

Abstract
AIM: To evaluate the proinflammatory effects and molecular mechanisms of interleukin (IL)-17 in intestinal epithelial cell line HT-29.
METHODS: HT-29 cells were cultured with IL-17, tumor necrosis factor (TNF)-α, or the combination of both IL-17 and TNF-α. Real-time PCR and Western blot were used to measure the gene expression levels of neutrophil chemokines CXCL1, CXCL2, CXCL5, CXCL6, IL-8 and TH-17 cell chemokine CCL20, the phosphorylation levels of p38 and TNF-α, and the expression level of IL-8, after using the p38 inhibitor in HT-29 cells. The stable Act1 knockdown HT-29 cell line was established to further test the phosphorylation changes of p38, after using IL-17 and TNF-α.
RESULTS: After HT-29 cells were cultured with IL-17 and TNF-α, the expression levels of neutrophil chemokines (CXCL1, CXCL2, CXCL5, CXCL6, IL-8) and Th17 chemokine (CCL20) significantly improved (24.96 ± 2.53, 28.47 ± 2.87, 38.08 ± 2.72, 33.47 ± 2.41, 31.7 ± 2.38, 44.37 ± 2.73, respectively), and the differences were all statistically significant (P < 0.01). Western blot results showed that IL-17 obviously enhanced the phosphorylation level of p38, which was induced by TNF-α. Compared with the control group, the expression level of IL-8 significantly declined (9.47 ± 1.36 vs 3.06 ± 0.67, P < 0.01) when TH-29 cells were cultured with IL-17 and TNF-α. p38 inhibition assay showed that the p38 pathway played an essential role in the inflammatory response induced by IL-17. p38 phosphorylation levels could not be changed after using IL-17 and TNF-α in the stable Act1 knockdown HT-29 cell line.
CONCLUSION: IL-17 significantly promoted the gene expression levels of TNF-α-induced neutrophil chemokines and Th17 cell chemokine. It is obvious that IL-17 and TNF-α have synergistic effects on p38.
Keywords: IL-17, HT-29, TNF-α, Inflammatory bowel disease
Core tip: Our study revealed that interleukin (IL)-17 significantly promoted the gene expression levels of tumor necrosis factor (TNF)-α-induced neutrophil chemokines and Th17 cell chemokines. It is obvious that IL-17 and TNF-α have synergistic effects on p38.
INTRODUCTION
Ulcerative colitis and Crohn’s disease are also known as inflammatory bowel disease (IBD)[1]. The disease refers to chronic inflammatory disorders of the gastrointestinal tract, which can easily recur. IBD occurs more often in males than females, and nowadays, the morbidity rate is showing an upward trend. IBD has become one of the most common digestive system diseases[2]. It is widely accepted that the interaction of genetic and environmental factors leads to the disease[3-5], but the pathogenesis is still unclear.
Hundorfean et al[6] found that the expression level of interlukin-17 (IL-17) significantly increased in the peripheral blood of patients with IBD, which implies that IL-17 may play an important role in the physiological and pathological processes of the disease. IL-17 and tumor necrosis factor (TNF) can accelerate inflammatory response by inducing various kinds of inflammatory cytokines in diseases such as IBD[7,8]; however, the molecular mechanism of the proinflammatory effects is still unknown. Wu et al[9] reported that IL-17 can induce neutrophil infiltration and related inflammatory cytokine expression, through the p38 pathway in myoblasts and fibroblasts; yet epithelial cell mechanisms were still rarely reported.
To deeply understand the pathogenesis of IBD, the molecular mechanisms of the proinflammatory effects of IL-17 and TNF-α in intestinal epithelial cell line HT-29 need to be studied. Since intestinal epithelial cell line HT-29 has normal colonic epithelial structures and functions, it has been the most common cell line used in laboratory to study the immunologic mechanisms of the intestinal mucosa[10,11]. Even though the IL-17 inhibitor has been proven to be ineffective in IBD treatments, blocking the other site of the pathway may prove to be hopeful in future studies. Therefore, it is of great importance to explore the mechanisms of action of IL-17.
MATERIALS AND METHODS
Materials
The following reagents were utilized for this study: McCoy’s 5A medium (hyClone), recombinant human IL-17 and TNF-α (eBiosciences), Eastep Universal RNA Extraction Kit (Promega), Fist Strand cDNA Synthesis Kit (Promega), SYBR Premix Ex TaqTM (Takara), p38 inhibitor SB203580 (Sigma), BCA protein assay kit (Thermo), Phospho-p38 MAPK antibody (Beyotime), HRP-labeled donkey anti-goat IgG (Beyotime), mouse anti-human Act1 antibody (Biolegend), and goat anti-mouse IgG (Biolegend).
The following instruments were utilized for this study: CO2 incubator (Thermo), PCR amplifier (Longgene), electrophoresis apparatus (Tanon), Gene Genius bioimaging system (BIO-RAD), polyacrylamide gel electrophoresis apparatus (Tanon), centrifugal machine (Debon), and microplate reader (Thermo).
Cell culture
Intestinal epithelial cell line HT-29 was cultured in McCoy’s 5A medium, supplemented with 10% fetal bovine serum, 100 mg/mL of streptomycin and 100 U/mL of penicillin, in a 5% CO2 humidified environment at 37 °C. The cells were cultured to the exponential phase for use.
Reverse transcription-PCR
TH-29 cells were seeded in 12-well plates (5 × 105 cells/well), the fetal bovine serum was reduced to 0.5% as they reached about 70% confluence, and the plates were cultured overnight. Then, rhIL-17 (50 ng/mL), rhTNF-α (0.5 ng/mL), or the combination of rhIL-17 and rhTNF-α was added into the plates. The cells were cultured for another 6 h, the samples were collected, and TRIzol (1 mL TRIzol per 5-10 × 106 cells) was added to lyse the cells. Before adding TRIzol, cells were not washed with a buffer solution, since the cellular RNA can be easily degraded. Total RNA was isolated using an Eastep Universal RNA Extraction kit and reverse transcribed with a Fist Strand cDNA Synthesis kit, according to the manufacturer’s instructions. Finally, real-time fluorescence quantitative PCR (SYBR Green I) was performed using 200 ng of cDNA. The reaction system was instructed by the SYBR Premix Ex Taq™ specifications purchased from Takara. The reaction conditions were as follows: initial denaturation at 95 °C for 10 min, followed by 40 cycles of denaturation at 95 °C for 15 s and annealing at 60 °C for 30 s. The fluorescence signal was then collected. After amplification, the melting curve from 65 °C to 95 °C was analyzed to exclude non-specific PCR products and amplified primer dimers. Agarose gel electrophoresis was then used to analyze the specificity of the product. GAPDH was amplified as a control for the amount of cDNA in each sample, and the expression level of each gene was computed. The gene levels of neutrophil chemokines and TH-17 cell chemokine (CXCL1, CXCL2, CXCL5, CXCL6, IL-8 and CCL20) were examined. All primers used in our study were designed according to the sequences published online (GenBank) and synthesized by Invitrogen (Table 1).
Table 1.
Primers used in RT-PCR
| Gene | Forward | Reverse |
| CXCL1 | AGATTCTATGTTAATATTTTAGGTGTAAAATAAT | AACTAACTTGGGGTTGACATTTC |
| CXCL2 | CTTCTATTTATTTATTTATTTATTTATTTGTTTGTTTT | GAACTAACTTGGGTTTGACCTAAA |
| CXCL5 | TGGCCCCTTTCACAGAGTAG | CTAAAAACCCGACAGGCATC |
| CXCL6 | AGTTTACAGCTCAGCTAATGAAGTACTAAT | CGGTAAGACTTTAAGGAATGTATGATA |
| IL-8 | GAATTGAATGGGTTTGCTAGA | CACTGTGAGGTAAGATGGTGG |
| CCL20 | CTGGCTGCTTTGATGTCAGT | CGTGTGAAGCCCACAATAAA |
IL-8: Interleukin-8.
Cells were cultured as described above, and rhIL-17, rhTNF-α, and p38 inhibitor SB203580 were added into the plate of the experiment groups. The same procedure was conducted to detect the gene expression level of IL-8.
Western blot
TH-29 cells were seeded in 6-well plates (8 × 105 cells/well), fetal bovine serum was reduced to 0.5% as they reached about 70% confluence, and the cells were cultured overnight. Then, rhIL-17 (50 ng/mL), rhTNF α (0.5 ng/mL), or the combination of rhIL-17 and rhTNF α was added into the plates. The samples were collected 10, 15 and 30 min later. At each time point, the medium was removed and the cells were washed with PBS solution; M2 was then added and the plate was left on ice for 30 min to lyse the cells. The cells were collected into a centrifuge tube with a cell scraper and lysed with ultrasonic irradiation for 3 s, 2 times. Cell debris was removed by lysate centrifugation at 12000 g for 2 min at 4 °C and protein concentration was determined using the BCA Protein assay kit. The working liquid was prepared according to the manufacturer’s instructions. The standard curve was made, the plate was incubated at 37 °C for 30 min, and the optical density (570) was tested with a microplate reader. Finally, protein concentration was computed according to the standard curve. All specimens were adjusted to have the same concentration. After mixing the protein supernate with the buffer, the liquid was boiled for 10 min, cooled, centrifuged, and resolved by 12% SDS-PAGE. The protein was then transferred to a nitrocellulose membrane. An enhanced chemiluminescence (ECL) based Western blot was performed using a primary anti-p38 antibody, followed by incubation with a horseradish peroxidase-labeled secondary antibody for chemiluminescence detection. Briefly, the 5% bovine serum albumin (BSA) blocked membrane was incubated for 12 h at 4 °C with the primary antibody (1:1000). Then, the membrane was washed and incubated with the horseradish peroxidase conjugated rabbit anti-mouse IgG for 60 min at room temperature. After washing, the protein was tested with an ECL Western blot analysis detection system.
Establishing the Act1 silenced cell line
HT-29 cells were cultured in 96-well plates. When the cells reached 80% confluence, Metafectene transfection reagents were used to transfer HT-29 targeted small interfering RNA into the HT-29 cells. The plasmid with the best knockdown performance (tested through transient transfection) was selected to establish a stable cell line, and the cells were cultured for subsequent experiments. Immunocytochemistry was performed to identify the transfected cells. The cells were briefly blocked with calf serum (diluted with PBS solution) for 15 min, the serum was removed, and the cells were incubated with a mouse anti-human Act1 antibody for 12 h at 4 °C. Unbounded antibodies were washed off with PBS solution, and cells were again incubated with a goat anti-mouse IgG for one hour at room temperature, followed by mounting and microscopic examination.
The transfected cells were seeded in 6-well plates (8 × 105 cells/well), fetal bovine serum was reduced to 0.5% as they reached about 80% confluence, and the cells were cultured overnight. Then, rhIL-17 (50 ng/mL), rhTNF-α (0.5 ng/mL) or the combination of rhIL-17 and rhTNF-α was added into the plates. The samples were collected 15 min later. The same method described above was used to test the expression levels of p-p38 and p-ERK.
Statistical analysis
SPSS 17.0 software was used for statistical analyses in this study. All data are expressed as mean ± SD, and t-test was applied in performing the statistical analysis. P < 0.05 was considered significant.
RESULTS
Neutrophil and TH-17 chemokine expression in HT-29 cells
Figure 1 showed that the chemokine RNA expression levels (A-E: neutrophil chemokines; F: TH-17 chemokine) were comparatively low, when IL-17 and TNF-α were used separately. However, when the chemokines were used together, the expression of mRNAs coding for all these mediators became strongly upregulated. The differences were statistically significant (P < 0.01).
Figure 1.
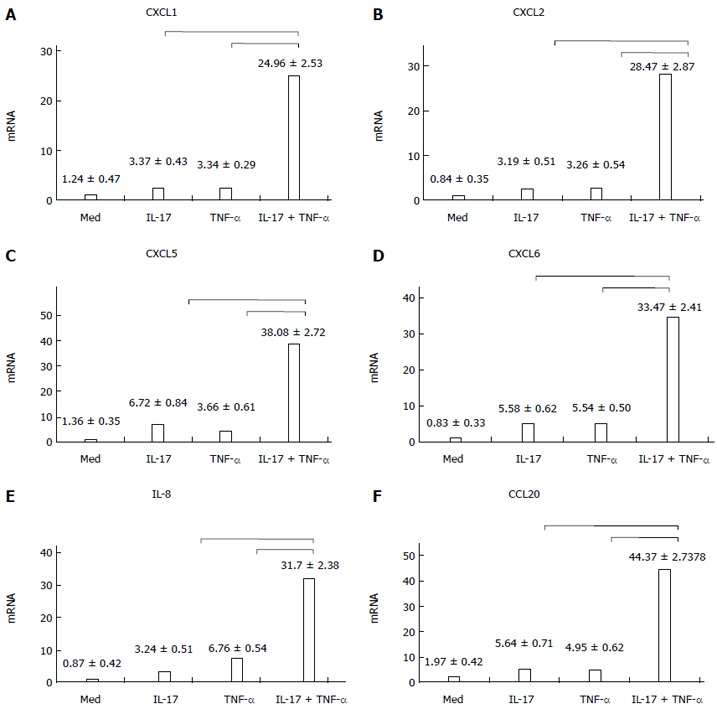
Chemokine expression when HT-29 cells were cultured with interleukin-17 and/or tumor necrosis factor-α. When interleukin (IL)-17 and tumor necrosis factor (TNF)-α were used separately, the expression levels of CXCL1, CXCL2, CXCL5, CXCL6, IL-8 and CCL20 in HT-29 cells were comparatively low. However, when IL-17 and TNF-α were used together, the expression levels became strongly upregulated; A: IL-17 + TNF-α vs IL-17, t = 30.424, P = 0.000; IL-17 + TNF-α vs TNF-α, t = 30.438, P = 0.000; B: IL-17 + TNF-α vs IL-17, t = 35.273, P = 0.000; IL-17 + TNF-α vs TNF-α, t = 35.125, P = 0.000; C: IL-17 + TNF-α vs IL-17, t = 85.718, P = 0.000; IL-17 + TNF-α vs TNF-α, t = 94.199, P = 0.000; D: IL-17 + TNF-α vs IL-17, t = 50.165, P = 0.000; IL-17 + TNF-α vs TNF-α, t = 50.224, P = 0.000; E: IL-17 + TNF-α vs IL-17, t = 54.585, P = 0.000; IL-17 + TNF-α vs TNF-α, t = 44.036, P = 0.000; F: IL-17 + TNF-α vs IL-17, t = 56.272, P = 0.000; IL-17 + TNF-α vs TNF-α, t = 57.410, P = 0.000. IL-8: Interleukin-8.
p38 and ERK phosphorylation levels
When IL-17 was used alone, no obvious phosphorylation occurred on p38 and ERK; and when TNF-α was used alone, the phosphorylation that occurred on p38 and ERK was very low. In contrast, when IL-17 and TNF-α were combined together, obvious phosphorylation can be easily observed; the longer IL-17 and TNF-α were combined together, the stronger the phosphorylation became (Figure 2).
Figure 2.
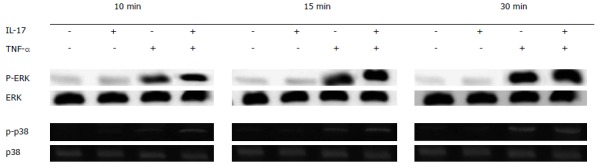
p38 and extracellular signal-regulated kinase phosphorylation levels. When interleukin (IL)-17 and tumor necrosis factor (TNF)-α were used separately, the phosphorylation levels of p38 and extracellular signal-regulated kinase (ERK) were very low. However, when IL-17 and TNF-α were combined together, the phosphorylation levels of p38 and ERK improved significantly, and the longer IL-17 and TNF-α were combined together, the stronger the phosphorylation became.
Expression level of IL-8 in the presence of p38 inhibitor
Whether or not the p38 inhibitor was added, the IL-17 and TNF-α combination could still significantly improve the expression level of IL-8; and compared with cells that were cultured without the p38 inhibitor, the expression level of IL-8 significantly reduced with the advent of the p38 inhibitor, in all the three groups (Figure 3).
Figure 3.
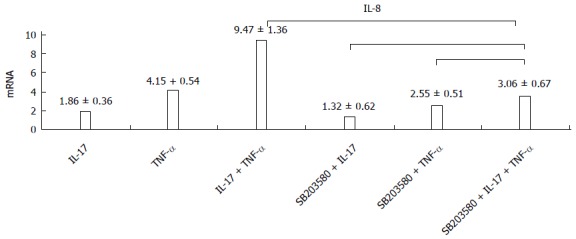
p38 inhibitor decreases the expression of interleukin-8. When p38 inhibitor was used, the interleukin (IL)-8 expression levels declined in all three groups. IL-17 + TNF-α vs S + IL-17 + TNF-α, t = 103.701, P = 0.000; S + IL-17 + TNF-α vs S + TNF-α, t = - 4.131, P = 0.07; S + IL-17 + TNF-α vs S + IL-17, t = -11.509, P = 0.000. IL-8: Interleukin-8; TNF-α: Tumor necrosis factor-α; S: SB203580.
Phosphorylation level of IL-8 in Act1 silenced HT-29 cell line
Immunocytochemistry was performed to identify the Act1 silenced cell. Fluorescent intensity greatly decreased as compared with the control group (Figure 4) and the positive rate reached up to 90%. In the control group, the IL-17 and TNF-α combination greatly elevated the phosphorylation level of p38; while in the Act1 silenced treatment group, no obvious change occurred (Figure 5).
Figure 4.
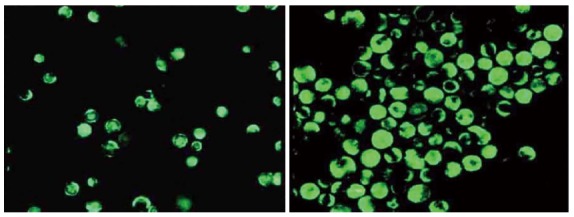
Act1 silenced HT-29 cells. Compared with the control group (A), the expression level of Act1 was comparatively low in Act1 silenced cells (B).
Figure 5.
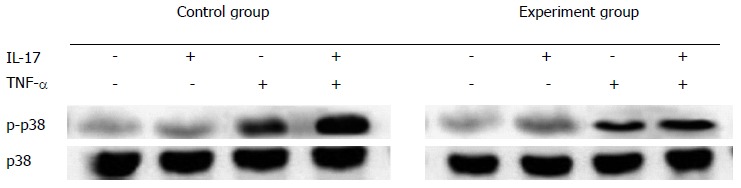
Phosphorylation levels of p38 in Act1 silenced HT-29 cell line. In the control group, interleukin (IL)-17 combined with tumor necrosis factor (TNF)-α could greatly improve the phosphorylation level of p38. In Act1 silenced cells, the phosphorylation level of p38 remained unchanged, when IL-17 and TNF-α were used together.
DISCUSSION
To deeply understand the pathogenesis of IBD, the molecular mechanisms of the proinflammatory effects of IL-17 and TNF-α in the intestinal epithelial cell line HT-29 were investigated in this study. We analyzed the expression levels of neutrophil and TH-17 cell chemokines, and the phosphorylation levels of p38 in HT-29 cells - when cultured with IL-17 and/or TNF-α. The results further confirmed that IL-17 could obviously enhance TNF-α induced p38 phosphorylation, and that the p38 pathway played an essential role in IL-17 induced inflammatory response.
Frontier of IL-17
Intestinal epithelium is the physical barrier between the intestinal tract and the external environment, playing an important role in the intestinal immune system. The cell line HT-29 used in our research is a human colonic epithelial cell line - the most common cell line used in laboratory to study the immunologic mechanisms of the intestinal mucosa. As the most representative member of the IL-17 family, IL-17 received much attention in recent years. Various studies suggested that multiple chemokines can be induced by IL-17 in epithelial cells to promote neutrophil infiltration in the lesion area[12,13]. It is currently known that IL-17 is involved in the pathological process of autoimmune diseases[14,15]. IL-17 monoclonal antibodies can be used for treating psoriasis, rheumatoid arthritis and uveitis, which are now in the clinical trial phase[16]. On the contrary, when the monoclonal antibodies were used in IBD treatment, the illness exacerbated[17,18]. This phenomenon suggested that IL-17 may have a special mode of action in IBD. TNF-α is another cytokine that plays an important role in the immune regulation of IBD[19]. Hence, in our study, we combined IL-17 with TNF-α, to investigate whether IL-17 could act alone or together with TNF-α in HT-29 cells.
IL-17 combined with TNF-α upregulates HT-29 chemokine expression levels
Chemokine factors CXCL1, CXCL2, CXCL5, CXCL6 and IL-8 play important roles in recruiting neutrophils in inflammation reactions; therefore, we detected their expression levels to monitor the proinflammatory effects of IL-17. We discovered that when IL-17 or TNF-α was used alone for cell stimulation, expression levels of chemokine factors were comparatively low, but when IL-17 and TNF-α were combined, the expression levels elevated significantly. The results suggest that there may be a synergy between IL-17 and TNF-α. The inflammatory response caused by IL-17 was achieved by the MAPK signal pathway, which mediates the stability of mRNAs[12,20]. TNF-α is a genetic transcription starter that can promote gene transcription[21,22]. The mRNAs regulated by IL-17 possess a similar sequence. There is an AU-rich sequence in the 3’ putative untranslated regions of the mRNA, which can be grasped by zinc finger proteins; and thus, the mRNA can be recognized and degraded by exosome complex[23]. For relatively stable mRNAs, the lack of the AU-rich sequence does not make it mediated by IL-17. Moreover, IL-17 can elevate the chemokine factor CCL20 through TNF-α, and CCL20 can combine with CCR6 on the cell surface to further recruit TH-17 cells, causing more IL-17 to be expressed. This would create a positive feedback and further promote inflammatory reactions[24,25]. Therefore, we can assume that IL-17 may improve the expression levels of related cytokines by stabilizing their mRNAs through the MAPK pathway together with TNF-α.
The proinflammatory mechanism of IL-17
In our study, p38 inhibitor could decrease IL-8 expression levels in HT-29 cells. This result suggests that the two cytokines may accelerate inflammatory response through the p38 pathway, which is similar to Guo’s report[26]. We also found that when IL-17 and TNF-α were used together to stimulate the Act1 silenced cells, p38 phosphorylation level significantly decreased, compared with the control group. The result further confirmed that the Act1-dependent p38 pathway is a very important factor for IL-17-mediated proinflammatory responses. Act1 is an important receptor protein in the IL-17 mediated signal pathway and its expression level affects the activation of the MARK pathway by IL-17[12,27]. Kang et al[28] discovered that inflammatory response could be reduced by lowering the expression level of Act1. It is well known that IL-17 can’t cause inflammatory response alone. It is commonly believed that several factors working together triggered the inflammatory response[29,30].
In conclusion, we discover that IL-17 can synergistically promote gene expression of TNF-α-mediated neutrophil chemokines and TH-17 cell chemokines. IL-17 obviously enhances the TNF-α-induced p38 phosphorylation, and the p38 pathway plays an essential role in IL-17-induced inflammatory response. In our study, we only examine the Act1-dependent p38 pathway, but it is still unclear whether other pathways (ERK, PI3K-AKT) or cytokines also participate in the function of IL-17; thus, further studies need to be performed.
COMMENTS
Background
Ulcerative colitis and Crohn’s disease are known as inflammatory bowel disease (IBD). It refers to chronic inflammatory disorders of the gastrointestinal tract, which can easily recur. IBD occurs more often in males than females, and nowadays, the morbidity rate is showing an upward trend. IBD has become one of the most common digestive system diseases. It is widely accepted that the interaction of genetic and environment factors leads to the disease, but the pathogenesis is still unclear.
Research frontiers
Previous studies show that interlukin-17 (IL-17) expression levels significantly increased in the peripheral blood of patients with IBD, which implies that IL-17 and tumor necrosis factor (TNF) can accelerate the inflammatory response through inducing various kinds of inflammatory cytokines in diseases such as IBD; however, the molecular mechanisms of the proinflammatory effects are still unknown. IL-17 can induce neutrophil infiltration and related inflammatory cytokine expression through the p38 pathway in myoblasts and fibroblasts, yet epithelial cell mechanisms were still rarely reported.
Innovations and breakthroughs
The authors discovered that IL-17 can synergistically promote gene expression of TNF-α-mediated neutrophil chemokines and TH-17 cell chemokine. IL-17 obviously enhanced TNF-α-induced p38 phosphorylation and the p38 pathway plays an essential role in IL-17-induced inflammatory response.
Applications
This study is helpful in deeply understanding the pathogenesis of IBD, and the molecular mechanisms of the proinflammatory effects of IL-17 and TNF-α on intestinal epithelial cell line HT-29. Even though the IL-17 inhibitor proved to be ineffective in IBD treatments, blocking the other site of the pathway may prove to be hopeful in future studies.
Peer review
This is a very interesting manuscript about the proinflammatory effects and molecular mechanisms of IL-17 in the intestinal epithelial cell line HT-29.
Footnotes
Supported by Minhang District Natural Science Foundation (to Wang YL) and the Science and Technology Commission in Shanghai, No. 10411968500; and National Natural Science Foundation of China, No. 81001324
P- Reviewer: Chandrakesan P S- Editor: Yu J L- Editor: Wang TQ E- Editor: Liu XM
References
- 1.Boland BS, Sandborn WJ, Chang JT. Update on Janus kinase antagonists in inflammatory bowel disease. Gastroenterol Clin North Am. 2014;43:603–617. doi: 10.1016/j.gtc.2014.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thia KT, Loftus EV, Sandborn WJ, Yang SK. An update on the epidemiology of inflammatory bowel disease in Asia. Am J Gastroenterol. 2008;103:3167–3182. doi: 10.1111/j.1572-0241.2008.02158.x. [DOI] [PubMed] [Google Scholar]
- 3.Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474:307–317. doi: 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knights D, Lassen KG, Xavier RJ. Advances in inflammatory bowel disease pathogenesis: linking host genetics and the microbiome. Gut. 2013;62:1505–1510. doi: 10.1136/gutjnl-2012-303954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, Bamba T, Fujiyama Y. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65–70. doi: 10.1136/gut.52.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hundorfean G, Neurath MF, Mudter J. Functional relevance of T helper 17 (Th17) cells and the IL-17 cytokine family in inflammatory bowel disease. Inflamm Bowel Dis. 2012;18:180–186. doi: 10.1002/ibd.21677. [DOI] [PubMed] [Google Scholar]
- 7.Cho JS, Pietras EM, Garcia NC, Ramos RI, Farzam DM, Monroe HR, Magorien JE, Blauvelt A, Kolls JK, Cheung AL, et al. IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. J Clin Invest. 2010;120:1762–1773. doi: 10.1172/JCI40891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Agache I, Ciobanu C, Agache C, Anghel M. Increased serum IL-17 is an independent risk factor for severe asthma. Respir Med. 2010;104:1131–1137. doi: 10.1016/j.rmed.2010.02.018. [DOI] [PubMed] [Google Scholar]
- 9.Wu Y, Zhu L, Liu L, Zhang J, Peng B. Interleukin-17A stimulates migration of periodontal ligament fibroblasts via p38 MAPK/NF-κB -dependent MMP-1 expression. J Cell Physiol. 2014;229:292–299. doi: 10.1002/jcp.24444. [DOI] [PubMed] [Google Scholar]
- 10.Hering NA, Andres S, Fromm A, van Tol EA, Amasheh M, Mankertz J, Fromm M, Schulzke JD. Transforming growth factor-β, a whey protein component, strengthens the intestinal barrier by upregulating claudin-4 in HT-29/B6 cells. J Nutr. 2011;141:783–789. doi: 10.3945/jn.110.137588. [DOI] [PubMed] [Google Scholar]
- 11.Sergent JA, Paget V, Chevillard S. Toxicity and genotoxicity of nano-SiO2 on human epithelial intestinal HT-29 cell line. Ann Occup Hyg. 2012;56:622–630. doi: 10.1093/annhyg/mes005. [DOI] [PubMed] [Google Scholar]
- 12.Roussel L, Houle F, Chan C, Yao Y, Bérubé J, Olivenstein R, Martin JG, Huot J, Hamid Q, Ferri L, et al. IL-17 promotes p38 MAPK-dependent endothelial activation enhancing neutrophil recruitment to sites of inflammation. J Immunol. 2010;184:4531–4537. doi: 10.4049/jimmunol.0903162. [DOI] [PubMed] [Google Scholar]
- 13.Fransen K, van Sommeren S, Westra HJ, Veenstra M, Lamberts LE, Modderman R, Dijkstra G, Fu J, Wijmenga C, Franke L, et al. Correlation of genetic risk and messenger RNA expression in a Th17/IL23 pathway analysis in inflammatory bowel disease. Inflamm Bowel Dis. 2014;20:777–782. doi: 10.1097/MIB.0000000000000013. [DOI] [PubMed] [Google Scholar]
- 14.Lin AM, Rubin CJ, Khandpur R, Wang JY, Riblett M, Yalavarthi S, Villanueva EC, Shah P, Kaplan MJ, Bruce AT. Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J Immunol. 2011;187:490–500. doi: 10.4049/jimmunol.1100123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weidlich S, Bulau AM, Schwerd T, Althans J, Kappler R, Koletzko S, Mayr D, Bufler P. Intestinal expression of the anti-inflammatory interleukin-1 homologue IL-37 in pediatric inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 2014;59:e18–e26. doi: 10.1097/MPG.0000000000000387. [DOI] [PubMed] [Google Scholar]
- 16.Rich P, Sigurgeirsson B, Thaci D, Ortonne JP, Paul C, Schopf RE, Morita A, Roseau K, Harfst E, Guettner A, et al. Secukinumab induction and maintenance therapy in moderate-to-severe plaque psoriasis: a randomized, double-blind, placebo-controlled, phase II regimen-finding study. Br J Dermatol. 2013;168:402–411. doi: 10.1111/bjd.12112. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Z, Hinrichs DJ, Lu H, Chen H, Zhong W, Kolls JK. After interleukin-12p40, are interleukin-23 and interleukin-17 the next therapeutic targets for inflammatory bowel disease? Int Immunopharmacol. 2007;7:409–416. doi: 10.1016/j.intimp.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 18.Towne E, Maxwell J, Zhang Y. O037 Differential roles for IL-23 and IL-17 in inflammatory bowel disease. Cytokine. 2012;59:515. [Google Scholar]
- 19.Strober W, Fuss IJ. Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology. 2011;140:1756–1767. doi: 10.1053/j.gastro.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Griffin GK, Newton G, Tarrio ML, Bu DX, Maganto-Garcia E, Azcutia V, Alcaide P, Grabie N, Luscinskas FW, Croce KJ, et al. IL-17 and TNF-α sustain neutrophil recruitment during inflammation through synergistic effects on endothelial activation. J Immunol. 2012;188:6287–6299. doi: 10.4049/jimmunol.1200385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiricozzi A, Guttman-Yassky E, Suárez-Fariñas M, Nograles KE, Tian S, Cardinale I, Chimenti S, Krueger JG. Integrative responses to IL-17 and TNF-α in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J Invest Dermatol. 2011;131:677–687. doi: 10.1038/jid.2010.340. [DOI] [PubMed] [Google Scholar]
- 22.Tanabe A, Konno J, Tanikawa K, Sahara H. Transcriptional machinery of TNF-α-inducible YTH domain containing 2 (YTHDC2) gene. Gene. 2014;535:24–32. doi: 10.1016/j.gene.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 23.Lee HH, Yoon NA, Vo MT, Kim CW, Woo JM, Cha HJ, Cho YW, Lee BJ, Cho WJ, Park JW. Tristetraprolin down-regulates IL-17 through mRNA destabilization. FEBS Lett. 2012;586:41–46. doi: 10.1016/j.febslet.2011.11.021. [DOI] [PubMed] [Google Scholar]
- 24.Li Q, Laumonnier Y, Syrovets T, Simmet T. Recruitment of CCR6-expressing Th17 cells by CCL20 secreted from plasmin-stimulated macrophages. Acta Biochim Biophys Sin (Shanghai) 2013;45:593–600. doi: 10.1093/abbs/gmt049. [DOI] [PubMed] [Google Scholar]
- 25.Meares GP, Ma X, Qin H, Benveniste EN. Regulation of CCL20 expression in astrocytes by IL-6 and IL-17. Glia. 2012;60:771–781. doi: 10.1002/glia.22307. [DOI] [PubMed] [Google Scholar]
- 26.Qu N, Xu M, Mizoguchi I, Furusawa J, Kaneko K, Watanabe K, Mizuguchi J, Itoh M, Kawakami Y, Yoshimoto T. Pivotal roles of T-helper 17-related cytokines, IL-17, IL-22, and IL-23, in inflammatory diseases. Clin Dev Immunol. 2013;2013:968549. doi: 10.1155/2013/968549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang SH, Park H, Dong C. Act1 adaptor protein is an immediate and essential signaling component of interleukin-17 receptor. J Biol Chem. 2006;281:35603–35607. doi: 10.1074/jbc.C600256200. [DOI] [PubMed] [Google Scholar]
- 28.Kang Z, Wang C, Zepp J, Wu L, Sun K, Zhao J, Chandrasekharan U, DiCorleto PE, Trapp BD, Ransohoff RM, et al. Act1 mediates IL-17-induced EAE pathogenesis selectively in NG2+ glial cells. Nat Neurosci. 2013;16:1401–1408. doi: 10.1038/nn.3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res. 2012;110:159–173. doi: 10.1161/CIRCRESAHA.111.243162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fabbri M, Paone A, Calore F, Galli R, Gaudio E, Santhanam R, Lovat F, Fadda P, Mao C, Nuovo GJ, et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc Natl Acad Sci USA. 2012;109:E2110–E2116. doi: 10.1073/pnas.1209414109. [DOI] [PMC free article] [PubMed] [Google Scholar]