

Abstract
Members of the transforming growth factor-β (TGF-β) family are potent regulatory cytokines that affect multiple cell types of the immune system mediating pro-inflammatory or anti-inflammatory responses. In the liver, TGF-β is produced by a multitude of non-parenchymal liver cells including hepatic stellate cells (HSCs), liver sinusoidal endothelial cells (LSECs), Kupffer cells (KCs), and dendritic cells (DCs) as well as natural killer (NK) T cells among other hepatic lymphocytes. The effect of TGF-β on other cells is highly versatile. In concert with other soluble factors, it controls the maturation, differentiation and activity of various T cell subsets that either prevent or actuate infections, graft-versus-host reactions, immune diseases, and cancer formation. During the last decades, it became evident that some TGFB1 polymorphisms are associated with the pathogenesis of hepatic disease and that plasma TGF-β is a suitable biomarker to detect liver lesions. Moreover, since TGF-β has capacity to influence the quantity and quality of T cell subsets as well as their activity, it is obvious that a well-balanced TGF-β activity is essential for liver homeostasis. In the present review, we highlight some pivotal functions of TGF-β in hepatic immunobiology. We discuss its regulatory function on adaptive immunity, the impact on differentiation of various T cell subsets, its crosstalk with Toll like receptor signaling, and its contribution to functional impairment of the liver.
Keywords: T cells, signaling, inflammation, Toll-like receptors (TLRs), hepatitis
The family of TGF-β contains the three closely related isoforms, namely TGF-β1, TGF-β2, and TGF-β3. They are all synthesized as large latent, inactive complexes in which proper folding, interaction with critical interacting partners such as the latent TGF-β binding proteins (LTBPs) or fibronectin and secretion/release from storage sites is controlled by disulfide bonds and many different activation factors (1-4). Synthesized as pre-pro-peptides, they form within the endoplasmic reticulum dimers that are subsequently processed in the trans-Golgi network by furin cleavage into the mature TGF-β dimer and the cleaved latency-associated peptide dimer (1,5,6). After cleavage, both TGF-β and the latency-associated peptide remain associated forming the small latent complex. In this complex TGF-β is masked and held in an inactive form. Moreover, this complex is covalently associated with members of the LTBPs that assist to localize TGF-β to the extracellular matrix (ECM) (1). The complex out of TGF-β/latency associated peptide and LTBPs is called large latent complex. TGF-β can be released from this high molecular latent protein complex by a number of activators (1). From all these factors, proper integrin-mediated TGF-β activation by dendritic cells (DCs) was found to be highly critical for preventing immune dysfunction (7).
Once released, the mature TGF-β dimer binds to specific receptors that drive the phosphorylation of regulatory Smad proteins (R-Smads) that interact with the common Smad4 and translocate into the nucleus where they interact with other transcription factors (TF) to activate or repress transcription of specific sensitive target genes (8). In the classical TGF-β signaling pathway (Figure 1), TGF-β first activates a TGF-β type II receptor by phosphorylation that in turn heterodimerizes with a TGF-β type I receptor (ALK5) that becomes transphosphorylated. Subsequently, the activation of ALK5 initiates the phosphorylation of the receptor associated Smad proteins Smad2 and Smad3 that interact with Smad4 and give rise to target gene expression. At the same time, an inhibitory Smad protein (i.e., Smad7) is transcriptionally induced by TGF-β forming a negative feedback inhibition loop that prevents unrestricted TGF-β signaling. In endothelial cells another type I receptor (ALK1) functions with the type II receptor and activates Smad1, Smad5 and Smad8. In addition to these two Smad branches, TGF-β can also activate alternative, so-called non-Smad pathways that transmit signals to a multitude of other pathways including the mitogen-activating protein kinases ERK, p38 and JNK as well as Rho-like GTPase and phosphatidylinositol-3-kinase/AKT signaling pathways (9,10). Moreover, the activity of TGF-β can be blunted by antagonistic acting cytokines (e.g., members of the bone morphogenetic proteins) or by a large number of different sequestering proteins that bind to TGF-β and prevent its binding to its cognitive receptors. Vice versa, several other proteins that lack an own recognizable signaling domain, such as the TGF-β type III receptor serve to enhance the binding of TGF-β to the type II receptor by capturing and presenting TGF-β to the signaling receptors (8). The effects of TGF-β further not only depend on the repertoire of receptors that are present on the target cell. The sensitivity of a specific cell type towards TGF-β is also modulated by the surface density of the different TGF-β receptors and a variety of posttranslational modifications such as including glycosylation or sumoylation (11,12). Moreover, the presence of auxiliary co-receptors such as Endoglin that interact with other affinity to the TGF-β signaling receptors, thereby influencing Smad-dependent and independent activities that might be crucial for the triggered biological effect of TGF-β (13,14).
Figure 1.
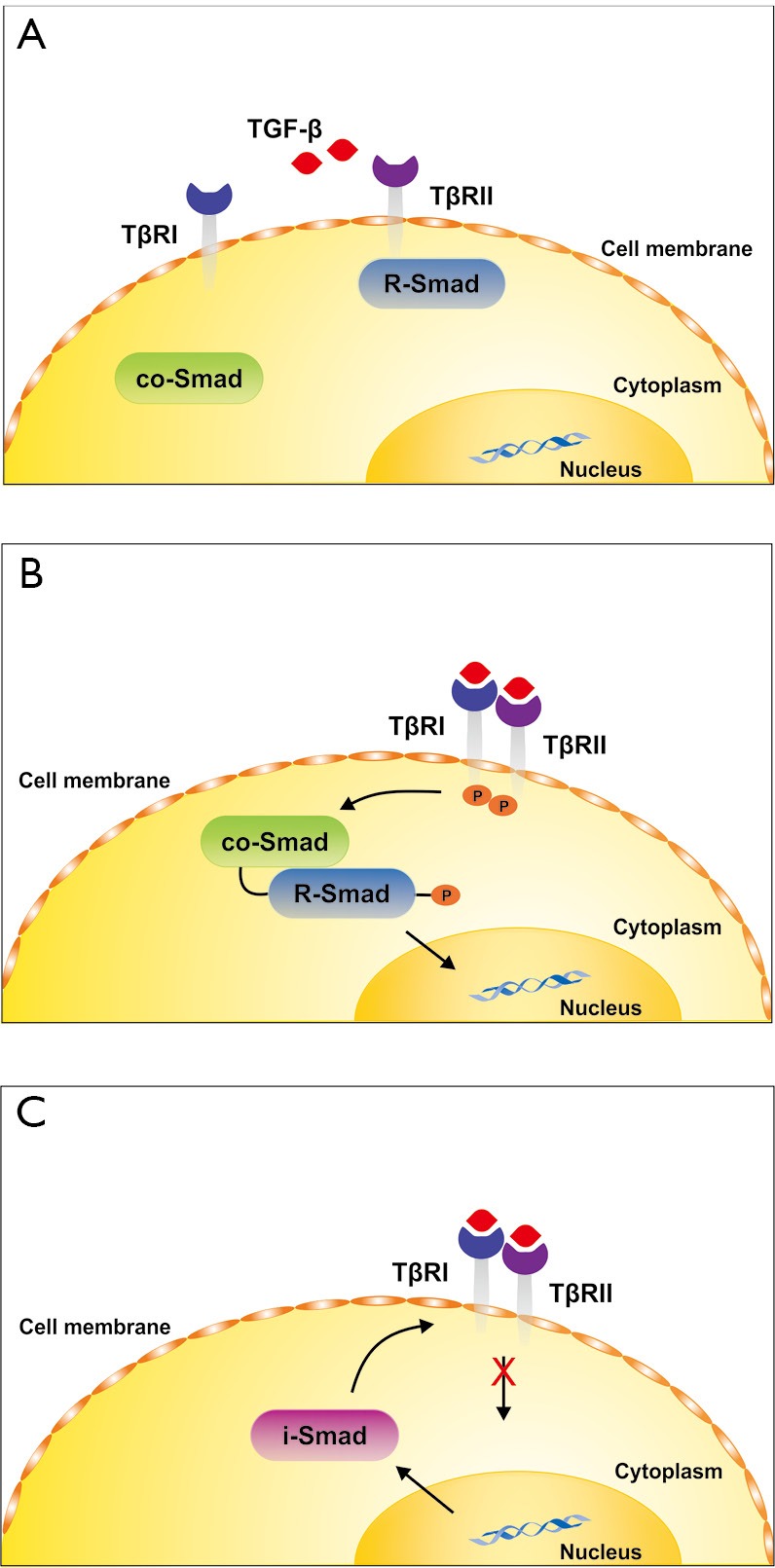
Simplified TGF-β signaling. (A) The classical TGF-β signaling cascade is formed by different types of surface receptors (TβRI, TβRII), receptor-associated R-Smads (Smad2, Smad3) and common Smad (Smad4); (B) upon binding of TGF-β to TβRII, a TβRI is recruited that becomes transphosphorylated by TβRII. The R-Smads become phosphorylated, dissociate from the receptors and interact with Smad4. The R-Smad/co-Smad complex translocates into the nucleus and drives activation or repression of specific Smad-sensitive target genes; (C) simultaneous to the intermediation of the signal, an inhibitory I-Smad (Smad7) is transcriptionally induced by TGF-β. This I-Smad predicts a negative feedback inhibition loop that associates with activated TβRI thereby blocking further R-Smad phosphorylation and TGF-β signaling. Beside this simple pathway, TGF-β mediates signals via many other signaling cascades. In addition, TGF-β activity in various TGF-β sensitive cells is modulated by many other factors that can bind to TGF-β or enhance or blunt its activity. For details see text. TGF-β, transforming growth factor-β.
During the last decades it has been shown that the expression or activity of some of these modulating factors or receptors in liver cells is differentially regulated during inflammatory insults (15). All these findings show that TGF-β is a versatile cytokine that has many options for regulating biological effects in various target cells. This was recently demonstrated by analysis of the dynamical transcriptional response to TGF-β treatment in different human and murine cell systems showing the existence of common and cell type-specific pathways (16). The degree of complexity is even worse. Many cell types have the capacity to synthesize TGF-β and there is an increasing list of known proteins, mechanisms and conditions that interfere with the regulation, processing and signaling of this cytokine (17,18).
In the present review, we will discuss the impact of TGF-β on immunobiology of the liver. In particular, we will summarize some aspects of TGF-β production in different immune and non-immune cells, its regulatory function on adaptive immunity and CD4+ T cell responses, the impact on differentiation of various regulatory T cells (Tregs), its crosstalk with Toll like receptor signaling, and its contribution to functional impairment of the liver.
Hepatic TGF-β production by immune and non-immune cells
Liver cells can be divided in parenchymal and non-parenchymal cells (Figure 2). Parenchymal cells are only represented by hepatocytes, whereas liver sinusoidal endothelial cells (LSECs), Kupffer cells (KCs), hepatic stellate cells (HSCs), also known as Ito or fat storing cells, and biliary epithelial cells are non-parenchymal cells (19,20). Furthermore, circulating intrahepatic lymphocytes and liver resident DCs complete the cell types that can be found within the liver (21).
Figure 2.
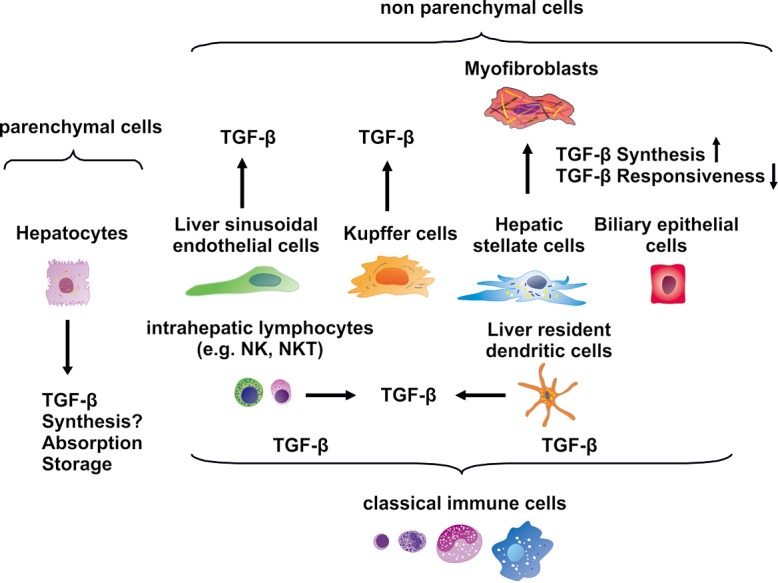
TGF-β expression in various liver cells. The liver is composed of parenchymal cells (hepatocytes) and a large number of non-parenchymal cells that include liver sinusoidal endothelial cells, Kupffer cells, hepatic stellate cells, biliary epithelial cells, a multitude of classical immune cells such as intrahepatic lymphocytes and liver resident dendritic cells. All non-parenchymal cells are reported to synthesize TGF-β, while hepatocytes are known to absorb and store TGF-β. It is presently still controversially discussed if these parenchymal cells have capacity to synthesize TGF-β. TGF-β, transforming growth factor-β.
Although one study performed in rat liver cells led to the conclusion that Tgfb1 gene expression is upregulated in hepatocytes following partial hepatectomy (22), other studies did not reveal any production of TGF-β1 by hepatocytes: examining TGFB1 gene expression in normal and fibrotic human liver revealed that apart from some hepatocytes originating from highly active cirrhosis, hepatocytes in general lacked TGFB1 gene expression (23). In another study TGFB1 gene expression could not be confirmed in hepatocytes that were derived from healthy and fibrotic rat livers (24). Even though there are contradictory results regarding TGFB1 gene expression in hepatocytes, they are at least assumed to absorb and store the latent form providing a major source of active cytokine that becomes released after hepatic injury (19). This assumption was first experimentally underpinned by the finding that primary hepatocytes although containing TGF-β and the LTBPs are deficient in respective mRNA (25) and the absence of LTBP mRNA in hepatocytes could also be confirmed in a subsequent study (26).
Quiescent HSC are located in the space of Disse, a zone between hepatocytes and sinusoids, and are normally attached to hepatocytes (27). In the quiescent state this mesenchymal cell type stores large amounts of vitamin A as retinyl palmitate and produces only small quantities of laminin and collagen type IV, both being important constituents of basement membranes (28,29). In response to soluble factors, such as TGF-β, platelet-derived growth factor (PDGF), and tumor necrosis factor (TNF)-α that are all released by injured hepatocytes and by activated KC, HSC themselves get activated, lose their lipid stores and morphologically change to cells with a myofibroblast-like phenotype (3,28). The most characteristic feature of these activated (or transdifferentiated) HSC is their capacity to synthesize extensive amounts of ECM constituents, especially collagen type I, and metalloproteases, which are necessary for the degradation of ECM in the parenchyma (29). As a consequence, activated HSC perform a predominant function in pathological processes such as liver fibrosis. On the other hand, TGF-β1 does not only belong to those factors that are necessary for the activation of HSC. It has been shown that activated HSC themselves manifest elevated levels of TGFB1 gene expression and that due to this autocrine stimulation the process of fibrogenesis is further stimulated (30). Recent findings further suggest that HSC are liver-resident antigen presenting cells (APC) that can activate T cells, thereby contributing to hepatic immunodefense (31).
LSEC together with KC and DCs are the classical hepatic APC. They flank the liver sinusoids discontinuously, thus leaving open small gaps in between, which can filter out antigens on the way to the parenchyma (32). Furthermore, LSEC remove antigens from the blood by means of receptor-mediated endocytosis. They are equipped with CD54, CD80, CD86, MHC class I and class II, and CD40 molecules on their surfaces to effectively present antigens to both CD4+ and CD8+ T cells (33). In response to endotoxin or lipopolysaccharides (LPS), a principle component of the outer membrane of gram-negative bacteria, LSEC release interleukin (IL)-10, TGF-β, and prostaglandin E2, thereby performing an immunosuppressive function and unleashing tolerance (32,33).
Within the sinusoids that form the small hepatic blood vessels, KC are attached to the layer of LSEC, which they can permeate using the small gaps between these endothelial cells to subsequently control pathogens (32). Their appearance is amoeboid and they constitute the resident macrophages of the liver (28). KC potently disables pathogens either by phagocytosing them or by affecting their pathogenic potential by releasing cytokines and chemokines such as IL-12 that activate other immune cells in the liver (32). After their activation by LPS or other bacterial components KC secrete IL-12 which in turn can activate liver natural killer (NK) cells and NK1.1 Ag+ T cells, both of which yield interferon (IFN)-γ, thus obtaining antibacterial and antitumoral properties (34). Correspondingly, the B7-2 antigen, i.e., CD86, has been found on the surfaces of KCs, which is a major T cell co-stimulatory antigen (32,35). In addition, activated KC also secrete TGF-β, PDGF, and TNF-α and other factors which also trigger the activation process of HSC (3,28).
In contrast to LSEC and KC, which both carry co-stimulatory molecules and have the capacity to release IL-1 and IFN-γ, hepatic DCs do not, which is why their immune state is also referred to as immature (32). DCs are predominantly located in the central veins, but, like KC, they can also pass the LSEC layer and infiltrate the parasinusoidal space or space of disse. They are classified as the most powerful APC (36) and originate from the bone marrow, from where they are scattered over all body tissues. In humans and in mice, they are further split into two subsets: myeloid or conventional DCs and plasmacytoid DCs (37). These immature, i.e., non-activated, DCs can gather and process antigens and subsequently begin to move to the lymph nodes, where they finish the process of maturation. Finally, the mature or activated DCs trigger the development of naïve CD4+ and CD8+ T cells into distinct effector and regulatory subsets or into cytotoxic T lymphocytes (CTLs), respectively (37). In addition, non-activated DCs promote immune tolerance by deletion of T cells with a small quantity of antigen or by an increase in Tregs (38). DCs as well as LSEC, KC, and HSC produce cytokines, including IL-10 and TGF-β that contribute to the implementation of the immunosuppressive effect (39). In one study, for example, it was demonstrated that both IL-10 and TGF-β levels were markedly elevated in bone marrow-derived DCs and in mouse DCs following stimulation with crude antigen originating from a liver fluke species, resulting in an anti-inflammatory response (40).
NK cells and NKT cells are the most prominent hepatic lymphocytes and they perform important functions in the defense against viruses and tumors (41). Hepatic NKT cells respond to cytokines, such as IL-12 and IL-18, as well as to lipid antigens on the surfaces of APC including DCs and HSC and thereby get activated. Subsequently, they release IFN-γ, IL-10, TGF-β and a host of additional cytokines, contributing to various effects (41). For example, weakly activated invariant NKT (iNKT) cells, i.e., type I CD1d-dependent NKT cells, suppress inflammation and prevent acute liver injury and fibrosis (41,42).
In summary, non-parenchymal liver cells, including HSC, LSEC, KC, and DCs as well as NKT cells among other hepatic lymphocytes produce TGF-β and various other cytokines. Upon secretion these cytokines may influence the properties of the originator cell in an autocrine fashion or act paracrine on other cell types, thereby promoting basic properties like the immune tolerance in the liver.
TGF-β effects on adaptive immunity and CD4+ T cell responses
Both CD4+ and CD8+ T cells develop in the thymus and are discriminated based on the expression of CD4 and CD8 antigens on their surfaces. The differentiation and maturation of T cells from these progenitor T cells is a rather complex (Figure 3). In an initial phase the original T cells neither express CD4 nor CD8 rendering them as double negative, followed by a double positive stadium in which both antigens are expressed and finally a third single positive stage (43). Double positive T cells are positively selected on major histocompatibility complex (MHC) molecules: those that respond to class I MHC molecules turn into CD4– CD8+, called CD8+ T cells and those that interact with class II MHC molecules grow into CD4+ CD8–, termed CD4+ T cells. These mature single positive T cells migrate from the thymus into the blood and to secondary lymphoid organs, where finally the portion of CD8+ T cells adds up to 30-40% and that of CD4+ T cells ranges between 60-70% (44). In response to antigen and APC and in the presence of specific cytokines naive CD4+ T cells further differentiate into distinct effector and regulatory subsets. Referring to their effects, the T cells of these subsets can be split into cells responsible for mediating a pro-inflammatory or an anti-inflammatory response: T helper (Th) 17 and Th9 cells belong to the first group and Th1, Th2 cells, and Tregs fall into the second category (45).
Figure 3.

Simplified scheme of T cell differentiation and maturation. Progenitor T cells neither express CD4 nor CD8. T cells that respond to class I MHC molecules turn into CD8+ T cells, while T cells that interact with class II MHC molecules grow into CD4+ cells. These mature T cells then migrate from the thymus into the blood and to secondary lymphoid organs. In response to antigen and antigen presenting cells and in the presence of specific cytokines naïve CD4+ T cells further differentiate into distinct effector and regulatory subsets that mediate various pro-inflammatory or anti-inflammatory responses. They all contribute to the outcome of immune reactions. For details see text. TGF-β, transforming growth factor-β.
Th17 cells play an important role in immune responses to extracellular bacteria and in inflammations associated with autoimmune reactions (44). Together with IL-6 or IL-21, TGF-β induces the development of Th17 cells, which express the retinoic acid receptor (RAR)-related orphan receptor γ isoform t (RORγt) and produce IL-17, IL-17F, and IL-22 (46). But in vivo also IL-23 in conjunction with IL-21 and TGF-β is necessary to cause Th17 cell differentiation via participation of RORγt and the TF STAT3 resulting in IL-17 production (47). Additionally, it has been reported that based on their cytokine production Th17 cells can also promote the proliferation on B cells in vitro and unleash the production of antibodies combined with class switching in vivo (48). The importance of TGF-β for Th17 cell development could be demonstrated by employing mice with T cells unable to respond to TGF-β. These mice were deficient in Th17 cells and did not show evidence of experimental autoimmune encephalomyelitis (49). On the other hand it was demonstrated that T cells exposed to both TGF-β and IL-6 did not develop the pro-inflammatory phenotype, even though the IL-17 production was augmented. Since the anti-inflammatory cytokine IL-10 was produced by these cells at the same time, the occurrence of pathogenic effects was prevented (50). Despite the fundamental role of TGF-β in Th17 differentiation the contribution of Smad proteins to signaling has not been proved beyond doubt yet: to the contrary, it was established that the absence of Smad2 and Smad3 in T cells did not alter the induction of RORγt by TGF-β (51) and that a Smad4 deficiency in naive Th cells did not affect the number of generated Th17 cells upon stimulation with TGF-β (52).
The second and latest group of the pro-inflammatory subset of CD4+ T cells is made up of Th9 cells, which owe their name due to the fact that their main characteristic is the production of the cytokine IL-9. Th9 cells appear to participate in allergic inflammation and autoimmune diseases (53). For the development into Th9 cells, both TGF-β and IL-4 are required and TGF-β directly induces the expression of the ETS family TF PU.1, which is an essential component in the Th9 differentiation process (54). IL-4 in turn contributes to the Th9 phenotype via the TF STAT6, leading to downregulation of the forkhead box P3 TF (Foxp3) expression, thereby preventing progress towards a Treg phenotype, and by STAT6-mediated repression of the T-box TF TBX21 (also known as T-bet) expression that would promote a Th1 cell phenotype (53). Besides, additional TFs and cytokines seem to be necessary for a potent IL-9 gene expression. Apart from Tregs, which will be described in detail later, Th1 and Th2 cells stimulate immune responses and are also belonging to the anti-inflammatory subset of CD4+ T cells, both often working side by side during immune responses, as Th1 cells fulfill functions in cell-mediated immune responses and Th2 cells, by contrast, take part in humoral and allergic processes (44).
Th1 cells feature expression of T-bet and produce a host of IFN-γ. Development towards the Th1 phenotype is propelled by IL-12 (55) and T-bet induces INFG gene expression by binding to a consensus sequence inside the IFNG promoter (56). Together with IL-12, IFN-γ enhances the expression of T-bet, thereby stimulating its own production by installing a positive feedback loop. Since T-bet also up-regulates IL12RB2 gene expression, more IL-12 receptor (IL-12R) β2 gene product is available for binding of IL-12 on the cell surface (56). Furthermore, T-bet addition to Th cells results in a decrease of IL-4, IL-5, and IL-13, cytokines typical of Th2 cells, and corresponding to this observation a T-bet deficiency triggers the development into Th2 cells in vitro and in vivo (57).
TGF-β efficiently suppresses differentiation into Th1 cells by blocking the production of T-bet (58). Additionally, TGF-β attenuates the expression of the IL-12R β2 subunit on CD4+ T cells, resulting in a decreased susceptibility of those cells to IL-12 and thereby counteracting the Th1 phenotype (59). This is the reason why impaired TGF-β signaling contributes to elevated Th1 cell differentiation in vivo.
The characteristic feature of Th2 cells is the expression of the trans-acting T cell-specific TF GATA-3, which is beside the cytokine IL-4 essential for the development of the Th2 phenotype (60). Th2 cells produce IL-4 which in turn enhances the expression of GATA-3 and GATA-3 again blocks the expression of IL-12R β2, thereby stopping the development towards Th1 cells. Conversely, IL-12 can disable GATA-3 expression, in this way cutting the progress towards Th2 cells and thus paving the way for the Th1 phenotype (61). As in the case of Th1 cells, TGF-β also negatively regulates the differentiation of Th2 cells. TGF-β achieves the inhibition of T cell differentiation by suppression of GATA-3 and this inhibitory effect can be reversed by ectopic expression of T-bet (60). Besides, it could be shown that TGF-β activates the TF SOX-4, which in turn disables GATA-3 by means of direct binding, thus preventing its functionality (62).
The implication of the T cell subsets in liver immunology covers the areas infections, especially virus infections, but also infections caused by sporozoa, liver cancer, graft-versus-host reaction following upon liver transplantation, and immune diseases. In most cases the endogenous immune defense does not succeed in putting a stop to HCV infections. This failure seems to be partly due to insufficient Th1 immunity: The HCV core protein appears to down regulate IL-12 gene expression, finally resulting in a decreased Th1-specific cytokine production, including IFN-γ and IL-2, and especially the lack of IFN-γ is responsible for incomplete lysis of cells infected with HCV or for a reduced effect on TGF-β-mediated prevention of hepatic fibrosis (63).
To determine their contribution to the pathogenesis of hepatitic B virus (HBV)-related liver diseases, the amounts of specific cytokines produced by Th1, Th2, Th17 cells, and Tregs were measured. Results indicate that inflated and disproportionate responses of Th1 and Th17 cells could lead to inflammation and liver damage, particularly Th17 cell activity can worsen inflammation and ultimately generate liver failure (64). Moreover, the rate of circulating Th17 cells relates to the progress of disease in patients infected with HBV, again confirming the promoting role of Th17 in the pathogenesis of liver damage (65). Further investigation in patients with HBV and cirrhosis also revealed elevated numbers of Th17 cells in the liver and in the periphery and Th17 cells also tended to directly amass in fibrotic zones of the liver. At least in vitro IL-17 secreted by Th17 cells can push on the activation of HSC, resulting in the increase of inflammation and liver fibrosis (66).
Concerning acute toxoplasmosis in the course of lethal infections in mice, it was shown that serum levels of cytokines typical of Th1 cells, such as IFN-γ, TNF-α, IL-12, and IL-18, were dramatically raised, with considerable liver damage co-occurring, whereas during nonlethal infection only moderate levels of Th1 cytokines were observable followed by minor tissue damage (67).
For hepatocellular carcinoma (HCC) cells it has been established that IL-17 fosters their invasiveness in vitro as well as tumor growth and the formation of new blood vessels in vivo. This seems to be achieved by IL-17-induced AKT signaling leading to the production of IL-6, whose induction of the JAK2/STAT3 pathway finally results in elevated IL-8, MMP2, and VEGF levels (68). Together these findings emphasize the importance of Th17 cells and their dominant cytokine IL-17 for the progress of HCC.
In short, naive CD4+ T cells can further differentiate into five subsets, each characterized by the occurrence of distinct TFs and cytokines, which together define their phenotype. Classified by their mediated effects, Th17 and Th9 cells trigger pro-inflammatory immune responses, whereas Th1 cells, Th2 cells, and Tregs promote anti-inflammatory responses. Therefore, these cells in sum critically contribute to the immune response of the liver. TGF-β suppresses differentiation of Th1 and Th2 cells and promotes development into the Th17, Th9, and the Treg phenotype. In the liver the different subsets contribute to the progress of infections and of HCC either by loss of T cells with anti-inflammatory phenotype or by the overrepresentation of cells with pro-inflammatory phenotype, resulting in a general imbalance and an associated liver damage.
Regulatory actions of TGF-β in the differentiation of Tregs
Tregs are circulating CD4+ immune cells that are well known for their ability to suppress physiological as well as pathological immune responses. Their immunosuppressive activity becomes evident when they are lacking, since a deficiency of Tregs leads to severe systemic autoimmunity (69). In addition, Tregs are subdivided into two groups: natural Tregs (nTregs) and adaptive or induced Tregs (iTregs).
nTregs make up 5-10% of the entire amount of CD4+ T cells in mice and humans and are selected by high-avidity interactions in the thymus (69,70). They express forkhead box P3 TF (Foxp3), which is characteristic of them (71). Since they also express the CD4 and CD25 antigens on their surfaces, they can be fully described as CD4+ CD25+ Foxp3+ Tregs. After their selection and development in the thymus, they move to peripheral tissues. For the generation of nTregs in the thymus a two-step model has been proposed: both T cell receptor signals and co-stimulatory molecules, such as the co-stimulatory receptor CD28 and the family of γc-dependent cytokine receptors, are necessary for the upregulation of CD25 and the development of a Treg precursor. Subsequent binding of IL-2 to the IL-2 receptor results in the induction of Foxp3 expression (72).
TGF-β protects nTregs from apoptosis during their development by two distinct mechanisms: firstly, it suppresses pro-apoptotic proteins and secondly through upregulation of the anti-apoptotic protein Bcl2 (73).
In contrast iTregs develop in secondary lymphoid organs and tissues from naive CD4+ T cells, also referred to as Th cells, due to stimulation with specific antigen (44). Their generation depends on the presence of TGF-β and IL-2, both inducing the upregulation of Foxp3 expression in those cells leading to the suppressive phenotype. Using knockout mice, it has been proved that both Smad2 and Smad3 are necessary to mediate the upregulation of Foxp3 expression (51) and to reach the stability of Foxp3 expression by demethylation of an evolutionarily conserved element (i.e., the Treg-specific demethylated region, TSDR) that is located upstream of exon 1 of the Foxp3 gene locus (74,75). In addition, it could be shown that the concentration of TGF-β determines the fate of CD4+ cells and thus is responsible for the cell type that will be generated.
Low concentrations of TGF-β inhibit the expression of the IL-23 receptor, which is crucial for the development into Th17 cells, a T cell type playing an importing role in pro-inflammatory immune responses, and foster Foxp3 expression, thereby determining the anti-inflammatory response of Tregs (76). On the other hand, high concentrations of TGF-β together with the cytokines IL-6 and IL-21 induce the upregulation of the IL-23 receptor, establishing the Th17 cell phenotype. Thus at least in vitro the concentration of TGF-β regulates the induction of functionally diverse T cell types such as iTregs and Th17 cells.
Furthermore, the presence of TGF-β seems to be important for maintaining a stable expression of Foxp3 and for retaining the regulatory function in iTregs, since the numbers of iTregs in young TGF-β1-deficient mice have been found to be significantly lessened (77), but for effector T cells, there also appears to be the need for the ability to react to TGF-β themselves in order to respond to iTreg mediated suppression (78). To prove the relative portion of nTregs and iTregs in immune responses, mice with a selective blockage in the differentiation of iTregs were used. The results of this study indicate that nTregs are sufficient to prevent systemic and tissue-specific autoimmunity, but not to avert inflammation at mucosal sites in the lung and the intestinal tract (79).
In regard to liver immunology, Tregs contribute to the pathogenesis of HCC and hepatitis B virus infection (HBV) by downregulation of host immune responses. In HCC, significantly increased numbers of Tregs are detected and moreover, Treg counts relate to disease progression (80). In addition, a correlation between the increased serum levels of IL-10 and TGF-β1 and the vitiated antitumor responses can be noticed (81). Generally, the response of the host immune system to cancer involves three distinct immune cell types: CD8+ cytotoxic T cells and NK cells, which both eliminate tumor cells, as well as Tregs (82). Comparing tumor tissue with healthy one of the liver revealed that Tregs amass in the area of the tumor and that the number of CD8+ T cells is significantly smaller (80). Tregs are attracted to cancer sites by chemokines like the chemokine (C-C motif) ligand 22 (CCL22), which is released by CCL22-secreting tumors (83) and binding of CCL22 to the chemokine (C-C motif) receptor 4 (CCR4), that is highly expressed on the surfaces of Tregs. Propagation and activation appear to be achieved via contact with tumor-associated antigens and normal self-antigen presented by the cancer cells (84). Once the number of Tregs is augmented in the affected areas of the liver, the effector function of CD8+ T cells becomes alleviated (80). There are three mechanisms by which Tregs can downregulate activation and proliferation of both CD4+ and CD8+ T cells in vitro and in vivo: firstly, effector T cells can be lysed with the help of granzyme B and perforin; secondly, apoptosis can be caused by loss of IL-2 by high-affinity CD25 and thirdly, inhibitory cytokines like TGF-β, IL-10, IL-35, and prostaglandin E2 can be set free to control immune responses of effector cells (85). On this basis, it can be concluded that a systemic removal of Tregs from cancer sites corrects natural and vaccine induced antitumor responses of T cells (84). Consequently, the elimination of Tregs with antibodies was conducted in HCC preclinical models, demonstrating that spreading of the tumor thus can be stopped (86). Similarly, Tregs foster the progress of HBV infections: Since Tregs exert the most decisive impact on HBV prognosis, they are brought into focus as a potential target for the application of immunotherapeutic approaches (87).
Overall, TGF-β plays an important role in suppressing immune responses by protecting nTregs from apoptosis and by inducing and maintaining Foxp3 expression in iTregs, thereby establishing their suppressive and anti-inflammatory phenotype. Depending on the concentration of TGF-β the generation of the pro-inflammatory Th17 cell phenotype can also occur. In HCC, augmented Tregs numbers in tumor areas down-regulate the amount and the cytotoxic action of CD8+ T cells, resulting in disease progression. Similarly, Tregs also determine the progress and prognosis of HBV infections.
Toll-like receptors (TLRs) and TGF-β signaling
As a result of an early investigation of the Toll gene that was already known to be important for the dorso-ventral shaping in embryos of drosophila, sequence analysis suggested that its gene product is a transmembrane protein (88). Later it was established that the Toll protein is involved in the regulation of Drosomycin, a peptide with antifungal properties in adult fruit flies (89). Shortly afterwards, five receptors were described in humans, showing a similar protein structure of the intra- and extracellular domains to that known from the Toll protein of Drosophila. Therefore these receptors were called TLRs 1-5 and due to this homology a potential role as constituents of the innate immunity in humans was deduced (90). In the meantime, 10 human TLRs and 11 TLRs in the mouse have been determined (91) and their importance for both innate immune reactions against pathogens and prompting reactions of the adaptive immunity has been verified (92).
TLRs belong to a group of receptors called pattern-recognition receptors (PRRs), that are characterized by their ability to specifically identify pathogen-associated molecular patterns (PAMPs), evolutionary conserved in microorganism and not present in host cells (91). PRRs can be split into three receptor families in respect of their function: soluble PRRs, as well as receptors involved in endocytosis and in signaling, respectively, which both are cellular receptors (91). Table 1 summarizes certain PRRs in humans, indicating their structure, their cellular localization, naturally occurring ligands, and origin as well as the TF activated by them (91,93).
Table 1. Overview of human pattern-recognition receptors.
| Receptor | Structure | Cellular localization | Ligand | Origin | Activated TF |
|---|---|---|---|---|---|
| TLR1 | Transmembrane | Cell membrane | Lipopeptides | Bacteria, mycobacteria | NF-κB |
| TLR2 | Transmembrane | Cell membrane | Lipopeptides | Bacteria, mycobacteria | NF-κB |
| TLR3 | Transmembrane | endosomes/lysosomes | dsRNA | Viruses | IRF-3 |
| TLR4 | Transmembrane | Cell membrane | LPS | Gram-negative bacteria | NF-κB |
| TLR5 | Transmembrane | Cell membrane | Flagellin | Bacteria | NF-κB |
| TLR6 | Transmembrane | Cell membrane | Lipopeptides | Mycoplasma | NF-κB |
| TLR7 | Transmembrane | Endosomes/lysosomes | ssRNA | Viruses | NF-κB |
| TLR8 | Transmembrane | Endosomes/lysosomes | ssRNA | Viruses | NF-κB |
| TLR9 | Transmembrane | Endosomes/lysosomes | CpGDNA | Bacteria | NF-κB |
| RIG-I | Soluble | Cytoplasm | ssRNA | Viruses | IRF-3 |
| NOD1 | Soluble | Cytoplasm | Peptidoglycans | Bacteria | NF-κB |
| NOD2 | Soluble | Cytoplasm | Peptidoglycans | Bacteria | NF-κB |
TLR, Toll like receptor; PRRs, pattern-recognition receptors; LPS, lipopolysaccharides.
Examples for soluble PRRs are the C reactive protein (CRP) and serum amyloid P (SAP), which both function as complement-activating factors (94) and are solely made in the liver (95). Scavenger receptors belong to those cellular receptors involved in endocytosis and can be found on macrophages, DCs, and some endothelial cells (96), including one member, the Scavenger receptor class B type I (SRB1), that is vital for the attachment of the hepatitis C virus to find its way into the cell (97). Signaling receptors comprise TLRs, nucleotide-binding oligomerization domain-containing (NOD) proteins, and helicases (91).
Since TLRs are transmembrane receptors they are either expressed on the cell surface (TLR1, 2, 4, 6) or in endosomes and lysosomes (TLR3, 7, 8, 9) (91). Another characteristic feature is the fact that they initiate signaling via the conserved myeloid differentiation factor 88 (MyD88)-adapter axis and through to the nuclear factor (NF)-κB and activating protein-1 (AP-1) TFs resulting in an inflammatory reaction (91).
In contrast to TLRs, NOD proteins track down pathogens intracellularly and both NOD1 and NOD2 detect different fragments of bacterial peptidoglycan within the cell: NOD1 identifies a structure mainly present in the peptidoglycan of Gram-negative bacteria, which is why NOD1 performs a specific function in the innate immune reactions directed against these pathogens. In comparison, NOD2 tracks down a component common to Gram-positive bacteria as well as to Gram-negative bacteria (98,99). Helicases are cellular proteins, such as retinoic acid-inducible gene I (RIG-I) and melanoma differentiation associated gene 5 (MDA5), which are able to ferret out viral infections within cells by using a helicase domain to identify viral RNA. Following recognition a signaling cascade is induced, finally leading to the expression of IFN-stimulated genes, whose gene products directly interfere and block viral replication (100). But viruses in turn produce inhibitors, which are able to disrupt this cellular defense: For example the nonstructural (NS) 3/4A protein produced by the hepatitis C virus suspends RIG-I-MDA5 signaling (100).
Upon contact with a pathogen, TLRs activate cells of the innate immunity, such as neutrophils, NK cells, monocytes and macrophages, and DCs, which then release antibacterial substances as well as chemokines and pro-inflammatory cytokines. Additionally, DCs absorb and process antigens and move to the lymph nodes, where they finish their maturation, featuring intensified production of co-stimulatory molecules, and then present antigens in MHC heterodimers to naive T cells, which in turn get activated and develop into specific effector cells and Tregs (91). In this way innate and adaptive immunity are interconnected and both stimulated by TLRs and TLR and TGF-β signaling come into contact: It has been found out that in vitro TGF-β1 negatively regulates the maturation process of DCs by blocking the expression of co-stimulatory molecules on their surface necessary for antigen presentation, thus controlling their capability of presenting antigens to T cells and unleashing a T cell-mediated immune reaction (101). Compared with these findings valid for DCs in lymphoid tissues, non-activated DCs in nonlymphoid tissues, such as precursors of Langerhans cells (LCs), a subpopulation of DCs in the skin, need TGF-β1 for their development and later for their performance as APCs (101). In a study using mice whose LCs were either unable to produce TGF-β1 or could not react to the cytokine, it could be demonstrated that TGF-β1 is vital for both evolution and survival of LCs and that TGF-β1 exerts influence on LCs in an autocrine and paracrine fashion, respectively (102). A recent investigation with mice featuring a Tgfbr2 gene knockout in DCs revealed lower Foxp3 expression levels in Tregs and an increased release of IFN-γ by DCs, the latter rendering them incapable of inducing Treg differentiation and could be changed back by adding anti-IFN-γ (103). Although the observed blocking of the expression of co-stimulatory molecules in DCs by TGF-β1 in vitro as well as its negative effect on DC-mediated Treg differentiation caused by IFN-γ in vivo provide two different explanations for the underlying mechanism, the result remains the same: TGF-β1 signaling in DCs exerts an immunosuppressive effect, resulting in Treg homeostasis and in immune tolerance, and its absence renders DCs more pro-inflammatory and in an extreme case leads to autoimmunity.
In the liver, TLR signaling is involved in wound healing and regeneration, but also contributes to pathological processes like chronic HBV and HCV infections, hepatic fibrosis, autoimmune liver disease, and alcoholic liver disease (104). During fibrogenesis, stimulation of TLR4 with bacterial LPS in quiescent HSC reduces levels of BMP and activin membrane-bound inhibitor homolog (BAMBI), a pseudo receptor lacking the kinase domain and therefore generating no signal, thus making HSC more susceptible to the fibrogenic signals of TGF-β (105). TLR4 signaling proceeds via the MyD88-adapter and the NF-κB pathway inducing secretion of chemokines by HSCs which in turn attract KC, releasing TGF-β and leading to unrestricted TGF-β signaling (106). Accordingly, the TLR4 inflammatory pathway enhances the fibrogenic effect of TGF-β signaling in the development of liver fibrosis by two distinct mechanisms. But it has also been reported that TGF-β1 prevents TLR2, 4, and 5-mediated NF-κB signaling and the secretion of TNF-α, achieved by both enhancing MyD88 decomposition in proteasomes as well as provoking MyD88 ubiquitination (107).
In addition, it was shown that the NF-κB pathway can promote the development of tumors and that blocking of this pathway results in a decreased emergence of inflammation-associated tumors (108). Since HCC normally arises from chronic inflammation, the investigation of the relationship between inflammation associated pathways (such as the NF-κB pathway) and hepatocarcinogenesis might yield new insights and therapeutic approaches (109).
In summary, TLRs are PRRs, which specifically identify PAMPs. TLR signaling results in innate immune reactions against pathogens as well as in prompting reactions of the adaptive immunity. In DCs, the connecting link between innate and adaptive immunity, TGF-β1 signaling exerts an immunosuppressive effect. Concerning liver fibrosis, TLR4 signaling in HSC enhances the fibrogenic effect of TGF-β signaling.
TGF-β in viral hepatitis
There is now a multitude of studies available linking TGF-β to the initiation, progression or resolution of viral hepatitis. The World Health Organization (WHO) defines viral hepatitis to be a health problem of global dimension, since the number of people with chronic HBV or HCV infections amounts to approximately 500 million people, with roughly 1 million people dying of consequences arising from viral hepatitis worldwide every year (110). Furthermore, 57% of cirrhosis and 78% of HCC are ascribable to infections with the B and C viruses on a global scale (111). A recent study indicates that the number of HCV infections is indeed lower than suggested by previous investigations, but this points to an increased fatality rate, because the number of deaths remains the same (112).
The HBV infection is caused by a partially double-stranded DNA virus, which is most frequently passed via sexual intercourse and by mother-to-child, i.e., vertical, transmission (113). The disease progresses from acute and chronic hepatitis via cirrhosis through to HCC (114). Already in 1980, HBV DNA could be established in DNA extracted from tissue samples of HCC (115) and later, it could be demonstrated that in transgenic mice constantly augmented levels of the HBV X (HBx) protein lead to neoplasia and finally to HCC (116). In vitro, HBx triggers signaling via the Ras-Raf-mitogen-activated protein kinase (MAPK) pathway to extracellular signal-regulated kinases (ERKs) and c-Jun N-terminal kinases (JNKs), ending in the induction of the proto-oncogene c-Jun by JNK that is associated with transformation and cell growth (117). Based on this early observations researcher tried to find out how TGF-β signaling might be affected during carcinogenesis in livers chronically infected with HBV (118). In hepatocytes of healthy liver TGF-β signaling leads to phosphorylation of Smad3 at its carboxyl terminus (pSmad3C) followed by downregulation of MYC gene expression and growth inhibition. In contrast, Smad3 can also be phosphorylated at the linker position between the two Mad-homology domains, thereby being converted to pSmad3L. It has been shown that linker phosphorylation can be induced by constitutively active Ras and is executed by JNK (119) and that pSmad3L solely concentrates in the nuclei of the employed cells (120). JNK-pSmad3L signaling finally results in lifting the downregulation of MYC gene expression and in proliferation, simultaneously changing from the tumor-suppressive effect mediated by TGF-β to a tumor-promoting impact (118). As a result of this study, it could be demonstrated that in normal hepatocytes both phosphorylation of the C-terminus and of the linker domain initially increases and then drops in response to TGF-β, but that in those hepatocytes expressing HBx the linker domain is constitutively phosphorylated upon constant stimulation with TGF-β (118). This reorientation towards oncogenesis can be inverted by selectively blocking the linker phosphorylation of Smad3, which recovers tumor suppressive TGF-β signaling via pSmad3C (118). These findings underline that HBV straightly promotes hepatocarcinogenesis owing to altered TGF-β signaling already in early stages of chronic infection in addition to the favoring effects mediated by immune reactions.
In contrast to the HBV, the hepatitis C virus exhibits positive-stranded RNA and the genome is translated into a polyprotein consisting of about 3,000 amino acids, afterwards cleaved into the structural protein core, E1, and E2 (E stands for envelope) and the non-structural proteins p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B, and at least the NS proteins play a role during replication (121,122). Being predominantly passed via blood, transmission as a consequence of sexual intercourse or from mother to child does less frequently occur (123) and only about a fifth of those people coming into contact with the virus are able to cope with the infection soon afterwards (124). HCV entry into hepatocytes is thought to be mediated by at least four molecules on their surface, namely glucosaminoglycans (GAGs), low-density lipoprotein receptors (LDLRs), SR-B1, and the tetraspanin CD81, which are able to bind to constituents of the virus envelope: Regarding SR-B1 and CD81, direct binding of the viral glycoprotein E2 has been confirmed (121). HCV uptake is accomplished by clathrin-mediated endocytosis and when it has been set free from early endosomes into the cytosol, translation and replication follow (123). TLR3 and RIG-I are involved in the innate reaction to HCV: TLR3 identifies dsRNA in endosomes compared to RIG-I, which detects viral RNA in the cytoplasm, together inducing signaling cascades that both lead to the phosphorylation of interferon regulatory factor 3 (IRF3) and its subsequent translocation into the nucleus, followed by the production of IFN-γ (123). Besides the disruption of RIG-I signaling by the NS3/4A protein (100), it could be shown that the HCV core protein also impairs innate responses by blocking the production of IFN-α in plasmacytoid DCs and by triggering apoptosis of these immune cells (125). In addition, NK cells from individuals with chronic HCV infections failed to activate DCs in vitro, presumably due to diminished synthesis of IL-10 and TGF-β (126), thereby also affecting adaptive immune reactions. As it holds true for the HBV, HCV infections also tend to develop into a chronic stage, often followed by cirrhosis and HCC and in the US, the infection represents the major cause of liver transplantations (127). Furthermore, the relation between HCV infection and autoimmunity is well attested (128). In one study the implication of HCV in autoimmune diseases and liver fibrosis has been investigated: Aminoacyl-tRNA synthetase complex-interacting multifunctional protein 1 (AIMP1/p43) steadies the E3 ubiquitin-protein ligase SMURF2, thus down-regulating TGF-β signaling and associated liver fibrosis, and also diminishes the expression of gp96 on the cell surface, thereby preventing Lupus-like autoimmune disease. As HCV E2 leads to decomposition of AIMP1/p43 by two distinct mechanisms, consequently TGF-β signaling as well as the surface expression of gp96 is enhanced (129). It has also been reported that the HCV core protein elevates the portion of active TGF-β in the liver of transgenic mice and in hepatoma cells, latter also capable of activating HSCs in the same culture shown to be mediated by TGF-β. In conclusion, the HCV core protein can positively regulate TGF-β signaling in hepatocytes and in a paracrine manner to contribute to liver fibrosis and hepatocarcinogenesis (130). But the core protein as well as NS3 are both able to prevent binding of Smad3 to DNA and thereby affecting transactivation of distinct TGF-β target genes: Whereas both HCV core and NS3 alleviate TGF-β/Smad3 induced apoptosis, only the core protein is able to curtail the activation of the p21 promoter mediated by TGF-β, thus impairing growth arrest in the G1 phase (131). Moreover, the core protein has been shown to directly induce TGFB1 gene expression via MAPK signaling in parenchymal cells, explaining how HCV infection might contribute to hepatic fibrosis (132). Also NS3 together with NS4A as cofactor can trigger TGFB1 gene expression in human hepatoma cells (133). Finally, it has also been demonstrated that the NS3 protease can bind to the TGF-β type I receptor simulating binding of TGF-β2, again promoting hepatic fibrosis (134). Based on the previously described observations indicating that TGF-β levels are enhanced by viral constituents together with the fact that TGF-β is essential for the induction and function of iTregs (77), the effect of HCV on T cell activity has been explored: HCV-infected hepatocytes in a co-culture with CD4+ T cells raised the number of Tregs and Treg induction was mediated by TGF-β (135). In addition, Tregs were able to blanket the effector T cell proliferation and levels of IFN-γ produced by CD4+ T cells significantly declined (135). Since Tregs are known to curb antiviral reactions of both CD8+ and CD4+ T cells, this immunosuppressive effect might weaken host immunity and further stabilize the chronic stage (135).
In a nutshell, the HBV infection is caused by a partial dsDNA virus, whose DNA integrates into the host DNA. Transmission occurs via sexual intercourse and from mother to child. The HBV infection tends to become chronic including cirrhosis and HCC. The HBx protein induces altered TGF-β signaling via pSmad3L associated with a tumor-promoting effect.
The HCV infection is connected with a positive-stranded RNA virus, which enters the cell by binding to molecules on the surface of hepatocytes followed by endocytosis. The virus is mainly transmitted by blood, transmission via sexual intercourse and from mother to child is less frequent. Chronic stages of infection with cirrhosis and HCC are common. Several mechanisms to evade host immunity mediated by viral proteins do exist.
TGF-β polymorphisms and liver immunity
In the mid-eighties the gene locus of TGFB1 has been determined and assigned to human chromosome 19q13.1-q13.3 (136). Over time, certain mutations or polymorphisms have been identified and this raised the question whether distinct genetic variants might contribute to differences in the progression of diseases, such as atopy and asthma (137), liver fibrosis and cirrhosis (138-140) or hepatic viral infections (141,142). The most frequently mentioned TGF-β1 polymorphisms are at base pairs (bp) -988 (cytosine → adenine), -800 (guanine → adenine), and -509 (cytosine → thymine) starting from the initiation site of transcription, which all belong to the promoter region, a further downstream at bp +72, being integral part of a region that is not translated, and two more at codon 10 (leucine → proline) and at codon 25 (arginine → proline), both within exon 1 (143). For example, position -509 is located within a promoter region important for the downregulation of transcriptional activity (137). The substitution of cytosine by thymine at bp -509 results in the formation of a Yin Yang-1 (YY1) activator sequence (144,145) and has been associated with heightened IgE levels (144). In Chinese affected by cirrhosis due to HBV infection, it was shown that the variant with a cytosine at position -509 displayed enhanced promoter activity and that an exchange for thymine impinged on binding of nuclear proteins to the binding site (146). In addition, the prevalence of both cytosine (C) at -509 and thymine (T) at codon 10 were in accordance with an increased degree of disease severity. Taken together, promoter activity analysis as well as the clinical results indicate that the variants with a C at bp -509 and with a T at codon 10 contribute to the progression and severity of liver cirrhosis in the Chinese population (146). Similarly, in another investigation that was performed in Caucasians, it was shown that TGFB1 genetic variants do occur more frequently than in Chinese and that a change to proline in codon 10 as well as in codon 25 promotes the development of much more pronounced liver fibrosis in the course of a HCV infection (146). However, in another study the -509C single nucleotide polymorphism (SNP) has been linked to lesser transcriptional activity and elevated elimination of the HCV (147), possibly due to the fact that reduced production of TGF-β1 abrogates its suppression of NK cell activity, thereby leading to a more efficient elimination of the HCV, primarily during the acute stage of infection (147). To assess whether genetic variants might differently promote the predisposition for a HCV infection, a research group investigated polymorphisms of various genes in healthy and in infected individuals: Whereas the statistical analysis for the codon 10 SNP of the TGFB1 gene yielded no significant result, it could be demonstrated that the prevalence of the G SNP of codon 25, i.e., the coding of the amino acid arginine, was significantly increased in the infected Brazilians, thus implying that the codon 25 SNP might predispose to HCV infections (141). Already earlier a relationship between genotype, TGF-β1 production, and hepatic fibrosis in the course of a chronic HCV infection has been detected: Patients with the arginine/arginine genotype at codon 25 of the TGFB1 gene produced higher amounts of TGF-β1 and were prone to extensive liver fibrosis as opposed to those featuring the heterozygous arginine/proline or the homozygous proline/proline genotypes (138,148). Although the aforementioned investigations all confirm links between TGFB1 polymorphisms and chronic liver diseases, a study aiming at revealing correlations between genetic variants and chronic HBV infections in Iran led to the conclusion that neither the -509C/T SNP nor the +915G/C SNP, i.e., the polymorphism at codon 25, were associated with a chronic HBV infection (149). To evaluate the effect of the T29C polymorphism on the predisposition for HBV infections among Egyptians, a preliminary study with a small number of participants has been conducted (142). The T29C polymorphism connotes a transition from T to C at nucleotide 29 of the TGFB1 gene, leading to a substitution of leucine by proline at amino acid 10 within the signal peptide sequence (150). The preliminary study revealed that the TT genotype prevailed in healthy participants, whereas the CC genotype dominated in HBV infected patients, indicating that the TT genotype might have a protective function and the CC genotype might promote HBV infections as a host genetic factor (142). To investigate whether the -509C/T polymorphism might affect the development of HCC in Chinese chronically infected with the HBV was the focus of another investigation (151). The chance of developing HCC was significantly heightened in patients featuring the CC genotype and in those already afflicted by HCC the plasma and mRNA levels of TGF-β1 were also significantly augmented in invalids with the homozygous CC genotype as contrasted with those exhibiting the TT genotype (151). Since there was no allelic variation within the HCC group itself, the implication of the -509C/T polymorphism might be confined to presence of HCC and genetic variants might not influence the progress of liver cancer (151).
Taken together, the best analyzed TGFB1 gene polymorphisms are the -509C/T SNP and those at codon 10 (Leu/Pro) and at codon 25 (Arg/Pro), which together are assumed to play a fundamental role in the predisposition for chronic liver diseases. Since the -509C/T polymorphism concerns the promoter region of TGFB1, different variants might influence the amount of TGF-β1 being produced, thereby altering the immunosuppressive and fibrogenic effect of the cytokine. Both codon 10 and codon 25 SNPs affect the signal peptide sequence and are presumably responsible for differences in the bioavailability of TGF-β1.
TGF-β as a diagnostic and prognostic marker of liver disease
In spite of the fact that insights into the pathogenesis of HCC have rapidly grown in recent years, prophylaxis and therapy still constitute a hitch, not least because HCC is the result of diverse etiologies with varying underlying genetic alterations (152). Thus there is no standard therapy available and cancer patients often die before the surgical removal of tumor tissue or before a liver transplantation, which are the only promising treatment modalities (153). In the majority of cases, HCC evolves on the basis of persistent inflammation, characteristic of chronic hepatitis and cirrhosis, which is why chronic HBV and HCV infections as well as intoxications due to alcohol abuse and Aflatoxin B1 rank among the major risk factors for HCC (152). Against this background, timely locating of fibrotic processes due to chronic hepatitis is required for early intervention (154) and thorough monitoring of molecular alterations provides the possibility of evaluating stage and progression of hepatic fibrosis, cirrhosis, and of HCC (155,156).
More than a decade ago, it could be proven in rats that the degree of fibrosis is related to both the portion of collagen and the TGF-β1 mRNA produced in the liver (157). In line, several inhibition strategies aiming at inhibition of TGF-β were highly successful when applied to experimental models of hepatic fibrogenesis (2). Similarly, in humans chronically infected with HCV plasma levels of TGF-β1 have been shown to relate to the scope of hepatic fibrosis as well as to the level of TGF-β1 found in the liver (158). Furthermore, it has been established that hepatic cirrhosis is accompanied by an increment of TGF-β1 levels in the plasma and that the plasma concentration might serve as a biomarker of the stage of fibrosis as well as of the functional impairment of the liver (159). The relationship between serum levels of TGF-β1 and chronic hepatitis could also be confirmed by another investigation: To assess the effect of an antiviral therapy for individuals suffering from chronic HCV infection both serum levels of TGF-β1 and mRNA levels produced in the liver have been determined before and after treatment (160). In those displaying a therapeutic success serum levels as well as mRNA levels of TGF-β1 significantly declined, whereas only mRNA levels diminished following a noneffective therapy, together leading to the appraisal that the serum and mRNA levels of TGF-β1 might be utilized as prognostic markers concerning an antiviral treatment of HCV infected individuals (160). Additionally, in one study TGF-β1 has been detected by immunostaining in tissue samples obtained from liver biopsies: The cytokine was present in samples derived from patients suffering from chronic hepatitis C, liver cirrhosis, and HCC, with a much more pronounced occurrence within the cirrhosis group, pointing at a synergistic action of TGF-β1, connective tissue growth factor (CTGF), and PDGF in hepatic cirrhosis (154). To evaluate the clinical significance of TGF-β1 with respect to the survival of individuals with unresectable HCC, plasma concentrations of TGF-β1 have been compared with the survival rate of the study participants: members of the group with higher TGF-β1 levels displayed a significantly shorter survival rate and lesser NK cell activity than those of the group with lower TGF-β1 plasma levels, indicating that the plasma concentration of TGF-β1 can predict the survival probability of patients with unresectable HCC and that the differences might be due to the immunosuppressive effect of the cytokine exerted on the anti-tumoral defense (161). Moreover, it has been found out that TGF-β1 outperforms alpha-fetoprotein (AFP) regarding the detection of small HCC: In patients with HCC plasma levels of TGF-β1 are augmented in contrast to those individuals only affected by cirrhosis and TGF-β1 mRNA is highly expressed particularly in small and marked-off HCC (162). Due to its higher sensitivity against AFP, TGF-β1 might be recommendable as a serum marker, especially advantageous for the early diagnosis of HCC and for the detection of small HCC (162). Since TGF-β1 induces and sustains a stable expression of Foxp3 in peripheral CD4+ CD25+ T cells and since it is necessary for retaining the regulatory function in these iTregs (77), the aim of one study was to reveal a potential relationship between TGF-β1 expression and the number of Tregs in HCC as well as to clarify whether their prevalence could forecast the progress of HCC (163). In comparison with a healthy liver significantly more Tregs have been found in tumor tissue and TGF-β1 expression in tumor cells seemed to be associated with the quantity of Tregs, thereby implying that TGF-β1 is responsible for the concentration of Tregs in HCC tissue (163). In conclusion, the rate of Tregs might be suitable for creating prognoses regarding HCC progression (163). Viral infections result in the activation of various signaling pathways and in the production of distinct pro-inflammatory factors, such as cytokines and cyclooxygenase-2 (COX-2) (164), the latter being one of two isoforms of COX or prostaglandin (PG) H synthase, which is essential for the formation of PGs (165). PGs again are involved in viral replication and in inflammatory reactions (164) and COX-2 facilitates angiogenesis and the proliferation of tumor cells (166), its gene expression is induced by both the HCV core and the NS5A protein and together with matrix metalloproteinases (MMPs) 2 and 9 it exacerbates fibrosis during chronic HCV infections (167). One research group tried to figure out how COX-2 together with TGF-β1 might affect chronic viral hepatitis and HCC (168). As a result, it could be demonstrated that the HCV stimulates COX-2 expression and that COX-2 can function as a marker for malignant transformation in the course of chronic viral hepatitis (168). Additionally, TGF-β1 expression has been linked to inflammation and cirrhosis and therefore TGF-β1 might be a potential marker indicating liver injury due to chronic viral hepatitis and might also be of importance in hepatocarcinogenesis (168). Finally, COX-2 and TGF-β1 might act synergistically during cancerogenesis (168).
In summary, plasma levels of TGF-β1 relate to the scope of hepatic fibrosis during the course of chronic hepatitis and are also heightened in hepatic cirrhosis and HCC, which is why TGF-β1 is appreciated to be of value as a prognostic marker. Together with other factors such as CTGF, PDGF, and COX-2, TGF-β1 might act synergistically on the manifestation of cirrhosis as well as of HCC. Due to its enhanced sensitivity, TGF-β1 might be useful as a serum marker for the detection of both small HCC and early stages of liver cancer.
Conclusions
During the last decades, it became evident that TGF-β is a highly versatile cytokine that affects liver immunology in many ways. TGF-β in concert with other soluble factors is important for the maturation and differentiation of many different immune cells in the liver that mediate pro-inflammatory or anti-inflammatory responses. It suppresses differentiation of Th1 and Th2 cells and promotes development into the Th17, Th9, and the Treg phenotype. In addition, TGF-β suppresses immune responses by protecting nTregs from apoptosis and by inducing and maintaining Foxp3 expression in iTregs that is necessary for their suppressive and anti-inflammatory phenotype. Since changes in the quantity of all these different cellular subsets contribute to the progression of infections and the formation of HCC, a well-balanced activity of TGF-β is essential for liver homeostasis. During the last decades, it became evident that some polymorphisms within the TGFB1 gene might predict or contribute to the pathogenesis of hepatic disease and that the concentrations of TGF-β1 in the plasma might be suitable as a biomarker for the functional impairment of the liver and to detect small HCC and early stages of liver cancer. A multitude of therapeutic strategies targeting overshooting TGF-β activities were already successfully tested in many independent experimental models. However, in regard to TGF-β there are still countless open questions and issues that need to be critically addressed in future studies. Many ongoing studies presently analyze the mechanisms that are responsible for the loss of T cell tolerance in models lacking individual components of the TGF-β signaling pathway, mechanisms responsible in TGF-β immune tolerance establishment, and the involvement of TGF-β signaling in regulating aspects of TLR signaling. Also the identification of the regulatory dynamics emanating from TGF-β and its consequences that might be specific for a certain immune cell subset open a number of new fields that needs to be experimentally addressed. Based on the fulminate role of TGF-β on liver immunology, it will be interesting to see how these strategies will be translated into clinical applications and how novel therapeutics targeting TGF-β pathways are suitable to modulate immunological features of diseases of the liver in humans.
Acknowledgements
The authors thank Sabine Weiskirchen for preparing the figures. Ralf Weiskirchen is supported by the German Research Foundation (DFG SFB/TRR57 P13) and the Interdisciplinary Centre for Clinical Research within the Faculty of Medicine at the RWTH Aachen University (IZKF E6-11).
Disclosure: The authors declare no conflict of interest.
References
- 1.Robertson IB, Rifkin DB. Unchaining the beast; insights from structural and evolutionary studies on TGFβ secretion, sequestration, and activation. Cytokine Growth Factor Rev 2013;24:355-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gressner AM, Weiskirchen R, Breitkopf K, et al. Roles of TGF-β in hepatic fibrosis. Front Biosci 2002;7:d793-807. [DOI] [PubMed] [Google Scholar]
- 3.Gressner AM, Weiskirchen R. Modern pathogenetic concepts of liver fibrosis suggest stellate cells and TGF-β as major players and therapeutic targets. J Cell Mol Med 2006;10:76-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weiskirchen R, Meurer SK. BMP-7 counteracting TGF-β1 activities in organ fibrosis. Front Biosci (Landmark Ed) 2013;18:1407-34. [DOI] [PubMed] [Google Scholar]
- 5.Dubois CM, Laprise MH, Blanchette F, et al. Processing of transforming growth factor β1 precursor by human furin convertase. J Biol Chem 1995;270:10618-24. [DOI] [PubMed] [Google Scholar]
- 6.Dubois CM, Blanchette F, Laprise MH, et al. Evidence that furin is an authentic transforming growth factor-β1-converting enzyme. Am J Pathol 2001;158:305-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Travis MA, Reizis B, Melton AC, et al. Loss of integrin alpha(v)beta8 on dendritic cells causes autoimmunity and colitis in mice. Nature 2007;449:361-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Massagué J, Chen YG. Controlling TGF-β signaling. Genes Dev 2000;14:627-44. [PubMed] [Google Scholar]
- 9.Moustakas A, Heldin CH. Non-Smad TGF-β signals. J Cell Sci 2005;118:3573-84. [DOI] [PubMed] [Google Scholar]
- 10.Zhang YE. Non-Smad pathways in TGF-β signaling. Cell Res 2009;19:128-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim YW, Park J, Lee HJ, et al. TGF-β sensitivity is determined by N-linked glycosylation of the type II TGF-β receptor. Biochem J 2012;445:403-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kang JS, Saunier EF, Akhurst RJ, et al. The type I TGF-β receptor is covalently modified and regulated by sumoylation. Nat Cell Biol 2008;10:654-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scherner O, Meurer SK, Tihaa L, et al. Endoglin differentially modulates antagonistic transforming growth factor-β1 and BMP-7 signaling. J Biol Chem 2007;282:13934-43. [DOI] [PubMed] [Google Scholar]
- 14.Meurer SK, Alsamman M, Scholten D, et al. Endoglin in liver fibrogenesis: Bridging basic science and clinical practice. World J Biol Chem 2014;5:180-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meurer SK, Tihaa L, Lahme B, et al. Identification of endoglin in rat hepatic stellate cells: new insights into transforming growth factor β receptor signaling. J Biol Chem 2005;280:3078-87. [DOI] [PubMed] [Google Scholar]
- 16.Abnaof K, Mallela N, Walenda G, et al. TGF-β stimulation in human and murine cells reveals commonly affected biological processes and pathways at transcription level. BMC Syst Biol 2014;8:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xie F, Zhang Z, van Dam H, et al. Regulation of TGF-β Superfamily Signaling by SMAD Mono-Ubiquitination. Cells 2014;3:981-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hawinkels LJ, ten Dijke P. Exploring anti-TGF-β therapies in cancer and fibrosis. Growth Factors 2011;29:140-52. [DOI] [PubMed] [Google Scholar]
- 19.Kmieć Z.Cooperation of liver cells in health and disease. Adv Anat Embryol Cell Biol 2001;161:III-XIII, 1-151. [DOI] [PubMed] [Google Scholar]
- 20.Chen Y, Sun R.Toll-like receptors in acute liver injury and regeneration. Int Immunopharmacol 2011;11:1433-41. [DOI] [PubMed] [Google Scholar]
- 21.Racanelli V, Rehermann B.The liver as an immunological organ. Hepatology 2006;43:S54-62. [DOI] [PubMed] [Google Scholar]
- 22.Bissell DM, Wang SS, Jarnagin WR, et al. Cell-specific expression of transforming growth factor-β in rat liver. Evidence for autocrine regulation of hepatocyte proliferation. J Clin Invest 1995;96:447-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Milani S, Herbst H, Schuppan D, et al. Transforming growth factors β1 and β2 are differentially expressed in fibrotic liver disease. Am J Pathol 1991;139:1221-9. [PMC free article] [PubMed] [Google Scholar]
- 24.De Bleser PJ, Niki T, Rogiers V, et al. Transforming growth factor-beta gene expression in normal and fibrotic rat liver. J Hepatol 1997;26:886-93. [DOI] [PubMed] [Google Scholar]
- 25.Roth-Eichhorn S, Kühl K, Gressner AM. Subcellular localization of (latent) transforming growth factor beta and the latent TGF-β binding protein in rat hepatocytes and hepatic stellate cells. Hepatology 1998;28:1588-96. [DOI] [PubMed] [Google Scholar]
- 26.Breitkopf K, Lahme B, Tag CG, et al. Expression and matrix deposition of latent transforming growth factor beta binding proteins in normal and fibrotic rat liver and transdifferentiating hepatic stellate cells in culture. Hepatology 2001;33:387-96. [DOI] [PubMed] [Google Scholar]
- 27.Baxter MA, Rowe C, Alder J, et al. Generating hepatic cell lineages from pluripotent stem cells for drug toxicity screening. Stem Cell Res 2010;5:4-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cubero FJ, Nieto N. Kupffer cells and alcoholic liver disease. Rev Esp Enferm Dig 2006;98:460-72. [DOI] [PubMed] [Google Scholar]
- 29.Sato M, Suzuki S, Senoo H.Hepatic stellate cells: unique characteristics in cell biology and phenotype. Cell Struct Funct 2003;28:105-12. [DOI] [PubMed] [Google Scholar]
- 30.Gressner AM. Cytokines and cellular crosstalk involved in the activation of fat-storing cells. J Hepatol 1995;22:28-36. [PubMed] [Google Scholar]
- 31.Winau F, Hegasy G, Weiskirchen R, et al. Ito cells are liver-resident antigen-presenting cells for activating T cell responses. Immunity 2007;26:117-29. [DOI] [PubMed] [Google Scholar]
- 32.Hsu W, Shu SA, Gershwin E, et al. The current immune function of hepatic dendritic cells. Cell Mol Immunol 2007;4:321-8. [PubMed] [Google Scholar]
- 33.Knolle PA, Gerken G. Local control of the immune response in the liver. Immunol Rev 2000;174:21-34. [DOI] [PubMed] [Google Scholar]
- 34.Seki S, Habu Y, Kawamura T, et al. The liver as a crucial organ in the first line of host defense: the roles of Kupffer cells, natural killer (NK) cells and NK1.1 Ag+ T cells in T helper 1 immune responses. Immunol Rev 2000;174:35-46. [DOI] [PubMed] [Google Scholar]
- 35.Inaba K, Witmer-Pack M, Inaba M, et al. The tissue distribution of the B7-2 costimulator in mice: abundant expression on dendritic cells in situ and during maturation in vitro. J Exp Med 1994;180:1849-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Doherty DG, O’Farrelly C. Dendritic cells: regulators of hepatic immunity or tolerance? J Hepatol 2001;34:156-60. [DOI] [PubMed] [Google Scholar]
- 37.Palucka K, Banchereau J.Cancer immunotherapy via dendritic cells. Nat Rev Cancer 2012;12:265-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol 2003;21:685-711. [DOI] [PubMed] [Google Scholar]
- 39.Tiegs G, Lohse AW. Immune tolerance: what is unique about the liver. J Autoimmun 2010;34:1-6. [DOI] [PubMed] [Google Scholar]
- 40.Jin Y, Wi HJ, Choi MH, et al. Regulation of anti-inflammatory cytokines IL-10 and TGF-β in mouse dendritic cells through treatment with Clonorchis sinensis crude antigen. Exp Mol Med 2014;46:e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao B, Radaeva S, Park O.Liver natural killer and natural killer T cells: immunobiology and emerging roles in liver diseases. J Leukoc Biol 2009;86:513-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park O, Jeong WI, Wang L, et al. Diverse roles of invariant natural killer T cells in liver injury and fibrosis induced by carbon tetrachloride. Hepatology 2009;49:1683-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang R, Xie H, Huang Z, et al. Developing and activated T cell survival depends on differential signaling pathways to regulate anti-apoptotic Bcl-x(L). Clin Dev Immunol 2012;2012:632837. [DOI] [PMC free article] [PubMed]
- 44.Chaplin DD. Overview of the immune response. J Allergy Clin Immunol 2010;125:S3-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Travis MA, Sheppard D. TGF-β activation and function in immunity. Annu Rev Immunol 2014;32:51-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Korn T, Bettelli E, Oukka M, et al. IL-17 and Th17 cells. Annu Rev Immunol 2009;27:485-517. [DOI] [PubMed] [Google Scholar]
- 47.Zhou L, Ivanov II, Spolski R, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol 2007;8:967-74. [DOI] [PubMed] [Google Scholar]
- 48.Mitsdoerffer M, Lee Y, Jäger A, et al. Proinflammatory T helper type 17 cells are effective B-cell helpers. Proc Natl Acad Sci USA 2010;107:14292-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Veldhoen M, Hocking RJ, Flavell RA, et al. Signals mediated by transforming growth factor-β initiate autoimmune encephalomyelitis, but chronic inflammation is needed to sustain disease. Nat Immunol 2006;7:1151-6. [DOI] [PubMed] [Google Scholar]
- 50.McGeachy MJ, Bak-Jensen KS, Chen Y, et al. TGF-β and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol 2007;8:1390-7. [DOI] [PubMed] [Google Scholar]
- 51.Takimoto T, Wakabayashi Y, Sekiya T, et al. Smad2 and Smad3 are redundantly essential for the TGF-beta-mediated regulation of regulatory T plasticity and Th1 development. J Immunol 2010;185:842-55. [DOI] [PubMed] [Google Scholar]
- 52.Yang XO, Nurieva R, Martinez GJ, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity 2008;29:44-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jabeen R, Kaplan MH. The symphony of the ninth: the development and function of Th9 cells. Curr Opin Immunol 2012;24:303-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chang HC, Sehra S, Goswami R, et al. The transcription factor PU.1 is required for the development of IL-9-producing T cells and allergic inflammation. Nat Immunol 2010;11:527-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lederer JA, Perez VL, DesRoches L, et al. Cytokine transcriptional events during helper T cell subset differentiation. J Exp Med 1996;184:397-406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oh S, Hwang ES. The role of protein modifications of T-bet in cytokine production and differentiation of T helper cells. J Immunol Res 2014;2014:589672. [DOI] [PMC free article] [PubMed]
- 57.Szabo SJ, Kim ST, Costa GL, et al. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 2000;100:655-69. [DOI] [PubMed] [Google Scholar]
- 58.Gorelik L, Constant S, Flavell RA. Mechanism of transforming growth factor β-induced inhibition of T helper type 1 differentiation. J Exp Med 2002;195:1499-505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gorham JD, Güler ML, Fenoglio D, et al. Low dose TGF-β attenuates IL-12 responsiveness in murine Th cells. J Immunol 1998;161:1664-70. [PubMed] [Google Scholar]
- 60.Gorelik L, Fields PE, Flavell RA. Cutting edge: TGF-β inhibits Th type 2 development through inhibition of GATA-3 expression. J Immunol 2000;165:4773-7. [DOI] [PubMed] [Google Scholar]
- 61.Ouyang W, Ranganath SH, Weindel K, et al. Inhibition of Th1 development mediated by GATA-3 through an IL-4-independent mechanism. Immunity 1998;9:745-55. [DOI] [PubMed] [Google Scholar]
- 62.Kuwahara M, Yamashita M, Shinoda K, et al. The transcription factor Sox4 is a downstream target of signaling by the cytokine TGF-β and suppresses T(H)2 differentiation. Nat Immunol 2012;13:778-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cecere A, Marotta F, Vangieri B, et al. Progressive liver injury in chronic hepatitis C infection is related to altered cellular immune response and to different citokine profile. Panminerva Med 2004;46:171-87. [PubMed] [Google Scholar]
- 64.Ye Y, Xie X, Yu J, et al. Involvement of Th17 and Th1 effector responses in patients with Hepatitis B. J Clin Immunol 2010;30:546-55. [DOI] [PubMed] [Google Scholar]
- 65.Wu W, Li J, Chen F, et al. Circulating Th17 cells frequency is associated with the disease progression in HBV infected patients. J Gastroenterol Hepatol 2010;25:750-7. [DOI] [PubMed] [Google Scholar]
- 66.Sun HQ, Zhang JY, Zhang H, et al. Increased Th17 cells contribute to disease progression in patients with HBV-associated liver cirrhosis. J Viral Hepat 2012;19:396-403. [DOI] [PubMed] [Google Scholar]
- 67.Mordue DG, Monroy F, La Regina M, et al. Acute toxoplasmosis leads to lethal overproduction of Th1 cytokines. J Immunol 2001;167:4574-84. [DOI] [PubMed] [Google Scholar]
- 68.Gu FM, Li QL, Gao Q, et al. IL-17 induces AKT-dependent IL-6/JAK2/STAT3 activation and tumor progression in hepatocellular carcinoma. Mol Cancer 2011;10:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Loser K, Beissert S.Regulatory T cells: banned cells for decades. J Invest Dermatol 2012;132:864-71. [DOI] [PubMed] [Google Scholar]
- 70.Curotto de Lafaille MA, Lafaille JJ. Natural and adaptive foxp3+ regulatory T cells: more of the same or a division of labor? Immunity 2009;30:626-35. [DOI] [PubMed] [Google Scholar]
- 71.Hoffmann P, Boeld TJ, Eder R, et al. Loss of FOXP3 expression in natural human CD4+CD25+ regulatory T cells upon repetitive in vitro stimulation. Eur J Immunol 2009;39:1088-97. [DOI] [PubMed] [Google Scholar]
- 72.Burchill MA, Yang J, Vang KB, et al. Linked T cell receptor and cytokine signaling govern the development of the regulatory T cell repertoire. Immunity 2008;28:112-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ouyang W, Beckett O, Ma Q, et al. Transforming growth factor-β signaling curbs thymic negative selection promoting regulatory T cell development. Immunity 2010;32:642-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Polansky JK, Kretschmer K, Freyer J, et al. DNA methylation controls Foxp3 gene expression. Eur J Immunol 2008;38:1654-63. [DOI] [PubMed] [Google Scholar]
- 75.Floess S, Freyer J, Siewert C, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol 2007;5:e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhou L, Lopes JE, Chong MM, et al. TGF-β-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORβ function. Nature 2008;453:236-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Marie JC, Letterio JJ, Gavin M, et al. TGF-β1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med 2005;201:1061-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fahlén L, Read S, Gorelik L, et al. T cells that cannot respond to TGF-β escape control by CD4(+)CD25(+) regulatory T cells. J Exp Med 2005;201:737-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Josefowicz SZ, Niec RE, Kim HY, et al. Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature 2012;482:395-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fu J, Xu D, Liu Z, et al. Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology 2007;132:2328-39. [DOI] [PubMed] [Google Scholar]
- 81.Feng X, Li B, Ye H, et al. Increased frequency of CD4+CD25(high)FoxP3+ regulatory T cells in patients with hepatocellular carcinoma. Arch Immunol Ther Exp (Warsz) 2011;59:309-14. [DOI] [PubMed] [Google Scholar]
- 82.Kobayashi N, Hiraoka N, Yamagami W, et al. FOXP3+ regulatory T cells affect the development and progression of hepatocarcinogenesis. Clin Cancer Res 2007;13:902-11. [DOI] [PubMed] [Google Scholar]
- 83.Chang DK, Sui J, Geng S, et al. Humanization of an anti-CCR4 antibody that kills cutaneous T-cell lymphoma cells and abrogates suppression by T-regulatory cells. Mol Cancer Ther 2012;11:2451-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nishikawa H, Sakaguchi S.Regulatory T cells in tumor immunity. Int J Cancer 2010;127:759-67. [DOI] [PubMed] [Google Scholar]
- 85.Zhao HQ, Li WM, Lu ZQ, et al. Roles of Tregs in development of hepatocellular carcinoma: a meta-analysis. World J Gastroenterol 2014;20:7971-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cany J, Tran L, Gauttier V, et al. Immunotherapy of hepatocellular carcinoma: is there a place for regulatory T-lymphocyte depletion? Immunotherapy 2011;3:32-4. [DOI] [PubMed] [Google Scholar]
- 87.Aalaei-Andabili SH, Alavian SM. Regulatory T cells are the most important determinant factor of hepatitis B infection prognosis: a systematic review and meta-analysis. Vaccine 2012;30:5595-602. [DOI] [PubMed] [Google Scholar]
- 88.Hashimoto C, Hudson KL, Anderson KV. The Toll gene of Drosophila, required for dorsal-ventral embryonic polarity, appears to encode a transmembrane protein. Cell 1988;52:269-79. [DOI] [PubMed] [Google Scholar]
- 89.Lemaitre B, Nicolas E, Michaut L, et al. The dorsoventral regulatory gene cassette spätzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 1996;86:973-83. [DOI] [PubMed] [Google Scholar]
- 90.Rock FL, Hardiman G, Timans JC, et al. A family of human receptors structurally related to Drosophila Toll. Proc Natl Acad Sci USA 1998;95:588-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Delneste Y, Beauvillain C, Jeannin P.Innate immunity: structure and function of TLRs. Med Sci (Paris) 2007;23:67-73. [DOI] [PubMed] [Google Scholar]
- 92.Kawai T, Akira S.TLR signaling. Cell Death Differ 2006;13:816-25. [DOI] [PubMed] [Google Scholar]
- 93.Nakamoto N, Kanai T.Role of toll-like receptors in immune activation and tolerance in the liver. Front Immunol 2014;5:221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sörman A, Zhang L, Ding Z, et al. How antibodies use complement to regulate antibody responses. Mol Immunol 2014;61:79-88. [DOI] [PubMed] [Google Scholar]
- 95.Garlanda C, Bottazzi B, Bastone A, et al. Pentraxins at the crossroads between innate immunity, inflammation, matrix deposition, and female fertility. Annu Rev Immunol 2005;23:337-66. [DOI] [PubMed] [Google Scholar]
- 96.Peiser L, Mukhopadhyay S, Gordon S.Scavenger receptors in innate immunity. Curr Opin Immunol 2002;14:123-8. [DOI] [PubMed] [Google Scholar]
- 97.Kim CW, Chang KM. Hepatitis C virus: virology and life cycle. Clin Mol Hepatol 2013;19:17-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Girardin SE, Boneca IG, Carneiro LA, et al. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science 2003;300:1584-7. [DOI] [PubMed] [Google Scholar]
- 99.Girardin SE, Travassos LH, Hervé M, et al. Peptidoglycan molecular requirements allowing detection by Nod1 and Nod2. J Biol Chem 2003;278:41702-8. [DOI] [PubMed] [Google Scholar]
- 100.Yoneyama M, Kikuchi M, Matsumoto K, et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol 2005;175:2851-8. [DOI] [PubMed] [Google Scholar]
- 101.Strobl H, Knapp W.TGF-β1 regulation of dendritic cells. Microbes Infect 1999;1:1283-90. [DOI] [PubMed] [Google Scholar]
- 102.Kaplan DH, Li MO, Jenison MC, et al. Autocrine/paracrine TGFβ1 is required for the development of epidermal Langerhans cells. J Exp Med 2007;204:2545-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ramalingam R, Larmonier CB, Thurston RD, et al. Dendritic cell-specific disruption of TGF-β receptor II leads to altered regulatory T cell phenotype and spontaneous multiorgan autoimmunity. J Immunol 2012;189:3878-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Broering R, Lu M, Schlaak JF. Role of Toll-like receptors in liver health and disease. Clin Sci 2011;121:415-26. [DOI] [PubMed] [Google Scholar]
- 105.Seki E, De Minicis S, Osterreicher CH, et al. TLR4 enhances TGF-β signaling and hepatic fibrosis. Nat Med 2007;13:1324-32. [DOI] [PubMed] [Google Scholar]
- 106.Luedde T, Trautwein C.A molecular link between inflammation and fibrogenesis: the bacterial microflora influences hepatic fibrosis via toll-like receptor 4-dependent modification of transforming growth factor-β signaling in hepatic stellate cells. Hepatology 2008;47:1089-91. [DOI] [PubMed] [Google Scholar]
- 107.Naiki Y, Michelsen KS, Zhang W, et al. Transforming growth factor-β differentially inhibits MyD88-dependent, but not TRAM- and TRIF-dependent, lipopolysaccharide-induced TLR4 signaling. J Biol Chem 2005;280:5491-5. [DOI] [PubMed] [Google Scholar]
- 108.Greten FR, Eckmann L, Greten TF, et al. IKK links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004;118:285-96. [DOI] [PubMed] [Google Scholar]
- 109.Nakagawa H, Maeda S.Inflammation- and stress-related signaling pathways in hepatocarcinogenesis. World J Gastroenterol 2012;18:4071-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.World Health Organization. Viral hepatitis. Report by the Secretariat. Available online: http://apps.who.int/gb/ebwha/pdf_files/EB126/B126_15-en.pdf. Last accessed on 6 December 2014.
- 111.Perz JF, Armstrong GL, Farrington LA, et al. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J Hepatol 2006;45:529-38. [DOI] [PubMed] [Google Scholar]
- 112.Gower E, Estes C, Blach S, et al. Global epidemiology and genotype distribution of the hepatitis C virus infection. J Hepatol 2014;61:S45-57. [DOI] [PubMed] [Google Scholar]
- 113.Franco E, Bagnato B, Marino MG, et al. Hepatitis B: Epidemiology and prevention in developing countries. World J Hepatol 2012;4:74-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Trehanpati N, Shrivastav S, Shivakumar B, et al. Analysis of Notch and TGF-β signaling expression in different stages of disease progression during Hepatitis B virus infection. Clin Transl Gastroenterol 2012;3:e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Brechot C, Pourcel C, Louise A, et al. Presence of integrated hepatitis B virus DNA sequences in cellular DNA of human hepatocellular carcinoma. Nature 1980;286:533-5. [DOI] [PubMed] [Google Scholar]
- 116.Koike K, Moriya K, Iino S, et al. High-level expression of hepatitis B virus HBx gene and hepatocarcinogenesis in transgenic mice. Hepatology 1994;19:810-9. [PubMed] [Google Scholar]
- 117.Benn J, Su F, Doria M, et al. Hepatitis B virus HBx protein induces transcription factor AP-1 by activation of extracellular signal-regulated and c-Jun N-terminal mitogen-activated protein kinases. J Virol 1996;70:4978-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Murata M, Matsuzaki K, Yoshida K, et al. Hepatitis B virus X protein shifts human hepatic transforming growth factor (TGF)-β signaling from tumor suppression to oncogenesis in early chronic hepatitis B. Hepatology 2009;49:1203-17. [DOI] [PubMed] [Google Scholar]
- 119.Sekimoto G, Matsuzaki K, Yoshida K, et al. Reversible Smad-dependent signaling between tumor suppression and oncogenesis. Cancer Res 2007;67:5090-6. [DOI] [PubMed] [Google Scholar]
- 120.Yamagata H, Matsuzaki K, Mori S, et al. Acceleration of Smad2 and Smad3 phosphorylation via c-Jun NH(2)-terminal kinase during human colorectal carcinogenesis. Cancer Res 2005;65:157-65. [PubMed] [Google Scholar]
- 121.Rice CM. New insights into HCV replication: potential antiviral targets. Top Antivir Med 2011;19:117-20. [PMC free article] [PubMed] [Google Scholar]
- 122.Spengler U, Nattermann J.Immunopathogenesis in hepatitis C virus cirrhosis. Clin Sci 2007;112:141-55. [DOI] [PubMed] [Google Scholar]
- 123.Rehermann B.Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J Clin Invest 2009;119:1745-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rosen HR. Emerging concepts in immunity to hepatitis C virus infection. J Clin Invest 2013;123:4121-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Dolganiuc A, Chang S, Kodys K, et al. Hepatitis C virus (HCV) core protein-induced, monocyte-mediated mechanisms of reduced IFN-alpha and plasmacytoid dendritic cell loss in chronic HCV infection. J Immunol 2006;177:6758-68. [DOI] [PubMed] [Google Scholar]
- 126.Jinushi M, Takehara T, Tatsumi T, et al. Negative regulation of NK cell activities by inhibitory receptor CD94/NKG2A leads to altered NK cell-induced modulation of dendritic cell functions in chronic hepatitis C virus infection. J Immunol 2004;173:6072-81. [DOI] [PubMed] [Google Scholar]
- 127.Seeff LB, Hoofnagle JH. Appendix: The National Institutes of Health consensus development conference management of Hepatitis C 2002. Clin Liver Dis 2003;7:261-87. [DOI] [PubMed] [Google Scholar]
- 128.Vergani D, Mieli-Vergani G.Autoimmune manifestations in viral hepatitis. Semin Immunopathol 2013;35:73-85. [DOI] [PubMed] [Google Scholar]
- 129.Kim MS, Kim S, Myung H. Degradation of AIMP1/p43 induced by hepatitis C virus E2 leads to upregulation of TGF-β signaling and increase in surface expression of gp96. PLoS One 2014;9:e96302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Benzoubir N, Lejamtel C, Battaglia S, et al. HCV core-mediated activation of latent TGF-β via thrombospondin drives the crosstalk between hepatocytes and stromal environment. J Hepatol 2013;59:1160-8. [DOI] [PubMed] [Google Scholar]
- 131.Cheng PL, Chang MH, Chao CH, et al. Hepatitis C viral proteins interact with Smad3 and differentially regulate TGF-beta/Smad3-mediated transcriptional activation. Oncogene 2004;23:7821-38. [DOI] [PubMed] [Google Scholar]
- 132.Taniguchi H, Kato N, Otsuka M, et al. Hepatitis C virus core protein upregulates transforming growth factor-β1 transcription. J Med Virol 2004;72:52-9. [DOI] [PubMed] [Google Scholar]
- 133.Presser LD, Haskett A, Waris G. Hepatitis C virus-induced furin and thrombospondin-1 activate TGF-β1: role of TGF-β1 in HCV replication. Virology 2011;412:284-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sakata K, Hara M, Terada T, et al. HCV NS3 protease enhances liver fibrosis via binding to and activating TGF-β type I receptor. Sci Rep 2013;3:3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hall CH, Kassel R, Tacke RS, et al. HCV+ hepatocytes induce human regulatory CD4+ T cells through the production of TGF-β. PLoS One 2010;5:e12154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Fujii D, Brissenden JE, Derynck R, et al. Transforming growth factor β gene maps to human chromosome 19 long arm and to mouse chromosome 7. Somat Cell Mol Genet 1986;12:281-8. [DOI] [PubMed] [Google Scholar]
- 137.Buckova D, Izakovicová Hollá L, Benes P, et al. TGF-β1 gene polymorphisms. Allergy 2001;56:1236-7. [DOI] [PubMed] [Google Scholar]
- 138.Bataller R, North KE, Brenner DA. Genetic polymorphisms and the progression of liver fibrosis: a critical appraisal. Hepatology 2003;37:493-503. [DOI] [PubMed] [Google Scholar]
- 139.Wang H, Mengsteab S, Tag CG, et al. Transforming growth factor-β1 gene polymorphisms are associated with progression of liver fibrosis in Caucasians with chronic hepatitis C infection. World J Gastroenterol 2005;11:1929-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wu XD, Zeng K, Gong CS, et al. Transforming growth factor-β genetic polymorphisms on development of liver cirrhosis in a meta-analysis. Mol Biol Rep 2013;40:535-43. [DOI] [PubMed] [Google Scholar]
- 141.Pereira FA, Pinheiro da Silva NN, Rodart IF, et al. Association of TGF-β1 codon 25 (G915C) polymorphism with hepatitis C virus infection. J Med Virol 2008;80:58-64. [DOI] [PubMed] [Google Scholar]
- 142.Talaat RM, Dondeti MF, El-Shenawy SZ, et al. Transforming growth factor- β1 gene polymorphism (T29C) in Egyptian patients with Hepatitis B virus infection: A preliminary study. Hepat Res Treat 2013;2013:293274. [DOI] [PMC free article] [PubMed]
- 143.Cambien F, Ricard S, Troesch A, et al. Polymorphisms of the transforming growth factor-β1 gene in relation to myocardial infarction and blood pressure. The Etude Cas-Témoin de l’Infarctus du Myocarde (ECTIM) Study. Hypertension 1996;28:881-7. [DOI] [PubMed] [Google Scholar]
- 144.Hobbs K, Negri J, Klinnert M, et al. Interleukin-10 and transforming growth factor-β promoter polymorphisms in allergies and asthma. Am J Respir Crit Care Med 1998;158:1958-62. [DOI] [PubMed] [Google Scholar]
- 145.Shrivastava A, Calame K.An analysis of genes regulated by the multi-functional transcriptional regulator Yin Yang-1. Nucleic Acids Res 1994;22:5151-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wang H, Zhao YP, Gao CF, et al. Transforming growth factor beta 1 gene variants increase transcription and are associated with liver cirrhosis in Chinese. Cytokine 2008;43:20-5. [DOI] [PubMed] [Google Scholar]
- 147.Kimura T, Saito T, Yoshimura M, et al. Association of transforming growth factor-beta 1 functional polymorphisms with natural clearance of hepatitis C virus. J Infect Dis 2006;193:1371-4. [DOI] [PubMed] [Google Scholar]
- 148.Powell EE, Edwards-Smith CJ, Hay JL, et al. Host genetic factors influence disease progression in chronic hepatitis C. Hepatology 2000;31:828-33. [DOI] [PubMed] [Google Scholar]
- 149.Hosseini Razavi A, Azimzadeh P, Mohebbi SR, et al. Lack of association between transforming growth factor β1-509C/T and +915G/C polymorphisms and chronic Hepatitis B in Iranian patients. Hepat Mon 2014;14:e13100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yokota M, Ichihara S, Lin TL, et al. Association of a T29-->C polymorphism of the transforming growth factor-β1 gene with genetic susceptibility to myocardial infarction in Japanese. Circulation 2000;101:2783-7. [DOI] [PubMed] [Google Scholar]
- 151.Qi P, Chen Y, Wang H, et al. -509C>T polymorphism in the TGF-beta1 gene promoter, impact on the hepatocellular carcinoma risk in Chinese patients with chronic hepatitis B virus infection. Cancer Immunol Immunother 2009;58:1433-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Jain S, Singhal S, Lee P, et al. Molecular genetics of hepatocellular neoplasia. Am J Transl Res 2010;2:105-18. [PMC free article] [PubMed] [Google Scholar]
- 153.Wang L, Yao M, Dong Z, et al. Circulating specific biomarkers in diagnosis of hepatocellular carcinoma and its metastasis monitoring. Tumour Biol 2014;35:9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.El-Bassiouni NE, Nosseir MM, Madkour ME, et al. Role of fibrogenic markers in chronic hepatitis C and associated hepatocellular carcinoma. Mol Biol Rep 2012;39:6843-50. [DOI] [PubMed] [Google Scholar]
- 155.Mas VR, Maluf DG, Stravitz R, et al. Hepatocellular carcinoma in HCV-infected patients awaiting liver transplantation: genes involved in tumor progression. Liver Transpl 2004;10:607-20. [DOI] [PubMed] [Google Scholar]
- 156.Shao RX, Hoshida Y, Otsuka M, et al. Hepatic gene expression profiles associated with fibrosis progression and hepatocarcinogenesis in hepatitis C patients. World J Gastroenterol 2005;11:1995-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Jung SA, Chung YH, Park NH, et al. Experimental model of hepatic fibrosis following repeated periportal necrosis induced by allylalcohol. Scand J Gastroenterol 2000;35:969-75. [DOI] [PubMed] [Google Scholar]
- 158.Tsushima H, Kawata S, Tamura S, et al. Reduced plasma transforming growth factor-beta1 levels in patients with chronic hepatitis C after interferon-alpha therapy: association with regression of hepatic fibrosis. J Hepatol 1999;30:1-7. [DOI] [PubMed] [Google Scholar]
- 159.Flisiak R, Pytel-Krolczuk B, Prokopowicz D.Circulating transforming growth factor beta(1) as an indicator of hepatic function impairment in liver cirrhosis. Cytokine 2000;12:677-81. [DOI] [PubMed] [Google Scholar]
- 160.Marek B, Kajdaniuk D, Mazurek U, et al. TGF-β1 mRNA expression in liver biopsy specimens and TGF-β1 serum levels in patients with chronic hepatitis C before and after antiviral therapy. J Clin Pharm Ther 2005;30:271-7. [DOI] [PubMed] [Google Scholar]
- 161.Okumoto K, Hattori E, Tamura K, et al. Possible contribution of circulating transforming growth factor-β1 to immunity and prognosis in unresectable hepatocellular carcinoma. Liver Int 2004;24:21-8. [DOI] [PubMed] [Google Scholar]
- 162.Song BC, Chung YH, Kim JA, et al. Transforming growth factor-β1 as a useful serologic marker of small hepatocellular carcinoma. Cancer 2002;94:175-80. [DOI] [PubMed] [Google Scholar]
- 163.Lin GH, Wang J, Li SH, et al. Relationship and clinical significance of TGF-beta1 expression with Treg cell infiltration in hepatocellular carcinoma. Chin J Cancer 2010;29:403-7. [DOI] [PubMed] [Google Scholar]
- 164.Steer SA, Corbett JA. The role and regulation of COX-2 during viral infection. Viral Immunol 2003;16:447-60. [DOI] [PubMed] [Google Scholar]
- 165.Hla T, Neilson K.Human cyclooxygenase-2 cDNA. Proc Natl Acad Sci USA 1992;89:7384-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Tortora G, Caputo R, Damiano V, et al. Combination of a selective cyclooxygenase-2 inhibitor with epidermal growth factor receptor tyrosine kinase inhibitor ZD1839 and protein kinase A antisense causes cooperative antitumor and antiangiogenic effect. Clin Cancer Res 2003;9:1566-72. [PubMed] [Google Scholar]
- 167.Núñez O, Fernández-Martínez A, Majano PL, et al. Increased intrahepatic cyclooxygenase 2, matrix metalloproteinase 2, and matrix metalloproteinase 9 expression is associated with progressive liver disease in chronic hepatitis C virus infection: role of viral core and NS5A proteins. Gut 2004;53:1665-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.El-Bassiouny AE, Zoheiry MM, Nosseir MM, et al. Expression of cyclooxygenase-2 and transforming growth factor-β1 in HCV-induced chronic liver disease and hepatocellular carcinoma. MedGenMed 2007;9:45. [PMC free article] [PubMed] [Google Scholar]