

Abstract
The lymph nodes are an essential part of the body’s immune system and as such are affected in many infectious, autoimmune, metabolic and malignant diseases. The cervical lymph nodes are particularly important because they are the first drainage stations for key points of contact with the outside world (mouth/throat/nose/eyes/ears/respiratory system) – a critical aspect especially among children – and can represent an early clinical sign in their exposed position on a child’s slim neck.
Involvement of the lymph nodes in multiple conditions is accompanied by a correspondingly large number of available diagnostic procedures. In the interests of time, patient wellbeing and cost, a careful choice of these must be made to permit appropriate treatment. The basis of diagnostic decisions is a detailed anamnesis and clinical examination. Sonography also plays an important role in differential diagnosis of lymph node swelling in children and is useful in answering one of the critical diagnostic questions: is there a suspicion of malignancy? If so, full dissection of the most conspicuous lymph node may be necessary to obtain histological confirmation.
Diagnosis and treatment of childhood cervical lymph node disorders present the attending pediatric and ENT physicians with some particular challenges. The spectrum of differential diagnoses and the varying degrees of clinical relevance – from banal infections to malignant diseases – demand a clear and considered approach to the child’s individual clinical presentation. Such an approach is described in the following paper.
1 Introduction
Neck masses are a symptom ENT physicians in hospitals and surgeries are frequently confronted with among children. If the mass originates from the lymph nodes, the first step should be to establish whether the lymph node itself is enlarged: this is the case above a diameter of >1 cm (in the angle of the mandible >1.5 cm) and is defined as lymphadenopathy. A distinction is made between an acute (<2 weeks), subacute (2–6 weeks) and chronic (>6 weeks) course of the lymphadenopathy [1]. Enlarged lymph nodes must also be differentiated from other possible causes of neck masses, such as midline thyroglossal cysts and branchial cysts, lipoma, vascular malformations (e.g. hemangioma), paraganglioma/neurinoma, lesions of the salivary or thyroid glands, lymphangioma, teratoma, (epi-)dermoid cysts, and ectopic thyroid tissue [2] (see Figure 1 (Fig. 1), Figure 2 (Fig. 2), Figure 3 (Fig. 3), Figure 4 (Fig. 4)).
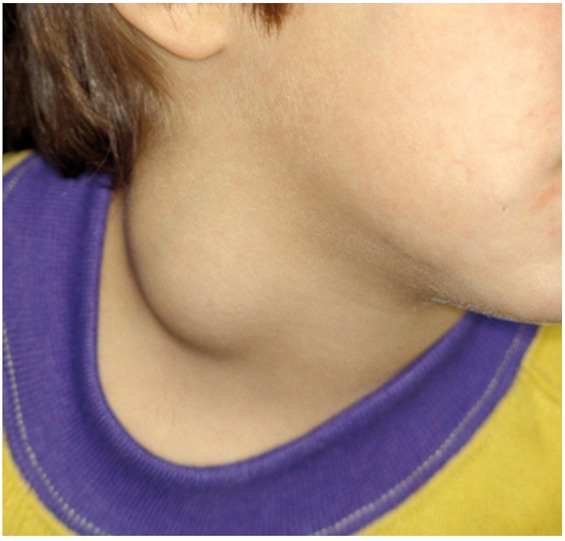
Figure 1. Ranula.
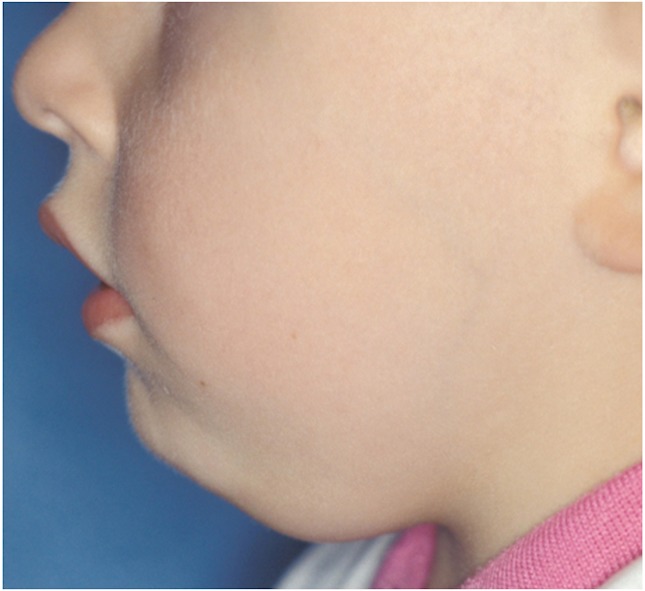
Figure 2. Mononucleosis.
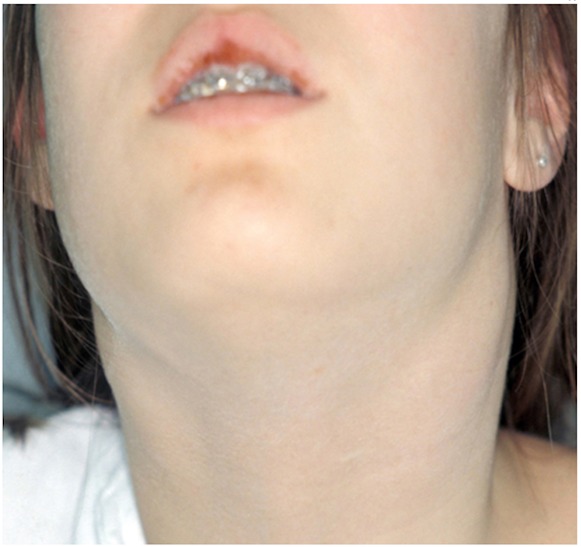
Figure 3. Submandibular abscess.
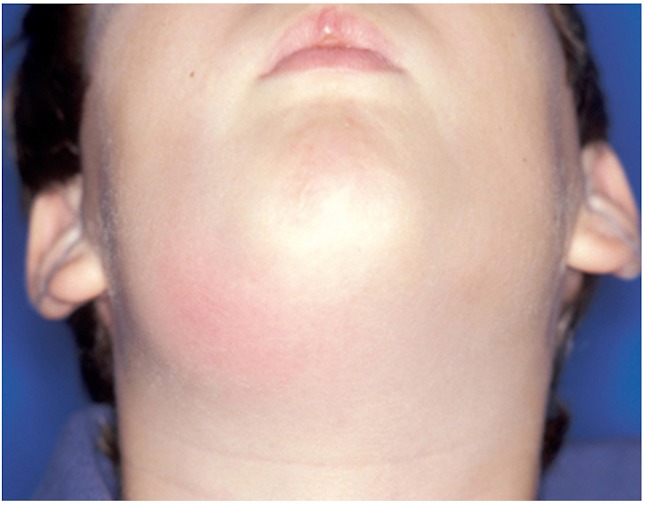
Figure 4. Branchial cyst.
As Table 1 (Tab. 1) shows, there are many possible causes of lymph node enlargement. For this reason, anamnesis and thorough clinical examination are crucial steps towards securing a diagnosis [3].
Table 1. Possible causes of cervical lymphadenopathy.

2 Anamnesis
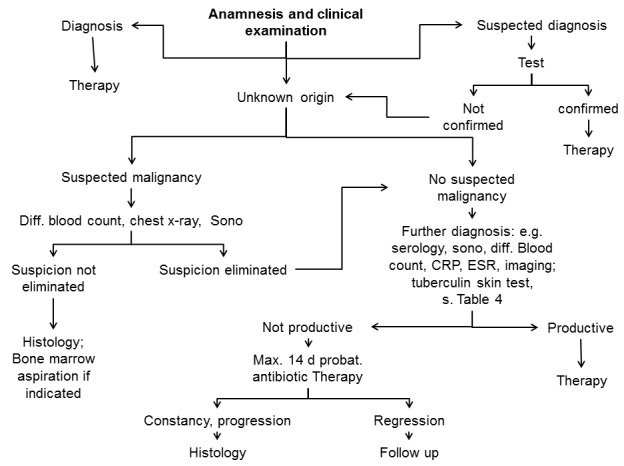
The more varied the possible differential diagnoses are, the greater the importance of the primary diagnostic instruments, namely anamnesis and clinical examination, available to the physician. The course of the illness is one of the first aspects to be considered in diagnosis: the majority of cases of acute lymphadenitis, which subside after 2 weeks, are of infectious origin; this contrasts with chronic lymph node enlargement, which is more likely to originate from neoplastic or metabolic disorders or opportunistic infections. Subacute lymph node enlargement has various possible causes, and additional criteria therefore need to be drawn on before determining the subsequent diagnostic and therapeutic steps (Figure 5 (Fig. 5)).
Figure 5. Algorithm of lymph node diagnosis.
The anamnesis must include any reference to a focus, including in the past, such as a sore throat, earache or toothache, insect bites or injuries. Local tenderness suggests an inflammatory component, while fever can occur equally in infections and as part of the B symptoms. Other B symptoms include night sweats (drenching) and weight loss (>10% of body weight in a period of 6 months). Positive B symptoms typically occur in malignant lymphomas (classification as A/B symptoms according to Ann Arbor), but may also accompany infectious diseases (tuberculosis, HIV, parasitosis). The same applies to symptoms such as bone/joint pain, weakness and lowered resilience or anorexia.
Details of foreign travel and possible contact with animals (in particular cats, rabbits, rodents, tick bites) and equally of all vaccinations must also be obtained in the anamnesis. Other environmental influences and noxa (including drugs) may more rarely be the cause of lymphadenopathy, but they should nevertheless always be considered. A family anamnesis not only includes acute infections but also chronic and systemic diseases such as sarcoidosis or tuberculosis.
3 Clinical examination
Clinical examination of the young patients initially comprises inspection, palpation, ENT examination and more extensive examinations as indicated, including palpation of other lymph node regions (axillary, inguinal etc.) or of the liver and spleen.
Inspection
Inspection must never focus solely on the region of the lymph node itself but also extend to the drainage regions and in particular local portals of entry (tonsils, scratches or skin lesions e.g. associated with an allergic exanthema).
Palpation
Palpation is similarly not restricted to the conspicuous lymph nodes and includes thorough examination of the head and neck region. It is advisable to work in a fixed order. The lymph nodes and diseases associated with them are listed in Table 2 (Tab. 2). One after the other, the submental and submandibular lymph nodes, the lymph nodes at the angle of the mandible, the pre- and post-auricular lymph nodes and the (sub-)occipital lymph nodes are palpated. These are followed by the group of vertical lymph nodes of the neck along the jugular vein and the posterior cervical lymph nodes along the anterior edge of the trapezius muscle and the supraclavicular lymph nodes in the supraclavicular fossa. Examination must also include other regions, in particular the axillary and inguinal lymph nodes, liver and spleen, in order to assess systemic involvement. Palpation of the deep cervical lymph nodes is facilitated by the patient inclining their head forwards slightly to relax the strong muscles that support the head [4].
Table 2. Overview of lymph node regions of the neck.

According to the AJCC Classification of cervical lymph nodes: A = Level Ia, B = Level Ib, F = Level II-IV; G = Level V; I = Level VI
The conspicuous lymph nodes are palpated for tenderness, consistency and mobility in relation to the surrounding tissue/adjacent lymph nodes. For example, swollen lymph nodes originating from an infection are generally painful, soft, fluctuant if abscessed, while nodes associated with lymphoma are typically firm, rubbery and painless, and lymph node metastases from carcinomas hard and seldom mobile [5], [6].
If not just a single lymph node is conspicuous and enlarged, but several – possibly also on both sides – are affected, other aspects must be taken into account for differential diagnosis. If infectious in origin, bilateral enlargement tends to suggest a viral (e.g. Epstein-Barr virus = EBV) or group A streptococcal infection (e.g. in tonsillitis), while rubella or Erythema infectiosum (fifth disease) are more rarely the cause. Table 3 (Tab. 3) provides an overview of distribution.
Table 3. Origin, localization and course of disease.

A = submental, B = submandibular, C = pre-auricular, D = post-auricular, E = (sub-)occipital, F = jugular, G = posterior cervical, H = supraclavicular (s. also Table 2)
The location of the lymph node may play an additional role in securing diagnosis: in infections of the scalp, the (sub-)occipital lymph nodes are often affected and therefore require careful inspection of the skin (beneath the hair). The (sub-)occipital lymph nodes are also enlarged in toxoplasmosis, inflammation of the outer ear and rubella. Enlarged pre-auricular lymph nodes may indicate infections of the eyes (e.g. lid infections, conjunctivitis), ears, teeth or the parotid gland. Submental/submandibular lymph nodes are often conspicuous in infectious processes of the oral cavity, the nose, the maxillary sinus or the face. Lymphomas may appear practically anywhere but nevertheless often chiefly affect the lymph nodes along the jugular vein and in the occipital or supraclavicular regions. In relation to the supraclavicular lymph nodes, differential diagnosis should consider sarcoidosis and the – albeit rare – existence of lesions in the thorax and gastrointestinal tract [6].
The age of the patient is a further criterion to be considered alongside location and palpation. Generally, the cause of lymph node swelling in young patients – from babies to infants and adolescents – is benign in over 80% of cases. This figure decreases considerably with age, to the extent that a malignant cause is found in over 60% of patients age 50 and above [7]. However, if there is a suspicion of malignancy in a young patient, the most frequent causes in children under 6 years of age are acute leukemia, neuroblastoma, rhabdomyosarcoma and non-Hodgkin lymphoma. Between the ages of 7 and 13, non-Hodgkin lymphoma and Hodgkin’s lymphoma are roughly equal, with rhabdomyosarcoma and thyroid cancer occurring more rarely. From the age of 13, Hodgkin’s disease is the leading malignant cause of neck masses during childhood and adolescence [8].
Clarifying whether a suspected malignancy exists is crucial to the further diagnostic process: a well thought-out and considerate approach to dealing with the concerned parents and the patient is essential in order to spare them any unnecessary (additional) worry while not trivializing a manifest suspicion. The doctor should assess the parents’ and patient’s need for information and be able to justify and communicate the next steps in the process. Conclusive evidence of malignancy is only possible by histological examination. In a study by Torsiglieri et. al, histological examination of 445 neck masses in children showed a malignant disease in 11% (n=48) of the cases [9]. The following criteria can be used to distinguish between malignant and benign:
Criteria more consistent with malignancy:
over 2/2.5 cm in size
firm/hard consistency
supraclavicular/axillary localization, posterior edge of the sternocleidomastoid muscle
absence of tenderness
low mobility
progressive course
B symptoms
Criteria less consistent with malignancy:
under 1.5 cm in size
soft consistency
tender
erythema
fluctuation
mobility
regressive course
Conclusion
Anamnesis and clinical examination raise the following cardinal questions, the answers to which are instrumental in determining subsequent procedure:
Is the lymph node swelling clearly pathological?
How long has the lymphadenopathy been apparent?
How old is the child?
Is the course progressive, constant, regressive or intermittent?
What is the localization?
Is the lymph node swelling on one or both sides, uni- or multifocal?
Apart from “tumor”, are there other signs of inflammation such as “rubor”, “calor”, “dolor”, “functio laesa”?
Are there any additional significant clinical findings?
Is there any suspicion of malignancy?
4 Further diagnosis
If the anamnesis and clinical examination prove inconclusive, or if confirmation of a suspected diagnosis is required, further diagnostic means are available, including serological tests, sonography as the main imaging technique, and for certain special indications also MRI and CT. As a means of obtaining histological confirmation, full dissection of a suspicious lymph node should take preference over fine-needle aspiration cytology (FNAC), compare 4.4.
Clinically it is neither reasonable nor cost-effective to routinely perform every one of the available serological and microbiological tests for every patient with lymph node swelling of uncertain origin. The same is true of the imaging and histological procedures. It is therefore a question of selecting a set of diagnostic measures that best meet the individual’s clinical presentation with the highest possible sensitivity and specificity. Table 4 (Tab. 4) gives an overview of clinical indications from anamnesis and examination, together with the possible cause and accompanying further diagnosis in each case.
Table 4. Clinical parameters and steps in differential diagnosis.

4.1 Laboratory
Laboratory diagnosis is widely used in lymphadenopathy of uncertain origin. Parameters such as the erythrocyte sedimentation rate (ESR), C-reactive protein (CRP) and lactatedehydrogenase (LDH) are elevated in many infectious but also neoplastic and immunologic disorders and therefore tend to be nonspecific. They are nevertheless often determined in order to add a further aspect to the diagnosis.
A blood count including microscopic differential hemogram can further help to confirm a suspected malignancy. Once the diagnosis has been established, the laboratory tests are used to establish which organs are involved in systemic diseases (uric acid, creatinine levels for renal involvement e.g. in SLE; GOT, GGT for liver involvement e.g. in mononucleosis; LDH and muscle enzymes e.g. in dermatomyositis) and as part of the follow-up and clinical monitoring (e.g. blood count for malignant tumors).
4.2 Microbiology
The success of microbiological diagnosis depends to a critical degree on the quality of the material submitted for testing. If this kind of diagnosis is not performed routinely on a regular basis and there is any doubt as to the sampling procedure, it is advisable to first contact the experts for further information. How the samples are transported (temperature, culture medium, aerobic/anaerobic milieu) likewise has a decisive influence on diagnosis.
Serology
Serology is primarily used to diagnose virus-associated diseases. Pathogens such as rubella, measles, CMV and EBV are diagnosed serologically (in combination with the corresponding clinical assessment). Here it is always necessary to obtain the patient’s vaccination anamnesis.
(Swab) samples
Samples must be placed in a transport medium. The time and site of sample removal must be recorded, together with details of any antibiotic treatment already initiated. The role of swabs in cervical lymphadenitis is primarily to diagnose abscessed lymph nodes. Staphylococcus aureus (S. aureus) and Streptococcus pyogenes (S. pyogenes) are found in the majority of cases (40–80%) [10], [11], [12], [13], [14]. The proportion of methicillin-resistant S. aureus (MRSA) bacteria in childhood ENT infections is 10–30% [15], [16]. In a study by Neff and colleagues, swabs were taken intraoperatively from 277 immunocompetent children and cultures set up for aerobic, anaerobic, acid-fast pathogens and fungi [17]. The results showed none of the fungal cultures, 1% of the anaerobic and 2% of the acid-fast cultures to be positive. In the study, the cost of the swab cultures was put at 402 $ US dollars per patient (aerobic 94 $, anaerobic 108 $, acid-fast 100 $, fungal 96 $). MRSA pathogens were found in 38 patients (14%), 100% of which were sensitive to clindamycin. Neff and colleagues conclude that only aerobic cultures should be employed and acid-fast pathogens only cultivated if a strong clinical suspicion exists. The authors recommend empiric (single) antibiotic therapy effective against S. aureus and S. pyogenes.
Blood culture and other means of pathogen detection
Blood cultures are considerably less reliable if antibiotic treatment has already begun, since antibiotic substances continue to work in the culture bottles and may hinder pathogen growth. Full diagnosis includes cultures in an aerobic and an anaerobic medium and should be repeated at intervals of several hours to raise their diagnostic value. Blood cultures are primarily indicated in septic patients, fever of unknown origin and serious infectious diseases such as meningitis.
Other means of detecting pathogens include indirect material sampling (e.g. bronchoalveolar lavage, BAL) or testing secretions (sputum, gastric juice etc.), where appropriate combined with a polymerase chain reaction (PCR).
Tuberculin skin test
There are various methods of tuberculin testing:
The percutaneous test is suitable for children under 10 years of age. It involves applying a salve containing tuberculin to the skin; the test is read after 72 hours [18].
In the Tine test, four needles treated with tuberculin are pressed into the skin for three seconds and moved briefly. This test is also read at the earliest 72 h after administration. Both tests are approximate screening methods – the percutaneous test in particular is considered unreliable – and are therefore not recommended [18].
The intracutaneous Mendel-Mantoux test is used to screen for suspected TBC by intracutaneously injecting tuberculin. The test is read after 72 hours; the induration must have a diameter of at least 6 mm to count as positive.
What is important is that all the tuberculin tests make no distinction between an acute or past infection and only confirm exposure to the disease. The test is also positive after BCG vaccination. Infection with non-tuberculous mycobacteria (NTM) may produce a false-positive test.
Another diagnostic test for tuberculosis is the quantiferon test. It is a serological investigation and – depending on the interferon-γ levels – evaluated as positive or negative. The advantages of the quantiferon test are its higher sensitivity (75–80%) and its independence from the vaccination status [19].
4.3 Imaging
Imaging is used in cervical lymph node swelling in children to confirm or eliminate a specific diagnosis (e.g. abscess) or to assess the location and number of affected structures and can significantly contribute to diagnosis where clinical assessment and anamnesis are inconclusive. Particular attention must be paid here to the age of the patients, some of whom are very young, especially in terms of their exposure to radiation, e.g. during CT, the practicability and difficulty of the procedure, and stress.
Sonography
Sonography is a very valuable way of assessing lymph node swelling in children. In inflamed processes, sonography is the instrument of choice for determining localization, number, size and characteristics of the affected structures [17], [20], [21]. In the ultrasound, inflamed lymph nodes appear enlarged and are hypoechoic compared to the adjoining connective and muscle tissue [22]. Both abscess-forming lymph nodes and tuberculous lymphadenitis are characterized by central cystic changes with loss of the echogenic hilum [23].
Size, form, structure, intranodal necrosis, hilum structure, calcification and edema of the surrounding connective tissue are also assessed in non-infectious processes [24]. Doppler sonography additionally permits imaging of the vascularization of the hilum structure and the node. Ultrasound is not only valuable in helping to identify inflammatory, abscess-forming conditions but also and above all in assessing malignant processes. Here it is important to differentiate between suspected metastases from a solid tumor and – particularly in children – manifestations of other entities, such as lymphomas.
Ultrasound criteria that suggest a malignant process are changes in size and form [25]. The specificity and sensitivity here essentially depend on the defined limits [26]. In general, a L/D ratio (length/depth) of <2 is seen as an indication of a malignant process [24], although this is an unreliable criterion especially in the case of small lymph nodes [27].
The situation is similar with the hilum echogenity: 84–92% of benign lymph nodes have an echogenic hilum, while the hilum structures in 76–96% of malignant lymph nodes cannot be detected on account of intranodal processes [28]. It is essential to note in sonographic assessment that this is rather to be seen as a late sign and that the negative predictive value of early involvement of the lymph nodes is low [29].
The cortex configuration of the lymph node similarly allows to draw conclusions as to the dignity of the lymph node. It is considered in relation to the hilum structure and is suspicious if the cortex diameter is greater than half the diameter of the hilum [29]. A distinction must be made between concentric (bulging into the hilum) thickening of the cortex, which is considered nonspecific and may also occur in reactive changes in the lymph nodes [29], and eccentric (bulging outwards) cortical thickening, which is more likely an early sign of malignancy with a high positive predictive value [30].
The internal structure of the lymph nodes may also help to identify origin. For example, microcalcification and necrosis can be caused by infections (e.g. tuberculosis or other inflammatory granulomatous conditions) and neoplastic diseases (e.g. metastases from thyroid carcinomas). At the same time, the existence of necrosis is less consistent with lymphoma [31].
A valuable contribution to differential diagnosis has been made with ongoing improvements in the quality of resolution by intranodal angiography using Doppler sonography: neoplastic lymph nodes often exhibit pronounced hypervascularization, i.e. formation of new vessels, chiefly in the periphery of the lymph node but in some cases also beyond the lymph node capsule, which is characterized by irregular distribution and flow properties [32]. In lymphatic neoplasms, the periphery is rarely affected on its own due to their intranodal origin [33]. In the literature, the measurement of vascular resistance and contrast agent-supported sonographic imaging are still subjects of controversial debate in relation to sensitivity and specificity in differential diagnosis [27].
Computer tomography and magnetic resonance imaging
On a CT scan, inflamed lymph nodes appear enlarged, absorb contrast agent and are oval in shape, with purulent abscesses exhibiting a ring with no contrast agent in the centre and thickening on the periphery. By contrast with tuberculous lymphadenitis, fatty deposits can often be observed in the nodes [34], [35]. This ring-shaped accumulation of contrast agent is often referred to in eliminating a suspected abscess, but since it is apparent in over 60% of lymphadenitis cases, it cannot be considered highly sensitive [17]. Alongside exposure to radiation, this is a further reason why sonography should always be the preferred method of primary examination [20], [21].
A number of authors recommend computer tomography as the diagnostic instrument of choice in deep neck infections in order to achieve the best possible picture of the spread and origin of infection, monitor its course and help to assess the endangered mediastinal compartments [36]. A CT scan provides morphological evidence to verify a suspected malignancy, primarily by size, shape and, in advanced stages, infiltration of the surrounding structures.
Magnetic resonance imaging (MRI) is a further procedure available as part of more extensive diagnosis and can above all help to identify neck masses not originating in the lymph nodes. In this case – where necessary and possible – sedation is preferable to endotracheal anesthesia in children.
4.4 Histological examination
There are two main examination techniques to obtain histological confirmation of a diagnosis: Lymph node dissection and fine-needle aspiration cytology (FNAC).
Full dissection of the affected lymph node is considered the standard procedure of choice when it comes to obtaining histological confirmation of a diagnosis, as it permits assessment of the histopathological structures and their distribution (e.g. vascular growth beyond the capsule). It also supplies sufficient tissue for pathological and microbiological examination. This makes it possible to perform more extensive immunohistochemical staining, which is essential to differential diagnosis of lymphomas in particular, and more detailed analysis of the suspected pathogen DNA by PCR.
Varying levels of sensitivity and specificity are also discussed in the literature in relation to fine-needle biopsy. In combination with sonography, this method produces accurate results with a high sensitivity (89–98%) and specificity (95–100%) in diagnosing malignant conditions [37], [38], [39]. Since the negative predictive value is approx. 80%, diagnosis is incorrectly eliminated in a fifth of cases [40]! This is particularly significant if only a single lymph node is affected, and FNAC causes a hemorrhagic lesion that hinders histological examination if dissection of the lymph node becomes necessary [6]. In this case, the experience and expertise of the examining physician and the cytopathologist are crucial [41]. Performing FNAC can be difficult anyway, particularly so in small children, and this is another consideration when deciding on procedure.
A fine-needle biopsy is an option if (i) several lymph nodes are suspicious, (ii) surgical dissection is associated with a high risk that can be eliminated by FNAC, (iii) sufficient expertise is available, and (iiii) the patient is in compliance (age of the patient!).
Conclusion
Further diagnosis essentially builds on the anamnesis and clinical examination. Special laboratory tests should be conducted in particular if a specific clinical suspicion exists. Sonography is the first and standard choice of imaging procedure and provides answers to a range of questions in differential diagnosis. If a condition persists or malignancy is suspected, histological examination is warranted without too much delay, in which case the gold standard is full dissection of the affected lymph node.
5 Causes
The following chapter looks at the causes of cervical lymphadenopathy, divided according to infectious, immunologic, metabolic and neoplastic origin.
5.1 Infectious
Indications of lymphadenopathy of infectious origin may be specific (pathogen entry site, concurrent tonsillitis/pharyngitis) and unspecific (signs of inflammation, fever, elevated CRP, ESR). Viral and bacterial infections are the most common causes in childhood, while parasites or fungal infections tend to play a less frequent role. Generalized/bilateral lymphadenitis is more consistent with viral infection, unilateral with bacterial.
5.1.1 Viral pathogens
Pathogenesis and etiology: Viral infection is the most frequent cause of lymphadenitis in childhood [42]. Precisely the recurring viral pathogens that are associated with infections of the upper respiratory tract (RS viruses, influenza and parainfluenza viruses, rhino-, adeno- and acute corona viruses) [43], occur repeatedly in childhood and often cause acute cervical lymphadenitis. Other common viral agents, which can also cause chronic lymphadenitis, are the Epstein Barr virus, particularly in adolescence, and cytomegalovirus (CMV) [1]. Less frequently lymphadenopathy is also caused by mumps, measles, rubella, varicella, Erythema infectiosum or fifth disease, herpes simplex and Coxsackie viruses [44]. HIV-associated lymphadenopathy is far less common among children than adults, but depending on the risk profile it should also be considered as a potential cause in children and especially adolescents.
Clinical presentation: The main symptoms are defined by the infection of the upper respiratory tract: fever, pharyngitis accompanied by odynophagia, occasionally otitis/sialadenitis. These may be attended by cutaneous symptoms typical of the relevant viral infection.
Lymph nodes: The involved lymph nodes are usually affected multiply on both sides; without erythema/fluctuation if there is no bacterial superinfection.
Diagnostics: If a specific suspicion exists, the diagnosis may additionally include virus serology for the relevant pathogen. If viruses that also affect the spleen and liver (e.g. EBV, CMV) are suspected, an ultrasound should always be performed on the abdomen to eliminate the possibility of hepatosplenomegaly.
Therapy: Immunocompetent patients receive supportive therapy, while prophylactic antibiotic or antiviral therapy is only indicated for immunocompromised patients or a severe clinical course [45]. If the symptoms persist, reevaluation is necessary.
5.1.2 Bacterial pathogens
Pathogenesis and etiology: Bacterial infections also affecting the lymph nodes can likewise be divided into acute and chronic infections. The majority (40–80%) of acute bacterial infections are caused by staphylococci and beta hemolytic serogroup A streptococci [10], [11], [12], [13], [14]. Francisella tularensis (tularemia), pasteurella, Haemophilus influenzae type B and anaerobic organisms such as propionibacteria, fusobacteria and peptostreptococci are more rarely the cause of cervical lymphadenopathy [1]. Chronic bacterial infections of the lymph nodes are chiefly caused by non-tuberculous mycobacteria (NTM) [46], followed by other agents such as bartonella (cat-scratch disease), tuberculosis bacteria and brucella (brucellosis), cf. Table 5 (Tab. 5).
Table 5. Overview of bacterial pathogens in cervical lymphadenopathy.

Clinical symptoms: The clinical symptoms are often comparable to an acute viral infection of the upper respiratory tract, accompanied by fever, odynophagia, otitis media or cough. Anaerobes may be apparent in connection with dental/gingival infections.
Lymph nodes: The lymph nodes in acute bacterial infections are typically painful, enlarged on one side and exhibit progressive growth. The predilection sites are the submental and submandibular, jugular and occipital lymph nodes [44].
Diagnostics: Diagnosis of bacterial lymphadenitis is based on the clinical symptoms and detection of pathogens in swabs/secretion by microscope, culture or PCR, serological procedures and evidence of antigens.
Therapy: The therapy is adapted to the relevant pathogen. The majority of acute bacterial infections are sensitive to a combination of benzylpenicillins + beta lactamase inhibitors. If empiric therapy does not lead to regression, other pathogens must be considered. In very young or immunosuppressed patients in particular, therapy may initially need to take place intravenously under inpatient supervision to avoid any serious complications.
5.1.3 Parasites/protozoa
Pathogenesis and etiology: If a parasitic or protozoa-associated lymphadenopathy is suspected, an anamnesis of foreign travel and environmental factors is of primary importance. Pathogens enter the body in food (unpasteurized milk, meat, contaminated water) or through wounds and close contact with animals. The neck and throat region, as the area associated with food intake, and the draining lymph node regions are predilection sites for manifestation of the diseases.
Clinical presentation: Parasitic infections are characterized by a deterioration in the patient’s general condition, organ involvement (toxocara (roundworm) → lungs, eyes; toxoplasmosis → eyes, lymphoreticular system; trypanosoma → heart, nerve cells; leishmaniasis → kidneys, intestine, skin; microfilaria → lymphoreticular system, heart, nerve cells) [47].
Lymph nodes: As part of the lymphoreticular system, the lymph nodes may be affected in parasitic infections.
Diagnostics: Serological procedure to determine antibodies or detect pathogens in a film of blood/secretion.
Therapy: Therapy is adapted to the relevant pathogen. Prophylaxis is important, particularly in preparing food and contact with animals. The recommended drug treatment for toxoplasmosis is a combination of pyrimethamine, sulfadiazine and folinic acid as well as corticosteroids [48]. Toxocara infections may be treated with Anthelmintics, if indicated by the symptoms [49]. There are several therapy options in the treatment of leishmaniasis, including N-methylglucamine antimonate, miltefosine, allopurinol, paromomycin, although a completely curative therapy is often not possible here [50].
5.1.4 Fungi
Pathogenesis and etiology: Histoplasmosis, coccidioidomycosis, blastomycosis and tinea are fungal infections caused by environmental contamination through the skin or the respiratory tract.
Clinical presentation: Characteristic symptoms are skin manifestations and lung involvement.
Lymph nodes: The lymph nodes are frequently affected as a result of organ involvement (lungs → supraclavicular; scalp → suboccipital [51])
Diagnostics: Serological tests, microscopic examination of swab and cultures are available means of diagnosis.
Therapy: The majority of infections require no treatment; an antifungal therapy may be necessary in the event of systemic complications and in immunosuppressed patients.
5.2 Immunologic
5.2.1 Granulomatous diseases
Sarcoidosis
Pathogenesis and etiology: Sarcoidosis (also known as Boeck’s disease, Schaumann-Besnier syndrome) is a systemic disease of the connective tissue. An increase in inflammatory activity with increased cellular immune response leads to the formation of non-caseating granulomas. The age peak lies between 20 and 40 [52], although the disease can occur in childhood. The cause of the increased immune response is not yet clear, but in epidemiological terms a genetic cause (BTLN2 mutation) appears at least to play a role [53]. Incidence of the disease in Germany is 10–12:100,000 inhabitants [54].
Clinical presentation: A distinction is made between the chronic and acute form (Löfgren syndrome) of the disease. Löfgren syndrome is characterized by the typical triad of symptoms: bilateral lymphadenopathy, polyarthritis and erythema nodosum [55]. General symptoms such as fever, fatigue and arthralgia are also typical. After the lungs (90%) and the lymph nodes (90%), the liver (60–90%) and spleen (50–60%) are the most frequently affected organs. Others include the eyes (25%), heart (5%), the skeleton (25–50%), nerve tissue and bone marrow (15–40%) [52]. The clinical course of the disease depends on which organs are affected and the pulmonary stage. Scadding [56] distinguishes between 5 different clinical stages of pulmonary sarcoidosis:
Stage 0: Lungs normal, other organ possibly affected (reversible)
Stage I: Symmetrical lymph node enlargement, no involvement of lung tissue (reversible)
Stage II: Bilateral lymph node enlargement with diffuse granulomas in lung tissue (reversible)
Stage III: Lungs affected without lymph node enlargement (reversible)
Stage IV: Pulmonary fibrosis with loss of function (irreversible)
In early stages and in the acute course of the disease, patients have a very good clinical prognosis with up to 90% spontaneous remission. However, around 5% of patients experience chronification with a potentially fatal outcome, chiefly as a result of pulmonary or cardiac involvement [57].
Lymph nodes: In children, peripheral lymphadenopathy is the main symptom of sarcoidosis. Typically, the lymph nodes on both sides are fixed and firm on palpation. The supraclavicular lymph nodes are affected in over 80% of cases [1].
Diagnostics: The clinically asymptomatic course of the disease often means that sarcoidosis is discovered as an incidental finding of a chest x-ray. Depending on the clinical symptoms, further imaging may be necessary to ascertain or eliminate organ involvement. The available procedures are sonography, CT, PET-CT, MRI and the now rarely indicated scintigraphy [58].
Most laboratory parameters are unspecific. Elevated levels of angiotensin converting enzyme (ACE), erythrocyte sedimentation rate (ESR), immunoglobulins, calcium and calcitriol and a left shift in the blood count may be detected [59].
Diagnosis of lung function is worthwhile, and if the diagnosis is unclear, bronchoalveolar lavage with cytological examination to detect lymphocytic alveolitis with an increased CD4/CD8 ratio (>5 in acute sarcoidosis; normal 2).
Histological examination following lymph node dissection or transbronchial ultrasound-controlled biopsy is relatively unspecific, typically revealing non-caseating, epitheloid cell granulomas with Langhans giant cells and periphery (monocytes, lymphocytes and fibroblasts) [60]. Diagnosis is therefore based on a combination of clinical signs and additive examinations.
Therapy: Symptomatic therapy is provided if organs are affected. Corticosteroids are the first choice of immunosuppressive medication for pharmacological treatment. Should they prove ineffective, azathioprine and chloroquine may also be used [61]. Therapy should not begin too early, since there is discussion of a higher recurrence rate following immunosuppressive therapy [62]. Tetracycline can be used for cutaneous symptoms [63]. Anti-inflammatory, analgesic and in some cases antipyretic drugs (aspirin, ibuprofen, diclofenac) are the first choice of medication in treating Löfgren syndrome [64].
Common variable immunodeficiency
Pathogenesis and etiology: Common variable immunodeficiency (CVID) is a congenital immune defect with hypogammaglobulinemia and a resulting defective antibody response leading to recurring infections [65], [66], [67]. It is thought to comprise a heterogeneous group of genetic defects with effects on dendritic cells and on B and T cell function [68], [69].
Clinical presentation: Patients present with recurring infections, particularly of the upper respiratory tract. Many also develop organ-granulomas, disorders of the gastrointestinal tract (diarrhea e.g. caused by Giardia lamblia infections, entoviruses) and skin manifestations (hair loss, vitiligo) [70]. Autoimmune phenomena such as idiopathic thrombocytopenic purpura (ITP) or autoimmune hemolytic anemia (AIHA) likewise occur with greater frequency [71].
Lymph nodes: Around one fifth of patients with CVID develop granulomas, 42% of them in lymph nodes [72]. Histological examination reveals non-caseating granulomas with multinucleated macrophages [73]. Differential diagnosis from sarcoidosis is considerably more difficult here, and particular attention must therefore be paid in the anamnesis to recurring infections, as these are typical of CVID [74].
Diagnostics: A serum protein electrophoresis is indicated if CVID is suspected. In the case of hypogammaglobulinemia, other possible causes should subsequently be eliminated (e.g. nephrotic syndrome, exsudative enteropathy).
Therapy: The pathogenesis of the disease is such that only symptomatic therapy options are available, e.g. substitution of immunoglobulins, antibiotics, immunosuppressive therapy and bone marrow transplantation [75].
Hyper-IgM syndrome
Pathogenesis and etiology: Hyper-IgM syndrome is a rare (incidence 1:1,000,000 [76]) immune system disorder in which a malfunction prevents IgM switching to IgG [77]. This results in elevated (or normal) IgM levels and reduced IgG levels [78]. To date, five different genetic defects have been identified [79].
Clinical presentation: Recurring infections of the upper respiratory tract, paranasal sinuses and the ears are frequently exhibited in affected patients. In 40% of cases, the initial symptom is an opportunistic lung infection with pneumocystis carinii [76].
Lymph nodes: Swelling of the lymph nodes occurs frequently in the course of the recurrent infections of the upper respiratory tract. Some case descriptions refer to necrotizing granulomas and affected lymph nodes [80].
Diagnostics: Serum protein electrophoresis and genetic analysis are used in diagnosis.
Therapy: Possible therapy involves treatment of the opportunistic infections, immunoglobulin substitution and bone marrow transplantation [81].
Chronic granulomatous disease
Pathogenesis and etiology: Chronic granulomatous disease (CDG) is a rare genetic immune disorder (incidence 1:200,000 [82]) with dysfunction of the neutrophil granulocytes (NADPH oxidase) [83]. Chronic granulomatous disease has so far been divided into five different forms depending on the location of the genetic defect. The majority of patients present clinically in the first two years of life [84].
Clinical presentation: Frequent bacterial infections and fungal infection of the internal organs, particularly of the lungs, osteomyelitis, pulmonary inflammation and gastrointestinal complaints frequently manifest during childhood due to granuloma formation.
Lymph nodes: Over 50% of cases result in purulent inflammation of the lymph nodes (predominantly associated with staphylococci [82]).
Diagnostics: The differential blood count is normal, diagnosis is obtained by granulocyte function testing and genetic analysis. Imaging (sonography, CT) is necessary to eliminate an abscess or assess the affected organs (lungs, abdomen) [85].
Therapy: Exposure prophylaxis is important, particularly to mold spores. Surgical intervention is necessary if abscess formation occurs, combined with a pathogen-adapted antibiotic therapy. Granulocyte transfusions are only indicated in severe infections [86].
5.2.2 Rheumatic diseases
Juvenile idiopathic arthritis
Pathogenesis and etiology: Juvenile idiopathic arthritis (JIA) is a chronic inflammatory disease of unknown origin that affects the joints and begins in childhood. There is discussion as to whether it is triggered by e.g. an acute infection that elicits a chronic inflammatory response in patients with a polygenic predisposition. There are several different forms of juvenile idiopathic arthritis [87]:
Juvenile chronic polyarthritis, adult type (positive rheumatoid factor)
Juvenile ankylosing spondylitis
Juvenile chronic arthritis, systemic onset (Still’s disease)
Juvenile chronic arthritis (negative rheumatoid factor), polyarticular onset
Juvenile chronic arthritis, oligoarticular onset
(Juvenile) psoriatic arthropathy
Other juvenile arthritis
Clinical presentation: The symptoms are common to all subforms of the disease: inflammation of one or more of the joints for longer than six weeks without identifiable cause, accompanied by rubor (rare), calor, dolor, tumor (swelling, effusion) and functio laesa (restricted mobility) [88].
Lymph nodes: Although joint problems are the main symptom, lymph node enlargement occurs in the majority of cases, particularly in the systemic forms of the disease [89], [90].
Diagnostics: Clinical findings, laboratory parameters and imaging are productive in diagnosis of JIA. In 2010, a set of common criteria of the rheumatological associations in the US (American College of Rheumatology; ACR) and Europe (European League Against Rheumatism; EULAR) was established in order to simplify early detection of rheumatic diseases [91]. Depending on the number of joints affected, the acute phase reaction (ESR or CRP) and other serological parameters (rheumatoid factors, anti-citrullinated protein/peptide antibodies; ACPA) and the duration of the arthritis, points are awarded (up to a maximum of 10). Rheumatoid arthritis is diagnosed at a total of six points and above.
Therapy: Treatment of rheumatoid arthritis involves dietary measures, physiotherapy, medication and interventions. A low-meat diet and regular exercise have a positive impact on the clinical course [92]. Four different groups of therapeutic agents are used:
Analgesics
Non-steroidal anti-inflammatory drugs (NSAIDs)
Glucocorticoids
Basic therapeutic agents, disease modifying anti-rheumatic drugs (DMARD)
Intervention options include (partial) arthrectomy, endoprosthesis, arthrodesis, and radiation synoviorthesis.
Systemic lupus erythematosus
Pathogenesis and etiology: Systemic lupus erythematosus (SLE) is a subform of lupus erythematosus, a collagen disease caused by autoantibodies and immune complexes (incidence 20–70:100,000) [93]. Many patients have a genetic predisposition, which may be triggered by factors such as EBV infection [94].
Clinical presentation: The clinical signs vary considerably depending on which organ is affected (kidneys, cardiac valves, bones, vessels, PNS and ZNS, lungs). General symptoms are fatigue, joint pain and sensitivity to light [95].
Lymph nodes: Lymphadenopathy presents in around 15% of cases of childhood SLE [96]. Histological examination shows zones of necrosis with haematoxylin inclusion bodies [97].
Diagnostics: The ACR criteria are drawn on in the diagnosis of SLE. If four or more of the following criteria are detected, an SLE diagnosis is 80–90% reliable:
1. Butterfly rash, 2. typical skin changes, 3. photosensitivity, 4. erosion of the oral mucosa, 5. joint pains and joint effusion, 6. serositis, 7. renal involvement, 8. CNS involvement, 9. hematalogical symptoms, 10. immunologic findings, 11. antinuclear antibodies (ANA) [98].
Therapy: Treatment includes protecting the patient against trigger factors (e.g. UV protection), drug therapy (NSAIDs, chloroquine, cortisone, azathioprine, cyclosporin A and mycophenolate mofetil, cyclophosphamide or methotrexate) and other forms of therapy (immunoadsorption, plasmapheresis, stem cell transplantation, immunoglobulins). How aggressive the therapy is must be adapted to the patient’s symptoms [99].
Juvenile dermatomyositis
Pathogenesis and etiology: Dermatomyositis is a rare form of vasculitis of unknown origin and belongs to the collagen diseases. The juvenile form of the disease typically manifests between the ages of 4 and 12 [100].
Clinical presentation: Children present with symmetric, proximal muscle weakness, pain and skin alterations (livid discoloration, dermatrophy/Gottron’s sign, teleangiectasia). Other organs may additionally be affected, particularly the heart, intestine, lungs, liver, spleen and CNS [100].
Lymph nodes: Lymphadenopathy can be part of the underlying disease but in rare cases also occur in dermatomyositis of paraneoplastic origin [101], [102].
Diagnostics: Diagnosis is based on clinical assessment, laboratory tests (muscle enzymes, LDH, transaminases increased), myography and – if necessary – biopsy. The likelihood of the diagnosis is determined according to Bohan and Peter [103], [104].
Therapy: Basic therapeutic options are physiotherapy and drug therapy with corticosteroids (e.g. prednisolone), cytostatics (e.g. methotrexate) and immunosuppressive agents (e.g. cyclosporin A) [105].
5.2.3 Lymphoproliferative and histiocytic disorders
Sinus histiocytosis
Pathogenesis and etiology: Sinus histiocytosis (Rosai-Dorfman disease) is a rare disorder with severe lymphadenopathy, typically presenting up to the age of 10 and entailing an accumulation of histiocytes in the region of the lymph node sinus [106].
Clinical presentation: The onset of the disease is associated with significant lymphadenopathy, usually of the cervical lymph node regions. Other lymph node stations may be affected in the course of the disease, and extranodal manifestations (such as the skin, the head and neck region, the kidneys, the skeletal system and the soft tissue [107]) are further symptoms. Occasionally patients exhibit fever and weight loss. The symptoms usually subside after around 6–9 months.
Lymph nodes: Lymph node involvement is obligate, histological examination shows plasmo- and histiocytosis (S100 positive) with florid hyperplasia and emperipolesis (envelopment of intact erythrocytes and lymphocytes) [108].
Diagnostics: Diagnosis is based on histological evidence. Laboratory tests can detect leucocytosis, an elevated ESR and hypergammaglobulinemia.
Therapy: Symptomatic therapy usually suffices; surgical, chemotherapy or radiotherapy options are available for severe site-specific symptoms (e.g. base of the skull, paratracheal) [109].
Langerhans cell histiocytosis
Pathogenesis and etiology: Langerhans cell histiocytosis (histiozytosis X) describes a proliferation of dendritic cells from the bone marrow.
Clinical presentation: A distinction is made between three different clinical forms. The first – eosinophilic granuloma – describes the localized form of the disease. It is the most frequent manifestation of histiocytosis X (70%) and is characterized by its uncomplicated course. The acute and disseminated form of histiocytosis X, Abt-Letterer-Siwe syndrome, is the most severe (10%). Patients are usually under the age of 2, and without treatment the disease has 90% mortality. The third form, Hand-Schüller-Christian syndrome, may be localized or generalized and occurs more frequently over the age of 2. The clinical course of the disease depends on which organs are affected.
Lymph nodes: It is not unusual for lymphadenopathy to be the initial symptom of histiocytosis X [110]. Histological examination detects the Langerhans cells, which immunohistochemically stain positive for S100, vimentin and CD1a. X-shaped granula known as Birbeck granules are often detected by electron microscope [111].
Diagnostics: Diagnosis is confirmed histologically.
Therapy: Depending on the course the disease, aggressive chemotherapy or bone marrow transplantation may be necessary in some cases to improve prognosis [112].
Hemophagocytic syndrome
Pathogenesis and etiology: Hemophagocytic syndrome (HPS), or reactive hemophagcytic syndrome (RHS); macrophage activation syndrome (MAS); or lymphohistiocytic syndrome (LHS), occurs in a familial form of genetic origin and a secondary form in reaction to an infection, neoplasia or autoimmune disorder. Hypercytokinemia with multiple organ involvement is common to all forms [113].
Clinical presentation: Patients exhibit bouts of high fever, hepatosplenomegaly and lymphadenopathy, cutaneous manifestations and polyserositis. The fatality rate is high at up to 50% [114].
Lymph nodes: Moderate lymphadenopathy is not obligatory for diagnosis, but together with icterus, edema, pericardial or pleural effusions is a supportive clinical symptom [115].
Diagnostics: Diagnosis of the syndrome should consider a possible genetic origin (some forms may be hereditary) and – usually infectious – trigger factors, as these affect both course and prognosis [116]. Henter and colleagues developed a revised list of criteria. Diagnosis is considered probable if five or more of the following clinical criteria are met: fever, splenomegaly, cytopenia of at least two cell lines, decreased hemoglobin, thrombocytopenia <100×109/l, neutropenia <1×109/l, hypertriglyceridemia (≥3 mmol/l) or hypofibrinogenemia (<1.5 g/l), hyperferritinemia (≥500 µg/l), soluble CD25 >2400 U/ml, hemophagocytosis of the bone marrow, the spleen or in lymph nodes, low or zero cytotoxicity of NK cells [117].
Therapy: The type of therapy depends on the cause and manifestation of the syndrome. In the primary form, curative therapy is possible by stem cell transplantation. In the secondary, the trigger factor must be treated. A possibility of immunosuppressive therapy with corticosteroids, cyclosporin A and the cytostatic etoposide also exists [115].
Autoimmune lymphoproliferative syndrome
Pathogenesis and etiology: Autoimmune lymphoproliferative syndrome (ALPS) is a disease affecting the reticuloendothelial system (RES) with a dysfunction of the FAS receptors and consecutive defect in apoptosis [118]. This results in increased survival time of the lymphocytes, which collect in the RES and produce clinical symptoms.
Clinical presentation: The main clinical symptoms are triggered by the increased lymphoproliferation: lymphadenopathy and hepatosplenomegaly are present in the majority of patients and manifest early. The average age of clinical presentation is approx. 1 year. ALPS is often associated with autoimmune disorders or cancer (10%).
Lymph nodes: The lymph nodes are affected in 90% of ALPS. Histologically the lymph nodes exhibit paracortical hyperplasia with expansion of the interfollicular regions and population with CD3+CD4-CD8- (double negative) T-cells [119].
Diagnostics: The diagnostic criteria were established by Straus and colleagues and distinguish between obligatory and supportive criteria [120].
Therapy: Therapy depends on the clinical symptoms and predominantly treats the associated autoimmune or malignant disorders. The first choice of therapy is with corticosteroids or immunoglobulins, the second mycophenolate mofetil, sirolimus or mercaptopurine [118].
Castleman’s disease
Pathogenesis and etiology: Castleman’s disease (or giant lymph node hyperplasia, angiofollicular lymph node hyperplasia) is a rare (incidence <1:100,000) disorder of uncertain origin with angiofollicular lymph node hyperplasia [121].
Clinical presentation: A distinction is made between the localized and the multicentric form. Half of patients with the localized form of the disease are clinically asymptomatic. Possible symptoms are fever, weight loss, fatigue and pain. Multicentric forms of the disease may also exhibit hepato- and/or splenomegaly or the so-called POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, monclonal gammopathy, skin alterations) [122].
Lymph nodes: The lymph nodes are affected in >80% of cases, the main manifestations are abdominal, thoracic and cervical. A distinction is made pathogenetically and histologically between two subtypes: the hyaline vascular type with the onion-skin structure of the hyaline lymph nodes, usually occurring at a single site, and the plasmacytic type (rarer, multifocal) with plasma cell proliferation [123].
Diagnostics and Therapy: Diagnosis is confirmed histologically. The affected regions imitate both benign and malignant entities in imaging [124]. Dissection is simultaneously the therapy.
Kawasaki syndrome (mucocutaneous lymph node syndrome)
Pathogenesis and etiology: Kawasaki syndrome (also known as mucocutaneous lymph node syndrome, MCLS) is a necrotizing vasculitis (incidence in Germany 9:100,000 [125]). It predominantly affects small children (<5 years); it is thought to be of infectious origin from an unknown pathogen and affects patients with a genetic predisposition.
Clinical presentation: The disease typically progresses in three phases: feverish, subacute and reconvalescent. The main symptoms are persistent high fever over a period of 5–10 days, rhagades in the oral cavity and on the lips, swelling, redness, eye involvement with conjunctival injection, skin involvement (exanthema), skin rash predominantly on the trunk, painful swelling of the hands and feet, and enlargement of the cervical lymph nodes [126].
The most serious complications are caused by secondary symptoms and are associated with cardiac involvement and the formation of aneurysms and thromboses of the coronary arteries on the one hand [127], and neurological symptoms including non-infectious meningitis and central hearing loss on the other. The lungs, intestine and joints may also be affected by the vasculitis [128].
Lymph nodes: The lymph nodes in the anterior cervical triangle are typically affected in the majority of patients by acute, non-purulent and relatively painless swelling.
Diagnostics: The disease is diagnosed clinically (obligatory fever + 4/5 skin symptoms) and can be confirmed serologically. Echocardiography (cardiac involvement) and beginning therapy early are important [129].
Therapy: Initial treatment comprises high-dose intravenous immunoglobulins combined with high-dose aspirin. Further treatment options are available in the event of chronification, e.g. with pentoxifylline, abciximab, infliximab and cyclophosphamide [130], [131], [132].
Kikuchi-Fujimoto disease
Pathogenesis and etiology: The pathogenesis of Kikuchi Fujimoto disease (lymphadenitis) is unknown to date. It primarily affects younger women [133], [134].
Clinical presentation: The symptoms are a combination of fever, lymphocytosis and necrotizing lymphadenitis as a result of stimulated apoptosis, mainly affecting the cervical and axillary lymph nodes. The disease is self-limiting, with remission after 1–4 months [135], in isolated cases however persisting for years. Recurrence is observed in rare cases (4%) [136].
Lymph nodes: Histology following lymph node dissection is critical to diagnosis: typical paracortical lesions with confluent zones of necrosis, atypical mononuclear cells, T-lymphocytes and histiocytes without granulocytes are exhibited. The lymph nodes are usually affected bilaterally, located in the posterior cervical triangle and are hard and tender.
Therapy: Therapeutic options are limited to symptomatic treatment (lowering fever, analgesia, intake of fluids) [135].
PFAPA syndrome
Pathogenesis and etiology: PFAPA syndrome (periodic fever, aphthous stomatitis, pharyngitis and cervical (lymph) adenitis) is similarly a disorder of unknown origin. It chiefly affects children under the age of five [137].
Clinical presentation: The clinical manifestations after which the disease is named may recur periodically in a 2–9 week cycle, spontaneously subsiding again after a few days [138].
Lymph nodes: The lymph nodes are enlarged on both sides.
Diagnostics: Diagnosis is reached by exclusion; no conclusive serological tests are available to date.
Therapy: The prognosis is excellent; very severe symptoms are treated with corticosteroids [138].
5.3 Metabolic
5.3.1 Storage diseases
Niemann-Pick disease
Pathogenesis and etiology: Niemann-Pick disease is a rare, autosomal recessive inherited sphingolipidosis in which a sphingomyelinase defect causes an accumulation of sphingomyelin stored in lysosomes in the liver, spleen, lymph nodes and brain [139].
Clinical presentation: The disease is divided into several subtypes, the most common of which is type IA, also known as the acute infantile neuropathic form. Affected children present early (at three months of age) with sucking weakness, dystrophy, hepatosplenomegaly, lymph node swellings and skin alterations. Neurological symptoms such as blindness, deafness and spasticity progress rapidly, leading in most children to early death. The course of the chronic visceral form without CNS involvement is substantially milder. Type C can manifest from early childhood up to late adolescence, usually with icterus, supratrochlear gaze palsy and cerebellar ataxia [140].
Lymph nodes: The lymph nodes are frequently affected in particular in type 1; the lysosomes can be stained in the cells of the reticuloendothelial system.
Diagnostics: Diagnosis is reached by clinical assessment, histological and molecular genetic testing.
Therapy: No curative therapy is available for any form of the disease to date. Drug treatment with miglustat (imino sugar, substrate reduction therapy) reduced progression of the disease in the case of type C [141].
Gaucher’s disease
Pathogenesis and etiology: Gaucher’s disease is the most common lyposomal storage disease of the lipometabolism. A usually autosomal recessive inherited enzyme defect in the beta-glucosidase is responsible for an accumulation of glucocerebrosides in macrophages and monocytes. This leads to an increased release of cytokines [142].
Clinical presentation: The clinical manifestations differ according to the degree of enzyme damage. Hepatosplenomegaly, anemia and thrombopenia, osteolysis and failure to thrive may occur in Gaucher’s disease [142].
Lymph nodes: The lymph nodes are less frequently affected; predilection sites are the mesenteric drainage regions [143].
Diagnostics: Diagnosis is based on the blood count, the level of enzyme activity and other means (e.g. sonography to detect hepatosplenomegaly).
Therapy: Miglustat as referred to earlier or so-called chaperone therapies are employed to reduce progression [144].
Tangier disease
Pathogenesis and etiology: The rare autosomal recessive inherited enzyme defect in Tangier disease affects a cholesterol transporter protein. It results in an accumulation of cholesterol deposits in the cells of the reticuloendothelial system [145].
Clinical presentation: Typical manifestations are yellow tonsils, with cholesterol deposits also found in other organs of the reticuloendothelial system, e.g. liver, spleen, in muscles and the nerve cell system [146].
Lymph nodes: The fat-storing macrophages in the affected lymph nodes appear as foam cells.
Diagnostics: Genetic analysis can confirm a clinical suspicion.
Therapy: No curative therapy exists. A low-fat diet is recommended prophylactically to reduce cardiovascular risk [147].
Amyloidosis
Amyloidosis is an umbrella term for the pathological deposition of certain proteins in the stroma. The symptoms are very varied depending on the localization and proteins deposited [148]. The lymph nodes may be affected by these deposits; histological examination shows the typical green birefringence under polarized light [149]. The therapy is adapted to the clinical findings.
5.3.2 Hypersensitivity
Serum sickness
Serum sickness is a Type III hypersensitivity reaction (Arthus reaction), as classified by Coombs and Gell, in which immune complexes are formed against exogenous antigen proteins and lead to activation of the complement system and damage to the surrounding tissue [150]. As formation of the antibodies must first be completed, in contrast to anaphylactic shock (Type I allergic reaction) it is usual here for days and weeks to elapse between exposure to the antigens and allergic reaction. This must be taken into account in the anamnesis of lymph node swelling (e.g. vaccination in the preceding months [151]) for differential diagnosis. Possible clinical symptoms are: swelling of the lymph nodes, fever, exanthema, arthralgia, circulatory deficiency. The therapy is adapted to the symptoms, where applicable watchful waiting, NSAIDs and corticoids. Vaccinations are not the only trigger for the aforementioned reactions, which are also possible to drugs and in particular antibiotics such as amoxicillin, cefaclor, cephalexin and trimethoprim-sulfamethoxazole [152].
Adverse drug reactions must not necessarily be accompanied by the other symptoms of serum sickness but can also cause isolated lymphadenopathy [153]. The anamnesis should therefore include the following drugs: antiepilectics; heparin; tuberculostatics such as isoniazid, antibiotics such as cephalosporins, penicillins; phenytoin, hydralazine, procainamide, allopurinol, dapsones, carbamazepine, atenolol, captopril, gold, primidones, pyrimethamines, quinidines, sulfonamides, sulindac [153].
A special form of the drug-induced systemic reaction with lymphadenopathy is DRESS syndrome (drug reaction with eosinophilia and systemic symptoms). This severe form involves multiple organ failure with 20% mortality [154]. Drugs associated with DRESS syndrome are allopurinol, minocyclinhydrochloride, anticonvulsants, sulfonamides and antibiotics. Generalized lymphadenopathy is exhibited in these cases.
5.4 Neoplastic
5.4.1 Malignant lymphoma
Non-Hodgkin lymphoma
Pathogenesis and etiology: Non-Hodgkin lymphomas (NHL) are a heterogeneous group of neoplastic disorders of the lymphatic cell line which can affect both B- (approx. 80%) and T-lymphocytes (approx. 20%). Their cause is considered to be hereditary or acquired genetic defects leading to a mismatch of proliferation and apoptosis of cells in the lymphatic system [155], [156].
Clinical presentation: Lymph node swelling and general symptoms such as fever, weight loss and night sweats are the predominant manifestations. Patients additionally often exhibit fatigue, weakness and changes in blood count. The stages are:
Stage I: A single lymph node region above or below the diaphragm is affected,
Stage II: Two or more lymph node regions above or below the diaphragm are affected,
Stage III: Both sides of the diaphragm are affected,
Stage IV: Organs other than primarily lymphatic organs are affected,
and appended: A = no general symptoms, B = with general symptoms, S = spread to spleen, E = extranodal spread. The WHO divides NHL into precursor and mature B and T-cell lymphomas.
Lymph nodes: The swollen lymph nodes are usually firm and non-tender; swelling may occur on one or both sides.
Diagnostics: Histological procedures with special immune staining and bone marrow aspiration are necessary for diagnosis. Imaging is useful in staging the disease (chest x-ray, CT and sonography of neck, thorax and abdomen) [157].
Therapy: Treatment is according to the stage and entity and comprises chemo, radiation and antibody therapy [155].
Hodgkin’s lymphoma
Pathogenesis and etiology: Hodgkin’s lymphoma is similarly a neoplastic disease of the lymphatic cell line and is distinguished histologically from NHL by its characteristic Reed-Sternberg cells. There is discussion of the disease being of multifactor origin with genetic predisposition, associated with viral oncogenes (e.g. EBV) or following immunosuppressive therapies. It is possible to differentiate histologically between five different forms of the disease (nodular sclerosing, mixed cellularity, lymphocyte rich, lymphocyte depleted and lymphocyte predominant).
Clinical presentation: Hodgkin’s lymphoma patients often present with swollen lymph nodes and B symptoms. Hepatosplenomegaly or involvement of other organs (lungs, CNS) may additionally occur. The Ann Arbor classification distinguishes between four stages:
Stage I: A single lymph node region is affected,
Stage II: Two or more lymph node regions on one side of the diaphragm are affected,
Stage III: Two or more lymph node regions on both sides of the diaphragm are affected,
Stage IV: Disseminated involvement of several extralymphatic organs with or without involvement of the lymph nodes,
and appended: A = no general symptoms, B = with general symptoms, S = spread to the spleen, E = extranodal spread.
Lymph nodes: The swollen, rubbers, non-tender lymph nodes are frequently the initial noticeable symptom. Histological examination reveals Hodgkin and multinucleated Reed-Sternberg cells and multicolor reactive lymphocytic reactions [158].
Diagnostics: Anamnesis and clinical examination provide diagnostic indicators; staging is performed as in NHL with imaging, bone marrow aspiration and histological evidence.
Therapy: Chemotherapy, and if applicable radiotherapy, according to the stage of the disease are used [159], [160], [161].
5.4.2 Leukemias
Acute lymphoblastic leukemia
Pathogenesis and etiology: Acute lymphoblastic leukemia (ALL) is a neuroplastic disorder of the lymphatic precursor cells. This leads to suppression of the bone marrow with consecutive insufficiency of the hematopoietic system. The early age peak (four years) means that ALL is particularly relevant among children and the most common malignant disorder in childhood [162].
Clinical presentation: Children present with anemia, weakness and exhaustion. They additionally have an increased bleeding tendency or increased susceptibility to infections. Lymph node swelling, hepatosplenomegaly, meningeosis or bone pain may be responsible for symptoms [163].
Lymph nodes: Around 50% of cases present with swollen lymph nodes.
Diagnostics: The laboratory tests are important in establishing diagnosis. Immunophenotyping by fluorescence-activated cell sorting (FACS) makes it possible to assign different grades of maturity, with the relevant consequences for prognosis and therapy [164].
Therapy: Therapy for ALL is divided into different phases, the duration and intensity of which depends on certain risk factors (detection of specific genes, CNS involvement). A distinction is made between the induction therapy and the subsequent consolidation therapy and the re-induction therapy and maintenance therapy [165]. Antibody therapies and inhibitor therapies are also employed.
Acute myeloid leukemia
Pathogenesis and etiology: Acute myeloid leukemia (AML) is a neoplastic disorder of the myeloid precursor cells. As in ALL, this results in a bone marrow insufficiency. AML is relatively rare in childhood and is responsible for around 20% of leukemias in this age group [166].
Clinical presentation: The symptoms are chiefly caused by the bone marrow suppression and are comparable to those of ALL (anemia, weakness, exhaustion, bleeding tendency, susceptibility to infections). Lymph node swelling, hepatosplenomegaly, meningeosis, bone pain, unilateral testicular swelling and gingival hyperplasia [167] are other common signs.
Lymph nodes: Involvement of the lymph nodes is frequently also found in AML.
Diagnostics: Diagnosis is based on morphology, immunophenotyping and cytogenetics. Bone marrow aspiration is indicated in addition to laboratory testing [167].
Therapy: Chemotherapy (induction and post-remission phase) and, if this proves ineffective, hematopoietic stem cell transplantation are available in the treatment of AML [168].
5.4.3 Lymph node metastasis from solid tumors
Rhabdomyosarcoma
Pathogenesis and etiology: Rhabdomyosarcoma is a highly malignant tumor of mesenchymal origin and responsible for around 5% of childhood tumor diseases [169].
Clinical presentation: Swelling and pain are the cardinal symptoms; other symptoms are possible depending on localization (e.g. hematuria if the bladder is affected).
Lymph nodes: Lymph node metastasis must be distinguished from a primary neck mass. Characteristic histological indicators are eosinophilic cells and multinucleate giant cells [170].
Diagnostics: Diagnosis is established by imaging (sonography, MRI) and histology.
Therapy: Therapy for rhabdomyosarcoma involves surgical excision with adjuvant radiotherapy. Neoadjuvant chemotherapy may be indicated for preoperative size reduction [171].
Neuroblastoma
Pathogenesis and etiology: Neuroblastoma is the third most common malignant disease in childhood and consists of proliferating cells of the neural crest called neuroblasts. The head and neck region is a common predilection site.
Clinical presentation: Neurological symptoms, B symptoms or abdominal complaints may arise depending on location. Classification is according to clinical aspects, taking into account localization, spread, lymph node involvement and distant metastasis [172].
Lymph nodes: Neuroblastoma often metastasizes in regional and distant lymph nodes.
Diagnostics: Staging examinations, bone marrow aspiration and histological examination confirm diagnosis [173].
Therapy: Depending on the stage, therapy options include dissection, (neo-)adjuvant chemotherapy, radiotherapy and stem cell transplantation as well as retinoids, arsenic trioxide and antibody therapies. A number of therapy options are associated with high comorbidity/comortality [174].
Nasopharyngeal carcinoma
Pathogenesis and etiology: Nasopharyngeal carcinomas are a rare tumor entity among children of western industrial nations (approx. 1% of malignant tumors) [175]. By contrast, the incidence of the disease is rising rapidly among patients from Asiatic countries [176].
Clinical presentation: Nosebleeds and nasal breathing difficulties and neck masses from lymph node metastasis are classic symptoms of a nasopharyngeal carcinoma.
Lymph nodes: Metastasis from the carcinomas is lymphogenous, rarely hematogenous. A distinction is made between keratinizing, well differentiated squamous cell carcinomas, non-keratinizing squamous cell carcinomas, and undifferentiated lymphoepithelial carcinomas (Schmincke-Regaud).
Diagnostics: Diagnosis is confirmed by clinical examination, imaging (MRI) and histology.
Therapy: Therapy is dependent on the localization and staging of the disease, primary radiochemotherapy is generally indicated.
6 Conclusion
Cervical lymph node swelling of uncertain origin in childhood can pose a special challenge to the attending ENT and pediatric physicians. Faced with the spectrum of possible differential diagnoses and malignant diseases, they must follow a clearly structured algorithm in their diagnostic steps and therapeutic strategies to avoid unnecessary delays in further diagnosis and safeguard against overhasty, and possibly too invasive, diagnosis and therapy. This paper is intended to support them in these efforts.
Notes
Competing interests
The authors declare that they have no competing interests.
References
- 1.Gosche JR, Vick L. Acute, subacute, and chronic cervical lymphadenitis in children. Semin Pediatr Surg. 2006 May;15(2):99–106. doi: 10.1053/j.sempedsurg.2006.02.007. Available from: http://dx.doi.org/10.1053/j.sempedsurg.2006.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schwetschenau E, Kelley DJ. The adult neck mass. Am Fam Physician. 2002 Sep;66(5):831–838. [PubMed] [Google Scholar]
- 3.Creutzig U GPOH; DGKJ; DKG. AWMF S1 Leitlinie Lymphknotenvergrößerung. Available from: http://www.awmf.org/leitlinien/detail/ll/025-020.html.
- 4.Gobbi PG, Broglia C, Carnevale Maffè G, Ruga A, Molinari E, Ascari E. Lymphomatous superficial lymph nodes: limitations of physical examination for accurate staging and response assessment. Haematologica. 2002 Nov;87(11):1151–1156. [PubMed] [Google Scholar]
- 5.Vassilakopoulos TP, Pangalis GA. Application of a prediction rule to select which patients presenting with lymphadenopathy should undergo a lymph node biopsy. Medicine (Baltimore) 2000 Sep;79(5):338–347. doi: 10.1097/00005792-200009000-00007. Available from: http://dx.doi.org/10.1097/00005792-200009000-00007. [DOI] [PubMed] [Google Scholar]
- 6.Ghirardelli ML, Jemos V, Gobbi PG. Diagnostic approach to lymph node enlargement. Haematologica. 1999 Mar;84(3):242–247. [PubMed] [Google Scholar]
- 7.Isselbacher KJ, Braunwald E, Wilson JD, Martin JB, Fauci AS, Kasper DL. Harrisons Innere Medizin. 13th ed. Wien: Blackwell; 1995. [Google Scholar]
- 8.Brown RL, Azizkhan RG. Pediatric head and neck lesions. Pediatr Clin North Am. 1998 Aug;45(4):889–905. doi: 10.1016/S0031-3955(05)70052-3. Available from: http://dx.doi.org/10.1016/S0031-3955(05)70052-3. [DOI] [PubMed] [Google Scholar]
- 9.Torsiglieri AJ, Jr, Tom LW, Ross AJ, 3rd, Wetmore RF, Handler SD, Potsic WP. Pediatric neck masses: guidelines for evaluation. Int J Pediatr Otorhinolaryngol. 1988 Dec;16(3):199–210. doi: 10.1016/0165-5876(88)90031-6. Available from: http://dx.doi.org/10.1016/0165-5876(88)90031-6. [DOI] [PubMed] [Google Scholar]
- 10.Barton LL, Feigin RD. Childhood cervical lymphadenitis: a reappraisal. J Pediatr. 1974 Jun;84(6):846–852. doi: 10.1016/S0022-3476(74)80761-4. Available from: http://dx.doi.org/10.1016/S0022-3476(74)80761-4. [DOI] [PubMed] [Google Scholar]
- 11.Dajani AS, Garcia RE, Wolinsky E. Etiology of cervical lymphadenitis in children. N Engl J Med. 1963 Jun;268:1329–1333. doi: 10.1056/NEJM196306132682403. Available from: http://dx.doi.org/10.1056/NEJM196306132682403. [DOI] [PubMed] [Google Scholar]
- 12.Scobie WG. Acute suppurative adenitis in children: a review of 964 cases. Scott Med J. 1969 Oct;14(10):352–354. doi: 10.1177/003693306901401003. [DOI] [PubMed] [Google Scholar]
- 13.Yamauchi T, Ferrieri P, Anthony BF. The aetiology of acute cervical adenitis in children: serological and bacteriological studies. J Med Microbiol. 1980 Feb;13(1):37–43. doi: 10.1099/00222615-13-1-37. Available from: http://dx.doi.org/10.1099/00222615-13-1-37. [DOI] [PubMed] [Google Scholar]
- 14.Sundaresh HP, Kumar A, Hokanson JT, Novack AH. Etiology of cervical lymphadenitis in children. Am Fam Physician. 1981;24:147–51. [PubMed] [Google Scholar]
- 15.Inman JC, Rowe M, Ghostine M, Fleck T. Pediatric neck abscesses: changing organisms and empiric therapies. Laryngoscope. 2008 Dec;118(12):2111–2114. doi: 10.1097/MLG.0b013e318182a4fb. Available from: http://dx.doi.org/10.1097/MLG.0b013e318182a4fb. [DOI] [PubMed] [Google Scholar]
- 16.Guss J, Kazahaya K. Antibiotic-resistant Staphylococcus aureus in community-acquired pediatric neck abscesses. Int J Pediatr Otorhinolaryngol. 2007 Jun;71(6):943–948. doi: 10.1016/j.ijporl.2007.03.006. Available from: http://dx.doi.org/10.1016/j.ijporl.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 17.Neff L, Newland JG, Sykes KJ, Selvarangan R, Wei JL. Microbiology and antimicrobial treatment of pediatric cervical lymphadenitis requiring surgical intervention. Int J Pediatr Otorhinolaryngol. 2013 May;77(5):817–820. doi: 10.1016/j.ijporl.2013.02.018. Available from: http://dx.doi.org/10.1016/j.ijporl.2013.02.018. [DOI] [PubMed] [Google Scholar]
- 18.Hof H, Dörries R. Medizinische Mikrobiologie. 3rd ed. Stuttgart: Thieme; 2005. [Google Scholar]
- 19.Lagrange PH, Thangaraj SK, Dayal R, Deshpande A, Ganguly NK, Girardi E, Joshi B, Katoch K, Katoch VM, Kumar M, Lakshmi V, Leportier M, Longuet C, Malladi SV, Mukerjee D, Nair D, Raja A, Raman B, Rodrigues C, Sharma P, Singh A, Singh S, Sodha A, Kabeer BS, Vernet G, Goletti D. A toolbox for tuberculosis (TB) diagnosis: an Indian multicentric study (2006-2008). Evaluation of QuantiFERON-TB gold in tube for TB diagnosis. PLoS ONE. 2013;8(9):e73579. doi: 10.1371/journal.pone.0073579. Available from: http://dx.doi.org/10.1371/journal.pone.0073579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dulin MF, Kennard TP, Leach L, Williams R. Management of cervical lymphadenitis in children. Am Fam Physician. 2008 Nov;78(9):1097–1098. [PubMed] [Google Scholar]
- 21.Niedzielska G, Kotowski M, Niedzielski A, Dybiec E, Wieczorek P. Cervical lymphadenopathy in children--incidence and diagnostic management. Int J Pediatr Otorhinolaryngol. 2007 Jan;71(1):51–56. doi: 10.1016/j.ijporl.2006.08.024. Available from: http://dx.doi.org/10.1016/j.ijporl.2006.08.024. [DOI] [PubMed] [Google Scholar]
- 22.Ying M, Ahuja A. Sonography of neck lymph nodes. Part I: normal lymph nodes. Clin Radiol. 2003 May;58(5):351–358. doi: 10.1016/S0009-9260(02)00584-6. Available from: http://dx.doi.org/10.1016/S0009-9260(02)00584-6. [DOI] [PubMed] [Google Scholar]
- 23.Ahuja A, Ying M, Yuen YH, Metreweli C. Power Doppler sonography of cervical lymphadenopathy. Clin Radiol. 2001 Dec;56(12):965–969. doi: 10.1053/crad.2001.0717. Available from: http://dx.doi.org/10.1053/crad.2001.0717. [DOI] [PubMed] [Google Scholar]
- 24.Ahuja A, Ying M. Sonography of neck lymph nodes. Part II: abnormal lymph nodes. Clin Radiol. 2003 May;58(5):359–366. doi: 10.1016/S0009-9260(02)00585-8. Available from: http://dx.doi.org/10.1016/S0009-9260(02)00585-8. [DOI] [PubMed] [Google Scholar]
- 25.Solbiati L, Cioffi V, Ballarati E. Ultrasonography of the neck. Radiol Clin North Am. 1992 Sep;30(5):941–954. [PubMed] [Google Scholar]
- 26.van den Brekel MW, Castelijns JA, Stel HV, Luth WJ, Valk J, van der Waal I, Snow GB. Occult metastatic neck disease: detection with US and US-guided fine-needle aspiration cytology. Radiology. 1991 Aug;180(2):457–461. doi: 10.1148/radiology.180.2.2068312. Available from: http://dx.doi.org/10.1148/radiology.180.2.2068312. [DOI] [PubMed] [Google Scholar]
- 27.Esen G. Ultrasound of superficial lymph nodes. Eur J Radiol. 2006 Jun;58(3):345–359. doi: 10.1016/j.ejrad.2005.12.039. Available from: http://dx.doi.org/10.1016/j.ejrad.2005.12.039. [DOI] [PubMed] [Google Scholar]
- 28.Rubaltelli L, Proto E, Salmaso R, Bortoletto P, Candiani F, Cagol P. Sonography of abnormal lymph nodes in vitro: correlation of sonographic and histologic findings. AJR Am J Roentgenol. 1990 Dec;155(6):1241–1244. doi: 10.2214/ajr.155.6.2122673. Available from: http://dx.doi.org/10.2214/ajr.155.6.2122673. [DOI] [PubMed] [Google Scholar]
- 29.Vassallo P, Wernecke K, Roos N, Peters PE. Differentiation of benign from malignant superficial lymphadenopathy: the role of high-resolution US. Radiology. 1992 Apr;183(1):215–220. doi: 10.1148/radiology.183.1.1549675. Available from: http://dx.doi.org/10.1148/radiology.183.1.1549675. [DOI] [PubMed] [Google Scholar]
- 30.Esen G, Gurses B, Yilmaz MH, Ilvan S, Ulus S, Celik V, Farahmand M, Calay OO. Gray scale and power Doppler US in the preoperative evaluation of axillary metastases in breast cancer patients with no palpable lymph nodes. Eur Radiol. 2005 Jun;15(6):1215–1223. doi: 10.1007/s00330-004-2605-9. Available from: http://dx.doi.org/10.1007/s00330-004-2605-9. [DOI] [PubMed] [Google Scholar]
- 31.Ahuja A, Ying M. Grey-scale sonography in assessment of cervical lymphadenopathy: review of sonographic appearances and features that may help a beginner. Br J Oral Maxillofac Surg. 2000 Oct;38(5):451–459. doi: 10.1054/bjom.2000.0446. Available from: http://dx.doi.org/10.1054/bjom.2000.0446. [DOI] [PubMed] [Google Scholar]
- 32.Tschammler A, Heuser B, Ott G, Schmitt S, Hahn D. Pathological angioarchitecture in lymph nodes: underlying histopathologic findings. Ultrasound Med Biol. 2000 Sep;26(7):1089–1097. doi: 10.1016/S0301-5629(00)00221-0. Available from: http://dx.doi.org/10.1016/S0301-5629(00)00221-0. [DOI] [PubMed] [Google Scholar]
- 33.Giovagnorio F, Galluzzo M, Andreoli C, De CM, David V. Color Doppler sonography in the evaluation of superficial lymphomatous lymph nodes. J Ultrasound Med. 2002 Apr;21(4):403–408. doi: 10.7863/jum.2002.21.4.403. [DOI] [PubMed] [Google Scholar]
- 34.Burrill J, Williams CJ, Bain G, Conder G, Hine AL, Misra RR. Tuberculosis: a radiologic review. Radiographics. 2007 Sep-Oct;27(5):1255–1273. doi: 10.1148/rg.275065176. Available from: http://dx.doi.org/10.1148/rg.275065176. [DOI] [PubMed] [Google Scholar]
- 35.Hanck C, Fleisch F, Katz G. Imaging appearance of nontuberculous mycobacterial infection of the neck. AJNR Am J Neuroradiol. 2004 Feb;25(2):349. [PMC free article] [PubMed] [Google Scholar]
- 36.Boscolo-Rizzo P, Marchiori C, Zanetti F, Vaglia A, Da Mosto MC. Conservative management of deep neck abscesses in adults: the importance of CECT findings. Otolaryngol Head Neck Surg. 2006 Dec;135(6):894–899. doi: 10.1016/j.otohns.2006.05.013. Available from: http://dx.doi.org/10.1016/j.otohns.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 37.Knappe M, Louw M, Gregor RT. Ultrasonography-guided fine-needle aspiration for the assessment of cervical metastases. Arch Otolaryngol Head Neck Surg. 2000 Sep;126(9):1091–1096. doi: 10.1001/archotol.126.9.1091. Available from: http://dx.doi.org/10.1001/archotol.126.9.1091. [DOI] [PubMed] [Google Scholar]
- 38.Atula TS, Varpula MJ, Kurki TJ, Klemi PJ, Grénman R. Assessment of cervical lymph node status in head and neck cancer patients: palpation, computed tomography and low field magnetic resonance imaging compared with ultrasound-guided fine-needle aspiration cytology. Eur J Radiol. 1997 Sep;25(2):152–161. doi: 10.1016/S0720-048X(96)01071-6. Available from: http://dx.doi.org/10.1016/S0720-048X(96)01071-6. [DOI] [PubMed] [Google Scholar]
- 39.Krishnamurthy S, Sneige N, Bedi DG, Edieken BS, Fornage BD, Kuerer HM, Singletary SE, Hunt KK. Role of ultrasound-guided fine-needle aspiration of indeterminate and suspicious axillary lymph nodes in the initial staging of breast carcinoma. Cancer. 2002 Sep;95(5):982–988. doi: 10.1002/cncr.10786. Available from: http://dx.doi.org/10.1002/cncr.10786. [DOI] [PubMed] [Google Scholar]
- 40.Hafez NH, Tahoun NS. Reliability of fine needle aspiration cytology (FNAC) as a diagnostic tool in cases of cervical lymphadenopathy. J Egypt Natl Canc Inst. 2011 Sep;23(3):105–114. doi: 10.1016/j.jnci.2011.09.009. Available from: http://dx.doi.org/10.1016/j.jnci.2011.09.009. [DOI] [PubMed] [Google Scholar]
- 41.Pappa VI, Hussain HK, Reznek RH, Whelan J, Norton AJ, Wilson AM, Love S, Lister TA, Rohatiner AZ. Role of image-guided core-needle biopsy in the management of patients with lymphoma. J Clin Oncol. 1996 Sep;14(9):2427–2430. doi: 10.1200/JCO.1996.14.9.2427. [DOI] [PubMed] [Google Scholar]
- 42.Peters TR, Edwards KM. Cervical lymphadenopathy and adenitis. Pediatr Rev. 2000 Dec;21(12):399–405. [PubMed] [Google Scholar]
- 43.Leung AK, Robson WL. Childhood cervical lymphadenopathy. J Pediatr Health Care. 2004 Jan-Feb;18(1):3–7. doi: 10.1016/j.pedhc.2003.08.008. Available from: http://dx.doi.org/10.1016/j.pedhc.2003.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kelly CS, Kelly RE., Jr Lymphadenopathy in children. Pediatr Clin North Am. 1998 Aug;45(4):875–888. doi: 10.1016/S0031-3955(05)70051-1. Available from: http://dx.doi.org/10.1016/S0031-3955(05)70051-1. [DOI] [PubMed] [Google Scholar]
- 45.Huntington MK, VanKeulen S, Hoffman WW Sanford Health Antibiotic Utilization Physician Collaborative. What ever happened to the common cold? Improving antibiotic utilization. S D Med. 2013 Apr;66(4):136–9, 141. [PubMed] [Google Scholar]
- 46.Penn R, Steehler MK, Sokohl A, Harley EH. Nontuberculous mycobacterial cervicofacial lymphadenitis--a review and proposed classification system. Int J Pediatr Otorhinolaryngol. 2011 Dec;75(12):1599–1603. doi: 10.1016/j.ijporl.2011.09.018. Available from: http://dx.doi.org/10.1016/j.ijporl.2011.09.018. [DOI] [PubMed] [Google Scholar]
- 47.Robert Koch Institut. RKI-Ratgeber für Ärzte – Infektionskrankheiten A-Z. Available from: http://www.rki.de/DE/Content/InfAZ/InfAZ_marginal_node.html.
- 48.Saadatnia G, Golkar M. A review on human toxoplasmosis. Scand J Infect Dis. 2012 Nov;44(11):805–814. doi: 10.3109/00365548.2012.693197. Available from: http://dx.doi.org/10.3109/00365548.2012.693197. [DOI] [PubMed] [Google Scholar]
- 49.Barry MA, Weatherhead JE, Hotez PJ, Woc-Colburn L. Childhood parasitic infections endemic to the United States. Pediatr Clin North Am. 2013 Apr;60(2):471–485. doi: 10.1016/j.pcl.2012.12.011. Available from: http://dx.doi.org/10.1016/j.pcl.2012.12.011. [DOI] [PubMed] [Google Scholar]
- 50.Abadir A, Patel A, Haider S. Systemic therapy of New World cutaneous leishmaniasis: A case report and review article. Can J Infect Dis Med Microbiol. 2010;21(2):e79–e83. doi: 10.1155/2010/768645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kelly BP. Superficial fungal infections. Pediatr Rev. 2012 Apr;33(4):e22–e37. doi: 10.1542/pir.33-4-e22. Available from: http://dx.doi.org/10.1542/pir.33-4-e22. [DOI] [PubMed] [Google Scholar]
- 52.Newman LS, Rose CS, Maier LA. Sarcoidosis. N Engl J Med. 1997 Apr;336(17):1224–1234. doi: 10.1056/NEJM199704243361706. Available from: http://dx.doi.org/10.1056/NEJM199704243361706. [DOI] [PubMed] [Google Scholar]
- 53.Morais A, Lima B, Peixoto MJ, Alves H, Marques A, Delgado L. BTNL2 gene polymorphism associations with susceptibility and phenotype expression in sarcoidosis. Respir Med. 2012 Dec;106(12):1771–1777. doi: 10.1016/j.rmed.2012.08.009. Available from: http://dx.doi.org/10.1016/j.rmed.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 54.Scharkoff T. Epidemiologie der Sarkoidose. [Epidemiology of sarcoidosis]. Pneumologie. 1993 Oct;47(10):588–592. (Ger). [PubMed] [Google Scholar]
- 55.Gran JT, Bøhmer E. Acute sarcoid arthritis: a favourable outcome? A retrospective survey of 49 patients with review of the literature. Scand J Rheumatol. 1996;25(2):70–73. doi: 10.3109/03009749609069210. Available from: http://dx.doi.org/10.3109/03009749609069210. [DOI] [PubMed] [Google Scholar]
- 56.Scadding JG. The definition of sarcoidosis. Postgrad Med J. 1970 Aug;46(538):465–467. doi: 10.1136/pgmj.46.538.465. Available from: http://dx.doi.org/10.1136/pgmj.46.538.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dempsey OJ, Paterson EW, Kerr KM, Denison AR. Sarcoidosis. BMJ. 2009;339:b3206. doi: 10.1136/bmj.b3206. Available from: http://dx.doi.org/10.1136/bmj.b3206. [DOI] [PubMed] [Google Scholar]
- 58.Youssef G, Leung E, Mylonas I, Nery P, Williams K, Wisenberg G, Gulenchyn KY, Dekemp RA, Dasilva J, Birnie D, Wells GA, Beanlands RS. The use of 18F-FDG PET in the diagnosis of cardiac sarcoidosis: a systematic review and metaanalysis including the Ontario experience. J Nucl Med. 2012 Feb;53(2):241–248. doi: 10.2967/jnumed.111.090662. Available from: http://dx.doi.org/10.2967/jnumed.111.090662. [DOI] [PubMed] [Google Scholar]
- 59.Jordan DR, Anderson RL, Nerad JA, Scrafford DB. The diagnosis of sarcoidosis. Can J Ophthalmol. 1988 Aug;23(5):203–207. [PubMed] [Google Scholar]
- 60.Mehrotra R, Dhingra V. Cytological diagnosis of sarcoidosis revisited: a state of the art review. Diagn Cytopathol. 2011 Jul;39(7):541–548. doi: 10.1002/dc.21455. Available from: http://dx.doi.org/10.1002/dc.21455. [DOI] [PubMed] [Google Scholar]
- 61.Fazzi P. Pharmacotherapeutic management of pulmonary sarcoidosis. Am J Respir Med. 2003;2(4):311–320. doi: 10.1007/BF03256659. Available from: http://dx.doi.org/10.1007/BF03256659. [DOI] [PubMed] [Google Scholar]
- 62.Gottlieb JE, Israel HL, Steiner RM, Triolo J, Patrick H. Outcome in sarcoidosis. The relationship of relapse to corticosteroid therapy. Chest. 1997 Mar;111(3):623–631. doi: 10.1378/chest.111.3.623. Available from: http://dx.doi.org/10.1378/chest.111.3.623. [DOI] [PubMed] [Google Scholar]
- 63.Sapadin AN, Fleischmajer R. Tetracyclines: nonantibiotic properties and their clinical implications. J Am Acad Dermatol. 2006 Feb;54(2):258–265. doi: 10.1016/j.jaad.2005.10.004. Available from: http://dx.doi.org/10.1016/j.jaad.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 64.King CS, Kelly W. Treatment of sarcoidosis. Dis Mon. 2009 Nov;55(11):704–718. doi: 10.1016/j.disamonth.2009.06.002. Available from: http://dx.doi.org/10.1016/j.disamonth.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 65.Cunningham-Rundles C. Common variable immunodeficiency. Curr Allergy Asthma Rep. 2001 Sep;1(5):421–429. doi: 10.1007/s11882-001-0027-1. Available from: http://dx.doi.org/10.1007/s11882-001-0027-1. [DOI] [PubMed] [Google Scholar]
- 66.Geha RS, Notarangelo LD, Casanova JL, Chapel H, Conley ME, Fischer A, Hammarström L, Nonoyama S, Ochs HD, Puck JM, Roifman C, Seger R, Wedgwood J International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee. Primary immunodeficiency diseases: an update from the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee. J Allergy Clin Immunol. 2007 Oct;120(4):776–794. doi: 10.1016/j.jaci.2007.08.053. Available from: http://dx.doi.org/10.1016/j.jaci.2007.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, Fieschi C, Thon V, Abedi MR, Hammarstrom L. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008 Jul;112(2):277–286. doi: 10.1182/blood-2007-11-124545. Available from: http://dx.doi.org/10.1182/blood-2007-11-124545. [DOI] [PubMed] [Google Scholar]
- 68.Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol. 1999 Jul;92(1):34–48. doi: 10.1006/clim.1999.4725. Available from: http://dx.doi.org/10.1006/clim.1999.4725. [DOI] [PubMed] [Google Scholar]
- 69.North ME, Ivory K, Funauchi M, Webster AD, Lane AC, Farrant J. Intracellular cytokine production by human CD4+ and CD8+ T cells from normal and immunodeficient donors using directly conjugated anti-cytokine antibodies and three-colour flow cytometry. Clin Exp Immunol. 1996 Sep;105(3):517–522. doi: 10.1046/j.1365-2249.1996.d01-795.x. Available from: http://dx.doi.org/10.1046/j.1365-2249.1996.d01-795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sillevis Smitt JH, Kuijpers TW. Cutaneous manifestations of primary immunodeficiency. Curr Opin Pediatr. 2013 Aug;25(4):492–497. doi: 10.1097/MOP.0b013e3283623b9f. Available from: http://dx.doi.org/10.1097/MOP.0b013e3283623b9f. [DOI] [PubMed] [Google Scholar]
- 71.Mechanic LJ, Dikman S, Cunningham-Rundles C. Granulomatous disease in common variable immunodeficiency. Ann Intern Med. 1997 Oct;127(8 Pt 1):613–617. doi: 10.7326/0003-4819-127-8_Part_1-199710150-00005. Available from: http://dx.doi.org/10.7326/0003-4819-127-8_Part_1-199710150-00005. [DOI] [PubMed] [Google Scholar]
- 72.Ardeniz O, Cunningham-Rundles C. Granulomatous disease in common variable immunodeficiency. Clin Immunol. 2009 Nov;133(2):198–207. doi: 10.1016/j.clim.2009.05.001. Available from: http://dx.doi.org/10.1016/j.clim.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Morimoto Y, Routes JM. Granulomatous disease in common variable immunodeficiency. Curr Allergy Asthma Rep. 2005 Sep;5(5):370–375. doi: 10.1007/s11882-005-0008-x. Available from: http://dx.doi.org/10.1007/s11882-005-0008-x. [DOI] [PubMed] [Google Scholar]
- 74.Arnold DF, Wiggins J, Cunningham-Rundles C, Misbah SA, Chapel HM. Granulomatous disease: distinguishing primary antibody disease from sarcoidosis. Clin Immunol. 2008 Jul;128(1):18–22. doi: 10.1016/j.clim.2008.03.510. Available from: http://dx.doi.org/10.1016/j.clim.2008.03.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Abolhassani H, Sagvand BT, Shokuhfar T, Mirminachi B, Rezaei N, Aghamohammadi A. A review on guidelines for management and treatment of common variable immunodeficiency. Expert Rev Clin Immunol. 2013 Jun;9(6):561–574. doi: 10.1586/eci.13.30. Available from: http://dx.doi.org/10.1586/eci.13.30. [DOI] [PubMed] [Google Scholar]
- 76.Winkelstein JA, Marino MC, Ochs H, Fuleihan R, Scholl PR, Geha R, Stiehm ER, Conley ME. The X-linked hyper-IgM syndrome: clinical and immunologic features of 79 patients. Medicine (Baltimore) 2003 Nov;82(6):373–384. doi: 10.1097/01.md.0000100046.06009.b0. Available from: http://dx.doi.org/10.1097/01.md.0000100046.06009.b0. [DOI] [PubMed] [Google Scholar]
- 77.Montella S, Maglione M, Giardino G, Di Giorgio A, Palamaro L, Mirra V, Ursini MV, Salerno M, Pignata C, Caffarelli C, Santamaria F. Hyper IgM syndrome presenting as chronic suppurative lung disease. Ital J Pediatr. 2012;38:45. doi: 10.1186/1824-7288-38-45. Available from: http://dx.doi.org/10.1186/1824-7288-38-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Notarangelo LD, Duse M, Ugazio AG. Immunodeficiency with hyper-IgM (HIM) Immunodefic Rev. 1992;3(2):101–121. [PubMed] [Google Scholar]
- 79.Erdos M, Durandy A, Maródi L. Genetically acquired class-switch recombination defects: the multi-faced hyper-IgM syndrome. Immunol Lett. 2005 Feb;97(1):1–6. doi: 10.1016/j.imlet.2004.09.021. Available from: http://dx.doi.org/10.1016/j.imlet.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 80.Goldstein MF, Kornstein MJ, Talbot S, Levinson AI. Acquired hyper-IgM syndrome with necrotizing granuloma. J Allergy Clin Immunol. 1985 Apr;75(4):472–478. doi: 10.1016/S0091-6749(85)80020-8. Available from: http://dx.doi.org/10.1016/S0091-6749(85)80020-8. [DOI] [PubMed] [Google Scholar]
- 81.Levy J, Espanol-Boren T, Thomas C, Fischer A, Tovo P, Bordigoni P, Resnick I, Fasth A, Baer M, Gomez L, Sanders EA, Tabone MD, Plantaz D, Etzioni A, Monafo V, Abinun M, Hammarstrom L, Abrahamsen T, Jones A, Finn A, Klemola T, DeVries E, Sanal O, Peitsch MC, Notarangelo LD. Clinical spectrum of X-linked hyper-IgM syndrome. J Pediatr. 1997 Jul;131(1 Pt 1):47–54. doi: 10.1016/S0022-3476(97)70123-9. Available from: http://dx.doi.org/10.1016/S0022-3476(97)70123-9. [DOI] [PubMed] [Google Scholar]
- 82.Winkelstein JA, Marino MC, Johnston RB, Jr, Boyle J, Curnutte J, Gallin JI, Malech HL, Holland SM, Ochs H, Quie P, Buckley RH, Foster CB, Chanock SJ, Dickler H. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore) 2000 May;79(3):155–169. doi: 10.1097/00005792-200005000-00003. Available from: http://dx.doi.org/10.1097/00005792-200005000-00003. [DOI] [PubMed] [Google Scholar]
- 83.Moltyaner Y, Geerts WH, Chamberlain DW, Heyworth PG, Noack D, Rae J, Doyle JJ, Downey GP. Underlying chronic granulomatous disease in a patient with bronchocentric granulomatosis. Thorax. 2003 Dec;58(12):1096–1098. doi: 10.1136/thorax.58.12.1096. Available from: http://dx.doi.org/10.1136/thorax.58.12.1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Finn A, Hadzić N, Morgan G, Strobel S, Levinsky RJ. Prognosis of chronic granulomatous disease. Arch Dis Child. 1990 Sep;65(9):942–945. doi: 10.1136/adc.65.9.942. Available from: http://dx.doi.org/10.1136/adc.65.9.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Khanna G, Kao SC, Kirby P, Sato Y. Imaging of chronic granulomatous disease in children. Radiographics. 2005 Sep-Oct;25(5):1183–1195. doi: 10.1148/rg.255055011. Available from: http://dx.doi.org/10.1148/rg.255055011. [DOI] [PubMed] [Google Scholar]
- 86.Segal BH, Leto TL, Gallin JI, Malech HL, Holland SM. Genetic, biochemical, and clinical features of chronic granulomatous disease. Medicine (Baltimore) 2000 May;79(3):170–200. doi: 10.1097/00005792-200005000-00004. Available from: http://dx.doi.org/10.1097/00005792-200005000-00004. [DOI] [PubMed] [Google Scholar]
- 87.Petty RE, Southwood TR, Baum J, Bhettay E, Glass DN, Manners P, Maldonado-Cocco J, Suarez-Almazor M, Orozco-Alcala J, Prieur AM. Revision of the proposed classification criteria for juvenile idiopathic arthritis: Durban, 1997. J Rheumatol. 1998 Oct;25(10):1991–1994. [PubMed] [Google Scholar]
- 88.Seid M, Opipari L, Huang B, Brunner HI, Lovell DJ. Disease control and health-related quality of life in juvenile idiopathic arthritis. Arthritis Rheum. 2009 Mar;61(3):393–399. doi: 10.1002/art.24477. Available from: http://dx.doi.org/10.1002/art.24477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Naz S, Mushtaq A, Rehman S, Bari A, Maqsud A, Khan MZ, Ahmad TM. Juvenile rheumatoid arthritis. J Coll Physicians Surg Pak. 2013 Jun;23(6):409–412. [PubMed] [Google Scholar]
- 90.Munro R, Porter DR, Sturrock RD. Lymphadenopathy in a patient with systemic onset juvenile chronic arthritis. Ann Rheum Dis. 1998 Sep;57(9):513–517. doi: 10.1136/ard.57.9.513. Available from: http://dx.doi.org/10.1136/ard.57.9.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO, 3rd, Birnbaum NS, Burmester GR, Bykerk VP, Cohen MD, Combe B, Costenbader KH, Dougados M, Emery P, Ferraccioli G, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010 Sep;62(9):2569–2581. doi: 10.1002/art.27584. Available from: http://dx.doi.org/10.1002/art.27584. [DOI] [PubMed] [Google Scholar]
- 92.Müller H, de Toledo FW, Resch KL. Fasting followed by vegetarian diet in patients with rheumatoid arthritis: a systematic review. Scand J Rheumatol. 2001;30(1):1–10. doi: 10.1080/030097401750065256. Available from: http://dx.doi.org/10.1080/030097401750065256. [DOI] [PubMed] [Google Scholar]
- 93.Hahn BH. Belimumab for systemic lupus erythematosus. N Engl J Med. 2013 Apr;368(16):1528–1535. doi: 10.1056/NEJMct1207259. Available from: http://dx.doi.org/10.1056/NEJMct1207259. [DOI] [PubMed] [Google Scholar]
- 94.Boehm I, Bieber T. Wasp-sting-triggered LE. Allergy. 2001 Jan;56(1):87–88. doi: 10.1034/j.1398-9995.2001.00761.x. Available from: http://dx.doi.org/10.1034/j.1398-9995.2001.00761.x. [DOI] [PubMed] [Google Scholar]
- 95.Von Feldt JM. Systemic lupus erythematosus. Recognizing its various presentations. Postgrad Med. 1995 Apr;97(4):79, 83, 86 passim. [PubMed] [Google Scholar]
- 96.Iqbal S, Sher MR, Good RA, Cawkwell GD. Diversity in presenting manifestations of systemic lupus erythematosus in children. J Pediatr. 1999 Oct;135(4):500–505. doi: 10.1016/S0022-3476(99)70174-5. Available from: http://dx.doi.org/10.1016/S0022-3476(99)70174-5. [DOI] [PubMed] [Google Scholar]
- 97.Kojima M, Nakamura S, Itoh H, Yoshida K, Asano S, Yamane N, Komatsumoto S, Ban S, Joshita T, Suchi T. Systemic lupus erythematosus (SLE) lymphadenopathy presenting with histopathologic features of Castleman' disease: a clinicopathologic study of five cases. Pathol Res Pract. 1997;193(8):565–571. doi: 10.1016/S0344-0338(97)80015-5. Available from: http://dx.doi.org/10.1016/S0344-0338(97)80015-5. [DOI] [PubMed] [Google Scholar]
- 98.Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, Schaller JG, Talal N, Winchester RJ. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982 Nov;25(11):1271–1277. doi: 10.1002/art.1780251101. Available from: http://dx.doi.org/10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 99.Janssens P, Arnaud L, Galicier L, Mathian A, Hie M, Sene D, Haroche J, Veyssier-Belot C, Huynh-Charlier I, Grenier PA, Piette JC, Amoura Z. Lupus enteritis: from clinical findings to therapeutic management. Orphanet J Rare Dis. 2013;8:67. doi: 10.1186/1750-1172-8-67. Available from: http://dx.doi.org/10.1186/1750-1172-8-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rosa Neto NS, Goldenstein-Schainberg C. Juvenile dermatomyositis: review and update of the pathogenesis and treatment. Rev Bras Reumatol. 2010 May-Jun;50(3):299–312. doi: 10.1590/S0482-50042010000300010. Available from: http://dx.doi.org/10.1590/S0482-50042010000300010. [DOI] [PubMed] [Google Scholar]
- 101.Kojima M, Nakamura S, Yamane Y, Tanaka H, Masawa N. Autoimmune disease-associated lymphadenopathy from dermatomyositis. A case report. Pathol Res Pract. 2003;199(10):691–694. doi: 10.1078/0344-0338-00482. Available from: http://dx.doi.org/10.1078/0344-0338-00482. [DOI] [PubMed] [Google Scholar]
- 102.Martini G, Calabrese F, Biscaro F, Zulian F. A child with dermatomyositis and a suspicious lymphadenopathy. J Rheumatol. 2005 Apr;32(4):744–746. [PubMed] [Google Scholar]
- 103.Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts) N Engl J Med. 1975 Feb;292(7):344–347. doi: 10.1056/NEJM197502132920706. Available from: http://dx.doi.org/10.1056/NEJM197502132920706. [DOI] [PubMed] [Google Scholar]
- 104.Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts) N Engl J Med. 1975 Feb;292(8):403–407. doi: 10.1056/NEJM197502202920807. Available from: http://dx.doi.org/10.1056/NEJM197502202920807. [DOI] [PubMed] [Google Scholar]
- 105.Feldman BM, Rider LG, Reed AM, Pachman LM. Juvenile dermatomyositis and other idiopathic inflammatory myopathies of childhood. Lancet. 2008 Jun;371(9631):2201–2212. doi: 10.1016/S0140-6736(08)60955-1. Available from: http://dx.doi.org/10.1016/S0140-6736(08)60955-1. [DOI] [PubMed] [Google Scholar]
- 106.Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. A newly recognized benign clinicopathological entity. Arch Pathol. 1969 Jan;87(1):63–70. [PubMed] [Google Scholar]
- 107.Hindermann W, Katenkamp D. Extranodale Rosai-Dorfman-Erkrankung (Sinushistiozytose mit massiver Lymphadenopathie). Ein Bericht über fünf Fallbeobachtungen. [Extranodal Rosai Dorfman disease (sinus histiocytosis with massive lymphadenopathy). Report of 5 cases]. Pathologe. 2004 May;25(3):222–228. doi: 10.1007/s00292-003-0642-9. (Ger). Available from: http://dx.doi.org/10.1007/s00292-003-0642-9. [DOI] [PubMed] [Google Scholar]
- 108.Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990 Feb;7(1):19–73. [PubMed] [Google Scholar]
- 109.Tomio R, Katayama M, Takenaka N, Imanishi T. Complications of surgical treatment of Rosai-Dorfman disease: A case report and review. Surg Neurol Int. 2012;3:1. doi: 10.4103/2152-7806.92161. Available from: http://dx.doi.org/10.4103/2152-7806.92161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Williams JW, Dorfman RF. Lymphadenopathy as the initial manifestation of histiocytosis X. Am J Surg Pathol. 1979 Oct;3(5):405–421. doi: 10.1097/00000478-197910000-00003. Available from: http://dx.doi.org/10.1097/00000478-197910000-00003. [DOI] [PubMed] [Google Scholar]
- 111.Favara BE, Jaffe R. The histopathology of Langerhans cell histiocytosis. Br J Cancer Suppl. 1994 Sep;23:S17–S23. [PMC free article] [PubMed] [Google Scholar]
- 112.McClain KL. Drug therapy for the treatment of Langerhans cell histiocytosis. Expert Opin Pharmacother. 2005 Nov;6(14):2435–2441. doi: 10.1517/14656566.6.14.2435. Available from: http://dx.doi.org/10.1517/14656566.6.14.2435. [DOI] [PubMed] [Google Scholar]
- 113.Usmani GN, Woda BA, Newburger PE. Advances in understanding the pathogenesis of HLH. Br J Haematol. 2013 Jun;161(5):609–622. doi: 10.1111/bjh.12293. Available from: http://dx.doi.org/10.1111/bjh.12293. [DOI] [PubMed] [Google Scholar]
- 114.Rosado FG, Kim AS. Hemophagocytic lymphohistiocytosis: an update on diagnosis and pathogenesis. Am J Clin Pathol. 2013 Jun;139(6):713–727. doi: 10.1309/AJCP4ZDKJ4ICOUAT. Available from: http://dx.doi.org/10.1309/AJCP4ZDKJ4ICOUAT. [DOI] [PubMed] [Google Scholar]
- 115.Bode SF, Lehmberg K, Maul-Pavicic A, Vraetz T, Janka G, Stadt UZ, Ehl S. Recent advances in the diagnosis and treatment of hemophagocytic lymphohistiocytosis. Arthritis Res Ther. 2012;14(3):213. doi: 10.1186/ar3843. Available from: http://dx.doi.org/10.1186/ar3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lehmberg K, Ehl S. Diagnostic evaluation of patients with suspected haemophagocytic lymphohistiocytosis. Br J Haematol. 2013 Feb;160(3):275–287. doi: 10.1111/bjh.12138. Available from: http://dx.doi.org/10.1111/bjh.12138. [DOI] [PubMed] [Google Scholar]
- 117.Henter JI, Elinder G, Ost A. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society. Semin Oncol. 1991 Feb;18(1):29–33. [PubMed] [Google Scholar]
- 118.Worth A, Thrasher AJ, Gaspar HB. Autoimmune lymphoproliferative syndrome: molecular basis of disease and clinical phenotype. Br J Haematol. 2006 Apr;133(2):124–140. doi: 10.1111/j.1365-2141.2006.05993.x. Available from: http://dx.doi.org/10.1111/j.1365-2141.2006.05993.x. [DOI] [PubMed] [Google Scholar]
- 119.Lim MS, Straus SE, Dale JK, Fleisher TA, Stetler-Stevenson M, Strober W, Sneller MC, Puck JM, Lenardo MJ, Elenitoba-Johnson KS, Lin AY, Raffeld M, Jaffe ES. Pathological findings in human autoimmune lymphoproliferative syndrome. Am J Pathol. 1998 Nov;153(5):1541–1550. doi: 10.1016/S0002-9440(10)65742-2. Available from: http://dx.doi.org/10.1016/S0002-9440(10)65742-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Straus SE, Sneller M, Lenardo MJ, Puck JM, Strober W. An inherited disorder of lymphocyte apoptosis: the autoimmune lymphoproliferative syndrome. Ann Intern Med. 1999 Apr;130(7):591–601. doi: 10.7326/0003-4819-130-7-199904060-00020. Available from: http://dx.doi.org/10.7326/0003-4819-130-7-199904060-00020. [DOI] [PubMed] [Google Scholar]
- 121.Rabinowitz MR, Levi J, Conard K, Shah UK. Castleman disease in the pediatric neck: a literature review. Otolaryngol Head Neck Surg. 2013 Jun;148(6):1028–1036. doi: 10.1177/0194599813479931. Available from: http://dx.doi.org/10.1177/0194599813479931. [DOI] [PubMed] [Google Scholar]
- 122.Castleman B, Towne VW. Case records of the Massachusetts General Hospital: Case No. 40231. N Engl J Med. 1954 Jun;250(23):1001–1005. doi: 10.1056/NEJM195406102502308. Available from: http://dx.doi.org/10.1056/NEJM195406102502308. [DOI] [PubMed] [Google Scholar]
- 123.Lin CY, Chang YL. Castleman's disease in the head and neck region: Meta-analysis of reported cases in Taiwan and literature review. J Formos Med Assoc. 2010 Dec;109(12):913–920. doi: 10.1016/S0929-6646(10)60139-8. Available from: http://dx.doi.org/10.1016/S0929-6646(10)60139-8. [DOI] [PubMed] [Google Scholar]
- 124.Bonekamp D, Horton KM, Hruban RH, Fishman EK. Castleman disease: the great mimic. Radiographics. 2011 Oct;31(6):1793–1807. doi: 10.1148/rg.316115502. Available from: http://dx.doi.org/10.1148/rg.316115502. [DOI] [PubMed] [Google Scholar]
- 125.Cremer H, Rieger C. Epidemiology of Kawasaki syndrome in Germany (FRG) Prog Clin Biol Res. 1987;250:61–65. [PubMed] [Google Scholar]
- 126.Yim D, Curtis N, Cheung M, Burgner D. An update on Kawasaki disease II: clinical features, diagnosis, treatment and outcomes. J Paediatr Child Health. 2013 Aug;49(8):614–623. doi: 10.1111/jpc.12221. Available from: http://dx.doi.org/10.1111/jpc.12221. [DOI] [PubMed] [Google Scholar]
- 127.Mandal S, Pande A, Mandal D, Sarkar A, Kahali D, Panja M. Various coronary artery complications of Kawasaki disease: Series of 5 cases and review of literature. J Cardiovasc Dis Res. 2012 Jul;3(3):231–235. doi: 10.4103/0975-3583.98900. Available from: http://dx.doi.org/10.4103/0975-3583.98900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Alves NR, Magalhães CM, Almeida Rde F, Santos RC, Gandolfi L, Pratesi R. Prospective study of Kawasaki disease complications: review of 115 cases. Rev Assoc Med Bras. 2011 May-Jun;57(3):295–300. [PubMed] [Google Scholar]
- 129.Gerding R. Kawasaki disease: a review. J Pediatr Health Care. 2011 Nov-Dec;25(6):379–387. doi: 10.1016/j.pedhc.2011.07.007. Available from: http://dx.doi.org/10.1016/j.pedhc.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 130.Furukawa S, Matsubara T, Umezawa Y, Motohashi T, Ino T, Yabuta K. Pentoxifylline and intravenous gamma globulin combination therapy for acute Kawasaki disease. Eur J Pediatr. 1994 Sep;153(9):663–667. doi: 10.1007/BF02190688. Available from: http://dx.doi.org/10.1007/BF02190688. [DOI] [PubMed] [Google Scholar]
- 131.Newburger JW, Takahashi M, Gerber MA, Gewitz MH, Tani LY, Burns JC, Shulman ST, Bolger AF, Ferrieri P, Baltimore RS, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Pediatrics. 2004 Dec;114(6):1708–1733. doi: 10.1542/peds.2004-2182. Available from: http://dx.doi.org/10.1542/peds.2004-2182. [DOI] [PubMed] [Google Scholar]
- 132.Williams RV, Wilke VM, Tani LY, Minich LL. Does Abciximab enhance regression of coronary aneurysms resulting from Kawasaki disease? Pediatrics. 2002 Jan;109(1):E4. doi: 10.1542/peds.109.1.e4. [DOI] [PubMed] [Google Scholar]
- 133.Bosch X, Guilabert A. Kikuchi-Fujimoto disease. Orphanet J Rare Dis. 2006;1:18. doi: 10.1186/1750-1172-1-18. Available from: http://dx.doi.org/10.1186/1750-1172-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bosch X, Guilabert A, Miquel R, Campo E. Enigmatic Kikuchi-Fujimoto disease: a comprehensive review. Am J Clin Pathol. 2004 Jul;122(1):141–152. doi: 10.1309/YF08-1L4T-KYWV-YVPQ. Available from: http://dx.doi.org/10.1309/YF08-1L4T-KYWV-YVPQ. [DOI] [PubMed] [Google Scholar]
- 135.Lin HC, Su CY, Huang CC, Hwang CF, Chien CY. Kikuchi's disease: a review and analysis of 61 cases. Otolaryngol Head Neck Surg. 2003;128(5):650–653. doi: 10.1016/S0194-5998(02)23291-X. Available from: http://dx.doi.org/10.1016/S0194-5998(02)23291-X. [DOI] [PubMed] [Google Scholar]
- 136.Hutchinson CB, Wang E. Kikuchi-Fujimoto disease. Arch Pathol Lab Med. 2010 Feb;134(2):289–293. doi: 10.5858/134.2.289. [DOI] [PubMed] [Google Scholar]
- 137.Feder HM, Salazar JC. A clinical review of 105 patients with PFAPA (a periodic fever syndrome) Acta Paediatr. 2010 Feb;99(2):178–184. doi: 10.1111/j.1651-2227.2009.01554.x. Available from: http://dx.doi.org/10.1111/j.1651-2227.2009.01554.x. [DOI] [PubMed] [Google Scholar]
- 138.Lee WI, Yang MH, Lee KF, Chen LC, Lin SJ, Yeh KW, Huang JL. PFAPA syndrome (Periodic Fever, Aphthous stomatitis, Pharyngitis, Adenitis) Clin Rheumatol. 1999;18(3):207–213. doi: 10.1007/s100670050086. Available from: http://dx.doi.org/10.1007/s100670050086. [DOI] [PubMed] [Google Scholar]
- 139.Orth M, Bellosta S. Cholesterol: its regulation and role in central nervous system disorders. Cholesterol. 2012;2012:292598. doi: 10.1155/2012/292598. Available from: http://dx.doi.org/10.1155/2012/292598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Patterson MC. A riddle wrapped in a mystery: understanding Niemann-Pick disease, type C. Neurologist. 2003 Nov;9(6):301–310. doi: 10.1097/01.nrl.0000094627.78754.5b. Available from: http://dx.doi.org/10.1097/01.nrl.0000094627.78754.5b. [DOI] [PubMed] [Google Scholar]
- 141.Héron B, Valayannopoulos V, Baruteau J, Chabrol B, Ogier H, Latour P, Dobbelaere D, Eyer D, Labarthe F, Maurey H, Cuisset JM, de Villemeur TB, Sedel F, Vanier MT. Miglustat therapy in the French cohort of paediatric patients with Niemann-Pick disease type C. Orphanet J Rare Dis. 2012;7:36. doi: 10.1186/1750-1172-7-36. Available from: http://dx.doi.org/10.1186/1750-1172-7-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Grabowski GA. Gaucher disease and other storage disorders. Hematology Am Soc Hematol Educ Program. 2012;2012:13–18. doi: 10.1182/asheducation-2012.1.13. [DOI] [PubMed] [Google Scholar]
- 143.Lee BH, Kim DY, Kim GH, Cho KJ, Yoon HK, Yoo HW. Progressive mesenteric lymphadenopathy with protein-losing enteropathy; a devastating complication in Gaucher disease. Mol Genet Metab. 2012 Mar;105(3):522–524. doi: 10.1016/j.ymgme.2011.12.010. Available from: http://dx.doi.org/10.1016/j.ymgme.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 144.Cox TM, Amato D, Hollak CE, Luzy C, Silkey M, Giorgino R, Steiner RD Miglustat Maintenance Study Group. Evaluation of miglustat as maintenance therapy after enzyme therapy in adults with stable type 1 Gaucher disease: a prospective, open-label non-inferiority study. Orphanet J Rare Dis. 2012;7:102. doi: 10.1186/1750-1172-7-102. Available from: http://dx.doi.org/10.1186/1750-1172-7-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sampietro T, Puntoni M, Bigazzi F, Pennato B, Sbrana F, Dal Pino B, Azzarà A, Magagnini E, Perossini P, Cei G, Bacci G. Images in cardiovascular medicine. Tangier disease in severely progressive coronary and peripheral artery disease. Circulation. 2009 May;119(20):2741–2742. doi: 10.1161/CIRCULATIONAHA.108.812164. Available from: http://dx.doi.org/10.1161/CIRCULATIONAHA.108.812164. [DOI] [PubMed] [Google Scholar]
- 146.Puntoni M, Sbrana F, Bigazzi F, Sampietro T. Tangier disease: epidemiology, pathophysiology, and management. Am J Cardiovasc Drugs. 2012 Oct;12(5):303–311. doi: 10.1007/BF03261839. Available from: http://dx.doi.org/10.1007/BF03261839. [DOI] [PubMed] [Google Scholar]
- 147.Nofer JR, Remaley AT. Tangier disease: still more questions than answers. Cell Mol Life Sci. 2005 Oct;62(19-20):2150–2160. doi: 10.1007/s00018-005-5125-0. Available from: http://dx.doi.org/10.1007/s00018-005-5125-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Westermark P, Benson MD, Buxbaum JN, Cohen AS, Frangione B, Ikeda S, Masters CL, Merlini G, Saraiva MJ, Sipe JD. A primer of amyloid nomenclature. Amyloid. 2007 Sep;14(3):179–183. doi: 10.1080/13506120701460923. Available from: http://dx.doi.org/10.1080/13506120701460923. [DOI] [PubMed] [Google Scholar]
- 149.Sipe JD, Benson MD, Buxbaum JN, Ikeda S, Merlini G, Saraiva MJ, Westermark P. Amyloid fibril protein nomenclature: 2010 recommendations from the nomenclature committee of the International Society of Amyloidosis. Amyloid. 2010 Sep;17(3-4):101–104. doi: 10.3109/13506129.2010.526812. Available from: http://dx.doi.org/10.3109/13506129.2010.526812. [DOI] [PubMed] [Google Scholar]
- 150.Ponvert C, Scheinmann P. Vaccine allergy and pseudo-allergy. Eur J Dermatol. 2003 Jan-Feb;13(1):10–15. [PubMed] [Google Scholar]
- 151.Vessal S, Kravis LP. Immunologic mechanisms responsible for adverse reactions to routine immunizations in children. Clin Pediatr (Phila) 1976 Aug;15(8):688–696. doi: 10.1177/000992287601500805. Available from: http://dx.doi.org/10.1177/000992287601500805. [DOI] [PubMed] [Google Scholar]
- 152.Platt R, Dreis MW, Kennedy DL, Kuritsky JN. Serum sickness-like reactions to amoxicillin, cefaclor, cephalexin, and trimethoprim-sulfamethoxazole. J Infect Dis. 1988 Aug;158(2):474–477. doi: 10.1093/infdis/158.2.474. Available from: http://dx.doi.org/10.1093/infdis/158.2.474. [DOI] [PubMed] [Google Scholar]
- 153.Ferrer R. Lymphadenopathy: differential diagnosis and evaluation. Am Fam Physician. 1998 Oct;58(6):1313–1320. [PubMed] [Google Scholar]
- 154.Eshki M, Allanore L, Musette P, Milpied B, Grange A, Guillaume JC, Chosidow O, Guillot I, Paradis V, Joly P, Crickx B, Ranger-Rogez S, Descamps V. Twelve-year analysis of severe cases of drug reaction with eosinophilia and systemic symptoms: a cause of unpredictable multiorgan failure. Arch Dermatol. 2009 Jan;145(1):67–72. doi: 10.1001/archderm.145.1.67. Available from: http://dx.doi.org/10.1001/archderm.145.1.67. [DOI] [PubMed] [Google Scholar]
- 155.Bollard CM, Lim MS, Gross TG COG Non-Hodgkin Lymphoma Committee. Children's Oncology Group's 2013 blueprint for research: non-Hodgkin lymphoma. Pediatr Blood Cancer. 2013 Jun;60(6):979–984. doi: 10.1002/pbc.24416. Available from: http://dx.doi.org/10.1002/pbc.24416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ekström-Smedby K. Epidemiology and etiology of non-Hodgkin lymphoma--a review. Acta Oncol. 2006;45(3):258–271. doi: 10.1080/02841860500531682. Available from: http://dx.doi.org/10.1080/02841860500531682. [DOI] [PubMed] [Google Scholar]
- 157.Demurtas A, Accinelli G, Pacchioni D, Godio L, Novero D, Bussolati G, Palestro G, Papotti M, Stacchini A. Utility of flow cytometry immunophenotyping in fine-needle aspirate cytologic diagnosis of non-Hodgkin lymphoma: A series of 252 cases and review of the literature. Appl Immunohistochem Mol Morphol. 2010 Jul;18(4):311–322. doi: 10.1097/PAI.0b013e3181827da8. Available from: http://dx.doi.org/10.1097/PAI.0b013e3181827da8. [DOI] [PubMed] [Google Scholar]
- 158.Gupta S, Yeh S, Chami R, Punnett A, Chung C. The prognostic impact of tumour-associated macrophages and Reed-Sternberg cells in paediatric Hodgkin lymphoma. Eur J Cancer. 2013 Oct;49(15):3255–3261. doi: 10.1016/j.ejca.2013.05.024. Available from: http://dx.doi.org/10.1016/j.ejca.2013.05.024. [DOI] [PubMed] [Google Scholar]
- 159.Kelly KM. Management of children with high-risk Hodgkin lymphoma. Br J Haematol. 2012 Apr;157(1):3–13. doi: 10.1111/j.1365-2141.2011.08975.x. Available from: http://dx.doi.org/10.1111/j.1365-2141.2011.08975.x. [DOI] [PubMed] [Google Scholar]
- 160.Freed J, Kelly KM. Current approaches to the management of pediatric Hodgkin lymphoma. Paediatr Drugs. 2010 Apr;12(2):85–98. doi: 10.2165/11316170-000000000-00000. Available from: http://dx.doi.org/10.2165/11316170-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 161.Devita VT, Jr, Serpick AA, Carbone PP. Combination chemotherapy in the treatment of advanced Hodgkin's disease. Ann Intern Med. 1970 Dec;73(6):881–895. doi: 10.7326/0003-4819-73-6-881. Available from: http://dx.doi.org/10.7326/0003-4819-73-6-881. [DOI] [PubMed] [Google Scholar]
- 162.Bene MC, Castoldi G, Knapp W, Ludwig WD, Matutes E, Orfao A, van't Veer MB. Proposals for the immunological classification of acute leukemias. European Group for the Immunological Characterization of Leukemias (EGIL) Leukemia. 1995 Oct;9(10):1783–1786. [PubMed] [Google Scholar]
- 163.Pui CH, Relling MV, Downing JR. Acute lymphoblastic leukemia. N Engl J Med. 2004 Apr;350(15):1535–1548. doi: 10.1056/NEJMra023001. Available from: http://dx.doi.org/10.1056/NEJMra023001. [DOI] [PubMed] [Google Scholar]
- 164.Thalhammer-Scherrer R, Mitterbauer G, Simonitsch I, Jaeger U, Lechner K, Schneider B, Fonatsch C, Schwarzinger I. The immunophenotype of 325 adult acute leukemias: relationship to morphologic and molecular classification and proposal for a minimal screening program highly predictive for lineage discrimination. Am J Clin Pathol. 2002 Mar;117(3):380–389. doi: 10.1309/C38D-D8J3-JU3E-V6EE. Available from: http://dx.doi.org/10.1309/C38D-D8J3-JU3E-V6EE. [DOI] [PubMed] [Google Scholar]
- 165.Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006 Jan;354(2):166–178. doi: 10.1056/NEJMra052603. Available from: http://dx.doi.org/10.1056/NEJMra052603. [DOI] [PubMed] [Google Scholar]
- 166.Rubnitz JE, Inaba H. Childhood acute myeloid leukaemia. Br J Haematol. 2012 Nov;159(3):259–276. doi: 10.1111/bjh.12040. Available from: http://dx.doi.org/10.1111/bjh.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Creutzig U, van den Heuvel-Eibrink MM, Gibson B, Dworzak MN, Adachi S, de Bont E, Harbott J, Hasle H, Johnston D, Kinoshita A, Lehrnbecher T, Leverger G, Mejstrikova E, Meshinchi S, Pession A, Raimondi SC, Sung L, Stary J, Zwaan CM, Kaspers GJ, Reinhardt D AML Committee of the International BFM Study Group. Diagnosis and management of acute myeloid leukemia in children and adolescents: recommendations from an international expert panel. Blood. 2012 Oct;120(16):3187–3205. doi: 10.1182/blood-2012-03-362608. Available from: http://dx.doi.org/10.1182/blood-2012-03-362608. [DOI] [PubMed] [Google Scholar]
- 168.Rubnitz JE. Childhood acute myeloid leukemia. Curr Treat Options Oncol. 2008 Feb;9(1):95–105. doi: 10.1007/s11864-008-0059-z. Available from: http://dx.doi.org/10.1007/s11864-008-0059-z. [DOI] [PubMed] [Google Scholar]
- 169.Raney RB, Anderson JR, Barr FG, Donaldson SS, Pappo AS, Qualman SJ, Wiener ES, Maurer HM, Crist WM. Rhabdomyosarcoma and undifferentiated sarcoma in the first two decades of life: a selective review of intergroup rhabdomyosarcoma study group experience and rationale for Intergroup Rhabdomyosarcoma Study V. J Pediatr Hematol Oncol. 2001 May;23(4):215–220. doi: 10.1097/00043426-200105000-00008. Available from: http://dx.doi.org/10.1097/00043426-200105000-00008. [DOI] [PubMed] [Google Scholar]
- 170.Rudzinski ER. Histology and fusion status in rhabdomyosarcoma. Am Soc Clin Oncol Educ Book. 2013:425–428. doi: 10.1200/EdBook_AM.2013.33.425. Available from: http://dx.doi.org/10.1200/EdBook_AM.2013.33.425. [DOI] [PubMed] [Google Scholar]
- 171.Walterhouse D, Watson A. Optimal management strategies for rhabdomyosarcoma in children. Paediatr Drugs. 2007;9(6):391–400. doi: 10.2165/00148581-200709060-00006. Available from: http://dx.doi.org/10.2165/00148581-200709060-00006. [DOI] [PubMed] [Google Scholar]
- 172.Kendall JM. Designing a research project: randomised controlled trials and their principles. Emerg Med J. 2003 Mar;20(2):164–168. doi: 10.1136/emj.20.2.164. Available from: http://dx.doi.org/10.1136/emj.20.2.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Park JR, Eggert A, Caron H. Neuroblastoma: biology, prognosis, and treatment. Pediatr Clin North Am. 2008 Feb;55(1):97–120, x. doi: 10.1016/j.pcl.2007.10.014. Available from: http://dx.doi.org/10.1016/j.pcl.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 174.Zage PE, Louis CU, Cohn SL. New aspects of neuroblastoma treatment: ASPHO 2011 symposium review. Pediatr Blood Cancer. 2012 Jul;58(7):1099–1105. doi: 10.1002/pbc.24116. Available from: http://dx.doi.org/10.1002/pbc.24116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Afqir S, Ismaili N, Alaoui K, Ahid S, Lotz JP, Horn E, Bouhafa T, Abouqal R, Errihani H. Nasopharyngeal carcinoma in adolescents: a retrospective review of 42 patients. Eur Arch Otorhinolaryngol. 2009 Nov;266(11):1767–1773. doi: 10.1007/s00405-009-0911-1. Available from: http://dx.doi.org/10.1007/s00405-009-0911-1. [DOI] [PubMed] [Google Scholar]
- 176.Cannon T, Zanation AM, Lai V, Weissler MC. Nasopharyngeal carcinoma in young patients: a systematic review of racial demographics. Laryngoscope. 2006 Jun;116(6):1021–1026. doi: 10.1097/01.mlg.0000217243.08756.0c. Available from: http://dx.doi.org/10.1097/01.mlg.0000217243.08756.0c. [DOI] [PubMed] [Google Scholar]