2010;115:510–518. doi: 10.1182/blood-2009-07-232694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fang X, Wang H, Toccata H, et al. Transcription factor Foxp1 exerts essential cell-intrinsic regulation of the quiescence of naive T cells. Nat Immunol. 2011;12:544–550. doi: 10.1038/ni.2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kermises YM, Beginner DR, Tenneco R, et al. Foxo1 links homing and survival of naive T cells by regulating L-selecting, CCR7 and interlocking 7 receptor. Nat Immunol. 2009;10:176–184. doi: 10.1038/ni.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duncan AW, Rates FM, Dibasic LN, et al. Integration of Notch and Wnt signaling in hematopoietic stem cell maintenance. Nat Immunol. 2005;6:314–322. doi: 10.1038/ni1164. [DOI] [PubMed] [Google Scholar]
- 11.Fleming HE, Janzen V, Lo Celso C, et al. Wnt signaling in the niche enforces hematopoietic stem cell quiescence and is necessary to preserve self-renewal in vivo. Cell Stem Cell. 2008;2:274–283. doi: 10.1016/j.stem.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inoki K, Ouyang H, Zhu T, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126:955–968. doi: 10.1016/j.cell.2006.06.055. [DOI] [PubMed] [Google Scholar]
- 13.Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8:387–398. doi: 10.1038/nrc2389. [DOI] [PubMed] [Google Scholar]
- 14.Niehrs C. The complex world of WNT receptor signalling. Nat Rev Mol Cell Biol. 2012;13:767–779. doi: 10.1038/nrm3470. [DOI] [PubMed] [Google Scholar]
- 15.Angers S, Moon RT. Proximal events in Wnt signal transduction. Nat Rev Mol Cell Biol. 2009;10:468–477. doi: 10.1038/nrm2717. [DOI] [PubMed] [Google Scholar]
- 16.Mosimann C, Hausmann G, Basler K. Beta-catenin hits chromatin: regulation of Wnt target gene activation. Nat Rev Mol Cell Biol. 2009;10:276–286. doi: 10.1038/nrm2654. [DOI] [PubMed] [Google Scholar]
- 17.Ioannidis V, Beermann F, Clevers H, Held W. The beta-catenin--TCF-1 pathway ensures CD4(+)CD8(+) thymocyte survival. Nat Immunol. 2001;2:691–697. doi: 10.1038/90623. [DOI] [PubMed] [Google Scholar]
- 18.Xu Y, Banerjee D, Huelsken J, et al. Deletion of beta-catenin impairs T cell development. Nat Immunol. 2003;4:1177–1182. doi: 10.1038/ni1008. [DOI] [PubMed] [Google Scholar]
- 19.Yu Q, Sharma A, Oh SY, et al. T cell factor 1 initiates the T helper type 2 fate by inducing the transcription factor GATA-3 and repressing interferon-gamma. Nat Immunol. 2009;10:992–999. doi: 10.1038/ni.1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Notani D, Gottimukkala KP, Jayani RS, et al. Global regulator SATB1 recruits beta-catenin and regulates T(H)2 differentiation in Wnt-dependent manner. PLoS Biol. 2010;8:e1000296. doi: 10.1371/journal.pbio.1000296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ding Y, Shen S, Lino AC, et al. Beta-catenin stabilization extends regulatory T cell survival and induces anergy in nonregulatory T cells. Nat Med. 2008;14:162–169. doi: 10.1038/nm1707. [DOI] [PubMed] [Google Scholar]
- 22.van Loosdregt J, Fleskens V, Tiemessen MM, et al. Canonical Wnt signaling negatively modulates regulatory T cell function. Immunity. 2013;39:298–310. doi: 10.1016/j.immuni.2013.07.019. [DOI] [PubMed] [Google Scholar]
- 23.Shibata H, Toyama K, Shioya H, et al. Rapid colorectal adenoma formation initiated by conditional targeting of the Apc gene. Science. 1997;278:120–123. doi: 10.1126/science.278.5335.120. [DOI] [PubMed] [Google Scholar]
- 24.Wolfer A, Bakker T, Wilson A, et al. Inactivation of Notch 1 in immature thymocytes does not perturb CD4 or CD8T cell development. Nat Immunol. 2001;2:235–241. doi: 10.1038/85294. [DOI] [PubMed] [Google Scholar]
- 25.Aoki K, Taketo MM. Adenomatous polyposis coli (APC): a multi- functional tumor suppressor gene. J Cell Sci. 2007;120:3327–3335. doi: 10.1242/jcs.03485. [DOI] [PubMed] [Google Scholar]
- 26.Askham JM, Moncur P, Markham AF, Morrison EE. Regulation and function of the interaction between the APC tumour suppressor protein and EB1. Oncogene. 2000;19:1950–1958. doi: 10.1038/sj.onc.1203498. [DOI] [PubMed] [Google Scholar]
- 27.Kaplan KB, Burds AA, Swedlow JR, et al. A role for the Adenomatous Polyposis Coli protein in chromosome segregation. Nat Cell Biol. 2001;3:429–432. doi: 10.1038/35070123. [DOI] [PubMed] [Google Scholar]
- 28.Joslyn G, Richardson DS, White R, Alber T. Dimer formation by an N-terminal coiled coil in the APC protein. Proc Natl Acad Sci USA. 1993;90:11109–11113. doi: 10.1073/pnas.90.23.11109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawasaki Y, Senda T, Ishidate T, et al. Asef, a link between the tumor suppressor APC and G-protein signaling. Science. 2000;289:1194–1197. doi: 10.1126/science.289.5482.1194. [DOI] [PubMed] [Google Scholar]
- 30.Watanabe T, Wang S, Noritake J, et al. Interaction with IQGAP1 links APC to Rac1, Cdc42, and actin filaments during cell polarization and migration. Dev Cell. 2004;7:871–883. doi: 10.1016/j.devcel.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 31.Gu H, Marth JD, Orban PC, et al. Deletion of a DNA polymerase beta gene segment in T cells using cell type-specific gene targeting. Science. 1994;265:103–106. doi: 10.1126/science.8016642. [DOI] [PubMed] [Google Scholar]
- 32.de Alboran IM, O’Hagan RC, Gartner F, et al. Analysis of C-MYC function in normal cells via conditional gene-targeted mutation. Immunity. 2001;14:45–55. doi: 10.1016/s1074-7613(01)00088-7. [DOI] [PubMed] [Google Scholar]
- 33.Chen C, Liu Y, Liu R, et al. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J Exp Med. 2008;205:2397–2408. doi: 10.1084/jem.20081297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gounari FCR, Cowan J, Guo Z, Dose M, Gounaris E, Khazaie K. Loss of adenomatous polyposis coli gene function disrupts thymic development. Nat Immunol. 2005;8:800–809. doi: 10.1038/ni1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin S, Bean MJ. Antigen-specific and nonspecific deletion of immature cortical thymocytes caused by antigen injection. Eur J Immunol. 1997;27:2726–2736. doi: 10.1002/eji.1830271037. [DOI] [PubMed] [Google Scholar]
- 36.Sansom OJ, V, Meniel S, Muncan V, et al. Myc deletion rescues Apc deficiency in the small intestine. Nature. 2007;446:676–679. doi: 10.1038/nature05674. [DOI] [PubMed] [Google Scholar]
- 37.Shi Y, Glynn JM, Guilbert LJ, et al. Role for c-myc in activation-induced apoptotic cell death in T cell hybridomas. Science. 1992;257:212–214. doi: 10.1126/science.1378649. [DOI] [PubMed] [Google Scholar]
- 38.Wang R, Brunner T, Zhang L, Shi Y. Fungal metabolite FR901228 inhibits c-Myc and Fas ligand expression. Oncogene. 1998;17:1503–1508. doi: 10.1038/sj.onc.1202059. [DOI] [PubMed] [Google Scholar]
- 39.Xu M, Sharma A, Hossain MZ, et al. Sustained expression of pre-TCR induced beta-catenin in post-beta-selection thymocytes blocks T cell development. J Immunol. 2009;182:759–765. doi: 10.4049/jimmunol.182.2.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu M, Sharma A, Wiest DL, Sen JM. Pre-TCR-induced beta- catenin facilitates traversal through beta-selection. J Immunol. 2009;182:751–758. doi: 10.4049/jimmunol.182.2.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu Q, Xu M, Sen JM. Beta-catenin expression enhances IL-7 receptor signaling in thymocytes during positive selection. J Immunol. 2007;179:126–131. doi: 10.4049/jimmunol.179.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu Q, Sen JM. Beta-catenin regulates positive selection of thymocytes but not lineage commitment. J Immunol. 2007;178:5028–5034. doi: 10.4049/jimmunol.178.8.5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sharma A, Chen Q, Nguyen T, et al. T cell factor-1 and beta-catenin control the development of memory-like CD8 thymocytes. J Immunol. 2012;188:3859–3868. doi: 10.4049/jimmunol.1103729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wielenga VJ, Smits R, Korinek V, et al. Expression of CD44 in Apc and Tcf mutant mice implies regulation by the WNT pathway. Am J Pathol. 1999;154:515–523. doi: 10.1016/S0002-9440(10)65297-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dose M, Emmanuel AO, Chaumeil J, et al. beta-Catenin induces T-cell transformation by promoting genomic instability. Proc Natl Acad Sci USA. 2014;111:391–396. doi: 10.1073/pnas.1315752111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guo Z, Dose M, Kovalovsky D, et al. Beta-catenin stabilization stalls the transition from double-positive to single-positive stage and predisposes thymocytes to malignant transformation. Blood. 2007;109:5463–5472. doi: 10.1182/blood-2006-11-059071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sharma A, Sen JM. Molecular basis for the tissue specificity of beta-catenin oncogenesis. Oncogene. 2013;32:1901–1909. doi: 10.1038/onc.2012.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gounari F, Aifantis I, Khazaie K, et al. Somatic activation of beta-catenin bypasses pre-TCR signaling and TCR selection in thymocyte development. Nat Immunol. 2001;2:863–869. doi: 10.1038/ni0901-863. [DOI] [PubMed] [Google Scholar]
- 49.Wolfer A, Wilson A, Nemir M, et al. Inactivation of Notch1 impairs VDJbeta rearrangement and allows pre-TCR-independent survival of early alpha beta Lineage Thymocytes. Immunity. 2002;16:869–879. doi: 10.1016/s1074-7613(02)00330-8. [DOI] [PubMed] [Google Scholar]
- 50.King C, Ilic A, Koelsch K, Sarvetnick N. Homeostatic expansion of T cells during immune insufficiency generates autoimmunity. Cell. 2004;117:265–277. doi: 10.1016/s0092-8674(04)00335-6. [DOI] [PubMed] [Google Scholar]
- 51.Liu Y, Zheng P. CD24: a genetic checkpoint in T cell homeostasis and autoimmune diseases. Trend Immunol. 2007;28:315–320. doi: 10.1016/j.it.2007.05.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.