

Abstract
The goal for the treatment of patients with diabetes has today shifted from merely reducing glucose concentrations to preventing the natural decline in β-cell function and delay the progression of disease. Pancreatic β-cell dysfunction and decreased β-cell mass are crucial in the development of diabetes. The β-cell defects are the main pathogenesis in patients with type 1 diabetes and are associated with type 2 diabetes as the disease progresses. Recent studies suggest that human pancreatic β-cells have a capacity for increased proliferation according to increased demands for insulin. In humans, β-cell mass has been shown to increase in patients showing insulin-resistance states such as obesity or in pregnancy. This capacity might be useful for identifying new therapeutic strategies to reestablish a functional β-cell mass. In this context, therapeutic approaches designed to increase β-cell mass might prove a significant way to manage diabetes and prevent its progression. This review describes the various β-cell defects that appear in patients with diabetes and outline the mechanisms of β-cell failure. We also review common methods for assessing β-cell function and mass and methodological limitations in vivo. Finally, we discuss the current therapeutic approaches to improve β-cell function and increase β-cell mass.
Keywords: β-Cell function, β-Cell mass, Therapeutic agents
INTRODUCTION
Type 1 diabetes mellitus (T1DM) is characterized by an absolute deficit of pancreatic β-cells caused by autoimmune-mediated destruction, whereas insulin resistance and abnormal insulin secretion are central to the development of type 2 diabetes mellitus (T2DM). In patients with T2DM, the pancreas responds initially by expanding the mass of β-cells and increasing insulin secretion to compensate for the increased metabolic demand [1]. However, when this compensation is not enough, blood glucose concentrations increase, leading to decreased β-cell mass and dysfunction [1]. Thus, the reduction of β-cell mass is a common feature of both T1DM and T2DM. In general, pancreatic β-cell functions decrease up to approximately 50% of normal at the time of diagnosis of T2DM [2]. In autopsy studies on patients with T2DM, the β-cell mass was found to be only about 40% to 60% that of control subjects [1,3]. Previous studies have reported that β-cells have a capacity for increased proliferation in response to increased insulin demands [4,5,6]. This aspect of their physiology might be useful for identifying new therapeutic strategies to delay the progression of diabetes. Decreased β-cell mass in patients with diabetes might be improved through pancreatic transplantation or intraportal transplantation of isolated islets [7]. However, such procedures are associated with both surgical morbidity and the adverse effects of chronic immunosuppression [8]. Therefore, there is a need for alternative approaches for increasing functional β-cell mass in patients with diabetes.
In this review, we will outline the common defects in β-cell mass and potential mechanisms of β-cell failure. Commonly used methods for assessing β-cell function and mass will be described. Finally, we will discuss current therapeutic approaches for increasing β-cell mass.
DECREASED β-CELL FUNCTION AND MASS IN PATIENTS WITH DIABETES
T1DM is characterized by autoimmune-mediated β-cell destruction, whereas T2DM is characterized by insulin resistance and a progressive decline in β-cell function. Moreover, β-cell dysfunction is now considered to be a precipitating event in the development of T2DM [9]. Clearly, β-cell dysfunction is crucial in the development of diabetes because it cannot occur without an impairment in insulin secretion [9]. β-Cells are able to compensate for insulin resistance for many years before the onset of diabetes. However, if this compensation is inadequate, blood glucose levels increase and eventually diabetes develops.
In addition to β-cell dysfunction, it has also been suggested that deficits in β-cell mass are critical in the progression of both T1DM and T2DM. T1DM is clearly associated with autoimmune-mediated β-cells death and a reduction in β-cell mass [10]. However, it is controversial whether β-cell mass is decreased in patients with T2DM [11,12]. An autopsy study has shown that patients with T2DM have decreased β-cell mass compared with age- and body mass index-matched nondiabetic individuals [12].
The importance of reduced β-cell mass in the development of diabetes is supported by several lines of evidence. First, there is a ~99% deficit in mass in patients with long-standing T1DM [10] and a ~65% deficit in those with long-standing T2DM [1]. Second, surgical reduction of β-cell mass led to the development of diabetes in humans as well as in animal models [4,13]. Third, the typical metabolic defects of patients with T2DM such as impaired insulin secretion and insulin resistance can be mimicked in animals by a progressive loss of β-cells [14]. Fourth, despite the overt presence of insulin resistance, hyperglycemia can be improved by restoration of β-cell mass through pancreatic transplantation [15]. These findings support the idea that reduced β-cell mass as well as β-cell dysfunction both contribute to the development and progressive course of DM.
ASSESSING β-CELL MASS
There are several limitations in the medical visualization of β-cell mass. Pancreatic islets are small structures (200 µm in diameter) dispersed throughout the pancreas [16]. Endogenous β-cells constitute only 1% to 2% of the total cell volume in pancreatic tissue [16]. Furthermore, there are no significant differences in density and echogenicity between the exocrine and endocrine pancreatic tissues [16]. These obstacles have so far hampered the reliable quantification of β-cell mass.
There are few studies on human β-cell mass because it is difficult to obtain pancreatic tissues. Limited autopsy studies have demonstrated that β-cell mass is decreased in patients who died with T2DM [1]. One study revealed that the relative β-cell volume is decreased in both obese and lean human patients with T2DM compared with nondiabetic controls [1]. However, the measured β-cell mass at autopsy is not the functional β-cell mass because of the lack of measured correlations with glycaemia, insulinaemia, or β-cell function.
Differences in pancreatic volume and fat content between normal subjects and patients have been measured after different durations of T2DM [17]. Pancreatic volume and fat were measured by 64-slice multidetector-row computed tomography. The pancreatic volume was smaller and pancreatic fat content was increased in patients with T2DM than the normal subjects. The pancreatic volume and fat accumulation in the pancreas were positively associated with biochemical parameters reflecting pancreatic β-cell function [17].
Although obtaining human pancreatic tissue in humans is challenging, noninvasive imaging approaches have been applied to determine the functional mass of β-cells in living subjects. These approaches have relied on the specific labeling of β-cells using enzymes, cell receptors, or surface structures that are predominantly or exclusively expressed on β-cells [16]. A number of different β-cell markers, including glucagon-like peptide-1 (GLP-1) receptors, sulfonylurea receptors, vesicular amine transporter 2, and gangliosides have been used as targets for labeling [16]. In addition, these labeling can be detected by radionuclide imaging such as positron emission tomography or magnetic resonance imaging [16]. However, β-cell markers for clinical use require high specificity and stability as well as high-resolution imaging. To date, none of these markers has provided approximations of β-cell mass with sufficient sensitivity and specificity to justify their routine application in humans.
Because of the lack of reliable routine methods to determine β-cell mass in humans, indirect testing of β-cell function could be a suitable alternative. Functional tests of insulin secretion are based on the assumption that the amount of insulin secreted is proportional to the β-cell mass [16]. Although, insulin secretion might well change independently from β-cell mass, indirect testing of β-cell function currently represents the most reliable method of estimating β-cell mass in humans. In human experiments, the acute insulin response (AIR) to glucose, AIR to arginine, and, maximal AIR to arginine were significantly correlated with estimates of β-cell mass, with r values of ~0.80 [18].
POTENTIAL MECHANISMS IN DECREASED β-CELL MASS AND DYSFUNCTION
Regulation of β-cell mass appears to involve a balance between the dynamic process of β-cell mass expansion, through replication and neogenesis, and loss via apoptosis (Fig. 1) [19]. Disruption in the pathways of β-cell formation or an increased rate of β-cell death can result in a decrease in β-cell mass [1]. An autopsy study suggested that the frequency and rate of β-cell apoptosis was increased significantly in patients with T2DM, whereas the rate of replication of β-cells was normal [1]. This imbalance between apoptosis and regeneration of pancreatic β-cells is poorly understood, but several mechanisms have been proposed as triggers for the increased loss [20]. Thus, in an environment of metabolic overload and insulin resistance, β-cells initially activate compensatory pathways to improve insulin secretion, but they eventually initiate several pathological programs that synergistically promote β-cell dysfunction and apoptosis [20]. These pathological programs include glucotoxicity, lipotoxicity [21], and oxidative stress. Other potential factors leading to β-cell death are endoplasmic reticulum (ER) stress [22], inflammatory stress, amyloid stress, and loss of islet cell integrity [20]. However, these mechanisms and the timing of diabetes onset at a given levels of β-cell loss vary between individuals and are influenced by factors such as genetic susceptibility [20].
Fig. 1.

Regulation of the pancreatic β-cell mass dynamics.
THERAPEUTIC APPROACHES FOR MAINTAINING β-CELL MASS IN PATIENTS WITH DIABETES
Some evidence suggests that β-cells have capacity for increased proliferation in response to increased insulin demand. In rodents, β-cell replication increased by 2.8-fold after partial pancreatectomy [4] and by 2.5-fold during pregnancy [5]. The overall capacity for β-cell replication in humans is much lower than in rodents, but it can increase by 50% in subjects with obesity [1]. Both insulin secretion and β-cell mass increase during pregnancy [6]. Given the important role of β-cell mass in the development of diabetes, their capacity for regeneration could be used to identify a therapeutic agent for reestablishing a functional β-cell mass.
Current antidiabetic medications aim at decreasing glucose levels, either by improving insulin secretion in the remaining β-cells (using insulin secretagogues), by improving the response to insulin in target tissues (using insulin sensitizers), or by replacing the missing insulin (using exogenous insulin treatment). A major concern in the treatment of patients with T2DM is to identify a therapeutic agent that can prevent the gradual declines in β-cell function and mass. Debate is ongoing as to the potential effects of oral hypoglycemic agents on β-cell death and proliferation. Some drugs have been proposed to accelerate β-cell loss (e.g., glibenclamide and glyburide) [23], while other drugs such as peroxisome proliferator-activated receptor-gamma (PPARγ) agonists, GLP-1 receptor agonists, dipeptidyl peptidase-4 (DPP-4) inhibitors, glycogen synthase kinase 3β (GSK3β) inhibitors, or G protein-coupled receptor 40 (GPR40) agonists have been suggested to protect β-cell function [16]. The effects of current and future anti-diabetic drugs on β-cell function and mass in animal and human data have been summarized in the Table 1.
Table 1.
Therapeutic approaches for maintaining β-cell function and mass in animal and human data
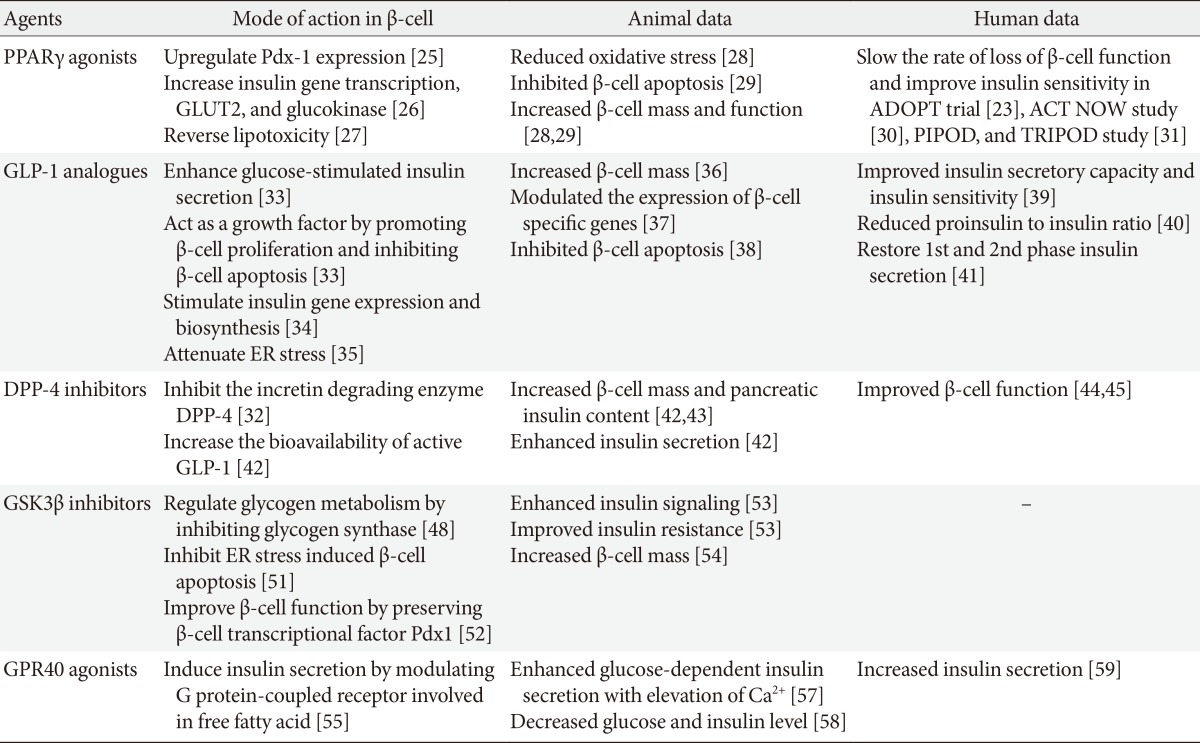
PPARγ, peroxisome proliferator-activated receptor-gamma; GLUT2, glucose transporter 2; ADOPT, A Diabetes Outcome Progression Trial; ACT NOW, Actos Now for Prevention of Diabetes; PIPOD, Pioglitazone In Prevention Of Diabetes; TRIPOD, TRoglitazone In the Prevention Of Diabetes; GLP-1, glucagon-like peptide-1; ER, endoplasmic reticulum; DPP-4, dipeptidyl peptidase-4; GSK3β, glycogen synthase kinase 3 β; GPR40, G protein-coupled receptor 40.
PPARγ agonists
Mode of action
Thiazolidinedione (TZD) is a synthetic ligand of PPARγ, which is a member of the nuclear hormone receptor family and contributes to many biological processes such as glucose homeostasis, lipid metabolism, and cellular proliferation and differentiation [24]. PPARγ regulates expression of the gene encoding prodifferentiation transcription-factor pancreas duodenum homeobox-1 (PDX-1) in β-cells [25]. TZDs are known to improve insulin sensitivity and preserve β-cell function, and several mechanisms have been proposed (Fig. 2). First, stimulation with PPARγ agonist upregulates PDX-1 expression and increases glucose transporter 2 (GLUT2) and glucokinase expression levels in β-cells [26]. Second, TZD reduces the intracellular levels of toxic lipid metabolites and reverses lipotoxicity [27]. TZD prevents the cytostatic effect of free fatty acids (FFAs) as well as FFA-induced inhibition of PPARγ and insulin mRNA expression levels, while restoring glucose-mediated insulin release and pancreatic islet hormone content [27]. Third, TZD increases insulin sensitivity in muscle and liver [24]. Thus, treatment with TZD can improve β-cell function via a variety of mechanisms.
Fig. 2.

Regulation of the pancreatic β-cell by peroxisome proliferator-activated receptor-gamma (PPARγ). RXR, retinoid X receptor; GLP-1R, glucagon-like peptide-1R; PI3K, phosphatidylinositol-3 kinase; IRS, insulin receptor substrate; PDX-1, pancreas duodenum homeobox-1; GLUT2, glucose transporter 2.
Animal studies
Several studies have demonstrated the effect of PPARγ agonists on reducing β-cell mass and alteration of β-cell function in animal models of T2DM. In a db/db mouse model, long-term treatment with pioglitazone reduced oxidative stress and thereby preserved β-cell mass [28]. In a Zucker diabetic fatty (ZDF) rat model, rosiglitazone reduced net β-cell death, and increased β-cell mass by levels almost twice that of the untreated control group [29]. These results support the idea that treatment with TZD has the potential to preserve β-cell mass.
Human studies
Several clinical studies have shown that TZD is effective in improving and preserving β-cell function. In the Actos Now for Prevention of Diabetes (ACT NOW) study, pioglitazone decreased the rate of conversion of subjects from having impaired glucose tolerance (IGT) to T2DM by 72% and significantly improved insulin sensitivity and β-cell function [30]. Development of diabetes mellitus in Hispanic women with gestational diabetes mellitus was decreased by 52% to 62% in the TRoglitazone In the Prevention Of Diabetes (TRIPOD) and Pioglitazone In Prevention Of Diabetes (PIPOD) studies [31]. In the A Diabetes Outcome Progression Trial (ADOPT) study, treatment with rosiglitazone slowed the rate of loss of β-cell function and improved insulin sensitivity more than did either metformin or glyburide [23]. In summary, TZD enhances β-cell function and insulin sensitivity in patients with diabetes.
GLP-1 agonists
Mode of action
GLP-1 is an incretin hormone secreted from enteroendocrine L cells in the distal ileum and colon [32]. GLP-1 has distinct actions on β-cells (Fig. 3). First, it enhances glucose-stimulated insulin secretion through activation of cyclic adenosine monophosphate (cAMP). Increased cAMP upregulates protein kinase A (PKA) and exchange protein activated by cAMP, which leads to rapid increases in intracellular calcium and insulin exocytosis in a glucose-dependent manner [33]. Second, GLP-1 acts as a growth factor by promoting β-cell proliferation and inhibiting β-cell apoptosis [33]. GLP-1 promotes epidermal growth factor receptor transactivation, which leads to the activation of phosphatidylinositol-3 kinase and its downstream effectors [34]. Activation of kinase cascades leads to the activation or repression of gene transcription and finally stimulating β-cell mass expansion [34]. Third, GLP-1 promotes insulin gene expression and biosynthesis, which increases the expression of the transcription factor Pdx-1 and the binding of this factor to the insulin gene promoter [34]. Fourth, GLP-1 attenuates ER stress via activation of PKA and preserves β-cell function and survival [35]. Therefore, these activities of GLP-1 suggest that it has the potential to reverse diabetes-associated defects in the failing β-cell.
Fig. 3.

Regulation of the pancreatic β-cell by glucagon-like peptide-1 (GLP-1). GLUT2, glucose transporter 2; K-ATP, ATP-sensitive potassium channel; TCA, tricarboxylic acid; EGFR, epidermal growth factor receptor; VDCC, voltage-dependent calcium channels; PI3K, phosphatidylinositol-3 kinase; IRS, insulin receptor substrate; PKC, protein kinase C; MAPK, mitogen-activated protein kinase; ER, endoplasmic reticulum; cAMP, cyclic adenosine monophosphate; PKA, protein kinase A; AC, adenylate cyclase; Epac, exchange protein activated by cAMP.
Animal studies
Several studies of rat models have suggested that GLP-1 agonists influence the development of β-cell mass and improve β-cell function. In glucose-intolerant Goto-Kakizaki rats, treatment with GLP-1 increased both the pancreatic insulin content and β-cell mass [36]. GLP-1 is also capable of modulating the expression of islet β-cell-specific genes [37]. In Wistar rats, the age-dependent decline in β-cell function and the subsequent impairment in glucose tolerance were reversed by GLP-1 administration [37]. In this animal model, GLP-1 treatment activated transcription of the genes encoding insulin and glucokinase, as well as GLUT2 [37]. This was associated with an expansion of β-cell mass via islet cell neogenesis. Furthermore, there is strong evidence to suggest that GLP-1 receptor activation can protect β-cells from apoptosis [38]. Thus, infusion of GLP-1 into ZDF rats promoted β-cell growth and inhibited apoptosis [38]. Islets of Langerhans from GLP-1-treated rats had significantly fewer apoptotic nuclei [38]. These results support the idea that treatment with GLP-1 has the potential to preserve the β-cell mass and function in animal models.
Human studies
A 6-week subcutaneous infusion of GLP-1 in patients with T2DM produced substantial improvements in insulin secretory capacity and insulin sensitivity, with a reduction in the hemoglobin A1c (HbA1c) level of 1.2% [39]. In another study of patients with T2DM, the addition of exenatide to classic oral antidiabetic agents resulted in an average 1.0% reduction in the HbA1c [40]. This study showed that an improvement in glycemic control was associated with a reduced proinsulin-to-insulin ratio, suggesting a beneficial effect on the β-cells [40]. Treatment with exenatide also restored first-phase insulin secretion and augmented second-phase insulin secretion when infused intravenously along with glucose in patients with T2DM [41]. In summary, treatment with GLP-1 enhances insulin secretory function as well as insulin sensitivity in patients with diabetes.
DPP-4 inhibitors
DPP-4 and mode of action of its inhibitors
Endogenous GLP-1 is rapidly degraded by the enzyme DPP-4, resulting in an active plasma half-life time of only a few minutes [32]. DPP-4 is a ubiquitous membrane-spanning cell-surface aminopeptidase widely expressed in many tissues [32]. DPP-4 preferentially cleaves peptides with a proline or alanine residue in the second amino terminal position. DPP-4 inhibitors block the incretin-degrading function of DPP-4, thereby increasing the bioavailability of active GLP-1, which results in enhanced consumption-related insulin secretion. DPP-4 inhibitors mimic many of the actions ascribed to GLP-1 receptor agonists, including stimulation of insulin production and inhibition of glucagon secretion, and preservation of β-cell mass by stimulating cell proliferation and inhibiting apoptosis [32].
Animal studies
Treatment with DPP-4 inhibitors improved glucose tolerance, stimulated insulin secretion, reduced glucagon secretion, and increased pancreatic β-cell mass in rodents [42,43]. Islets isolated from inhibitor-treated animals showed enhanced GLUT2 expression and glucose-stimulated insulin secretion after 8 weeks of treatment [42]. In that study, DPP-4 inhibitors caused an 8-fold increase in insulin content and an increase in the number of small islets and β-cells [42]. In another study using a model of high fat diet-induced obesity, insulin secretion was improved and prevented extensive peri-insulitis after 11 months of vildagliptin treatment. These results suggest that DPP-4 inhibitors improve β-cell function, promote β-cell mass expansion, and stimulate insulin biosynthesis.
Human studies
Assessment of the insulin response to a mixed meal by mathematical model analysis indicated that vildagliptin would improve β-cell function in the short (28 days) and long term (52 weeks) [44,45]. After 52 weeks of vildagliptin treatment, insulin secretion and insulin sensitivity during meal ingestion increased [44]. The early β-cell response, calculated as the insulinogenic index, and the postprandial area under the curve of insulin and glucose levels were improved by DPP-4 inhibitors in several trials [46,47]. In summary, evidence suggests that DPP-4 inhibitors improve pancreatic islet cell function in humans based on both static and dynamic parameters.
GSK3β inhibitors
Role of GSK3 in diabetes and mode of action of its inhibitors
GSK3β is a serine/threonine protein kinase involved in numerous cellular processes including glycogen metabolism [48]. Glycogen metabolism is important for glucose homeostasis. The level of blood glucose is largely determined by the rate at which glucose is taken up by skeletal muscle and liver and converted into glycogen, and by the rate at which it is produced by the liver [49]. During this process, GSK3β is a downstream target of insulin-activated signaling and is an important enzyme involved in the control of glycogen metabolism [48]. Glycogen synthase, the rate-limiting enzyme for glycogen synthesis, is a GSK3β substrate and GSK3β inhibits its activity by inducing its phosphorylation. Conversely, insulin promotes dephosphorylation and activation of glycogen synthase by suppressing GSK3 activity, and this is mediated through the activation of phosphatidylinositol kinase-3 and its downstream target, protein kinase B (Fig. 4) [50]. GSK3β is also involved in ER stress-induced β-cell apoptosis in the pancreas [51]. Moreover, GSK3β contributes to β-cell dysfunction by modulating the transcriptional factor Pdx-1 [52].
Fig. 4.

Insulin-induced inactivation of glycogen synthase kinase 3 β (GSK3β). IRS, insulin receptor substrate; PI3K, phosphatidylinositol-3 kinase.
Animal studies
In several experimental studies with animal models of diabetes, treatment with GSK3β inhibitors has shown to enhance insulin signaling and improve insulin resistance [53]. Furthermore, recent animal data showed that inhibiting GSK3β in the pancreas leads to increased β-cell mass [54]. Based on these findings, inhibiting GSK3β could be a novel therapeutic strategy for the treatment of patients with T2DM, not only by enhancing insulin sensitivity, but also by improving insulin secretion.
GPR40 agonists
Role of GPR40 in diabetes and mode of action of agonists
GPR40, or the FFA receptor 1, is a GPR involved in FFA-induced insulin secretion [55]. GPR40 is highly expressed in pancreatic β-cells [55]. It was found to play an important role in regulating glucose-stimulated insulin secretion [55]. GPR40 may be coupled with the α-subunit of the Gq family of G proteins (Gqα) and activates phospholipase C (PLC) (Fig. 5). In turn, PLC hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) into the inositol 1,4,5-triphophate (IP3) and diacylglycerol (DAG). Cytosolic IP3 triggers release of calcium, whereas DAG activates PKC [56]. Subsequently, GPR-mediated increase in intracellular calcium may be linked to insulin release [55]. GPR40 has drawn much attention as a potential therapeutic target in managing patients with diabetes.
Fig. 5.

Regulation of the pancreatic β-cell by G protein-coupled receptor 40 (GPR40). GLUT2, glucose transporter 2; K-ATP, ATP-sensitive potassium channel; TCA, tricarboxylic acid; VDCC, voltage-dependent calcium channels; PLC, phospholipase C; DAG, diacylglycerol; PKC, protein kinase C; IP3, 1,4,5-trisphosphate; PIP2, 4,5-bisphosphate; ER, endoplasmic reticulum; cAMP, cyclic adenosine monophosphate; PKA, protein kinase A; Epac, exchange protein activated by cAMP; AC, adenylate cyclase; GLP-1, glucagon-like peptide-1.
In vitro studies
In both human and rat islets, TAK-875, a GPR40 agonist, enhanced glucose-induced insulin secretion in a glucose-dependent manner [57]. This insulin-secretory effect was correlated with the elevation of Ca2+ concentrations in β-cells. Long-term treatment with low doses of GW-9508-another GPR40 agonist-decreased circulating glucose and insulin levels significantly, which resulted in improvements in glucose intolerance in high fat diet-induced diabetic mice [58].
In vivo studies
There have been two phase II clinical trials with GPR40 agonists. The first study showed that administration of TAK-875 to patients with T2DM improved glucose homeostasis by increasing insulin secretion [59]. The second study with 426 patients randomly assigned to TAK-875, placebo, or glimepiride treatment groups showed significant reductions in HbA1c levels from baseline in the TAK-875 (6.25 to 50 mg) and glimepiride (4 mg) treatment groups compared with the placebo group at week 12 [60]. These data suggest that GPR40 agonists might have a therapeutic role in preserving β-cell function. However, further clinical trials with TAK-875 have been recently suspended because it was found to be associated with elevated levels of liver enzymes.
CONCLUSIONS
The treatment of patients with T2DM should aim not only to control glucose levels, but also to prevent the gradual decline in β-cell function and decrease in β-cell mass. These alterations are common features of both T1DM and T2DM. Several studies have reported that β-cells have the capacity for increased proliferation in response to an increased demand for insulin. Given the important role and plasticity of the overall β-cell mass, therapeutic strategies aimed at preserving β-cell mass and function will have significant clinical impact on the severity of complications associated with both types of diabetes.
In this review, we have discussed potential therapeutic agents that maintain or restore β-cell mass and function and have summarized their modes of action based on studies in animals and humans. However, there are limitations in the visualization of β-cell mass in living humans and there are no direct imaging methods for quantifying β-cell mass. Therefore, methods for the indirect testing of β-cell function might offer suitable alternatives. In conclusion, the onset of both T1DM and T2DM is closely associated with insulin secretory dysfunction and significant loss of functional β-cell mass. A better understanding of the assessment of β-cell mass and protective mechanisms for improving β-cell function and mass will be helpful for modulating the natural progression of these diseases.
Footnotes
No potential conflict of interest relevant to this article was reported.
References
- 1.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 2.Ferrannini E, Gastaldelli A, Miyazaki Y, Matsuda M, Mari A, DeFronzo RA. Beta-cell function in subjects spanning the range from normal glucose tolerance to overt diabetes: a new analysis. J Clin Endocrinol Metab. 2005;90:493–500. doi: 10.1210/jc.2004-1133. [DOI] [PubMed] [Google Scholar]
- 3.Rahier J, Guiot Y, Goebbels RM, Sempoux C, Henquin JC. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab. 2008;10(Suppl 4):32–42. doi: 10.1111/j.1463-1326.2008.00969.x. [DOI] [PubMed] [Google Scholar]
- 4.Peshavaria M, Larmie BL, Lausier J, Satish B, Habibovic A, Roskens V, Larock K, Everill B, Leahy JL, Jetton TL. Regulation of pancreatic beta-cell regeneration in the normoglycemic 60% partial-pancreatectomy mouse. Diabetes. 2006;55:3289–3298. doi: 10.2337/db06-0017. [DOI] [PubMed] [Google Scholar]
- 5.Sorenson RL, Brelje TC. Adaptation of islets of Langerhans to pregnancy: beta-cell growth, enhanced insulin secretion and the role of lactogenic hormones. Horm Metab Res. 1997;29:301–307. doi: 10.1055/s-2007-979040. [DOI] [PubMed] [Google Scholar]
- 6.Van Assche FA, Aerts L, De Prins F. A morphological study of the endocrine pancreas in human pregnancy. Br J Obstet Gynaecol. 1978;85:818–820. doi: 10.1111/j.1471-0528.1978.tb15835.x. [DOI] [PubMed] [Google Scholar]
- 7.Robertson RP. Successful islet transplantation for patients with diabetes: fact or fantasy? N Engl J Med. 2000;343:289–290. doi: 10.1056/NEJM200007273430409. [DOI] [PubMed] [Google Scholar]
- 8.Robertson RP. Islet transplantation as a treatment for diabetes: a work in progress. N Engl J Med. 2004;350:694–705. doi: 10.1056/NEJMra032425. [DOI] [PubMed] [Google Scholar]
- 9.Bonora E. Protection of pancreatic beta-cells: is it feasible? Nutr Metab Cardiovasc Dis. 2008;18:74–83. doi: 10.1016/j.numecd.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 10.Meier JJ, Bhushan A, Butler AE, Rizza RA, Butler PC. Sustained beta cell apoptosis in patients with long-standing type 1 diabetes: indirect evidence for islet regeneration? Diabetologia. 2005;48:2221–2228. doi: 10.1007/s00125-005-1949-2. [DOI] [PubMed] [Google Scholar]
- 11.Guiot Y, Sempoux C, Moulin P, Rahier J. No decrease of the beta-cell mass in type 2 diabetic patients. Diabetes. 2001;50(Suppl 1):S188. doi: 10.2337/diabetes.50.2007.s188. [DOI] [PubMed] [Google Scholar]
- 12.Matveyenko AV, Butler PC. Relationship between beta-cell mass and diabetes onset. Diabetes Obes Metab. 2008;10(Suppl 4):23–31. doi: 10.1111/j.1463-1326.2008.00939.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kendall DM, Sutherland DE, Najarian JS, Goetz FC, Robertson RP. Effects of hemipancreatectomy on insulin secretion and glucose tolerance in healthy humans. N Engl J Med. 1990;322:898–903. doi: 10.1056/NEJM199003293221305. [DOI] [PubMed] [Google Scholar]
- 14.Matveyenko AV, Butler PC. Beta-cell deficit due to increased apoptosis in the human islet amyloid polypeptide transgenic (HIP) rat recapitulates the metabolic defects present in type 2 diabetes. Diabetes. 2006;55:2106–2114. doi: 10.2337/db05-1672. [DOI] [PubMed] [Google Scholar]
- 15.Nath DS, Gruessner AC, Kandaswamy R, Gruessner RW, Sutherland DE, Humar A. Outcomes of pancreas transplants for patients with type 2 diabetes mellitus. Clin Transplant. 2005;19:792–797. doi: 10.1111/j.1399-0012.2005.00423.x. [DOI] [PubMed] [Google Scholar]
- 16.Meier JJ. Beta cell mass in diabetes: a realistic therapeutic target? Diabetologia. 2008;51:703–713. doi: 10.1007/s00125-008-0936-9. [DOI] [PubMed] [Google Scholar]
- 17.Lim S, Bae JH, Chun EJ, Kim H, Kim SY, Kim KM, Choi SH, Park KS, Florez JC, Jang HC. Differences in pancreatic volume, fat content, and fat density measured by multidetector-row computed tomography according to the duration of diabetes. Acta Diabetol. 2014;51:739–748. doi: 10.1007/s00592-014-0581-3. [DOI] [PubMed] [Google Scholar]
- 18.Robertson RP. Estimation of beta-cell mass by metabolic tests: necessary, but how sufficient? Diabetes. 2007;56:2420–2424. doi: 10.2337/db07-0742. [DOI] [PubMed] [Google Scholar]
- 19.Hanley S. Pancreatic beta-cell mass as a pharmacologic target in diabetes. Mcgill J Med. 2009;12:51. [PMC free article] [PubMed] [Google Scholar]
- 20.Halban PA, Polonsky KS, Bowden DW, Hawkins MA, Ling C, Mather KJ, Powers AC, Rhodes CJ, Sussel L, Weir GC. Beta-cell failure in type 2 diabetes: postulated mechanisms and prospects for prevention and treatment. J Clin Endocrinol Metab. 2014;99:1983–1992. doi: 10.1210/jc.2014-1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poitout V, Amyot J, Semache M, Zarrouki B, Hagman D, Fontes G. Glucolipotoxicity of the pancreatic beta cell. Biochim Biophys Acta. 2010;1801:289–298. doi: 10.1016/j.bbalip.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laybutt DR, Preston AM, Akerfeldt MC, Kench JG, Busch AK, Biankin AV, Biden TJ. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia. 2007;50:752–763. doi: 10.1007/s00125-006-0590-z. [DOI] [PubMed] [Google Scholar]
- 23.Kahn SE, Haffner SM, Heise MA, Herman WH, Holman RR, Jones NP, Kravitz BG, Lachin JM, O'Neill MC, Zinman B, Viberti G ADOPT Study Group. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med. 2006;355:2427–2443. doi: 10.1056/NEJMoa066224. [DOI] [PubMed] [Google Scholar]
- 24.Lehrke M, Lazar MA. The many faces of PPARgamma. Cell. 2005;123:993–999. doi: 10.1016/j.cell.2005.11.026. [DOI] [PubMed] [Google Scholar]
- 25.Gupta D, Jetton TL, Mortensen RM, Duan SZ, Peshavaria M, Leahy JL. In vivo and in vitro studies of a functional peroxisome proliferator-activated receptor gamma response element in the mouse pdx-1 promoter. J Biol Chem. 2008;283:32462–32470. doi: 10.1074/jbc.M801813200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim HI, Cha JY, Kim SY, Kim JW, Roh KJ, Seong JK, Lee NT, Choi KY, Kim KS, Ahn YH. Peroxisomal proliferator-activated receptor-gamma upregulates glucokinase gene expression in beta-cells. Diabetes. 2002;51:676–685. doi: 10.2337/diabetes.51.3.676. [DOI] [PubMed] [Google Scholar]
- 27.Lupi R, Del Guerra S, Marselli L, Bugliani M, Boggi U, Mosca F, Marchetti P, Del Prato S. Rosiglitazone prevents the impairment of human islet function induced by fatty acids: evidence for a role of PPARgamma2 in the modulation of insulin secretion. Am J Physiol Endocrinol Metab. 2004;286:E560–E567. doi: 10.1152/ajpendo.00561.2002. [DOI] [PubMed] [Google Scholar]
- 28.Ishida H, Takizawa M, Ozawa S, Nakamichi Y, Yamaguchi S, Katsuta H, Tanaka T, Maruyama M, Katahira H, Yoshimoto K, Itagaki E, Nagamatsu S. Pioglitazone improves insulin secretory capacity and prevents the loss of beta-cell mass in obese diabetic db/db mice: possible protection of beta cells from oxidative stress. Metabolism. 2004;53:488–494. doi: 10.1016/j.metabol.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 29.Finegood DT, Topp BG. Beta-cell deterioration: prospects for reversal or prevention. Diabetes Obes Metab. 2001;3(Suppl 1):S20–S27. [PubMed] [Google Scholar]
- 30.Defronzo RA, Tripathy D, Schwenke DC, Banerji M, Bray GA, Buchanan TA, Clement SC, Gastaldelli A, Henry RR, Kitabchi AE, Mudaliar S, Ratner RE, Stentz FB, Musi N, Reaven PD, Study AN. Prevention of diabetes with pioglitazone in ACT NOW: physiologic correlates. Diabetes. 2013;62:3920–3926. doi: 10.2337/db13-0265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiang AH, Peters RK, Kjos SL, Marroquin A, Goico J, Ochoa C, Kawakubo M, Buchanan TA. Effect of pioglitazone on pancreatic beta-cell function and diabetes risk in Hispanic women with prior gestational diabetes. Diabetes. 2006;55:517–522. doi: 10.2337/diabetes.55.02.06.db05-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368:1696–1705. doi: 10.1016/S0140-6736(06)69705-5. [DOI] [PubMed] [Google Scholar]
- 33.Buteau J. GLP-1 receptor signaling: effects on pancreatic beta-cell proliferation and survival. Diabetes Metab. 2008;34(Suppl 2):S73–S77. doi: 10.1016/S1262-3636(08)73398-6. [DOI] [PubMed] [Google Scholar]
- 34.Buteau J, Roduit R, Susini S, Prentki M. Glucagon-like peptide-1 promotes DNA synthesis, activates phosphatidylinositol 3-kinase and increases transcription factor pancreatic and duodenal homeobox gene 1 (PDX-1) DNA binding activity in beta (INS-1)-cells. Diabetologia. 1999;42:856–864. doi: 10.1007/s001250051238. [DOI] [PubMed] [Google Scholar]
- 35.Yusta B, Baggio LL, Estall JL, Koehler JA, Holland DP, Li H, Pipeleers D, Ling Z, Drucker DJ. GLP-1 receptor activation improves beta cell function and survival following induction of endoplasmic reticulum stress. Cell Metab. 2006;4:391–406. doi: 10.1016/j.cmet.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 36.Tourrel C, Bailbe D, Lacorne M, Meile MJ, Kergoat M, Portha B. Persistent improvement of type 2 diabetes in the Goto-Kakizaki rat model by expansion of the beta-cell mass during the prediabetic period with glucagon-like peptide-1 or exendin-4. Diabetes. 2002;51:1443–1452. doi: 10.2337/diabetes.51.5.1443. [DOI] [PubMed] [Google Scholar]
- 37.Wang Y, Perfetti R, Greig NH, Holloway HW, DeOre KA, Montrose-Rafizadeh C, Elahi D, Egan JM. Glucagon-like peptide-1 can reverse the age-related decline in glucose tolerance in rats. J Clin Invest. 1997;99:2883–2889. doi: 10.1172/JCI119482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Farilla L, Hui H, Bertolotto C, Kang E, Bulotta A, Di Mario U, Perfetti R. Glucagon-like peptide-1 promotes islet cell growth and inhibits apoptosis in Zucker diabetic rats. Endocrinology. 2002;143:4397–4408. doi: 10.1210/en.2002-220405. [DOI] [PubMed] [Google Scholar]
- 39.Zander M, Madsbad S, Madsen JL, Holst JJ. Effect of 6-week course of glucagon-like peptide 1 on glycaemic control, insulin sensitivity, and beta-cell function in type 2 diabetes: a parallel-group study. Lancet. 2002;359:824–830. doi: 10.1016/S0140-6736(02)07952-7. [DOI] [PubMed] [Google Scholar]
- 40.DeFronzo RA, Ratner RE, Han J, Kim DD, Fineman MS, Baron AD. Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care. 2005;28:1092–1100. doi: 10.2337/diacare.28.5.1092. [DOI] [PubMed] [Google Scholar]
- 41.Fehse F, Trautmann M, Holst JJ, Halseth AE, Nanayakkara N, Nielsen LL, Fineman MS, Kim DD, Nauck MA. Exenatide augments first- and second-phase insulin secretion in response to intravenous glucose in subjects with type 2 diabetes. J Clin Endocrinol Metab. 2005;90:5991–5997. doi: 10.1210/jc.2005-1093. [DOI] [PubMed] [Google Scholar]
- 42.Pospisilik JA, Martin J, Doty T, Ehses JA, Pamir N, Lynn FC, Piteau S, Demuth HU, McIntosh CH, Pederson RA. Dipeptidyl peptidase IV inhibitor treatment stimulates beta-cell survival and islet neogenesis in streptozotocin-induced diabetic rats. Diabetes. 2003;52:741–750. doi: 10.2337/diabetes.52.3.741. [DOI] [PubMed] [Google Scholar]
- 43.Mu J, Woods J, Zhou YP, Roy RS, Li Z, Zycband E, Feng Y, Zhu L, Li C, Howard AD, Moller DE, Thornberry NA, Zhang BB. Chronic inhibition of dipeptidyl peptidase-4 with a sitagliptin analog preserves pancreatic beta-cell mass and function in a rodent model of type 2 diabetes. Diabetes. 2006;55:1695–1704. doi: 10.2337/db05-1602. [DOI] [PubMed] [Google Scholar]
- 44.Ahren B, Pacini G, Foley JE, Schweizer A. Improved meal-related beta-cell function and insulin sensitivity by the dipeptidyl peptidase-IV inhibitor vildagliptin in metformin-treated patients with type 2 diabetes over 1 year. Diabetes Care. 2005;28:1936–1940. doi: 10.2337/diacare.28.8.1936. [DOI] [PubMed] [Google Scholar]
- 45.Mari A, Sallas WM, He YL, Watson C, Ligueros-Saylan M, Dunning BE, Deacon CF, Holst JJ, Foley JE. Vildagliptin, a dipeptidyl peptidase-IV inhibitor, improves model-assessed beta-cell function in patients with type 2 diabetes. J Clin Endocrinol Metab. 2005;90:4888–4894. doi: 10.1210/jc.2004-2460. [DOI] [PubMed] [Google Scholar]
- 46.Pratley RE, Schweizer A, Rosenstock J, Foley JE, Banerji MA, Pi-Sunyer FX, Mills D, Dejager S. Robust improvements in fasting and prandial measures of beta-cell function with vildagliptin in drug-naive patients: analysis of pooled vildagliptin monotherapy database. Diabetes Obes Metab. 2008;10:931–938. doi: 10.1111/j.1463-1326.2007.00835.x. [DOI] [PubMed] [Google Scholar]
- 47.Rosenstock J, Aguilar-Salinas C, Klein E, Nepal S, List J, Chen R CV181-011 Study Investigators. Effect of saxagliptin monotherapy in treatment-naive patients with type 2 diabetes. Curr Med Res Opin. 2009;25:2401–2411. doi: 10.1185/03007990903178735. [DOI] [PubMed] [Google Scholar]
- 48.Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol. 2001;65:391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- 49.Irimia JM, Meyer CM, Peper CL, Zhai L, Bock CB, Previs SF, McGuinness OP, DePaoli-Roach A, Roach PJ. Impaired glucose tolerance and predisposition to the fasted state in liver glycogen synthase knock-out mice. J Biol Chem. 2010;285:12851–12861. doi: 10.1074/jbc.M110.106534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kaidanovich O, Eldar-Finkelman H. The role of glycogen synthase kinase-3 in insulin resistance and type 2 diabetes. Expert Opin Ther Targets. 2002;6:555–561. doi: 10.1517/14728222.6.5.555. [DOI] [PubMed] [Google Scholar]
- 51.Tanabe K, Liu Y, Hasan SD, Martinez SC, Cras-Meneur C, Welling CM, Bernal-Mizrachi E, Tanizawa Y, Rhodes CJ, Zmuda E, Hai T, Abumrad NA, Permutt MA. Glucose and fatty acids synergize to promote B-cell apoptosis through activation of glycogen synthase kinase 3beta independent of JNK activation. PLoS One. 2011;6:e18146. doi: 10.1371/journal.pone.0018146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Boucher MJ, Selander L, Carlsson L, Edlund H. Phosphorylation marks IPF1/PDX1 protein for degradation by glycogen synthase kinase 3-dependent mechanisms. J Biol Chem. 2006;281:6395–6403. doi: 10.1074/jbc.M511597200. [DOI] [PubMed] [Google Scholar]
- 53.Dokken BB, Henriksen EJ. Chronic selective glycogen synthase kinase-3 inhibition enhances glucose disposal and muscle insulin action in prediabetic obese Zucker rats. Am J Physiol Endocrinol Metab. 2006;291:E207–E213. doi: 10.1152/ajpendo.00628.2005. [DOI] [PubMed] [Google Scholar]
- 54.Figeac F, Ilias A, Bailbe D, Portha B, Movassat J. Local in vivo GSK3beta knockdown promotes pancreatic beta cell and acinar cell regeneration in 90% pancreatectomized rat. Mol Ther. 2012;20:1944–1952. doi: 10.1038/mt.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fujiwara K, Maekawa F, Yada T. Oleic acid interacts with GPR40 to induce Ca2+ signaling in rat islet beta-cells: mediation by PLC and L-type Ca2+ channel and link to insulin release. Am J Physiol Endocrinol Metab. 2005;289:E670–E677. doi: 10.1152/ajpendo.00035.2005. [DOI] [PubMed] [Google Scholar]
- 56.Falkenburger BH, Dickson EJ, Hille B. Quantitative properties and receptor reserve of the DAG and PKC branch of G(q)-coupled receptor signaling. J Gen Physiol. 2013;141:537–555. doi: 10.1085/jgp.201210887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yashiro H, Tsujihata Y, Takeuchi K, Hazama M, Johnson PR, Rorsman P. The effects of TAK-875, a selective G protein-coupled receptor 40/free fatty acid 1 agonist, on insulin and glucagon secretion in isolated rat and human islets. J Pharmacol Exp Ther. 2012;340:483–489. doi: 10.1124/jpet.111.187708. [DOI] [PubMed] [Google Scholar]
- 58.Ou HY, Wu HT, Hung HC, Yang YC, Wu JS, Chang CJ. Multiple mechanisms of GW-9508, a selective G protein-coupled receptor 40 agonist, in the regulation of glucose homeostasis and insulin sensitivity. Am J Physiol Endocrinol Metab. 2013;304:E668–E676. doi: 10.1152/ajpendo.00419.2012. [DOI] [PubMed] [Google Scholar]
- 59.Araki T, Hirayama M, Hiroi S, Kaku K. GPR40-induced insulin secretion by the novel agonist TAK-875: first clinical findings in patients with type 2 diabetes. Diabetes Obes Metab. 2012;14:271–278. doi: 10.1111/j.1463-1326.2011.01525.x. [DOI] [PubMed] [Google Scholar]
- 60.Burant CF, Viswanathan P, Marcinak J, Cao C, Vakilynejad M, Xie B, Leifke E. TAK-875 versus placebo or glimepiride in type 2 diabetes mellitus: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet. 2012;379:1403–1411. doi: 10.1016/S0140-6736(11)61879-5. [DOI] [PubMed] [Google Scholar]