

Abstract
In regenerative medicine, human cord blood-derived multipotent mesenchymal stromal cells (CBMSCs) stand out for their biological peculiarities demonstrated in in vitro and in vivo preclinical studies. Here, we present our 9-year experience for the consistent isolation of CBMSCs. Although nearly one CB unit out of two retains the potential to give rise to MSC colonies, only 46% of them can be cultured till low passages (P≥4), but one-fourth of those reaches even higher passages (P≥8). Subsequent characterization for morphological, clonal, differentiation, and proliferation properties revealed two divergent CBMSC behaviors. In particular, a cumulative population doublings cut-off (CPD=15) was identified that undoubtedly distinguishes two growth curves, and different degrees of commitment toward osteogenesis were observed. These data clearly show the existence of at least two distinct CBMSC subsets: one mainly short-living and less proliferative (SL-CBMSCs), the other long-living, with higher growth rate, and, very importantly, with significantly (P≤0.01) longer telomere (LL-CBMSCs). Moreover, significant differences in the immunoprofile before seeding were found among CB units giving rise to LL-CBMSCs or SL-CBMSCs or showing no colony formation. Finally, all the aforementioned results provided a peculiar and useful set of parameters potentially predictive for CBMSC culture outcome.
Introduction
In the past decades, multipotent mesenchymal stromal cells (MSCs) have been widely studied for their therapeutic properties and have gained a primary role in the field of cell therapy and regenerative medicine (http://clinicaltrials.gov/ct2/results?term=MSC&Search=Search, 03/11/2014). In particular, they have been proposed as a potent tool for the treatment of several diseases characterized by degeneration of affected tissues, but also in other innovative approaches, due to their homing capabilities, immunomodulatory effects, drugstore features, and differentiative potential [1]. These cells were first described in adult tissues [2–4], but in recent years, they have been found in many fetal tissues as well [5–7]. Thus, even if a definition for MSCs has been provided by the International Society for Cellular Therapy (ISCT) [8], this acronym comprises a wide range of cells obtained from various sources that frequently possess different biological characteristics [9–11].
In this context, cord blood-derived multipotent mesenchymal stromal cells (CBMSCs) [6,12] represent a peculiar population since its source is blood and not a solid tissue, and they show higher proliferative capabilities compared with other MSCs [9] and peculiar morphological, differentiative, and trophic properties [13]. Another unique feature of CBMSCs is their low immunogenicity, which makes them suitable for allogenic cell therapies [14–16].
Despite all the promising and intriguing aspects of CBMSCs, translation to the clinic has to face one major obstacle, that is inconsistency of isolation protocols due to the low frequency of these cells in cord blood if compared to other sources [17]. Although a growing number of reports point out experimental and sample conditions for optimal isolation of CBMSCs, the success rate is still controversial and they often require very fresh and abundant cord blood units [18–21]. A further issue that could be related to the low isolation rate is the origin of this stromal population in cord blood. For instance, one of the theories trying to explain the presence of MSCs in various tissues considers pericytes as their direct precursors and a sort of in vivo counterpart [22]. Yet, it is not clear why perivascular cells should abandon their niche and should be circulating in cord blood. In addition, cord blood was reported to give rise to distinct stromal populations in terms of morphology, isolation frequency, proliferation rate, differentiation potentialities, and surface markers [23–26].
Starting from these premises, the goal of this work was first of all to summarize our 9-year experience in the field of CBMSCs. Moreover, we wanted to investigate if any parameter existed that could answer to the following questions: do all the CB units have the same potential to produce stromal cells? Does cord blood contain more than one stromal cell population? And finally, can we predict the cultural outcome of cord blood cell samples?
Materials and Methods
Isolation and culture of human cord blood multipotent MSCs
Umbilical cord blood was collected from normal deliveries in a multiple system bag (Machopharma, Mouvax, France) containing 29 mL of citrate phosphate dextrose as anticoagulant. The authors state that this study was performed according to the amended Declaration of Helsinki. In addition, written informed consent has been obtained for all the cord blood units used in the study. Different cell isolation approaches were carried out to obtain CBMSCs. When specified, lysis of red blood cells was performed: an aliquot of sample was diluted 1:9 in lysing buffer 1X (BD Pharm Lyse 10X; BD Biosciences, San Diego, CA), incubated at room temperature for 10 min, and then centrifuged at 1,200 rpm for 8 min. The approaches included:
(a) Samples were seeded as whole blood at 0.5×106 total nucleated cells (TNCs)/cm2.
(b) Alternatively, mononuclear cells (MNCs) from buffy coat were seeded at a density of 0.5×106 cells/cm2. Briefly, whole blood was centrifuged at 1,900 rpm for 15 min, the plasma subsequently discarded, and the buffy coat and a portion of red blood cells collected with a serological pipette, pelleted at 1,200 rpm for 8 min, and resuspended in the complete medium.
(c) Sepax S-100 system (Biosafe, Eysins, Switzerland) was exploited for automated cell separation. CB units were processed directly by the device without external operator manipulation. MNCs were automatically harvested in a bag and subsequently resuspended in the complete medium for seeding at 1×106 cells/cm2.
(d) Another isolation approach was to obtain MSC precursors by immunodepletion using a commercial kit (RosetteSep Mesenchymal Stem Cell enrichment cocktail; StemCell Technologies, Vancouver, BC, Canada) that negatively selects CD3+, CD14+, CD38+, CD19+, glycophorin A, and CD66b+ cells. In summary, we obtained the buffy coat as previously described (b), and the harvested MNCs and a portion of red blood cells were incubated with 50 μL/mL RosetteSep MSC enrichment cocktail for 20 min at room temperature. The sample was then diluted 1:2 with phosphate-buffered saline (PBS; Gibco, Grand Island, NY), ethylenediaminetetraacetic acid (Sigma-Aldrich, St. Louis, MO), and human serum albumin (Kedrion, Lucca, Italy) and separated under standard density gradient conditions (Ficoll Paque Plus 1.077±0.001 g/L; GE Healthcare Europe, Freiburg, Germany). One million cells was collected for the assessment of the immunophenotype, whereas the remaining cells were resuspended in the complete medium and then plated at 1×106 cells/cm2.
The complete medium consisted of alphaMEM-GlutaMAX (Invitrogen, Carlsdad, CA) supplemented with 20% fetal bovine serum (FBS; Biochrom, Berlin, Germany). Cultures were maintained at 37°C in a humidified atmosphere containing 5% CO2. After 48 h, nonadherent cells were removed and the fresh medium was added; the culture medium was changed every 3 days; nonadherent cells in suspension in the supernatants were collected for immunophenotype assessment. At 80% confluence, the cells were harvested using 25% TrypLE Select 1X (Gibco) and were washed with PBS (Gibco) and subcultured at a concentration of 2–4×103 cells/cm2. Adherent cells that did not give rise to colonies after 40 days were detached with a scraper and collected for the assessment of the immunophenotype (n=10). Bone marrow-derived (BM) MSCs were obtained as elsewhere reported [27].
Cumulative population doublings
Population doubling (PD) was calculated for each population using the following equation: PD=log10(N)/log10(2), where N is the number of cells harvested at the end of the culture/the number of seeded cells. Cumulative PD was calculated adding to the PD of the passage under analysis the PDs of the previous passages.
Morphology analysis
Images of cells in culture were taken with a Nikon Eclipse TS100 microscope. Measurements of the nucleus diameter and of major cell axis (n=10 for each cell type) were acquired and processed with the Media Cybernetics Image-Pro Plus 2 (Media Cybernetics, Rockville, MD). For each cell, the ratio between the two parameters (cell major axis/nucleus diameter) was calculated. This value was considered indicative of cell morphology and used to distinguish more spindle-like cells from those having a more compact shape.
Colony forming unit-fibroblast assay
Two hundred CBMSCs (P4) were plated per 100-mm Petri dish (BD Biosciences) in alphaMEM-GlutaMAX (Invitrogen) supplemented with 20% FBS (Biochrom). Four CBMSCs in duplicate were analyzed [two long-living CBMSCs (LL-CBMSCs) and two short-living CBMSCs (SL-CBMSCs)]. The medium was changed weekly, and after 2 weeks, the cells were washed with PBS (Gibco), fixed with methanol (154903; Sigma-Aldrich), and stained with the carbolic gentian violet solution (Ral diagnostics, Cedex, France). After two washing steps with milliQ-grade water, colonies with diameter bigger than 1–2 mm were counted.
Telomere length assessment
Pellets of CBMSCs at different passages were collected during cell culture and stored at −80°C for subsequent analysis. DNA was extracted with the QIAamp DNA Blood Mini Kit (51104; Qiagen, Hilden, Germany) following the instructions provided. A pool of cells from different CBMSC populations and at different passages was used to build the standard curve by 1:1 serial dilution (100–1.56 ng). Real-time quantitative polymerase chain reaction was performed on a Bio-Rad CFX96 preparing one 96-well plate for telomere and one for single copy reference gene (36B4). The experiment outline, the primers, and the reaction mixes were based on the methodology presented in a recent report [28]. The thermal profile was set up for optimal activity of SsoFast EvaGreen Supermix: enzyme activation at 98°C (30 s), 46 cycles of 95°C (5 s), 54°C (2 min), and a final stage at 62°C (5 s) for telomere assay plate; enzyme activation at 98°C (30 s), 46 cycles of 95°C (5 s), 58°C (1 min), and a final stage at 62°C (5 s) for reference gene assay plate. The analysis of RT data was carried out using Bio-Rad CFX Manager software.
Differentiation into mesodermal lineages and staining of specialized cells
To promote adipogenesis and osteogenesis, commercial media (Lonza, Basel, Switzerland) were used, and the manufacturer's protocols were followed or slightly modified. Five CBMSC populations at different passages (P3–P7) were tested for multipotent differentiative potential.
For adipogenesis, cells were seeded at a density of 2.1×104 per cm2 in alphaMEM-GlutaMAX (Invitrogen) 20% FBS (Biochrom); the medium was changed with fresh one every 2–3 days. When cells reached 100% confluence, three (standard protocol) or five to six (modified protocol) cycles of induction/maintenance were performed; induction step consisted of 7 days in the human MSC adipogenesis induction medium (PT-3102B), with a medium change after 3 days; for the maintenance step, the medium was switched to the human MSC adipogenesis maintenance medium (PT-3102A). The final maintenance cycle lasted 7 days, during which the medium was changed every 2–3 days. Adipogenic differentiation was detected by microscopic observation, and lipid vacuoles were stained with the fresh Oil Red O solution (O0625; Sigma-Aldrich) following the manufacturer's instructions.
For osteogenesis, 3.1×103 CBMSC/cm2 were plated in alphaMEM-GlutaMAX (Invitrogen) plus 20% FBS (Biochrom); the day after the medium was substituted with the human MSC osteogenesis induction medium (PT-3002). The cells were maintained in culture for 3 weeks, with medium changes every 3–4 days.
Calcium deposits were stained with Alizarin Red S (A5533; Sigma-Aldrich). Briefly, cells were washed with PBS (Gibco), fixed for 30 min at 4°C with EtOH (02870-1L-F; Sigma-Aldrich) 70% in water, washed with PBS (Gibco), stained for 15 min at RT with the Alizarin Red S solution, and finally multiple washing steps were performed with PBS (Gibco) putting the cells onto an orbital shaker, until extra staining solution and debris were cleaned out. Images of lipid vacuoles and calcium deposits were acquired on a Nikon Eclipse TS100 microscope.
Gene expression
Isolation of total RNA and real-time quantitative reverse transcription polymerase chain reaction assays for the assessment of adipogenic and osteogenic differentiation were conducted as previously reported [29]. The following primers were used:
FW 5′-CATTCCATTCACAAGAACAGAT-3′, RV 5′-GGCTTATTGTAGAGCTGAGT-3′ (PPARG); FW 5′-GGA GCAGTGGGTGCTGAG-3′, RV 5′-AGCTGTAGCAGCA GGAGGTC-3′ (CFD); FW 5′-GCTACAGACGAGGAC ATC-3′, RV 5′-ATATAATCTGGACTGCTTGTG-3′ (OPN); FW 5′-TACAAGGTGGTGGGCGGTGAACGA-3′, RV 5′-TGGCGCAGGGGCACAGCAGAC-3′ (ALP); FW 5′-CGG AAGACCCCAGTCCAGATCCA-3′, RV 5′-TGCCCTCGG GAGACCTGCAA-3′ (THY1); FW 5′-TGTGGGCTCCAA GCAGATGCA-3′, RV 5′-GCAGCAGTTTCTCCAGAGCT GGG-3′ (RPLP0); FW 5′-CATAGGAAGCTGGGAGC AAG-3′, RV 5′-GCCCTCCAATCAGTCTTCTG-3′ (RPL13alpha); FW 5′-GCCACGCCAGCTTCGGAGAG-3′, RV 5′-CCGCAGCAAACCGCTTGGGA-3′ (TBP); FW 5′-CCGCTGGTGATGACAAGAAAGGGAT-3′, RV 5′-AGGGCCAGACCCAGTCTGATAGGA-3′ (YHWAZ).
Flow cytometry
The previously described collected cell samples were characterized by flow cytometry. A total of three CB immunodepleted buffy coat samples for each possible cell culture outcome were analyzed, and at least three measurements per marker were taken for each sample. Cells were washed with PBS (Gibco), centrifuged at 1,200 rpm for 8 min, and resuspended in PBS (Gibco) plus 2% FBS (Biochrom) or washed in PBS (Gibco) for 20 min at RT and then incubated in the dark with the following mixes of directly coupled mouse anti-human fluorochrome-conjugated antibodies: CD45-FITC (21270453X2; Miltenyi Biotec, Bergisch Gladbach, Germany), CD73-PE (550257; BD Biosciences), CD34-PE-Cy5 (A07777; Beckman Coulter, Brea, CA), CD117-PE-Cy7 (PNIM3698; Beckman Coulter), CD90-APC (559869; BD Biosciences), CD146-APC-Cy7 (5050-B100T; BioCytex, Marseille, France), CD105-Pacific Blue (PB298T100; Exbio, Vestec, Czech Republic) for the first mix; CD31-FITC (555445; BD Biosciences), CD144-PE (PNA07481; Beckman Coulter), CD34-PE-Cy5 (Beckman Coulter), CD117-PE-Cy7 (Beckman Coulter), CD133-APC (130-092-880; Miltenyi Biotec), CD146-APC-Cy7 (BioCytex), CD45-Pacific Blue (130092880; Miltenyi Biotec) for the second mix; alphaSMA-FITC (F3777; Sigma-Aldrich), PDGFRB-PE (FAB1263P; R&D System, Minneapolis, MN), CD90-PE-Cy5 (PNIM3703; Beckman Coulter), CD34-PE-Cy7 (A21691; Beckman Coulter), CD45-APC (21270456X2; ImmunoTools, Friesoythe, Germany), CD146-APC-Cy7 (BioCytex), CD105-Pacific Blue (Exbio) for the third mix; NG2-PE (PNIM3454U; Beckman Coulter), CD56-PE-Cy5 (555517; BD Biosciences), CD117-PE-Cy7 (Beckman Coulter), CD133-APC (Miltenyi Biotec), CD146-APC-Cy7 (BioCytex), CD45-Pacific Blue (Miltenyi Biotec) for the fourth mix; CD31-FITC (BD Biosciences), CD146-APC-Cy7 (BioCytex), CD45-Pacific Blue (Miltenyi Biotec) for the fifth mix. After staining, the cells were washed once with PBS (Gibco) containing 0.1% bovine serum albumin (Kedrion). At least 100,000 events were acquired with a FACSCanto II (BD Biosciences), and isotype-matched mouse immunoglobulins were used as controls under the same conditions. Plots of CD45- cells were generated using FlowJo analysis software v8.8.7 (Tree Star, Ashland, OR).
Statistical analysis
A normality test based on Z-score formula (www.seeingstatistics.com, 03/11/2014) was used to calculate the probability for every data set to be normally distributed. The Student's t-test was applied to compare means (www.graphpad.com, 03/11/2014) only for groups with an assessed probability of ≥70%.
Results and Discussion
During the past 9 years, the R&D section of our facility (Cell Factory “Franco Calori”) has been extensively investigating and setting up protocols for efficient isolation, culture, and characterization of stromal cells from cord blood, given the large availability of discarded CB units not matching the requirements for transplantation purposes at the affiliated Milano Cord Blood Bank (MICB). In addition, cord blood is widely recognized as hematopoietic stem/progenitor source and the possible identification of CB stromal cells of mesodermal origin is an intriguing issue to address. Extensive characterization of MSCs obtained from CB as multipotent cells, including trilineage differentiation, immunophenotyping, and hematopoietic support, has been performed by various research groups [13].
A total of 264 CB samples were processed. Units were not manipulated if the time from delivery to sample collection was greater than 48 h. In the event of hemolysis or the presence of clots detected during any step of the isolation protocol, the procedure was not completed and the sample not included in the study. Importantly, to evaluate whether we had successfully processed a cord blood unit, we considered the appearance of colonies composed by at least 10 fibroblastic-like cells as the positive event, in accordance to previous studies [18]. This standard was adopted to facilitate the assessment of colony formation within the context of CB primary cell cultures, characterized by a mixed cell environment as described below.
Definition of a standard protocol for the isolation of stromal cells from human cord blood
In a first step of our study, three isolation strategies were implemented and 25 CB units were processed. We obtained some initial encouraging results but with a quite low rate of successful events. Nonetheless, we observed colony formation from unprocessed whole blood (2 positive events out of 10 processed units), untreated MNCs from buffy coat (3 out of 9), and Sepax-separated cord blood cells (1 out of 6). In five experiments with whole blood and four with buffy coat, we investigated if red blood cell lysis could be useful to improve MSC attachment [26,30]. In our case, the lysis did not facilitate the appearance of colonies as none of the isolations presented a positive result. It is possible that the treatment and subsequent centrifugations caused loss of crucial progenitor cells.
After these preliminary results, we adopted a fourth methodological approach consisting of the immunodepletion of nonmesenchymal populations from CB buffy coat. The ratio behind this choice was again to eliminate adherent and nonadherent “contaminant” cell types, which could make more difficult for a stromal progenitor cell to attach to the culture surface and to form a colony [21,30]. Among the adherent portion of “contaminant” cells, we observed spindle-shaped cells with short stretched extensions and osteoclast-like cells with very large cytoplasm, frequently showing more than one nuclei (Fig. 1A, B).
FIG. 1.
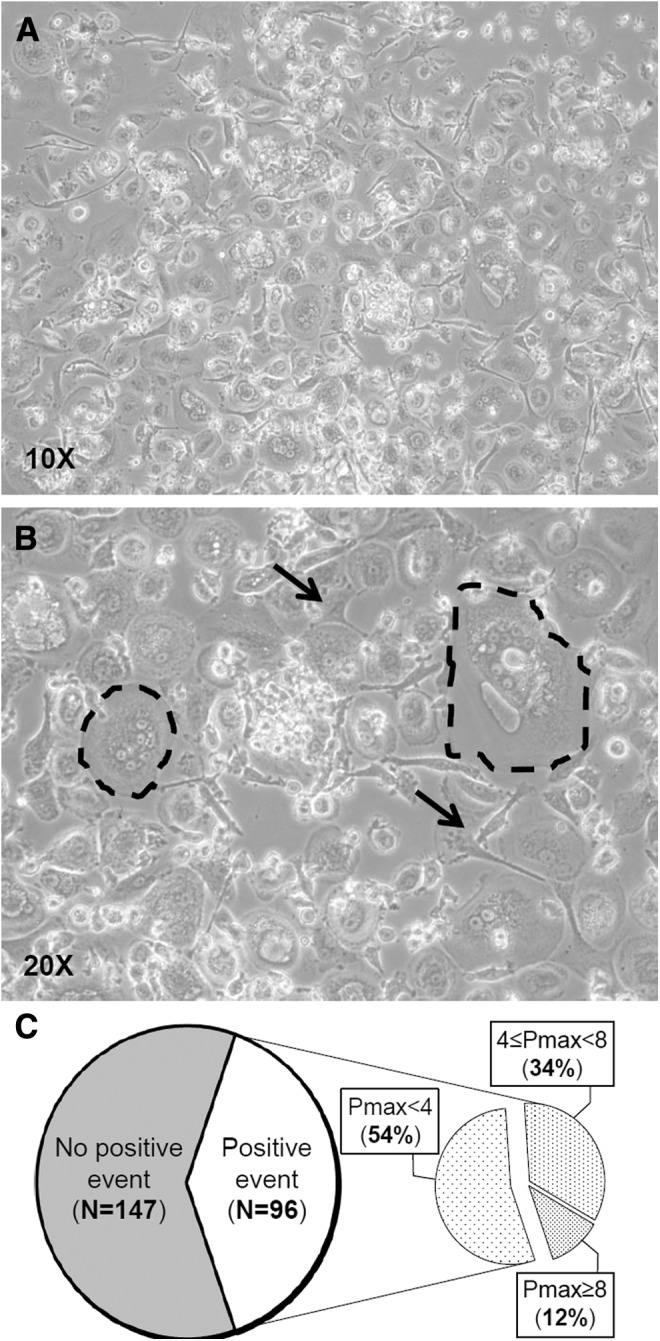
Establishment of CBMSC cell cultures. In T0 culture, it is frequent to observe adherent cells showing different morphologies that we do not recognize as MSCs (A). Among them, (B) nonproliferating fibroblast-like cells (indicated by arrows) and polynucleated (two to seven distinguishable nuclei) cells with a very large cytoplasm (dashed circles). (C) The pie chart at the left represents total isolation experiments relying on immunodepletion of nonmesenchymal cells; the right pie chart summarizes culture outcomes of CB units that gave rise to primary colonies of stromal cells (positive events). CBMSCs, cord blood-derived multipotent mesenchymal stromal cells; MSCs, mesenchymal stromal cells.
Isolation efficiency of stromal clones and establishment of primary culture
In the second group of experiments, 243 CB units were processed, obtaining colonies of stromal cells in nearly 40% of them (Fig. 1C). The appearance of colonies occurred with a median of 16 days after seeding, from a minimum of 4 days to a maximum of 64. The majority of them (90%) were detected within 4 weeks from seeding. It is worth noting that clones appeared after 40 days represented only 2% of total colonies and that they were not able to reach confluence at passage 1. Therefore, after these considerations, we defined 40 days as the detection time of colony appearance; adherent cells from CB cultures exceeding this detection time were used for the immunophenotype characterization. Notably, even if the morphology of the cells forming the colonies was fibroblastic-like and similar to that of bone marrow MSCs, CBMSCs were smaller and less spindle shaped. Upon appearance of a colony, we chose not to wait till high confluence before the first trypsinization in order not to cause stress to the “newly born” cells but to detach the cells when still actively dividing. Thus, the first passage came after a median of 22 days after seeding, approximately a week after the detection of the colony; CBMSC morphology is shown in Fig. 2A.
FIG. 2.

CBMSC morphology and growth kinetics. Adherent and proliferative cells isolated from processed CB units possess distinct morphology and cell shape. The images were taken from a representative CBMSC population and show subconfluent cells at early passages (P1, P4; A) and in long-term culture (P8, P12; B) at the indicated magnitudes. Representative growth trends of CBMSCs grouped by similar CPD values (vertical axis) and longevity (horizontal axis) are presented in (C, D). In the boxes, the means (black rectangle) and deviation standards (vertical bars) of CPDs for each passage are also shown. The values obtained fitted almost perfectly with a linear function in the case of LL-CBMSCs (y=3.0x−0.3), whereas in the case of SL-CBMSCs, the growth was slower at the beginning and the arrest was gradual starting from the last passages, so that the mean CPD values better fitted a polynomial curve (y=−0.1x3+1.1x2−0.9x+1.6). CPD, cumulative population doublings cut-off; LL-CBMSCs, long-living CBMSCs; SL-CBMSCs, short-living CBMSCs.
To pass subconfluent cells once a week, we fixed optimal seeding density from 2,000 to 4,000 cells/cm2. In addition, we observed that during the first trypsinization, a good portion of osteoclast-like cells remained attached to the plastic surface, showing spiky cytoplasm extroflessions. Thus, it is recommended to perform the first passage in a gentle manner in order not to detach unwished cells and favor the formation of as homogeneous as possible stromal populations. In fact, it is also important to underline that especially during the first passages (P1–P3) stromal cells present a high degree of heterogeneity [31,32]. Therefore, it is possible that a stochastic selection of a certain subset among this mixed population could alter the outcome of the long-term culture. As a consequence, cell suspensions should be handled with great attention to maintain them homogeneous, without altering their supposedly mixed composition.
Long-term follow-up of CBMSC populations
Focusing on long-term CBMSC culture, we obtained the most interesting and challenging results (Fig. 1C). Even if about 46% of clones gave rise to CBMSC populations that could reach at least passage 4, among them only one-fourth went on growing till, or beyond, passage 8 before entering a senescent state and arresting proliferation. More interestingly, we could visualize two main types of growth curves: the first one having higher CPD values, over 15 and in few cases more than 30 (Fig. 2C); the second one with lower CPD peaks (≤15) (Fig. 2D). The two groups start changing their growth trend around passage 5, after which cell populations belonging to the second group tend to reach plateau (Fig. 2D). These two behaviors in fact correlate in a certain extent with longevity and, in general, with “young” or nonstressed phenotype after long-term cell culture. Specifically, all the populations of the first group, which we decided to refer to as LL-CBMSCs, possess a healthy and prevalently nonsenescent morphology till high passages (P8 or over, till P13), as shown in Fig. 2B. In contrast, stromal populations showing lower CPD values, which we named SL-CBMSCs, arrest their growth around passage 5.
Unlike bone marrow MSCs, for which donor age heavily influence their growth and stemness properties [33,34], those isolated from cord blood are all of fetal, or neonatal, origin. Thus, the differences between LL-CBMSCs and SL-CBMSCs cannot be explained in terms of donor age, whereas a likely possibility is that the two represent indeed distinct stromal populations.
Biological characterization of LL-CBMSCs and SL-CBMSCs: morphology, cloning properties, and telomere length
Since we noted differences in cell shape during cell culture, we wondered if we could distinguish the different cell types by means of morphology parameters. We performed a comparative analysis between BMMSCs and CBMSCs that after long-term culture were recognized as LL-CBMSCs or SL-CBMSCs. Two parameters, cell major axis and nucleus diameter, were measured using images of the cultures at passage 4. This retrospective analysis revealed that CBMSCs present lower cell major axis/nucleus diameter ratio compared with BMMSCs and that within CBMSC populations LL-CBMSCs are the cells with the lowest value (Fig. 3A–D). In addition, the coefficient of variation (relative standard deviation) for calculated ratios in the case of LL-CBMSCs (33%) is the lowest, indicating that they are a more homogeneous population in terms of morphology characteristics if compared with SL-CBMSCs (49%) and BMMSCs (47%).
FIG. 3.

Cell shape comparison and clonogenic properties. Panels (A), (B), and (C) and magnified areas (black rectangles) represent some MSC morphologies at passage 4. BMMSCs (A) possess significantly higher cell major axis/nucleus diameter ratio compared with LL-CBMSCs (B) and SL-CBMSCs (C) (D, see table for mean values). LL-CBMSCs compared to SL-CBMSCs seem to have a lower ratio even if no statistical significance was found. (E) Table summarizes the mean number of colonies (CFU-Fs) and related clonogenic potential (percentage of clonogenic cells) for two LL-CBMSC and two SL-CBMSC populations at passage 4. (F, G) Images of LL-CBMSC- and SL-CBMSC-derived CFU-Fs, respectively. LL-CBMSC colonies are bigger and more easily detectable compared to SL-CBMSCs, whose colonies are very small and difficult to detect (indicated by arrows). *P≤0.05; **P≤0.01. CFU-Fs, colony forming unit-fibroblasts.
Concerning heterogeneity of MSC populations, we wondered if established LL-CBMSCs and SL-CBMSCs could also present differences in the clonogenic progenitor content. A retrospective analysis of colony forming unit-fibroblast assay data was performed to address this issue, and indeed, a strong difference in terms of number and morphology of colonies was observed: LL-CBMSCs retained a higher clonogenic potential, whereas SL-CBMSCs almost lacked it (Fig. 3, table E). In addition, colonies from LL-CBMSC populations covered larger areas and the cell density was clearly higher so that they were easily detected and counted (Fig. 3F, G).
From the observation that one major difference between LL-CBMSCs and SL-CBMSCs is lifespan, and since lifespan is related to telomere length [35], we investigated if longer telomeres could be a feature of LL-CBMSCs compared with SL-CBMSCs. Our results showed that passage 1 LL-CBMSCs possess telomeres that can be even 12 times longer than those of SL-CBMSCs (Fig. 4A). This significantly superior telomere length is shortened by a half during the first passage, whereas shorter SL-CBMSC telomeres do not change drastically their length during cell culture. Moreover, LL-CBMSC telomere length decreases gradually through various passages, and only at high passages becomes comparable to that of low passage SL-CBMSCs (Fig. 4A). Thus, telomere length could be a useful parameter to predict the culture outcome of a passage 1 CBMSC population. A longer telomere will indicate the probable establishment of LL-CBMSCs, whereas from a short telomere, SL-CBMSCs derivation is to be expected.
FIG. 4.

Mean telomere length at different passages for three LL-CBMSCs and three SL-CBMSCs with related SEM values (black bars) is shown in (A). In the box, representative passage 1 LL-CBMSCs and SL-CBMSCs show dramatic differences in telomere length (standard deviation represented with black bars). Differentiation properties of a representative CBMSC population are shown in (B, C). Oil Red O solution stains lipid vacuoles of mature adipocytes (B); Alizarin Red S stains calcium deposits produced by CBMSC-derived osteocytes (C). Molecular data confirmed the differentiation of induced compared with noninduced CBMSCs: increased expression of CFD (78-folds) and PPARgamma (26-folds) (house-keeping genes: TBP and YHWAZ) for adipogenesis (B), whereas high expression levels of ALP (216-folds) and OPN (32-folds) genes (house-keeping genes: RPLP0 and RPL13alpha) assessed osteogenic differentiation (C). Standard deviation represented with black bars when visible. **P≤0.01.
Differentiation into adipocytes and osteocytes
Previous reports [24,25,36] described differences in adipogenic and osteogenic potentials for distinct CB stromal populations. To verify if this could also distinguish LL-CBMSCs from SL-CBMSCs, both populations were induced to differentiate into mesodermal mature cells.
Adipogenic differentiation induced by the standard protocol failed to produce large fat deposits, with very few cells showing little cytoplasmic droplets (data not shown). Instead, following a modified protocol, lipid vacuoles began to appear between 3 and 4 weeks; nonetheless, also in this case, only few CBMSCs presented large Oil Red O-positive deposits, even if molecular analysis confirmed the presence of adipocytes in the culture (Fig. 4B). These experimental tricks may suggest that CBMSCs are less prone than MSCs from other sources to give rise to adipose tissue-differentiated cell types. In this regard, differences at molecular level between CBMSCs and BMMSCs that explain this behavior during adipogenic induction have been recently elucidated [10]. CBMSCs could lack in some measure adipogenic potential or either their being “fetal” cells implies also a more immature state that necessitates longer or ad hoc differentiation protocols. It has been proposed that DLK1, a gene encoding a protein involved in adipogenesis, could act as a molecular marker distinguishing CBMSCs from unrestricted somatic stem cells (USSCs) [36]. In this model, DLK1 is highly expressed in USSCs, inhibiting differentiation into adipocytes and correlating to high proliferative potential compared to less proliferative and adipogenesis-competent CBMSCs, for which DLK1 is less expressed or absent. We also analyzed this gene and found variability in relative expression between different batches of CBMSCs, even if with Ct values not reliable (>36), but no consistent differences were observed between SL- and LL-CBMSCs (data not shown). Moreover, we did not detect any major difference in adipogenic potential between CBMSCs, but a general lack of abundant lipid droplets, as others similarly reported [18]. This is also in contrast with the reports suggesting higher adipogenic properties for less frequent and at times more proliferative subsets of spindle-shaped CB stromal cells [24,25].
On the other hand, calcium deposits appeared very soon (7 days after switch to the differentiation medium) in cultured cells undergoing osteogenesis. The formation of Alizarin Red S-positive deposits and molecular analysis assessed the differentiation of both LL-CBMSCs and SL-CBMSCs into osteocytes (Fig. 4C). Macrodifferences in the extent of mineralization were observed, with larger and more strongly stained deposits in cells from LL-CBMSC populations.
Although all these data identify the isolated cells as multipotent MSCs, great discrepancies with previous reports concerning their precise differentiation potentials remain. These inconsistencies could be caused by differences in the isolation methodologies, differentiation protocols and also by the lack of unequivocal criteria or markers for the isolation and definition of the distinct subsets of stromal populations.
Characteristics of CB units
Cord blood unit characteristics were considered as potential predictive parameters of cell culture outcome and thus analyzed in terms of TNC content, time from collection to processing, and total volume (blood plus anticoagulant). Also gender and gestational age were considered, but this analysis did not show any interesting result, as already reported in the literature [18,21]. For this analysis, 146 blood units were analyzed: 65 presented positive events after the immunodepletion approach, whereas the other 81 did not.
The percentage of monocytes (median) in whole cord blood units giving rise to LL-CBMSCs was lower, but not statistically significant, if compared with those giving rise to SL-CBMSCs or not showing any positive event (Fig. 5A). As clearly evident from the wider range of monocyte percentages in CB units giving rise to SL-CBMSCs and no positive event, we can suggest that those samples having a monocyte percentage higher than 10% should not be processed, or effective methodologies for monocyte depletion should be considered. The fact that monocytes could act as a sort of inhibiting population in respect to colony formation and establishment of SL- and LL-CBMSCs is in accordance with the concept already discussed of steric hindrance exerted by “contaminant” adherent cell types. In fact, it has been demonstrated that monocytes/macrophages can fuse in vivo to form polynucleated cells spreading over large areas and recognized as osteoclast-like cells [37,38]. It is possible that the cocktail we use for immunodepletion of hematopoietic lineages fails to work properly when monocytes or macrophages are very abundant in the cord blood unit.
FIG. 5.

CB unit characteristics. (A) The percentage of monocytes was lower in CB units that gave rise to LL-CBMSCs (median=5.0; min=3.6; max=7.4; n=7), compared with those giving rise to SL-CBMSCs (median=6.0; min=3.1; max=30.4; n=49) or units for which no positive event was observed (median=6.5; min=2.9; max=34.4; n=81). The black rectangles cover the portion of values between the first and third quantiles, whereas the bars represent the range of values from minimum to maximum. No statistical significance was observed. (B, C) No correlation between total nucleated cells (TNCs) recovered after immunodepletion or maximum passage and time from collection to processing. Positivity of immunodepleted CB cells before seeding for single surface markers is shown in (D). Percentages of expression reported in the graph are the mean with associated SEM (black bars). *P≤0.05; **P≤0.01.
Relevance of time from collection to processing was considered in relation to other two parameters: TNC recovered postprocessing and maximum passage reached by CBMSCs when cell culture could be established (Fig. 5B, C). Even if there was no correlation between these parameters, we ascertained that the majority (90%) of the units that could not give rise to colonies had a median value for time from the collection of 18 h and 20 min, which dropped to 13 h and 9 min for CB units that had given rise to stromal colonies. This confirms previous reports [18,21] and indicates that it is advisable to process a CB unit when time from collection is less than 14 h. We can speculate that since CB is the only nonsolid MSC source, the few stromal progenitors circulating in this blood need to adhere to a solid substrate as soon as possible to activate molecular mechanisms for survival and maintenance of their MSC features [39–41].
We also evaluated the influence of sample volume on isolation of stromal cells. We found that among the 146 analyzed CB units, the median values varied from 89 mL (range: 34–127 mL) for units that presented no positive event to 100 mL (range: 87–129 mL) for units that formed SL-CBMSCs and 110 mL (range: 89–121 mL) for units giving rise to LL-CBMSCs. This suggests that sample volume could be a parameter to consider for efficient isolation of adherent clones, as already reported [18]. We believe that the importance of sample volume could be related to the stem/progenitor cell frequency in CB. Even if it is not yet clear and unequivocal which is the complete phenotype that identify the stromal cell circulating in cord blood, based on our herein reported experience we could assume that a higher blood volume contains a higher number of stromal stem/progenitor cells.
Immunophenotypic characterization
Samples at different steps were extensively characterized for the expression of hematopoietic, endothelial, perivascular, and mesenchymal surface markers: immunodepleted CB buffy coat before seeding, nonadherent cells after the first medium change, and nonclonogenic adherent cells after 40 days of culture.
In the case of immunodepleted buffy coat, the analysis revealed distinctive immunoprofiles associated with different cell culture outcomes. In fact, when considering positivity for a single cell marker, we detected some intriguing differences between samples not giving rise to positive event, SL-CBMSCs or LL-CBMSCs. The most evident and significant differences were found for NG2, highly expressed in samples giving rise to LL-CBMSCs, and for PDGFR-B, more expressed in samples for which no positive event was observed. Moreover, CD133 and CD146 were associated to samples giving rise to both LL-CBMSCs and SL-CBMSCs, whereas they were significantly less expressed in samples not showing positive events. In addition, alphaSMA+ and CD117+ cells were significantly more present in LL-CBMSC-related samples than no positive event and SL-CBMSC-related ones, respectively (Fig. 5D).
The immunophenotype of nonadherent contaminant cells did not give relevant results, whereas the nonclonogenic adherent fraction revealed high expression (with a maximum of 90%) of CD105 and CD45 and no presence of CD90-positive cells, both indicating the strong presence of mature endothelial cells.
Conclusion
All the reported data support, if still necessary, the concept that multipotent MSCs can be isolated from cord blood, even if at low frequency. In addition, we observed two alternative and specific behaviors of CBMSCs in cell culture, which could identify two distinct stromal populations circulating in cord blood, with different biological properties. We named them LL-CBMSCs and SL-CBMSCs based on differences in observed lifespan. Our findings induced us to consider LL-CBMSCs “better” CBMSCs, in reason of their nonsenescent morphology even after extensive culture, longer telomeres, higher proliferation rate, and osteogenic potential when compared with SL-CBMSCs. To sum up, LL-CBMSCs should possess a maximum CPD value over 15, pass the passage 5–6 limit, have a nonstressed and juvenile morphology till at least passage 8, with a steady and almost linear growth curve before entering a senescent state.
In conclusion, low frequency in cord blood of CBMSC progenitors drastically reduces isolation rate compared to other sources and could prevent from widening scientific knowledge about these cells. On the other hand, this low frequency could paradoxically shed lights on the reason why these stromal cells are present in cord blood. Our hypothesis is that it could be the result of flushing of blood coming from fetal organs. From this point of view, LL-CBMSCs and SL-CBMSCs could simply derive from different compartments of the fetus, that is, fetal liver or bone marrow, and for this reason, they show dissimilar biological properties. Otherwise, another interesting hypothesis could be that LL-CBMSC and SL-CBMSC behavior in culture may reflect different developmental stages of the same cell population. Moreover, these hypotheses concerning CBMSC derivation are not in contrast with the innovative theory of the perivascular origin of MSCs [22], but further in vivo studies should be done to unveil the origin of these populations [42].
Recently, it was hypothesized that replicative senescence of stem cells cultured in vitro could reflect their in vivo properties [43]. In fact, the aging process is characterized by impaired regenerative capabilities and it has been proposed that this is also related to less effective or absent adult stem cell functionalities [44,45]. This has prompted some researchers to consider lifespan shown during cell culture as a potency assay informative of stem cell therapeutic potentialities [46]. Moreover, if an allogeneic cell therapy context is envisioned for CBMSCs, there is strong need for healthy and proliferative cells able to sustain the requested scaling-up to reach clinically relevant cell doses [34]. All together, these considerations suggest that all the efforts in the CBMSC field should be addressed to isolate LL-CBMSCs, the most promising cord blood stromal population for regenerative medicine application, based on the presented results. Therefore, since we strongly believe in the high therapeutic potentialities of CBMSCs [47–49], herein we proposed a sort of “recipe” containing useful and effective parameters predictive of CBMSC culture outcome, defined through the dissection of the CB stromal cellular component.
And now back to our Introduction and to our questions. “Do all the CB units have the same potential to produce stromal cells?”: unfortunately they do not. “Does cord blood contain more than one stromal cell population?”: fortunately they do. “Can we predict the cultural outcome of cord blood cell samples?”: maybe now we can.
Acknowledgments
The authors thank all the researchers working at our facility Cell Factory for their critical advices during the laboratory meetings. We are also grateful to the Milano Cord Blood group for their help in providing cord blood units. In addition, special mention must be made to all the colleagues who have worked on this project over the last few years: Viviana Lo Cicero, Gabriella Andriolo, Gabriella Spaltro, and Francesca Chelli.
This work was partially supported by funds from Regione Lombardia (PB 0098), from Ministero della Salute Italiano (“Young Researchers” grants: R.F.G.R. 2010-2318448, R.F.G.R. 2010-2312573), and from European Union's Seventh Programme (Grant Agreement Number 241879).
Author Disclosure Statement
No competing financial interests exist. This work was part of the PhD project of Mario Barilani for the PhD School in Biosciences and Biotechnology - curriculum Cell Biology of the University of Padoa.
References
- 1.Di Trapani M, Bassi G, Ricciardi M, Fontana E, Bifari F, Pacelli L, Giacomello L, Pozzobon M, Féron F, et al. (2013). Comparative study of immune regulatory properties of stem cells derived from different tissues. Stem Cells Dev 22:2990–3002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Owen M. and Friedenstein AJ. (1988). Stromal stem cells: marrow-derived osteogenic precursors. Ciba Found Symp 136:42–60 [DOI] [PubMed] [Google Scholar]
- 3.Zuk PA, Zhu M, Mizuno H, Huang J, Futrell JW, Katz AJ, Benhaim P, Lorenz HP. and Hedrick MH. (2001). Multilineage cells from human adipose tissue: implications for cell-based therapies. Tissue Eng 7:211–228 [DOI] [PubMed] [Google Scholar]
- 4.Gronthos S, Mankani M, Brahim J, Robey PG. and Shi S. (2000). Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc Natl Acad Sci U S A 97:13625–13630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McElreavey KD, Irvine AI, Ennis KT. and Mc Lean WH. (1991). Isolation, culture and characterisation of fibroblast-like cells derived from the Wharton's jelly portion of human umbilical cord. Biochem Soc Trans 19:29S. [DOI] [PubMed] [Google Scholar]
- 6.Erices A, Conget P. and Minguell JJ. (2000). Mesenchymal progenitor cells in human umbilical cord blood. Br J Haematol 109:235–242 [DOI] [PubMed] [Google Scholar]
- 7.Igura K, Zhang X, Takahashi K, Mitsuru A, Yamaguchi S. and Takashi TA. (2004). Isolation and characterization of mesenchymal progenitor cells from chorionic villi of human placenta. Cytotherapy 6:543–553 [DOI] [PubMed] [Google Scholar]
- 8.Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop DJ. and Horwitz E. (2006). Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8:315–317 [DOI] [PubMed] [Google Scholar]
- 9.Jin HJ, Bae YK, Kim M, Kwon SJ, Jeon HB, Choi SJ, Kim SW, Yang YS, Oh W. and Chang JW. (2013). Comparative analysis of human mesenchymal stem cells from bone marrow, adipose tissue, and umbilical cord blood as sources of cell therapy. Int J Mol Sci 14:17986–18001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ragni E, Viganò M, Parazzi V, Montemurro T, Montelatici E, Lavazza C, Budelli S, Vecchini S, Rebulla P, Giordano R. and Lazzari L. (2013). Adipogenic potential in human mesenchymal stem cells strictly depends on adult or foetal tissue harvest. Int J Biochem Cell Biol 45:2456–2466 [DOI] [PubMed] [Google Scholar]
- 11.Hoogduijn MJ, Betjes MG. and Baan CC. (2014). Mesenchymal stromal cells for organ transplantation: different sources and unique characteristics?. Curr Opin Organ Transplant 19:41–46 [DOI] [PubMed] [Google Scholar]
- 12.Goodwin HS, Bicknese AR, Chien SN, Bogucki BD, Quinn CO. and Wall DA. (2001). Multilineage differentiation activity by cells isolated from umbilical cord blood: expression of bone, fat, and neural markers. Biol Blood Marrow Transplant 7:581–588 [DOI] [PubMed] [Google Scholar]
- 13.Flynn A, Barry F. and O'Brien T. (2007). UC blood-derived mesenchymal stromal cells: an overview. Cytotherapy 9:717–726 [DOI] [PubMed] [Google Scholar]
- 14.Stubbendorff M, Deuse T, Hua X, Phan TT, Bieback K, Atkinson K, Eiermann TH, Velden J, Schröder C, et al. (2013). Immunological properties of extraembryonic human mesenchymal stromal cells derived from gestational tissue. Stem Cells Dev 22:2619–2629 [DOI] [PubMed] [Google Scholar]
- 15.Wang M, Yang Y, Yang D, Luo F, Liang W, Guo S. and Xu J. (2009). The immunomodulatory activity of human umbilical cord blood-derived mesenchymal stem cells in vitro. Immunology 126:220–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Riordan NH, Chan K, Marleau AM. and Ichim TE. (2007). Cord blood in regenerative medicine: do we need immune suppression?. J Transl Med 5:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bieback K, Kern S, Kocaömer A, Ferlik K. and Bugert P. (2008). Comparing mesenchymal stromal cells from different human tissues: bone marrow, adipose tissue and umbilical cord blood. Biomed Mater Eng 18:S71–S76 [PubMed] [Google Scholar]
- 18.Zhang X, Hirai M, Cantero S, Ciubotariu R, Dobrila L, Hirsh A, Igura K, Satoh H, Yokomi I, et al. Isolation and characterization of mesenchymal stem cells from human umbilical cord blood: reevaluation of critical factors for successful isolation and high ability to proliferate and differentiate to chondrocytes as compared to mesenchymal stem cells from bone marrow and adipose tissue. J Cell Biochem 112:1206–1218 [DOI] [PubMed] [Google Scholar]
- 19.Laitinen A, Nystedt J. and Laitinen S. (2011). The isolation and culture of human cord blood-derived mesenchymal stem cells under low oxygen conditions. Methods Mol Biol 698:63–73 [DOI] [PubMed] [Google Scholar]
- 20.Sibov TT, Severino P, Marti LC, Pavon LF, Oliveira DM, Tobo PR, Campos AH, Paes AT, Amaro E, Gamarra L. and Moreira-Filho CA. (2012). Mesenchymal stem cells from umbilical cord blood: parameters for isolation, characterization and adipogenic differentiation. Cytotechnology 64:511–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bieback K, Kern S, Klüter H. and Eichler H. (2004). Critical parameters for the isolation of mesenchymal stem cells from umbilical cord blood. Stem Cells 22(4):625–634 [DOI] [PubMed] [Google Scholar]
- 22.Crisan M, Yap S, Casteilla L, Chen CW, Corselli M, Park TS, Andriolo G, Sun B, Zheng B, et al. (2008). A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 3:301–313 [DOI] [PubMed] [Google Scholar]
- 23.Sun HP, Zhang X, Chen XH, Zhang C, Gao L, Feng YM, Peng XG. and Gao L. (2012). Human umbilical cord blood-derived stromal cells are superior to human umbilical cord blood-derived mesenchymal stem cells in inducing myeloid lineage differentiation in vitro. Stem Cells Dev 21:1429–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Markov V, Kusumi K, Tadesse MG, William DA, Hall DM, Lounev V, Carlton A, Leonard J, Cohen RI, Rappaport EF. and Saitta B. (2007). Identification of cord blood-derived mesenchymal stem/stromal cell populations with distinct growth kinetics, differentiation potentials, and gene expression profiles. Stem Cells Dev 16:53–73 [DOI] [PubMed] [Google Scholar]
- 25.Chang YJ, Tseng CP, Hsu LF, Hsieh TB. and Hwang SM. (2006). Characterization of two populations of mesenchymal progenitor cells in umbilical cord blood. Cell Biol Int 30:495–499 [DOI] [PubMed] [Google Scholar]
- 26.Kögler G, Sensken S, Airey JA, Trapp T, Müschen M, Feldhahn N, Liedtke S, Sorg RV, Fischer J, et al. (2004). A new human somatic stem cell from placental cord blood with intrinsic pluripotent differentiation potential. J Exp Med 200:123–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ragni E, Montemurro T, Montelatici E, Lavazza C, Viganò M, Rebulla P, Giordano R. and Lazzari L. (2013). Differential microRNA signature of human mesenchymal stem cells from different sources reveals an “environmental-niche memory” for bone marrow stem cells. Exp Cell Res 319:1562–1574 [DOI] [PubMed] [Google Scholar]
- 28.Samsonraj RM, Raghunath M, Hui JH, Ling L, Nurcombe V. and Cool SM. (2013). Telomere length analysis of human mesenchymal stem cells by quantitative PCR. Gene 519:348–355 [DOI] [PubMed] [Google Scholar]
- 29.Ragni E, Viganò M, Rebulla P, Giordano R. and Lazzari L. (2013). What is beyond a qRT-PCR study on mesenchymal stem cell differentiation properties: how to choose the most reliable housekeeping genes. J Cell Mol Med 17:168–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laitinen A. and Laine J. (2007). Isolation of mesenchymal stem cells from human cord blood. Curr Protoc Stem Cell Biol 2: 2A.3. [DOI] [PubMed] [Google Scholar]
- 31.Phinney DG. (2012). Functional heterogeneity of mesenchymal stem cells: implications for cell therapy. J Cell Biochem 113:2806–2812 [DOI] [PubMed] [Google Scholar]
- 32.Ho AD, Wagner W. and Franke W. (2008). Heterogeneity of mesenchymal stromal cell preparations. Cytotherapy 10:320–330 [DOI] [PubMed] [Google Scholar]
- 33.Siegel G, Kluba T, Hermanutz-Klein U, Bieback K, Northoff H. and Schäfer R. (2013). Phenotype, donor age and gender affect function of human bone marrow-derived mesenchymal stromal cells. BMC Med 11:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bajek A, Czerwinski M, Olkowska J, Gurtowska N, Kloskowski T. and Drewa T. (2012). Does aging of mesenchymal stem cells limit their potential application in clinical practice?. Aging Clin Exp Res 24:404–411 [DOI] [PubMed] [Google Scholar]
- 35.Mikhelson VM. and Gamaley IA. (2012). Telomere shortening is a sole mechanism of aging in mammals. Curr Aging Sci 5:203–208 [DOI] [PubMed] [Google Scholar]
- 36.Kluth SM, Buchheiser A, Houben AP, Geyh S, Krenz T, Radke TF, Wiek C, Hanenberg H, Reinecke P, Wernet P. and Kögler G. (2010). DLK-1 as a marker to distinguish unrestricted somatic stem cells and mesenchymal stromal cells in cord blood. Stem Cells Dev 19:1471–1483 [DOI] [PubMed] [Google Scholar]
- 37.Udagawa N, Takahashi N, Akatsu T, Tanaka H, Sasaki T, Nishihara T, Koga T, Martin TJ. and Suda T. (1990). Origin of osteoclasts: mature monocytes and macrophages are capable of differentiating into osteoclasts under a suitable microenvironment prepared by bone marrow-derived stromal cells. Proc Natl Acad Sci U S A 87:7260–7264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim YG, Lee CK, Oh JS, Kim SH, Kim KA. and Yoo B. (2010). Effect of interleukin-32gamma on differentiation of osteoclasts from CD14+ monocytes. Arthritis Rheum 62:515–523 [DOI] [PubMed] [Google Scholar]
- 39.Zhong X. and Rescorla FJ. (2012). Cell surface adhesion molecules and adhesion-initiated signaling: understanding of anoikis resistance mechanisms and therapeutic opportunities. Cell Signal 24:393–401 [DOI] [PubMed] [Google Scholar]
- 40.Stupack DG. and Cheresh DA. (2002). Get a ligand, get a life: integrins, signaling and cell survival. J Cell Sci 115:3729–3738 [DOI] [PubMed] [Google Scholar]
- 41.Mathieu PS. and Loboa EG. (2012). Cytoskeletal and focal adhesion influences on mesenchymal stem cell shape, mechanical properties, and differentiation down osteogenic, adipogenic, and chondrogenic pathways. Tissue Eng Part B Rev 18:436–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murray IR, West CC, Hardy WR, James AW, Park TS, Nguyen A, Tawonsawatruk T, Lazzari L, Soo C. and Péault B. (2014). Natural history of mesenchymal stem cells, from vessel walls to culture vessels. Cell Mol Life Sci 71:1353–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wagner W, Bork S, Horn P, Krunic D, Walenda T, Diehlmann A, Benes V, Blake J, Huber FX, et al. (2009). Aging and replicative senescence have related effects on human stem and progenitor cells. PLoS One 4:e5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Behrens A, Deursen van JM, Rudolph KL. and Schumacher B. (2014). Impact of genomic damage and ageing on stem cell function. Nat Cell Biol 16:201–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.López-Otín C, Blasco MA, Partridge L, Serrano M. and Kroemer G. (2013). The hallmarks of aging. Cell 153:1194–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wagner W, Ho AD. and Zenke M. (2010). Different facets of aging in human mesenchymal stem cells. Tissue Eng Part B Rev 16:445–453 [DOI] [PubMed] [Google Scholar]
- 47.Pierro M, Ionescu L, Montemurro T, Vadivel A, Weissmann G, Oudit G, Emery D, Bodiga S, Eaton F, et al. (2013). Short-term, long-term and paracrine effect of human umbilical cord-derived stem cells in lung injury prevention and repair in experimental bronchopulmonary dysplasia. Thorax 68:475–484 [DOI] [PubMed] [Google Scholar]
- 48.Zanier ER, Montinaro M, Viganò M, Villa P, Fumagalli S, Pischiutta F, Longhi L, Leoni ML, Rebulla P, et al. (2011). Human umbilical cord blood mesenchymal stem cells protect mice brain after trauma. Crit Care Med 39:2501–2510 [DOI] [PubMed] [Google Scholar]
- 49.Morigi M, Rota C, Montemurro T, Montelatici E, Cicero Lo V, Imberti B, Abbate M, Zoja C, Cassis P, et al. (2010). Life-sparing effect of human cord blood-mesenchymal stem cells in experimental acute kidney injury. Stem Cells 28:513–522 [DOI] [PubMed] [Google Scholar]