

Significance
Inactivating mutations in the chromodomain helicase DNA binding protein 7 (CHD7) gene causes a severe developmental disorder known as CHARGE syndrome. Recently, several missense mutations in CHD7 have been reported in isolated gonadotropin-releasing hormone (GnRH) -deficiency (IGD) patients who lack full CHARGE features. However, the precise functional consequence of these IGD-associated missense mutations on the activity of CHD7 protein is not known. This study confirms the predominance of missense CHD7 alleles in 5% of IGD patients and provides, to our knowledge, first experimental evidence that functionally compromised CHD7 missense alleles contribute to the pathogenesis of both the anosmic and normosmic forms of IGD. These results imply a preferential sensitivity for CHD7 dysfunction in the developmental ontogeny as well as neuroendocrine regulation of GnRH neurons in humans.
Keywords: CHD7, Kallmann syndrome, idiopathic hypogonadotropic hypogondism, CHARGE syndrome, missense mutations
Abstract
Inactivating mutations in chromodomain helicase DNA binding protein 7 (CHD7) cause CHARGE syndrome, a severe multiorgan system disorder of which Isolated gonadotropin-releasing hormone (GnRH) deficiency (IGD) is a minor feature. Recent reports have described predominantly missense CHD7 alleles in IGD patients, but it is unclear if these alleles are relevant to causality or overall genetic burden of Kallmann syndrome (KS) and normosmic form of IGD. To address this question, we sequenced CHD7 in 783 well-phenotyped IGD patients lacking full CHARGE features; we identified nonsynonymous rare sequence variants in 5.2% of the IGD cohort (73% missense and 27% splice variants). Functional analyses in zebrafish using a surrogate otolith assay of a representative set of these CHD7 alleles showed that rare sequence variants observed in controls showed no altered function. In contrast, 75% of the IGD-associated alleles were deleterious and resulted in both KS and normosmic IGD. In two families, pathogenic mutations in CHD7 coexisted with mutations in other known IGD genes. Taken together, our data suggest that rare deleterious CHD7 alleles contribute to the mutational burden of patients with both KS and normosmic forms of IGD in the absence of full CHARGE syndrome. These findings (i) implicate a unique role or preferential sensitivity for CHD7 in the ontogeny of GnRH neurons, (ii) reiterate the emerging genetic complexity of this family of IGD disorders, and (iii) demonstrate how the coordinated use of well-phenotyped cohorts, families, and functional studies can inform genetic architecture and provide insights into the developmental biology of cellular systems.
Inactivating mutations in the gene encoding chromodomain helicase DNA binding protein 7 (CHD7, MIM 608892) cause a severe developmental disorder that affects multiple organ systems and is referred to as CHARGE syndrome (Coloboma, Heart defects, choanal Atresia, Retardation of growth and development, Gonadal defects, and Ear/hearing abnormalities, MIM 214800) (1–3). More than 90% of the reported mutations causing this syndrome are de novo severe truncating lesions that are equally distributed across CHD7’s 37 exons and various functional domains (3–6). The “gonadal defect” in CHARGE patients is in fact hypogonadotropic hypogonadism, which is typically responsive to exogenous gonadotropin-releasing hormone (GnRH) administration: that is, they represent isolated GnRH deficiency (IGD) (7–11).
IGD is an inherited reproductive disorder caused by defects in the secretion of GnRH and its action on the anterior pituitary gonadotrope (12). Around 60% of IGD patients also manifest a variety of nonreproductive defects, principally anosmia, which defines Kallmann syndrome (KS), whereas the remainder exhibit a pure nonsyndromic neuroendocrine phenotype, referred to as normosmic IGD (nIGD) (12, 13). In KS, a shared migratory defect of GnRH and olfactory axons produces IGD and anosmia, respectively, whereas other defects result from perturbation of other developmental pathways and processes (14). In contrast, the biological basis of nIGD cases resides in the failure of one or more hypothalamic neuroendocrine systems (12).
Several recent reports have suggested that rare sequence variants (RSVs) in CHD7 occur in ∼6% of IGD patients either with (9, 15) or without (8) any features of CHARGE syndrome. Although the initial report implicated CHD7 in both KS and nIGD (8), two subsequent studies detected putative pathogenic CHD7 RSVs in KS patients only (9, 16). However, the possible effects of these IGD-associated CHD7 variants on protein function have not been investigated to date (8, 9, 15, 16). Therefore, we sought to: (i) determine the frequency spectrum, phenotypes, genotypes, and mode of inheritance of CHD7 RSVs in a large, well-characterized cohort of IGD; (ii) characterize functionally a representative spectrum of these IGD- and CHARGE-associated RSVs; and (iii) consider whether differential activity of the protein, or loss thereof, contributes to KS/nIGD/CHARGE or other specific endophenotypes.
Results
RSVs in CHD7 Are Common in IGD Patients.
All 37 exons and intron–exon junctions of CHD7 were sequenced in 783 well-phenotyped patients with IGD. Following further detailed phenotypic review of patients with demonstrable mutations, four from the original cohort with an initial diagnosis of IGD were reclassified as CHARGE syndrome. Overall, we identified CHD7 RSVs in 41 IGD patients (5.2%) lacking full CHARGE features (Fig. 1 and Table S1), a number consistent with previous reports (8, 16). Only 9 of 41 of these CHD7+ IGD patients showed evidence of minor CHARGE features (Table S2). In keeping with a recent report (16), a striking feature of this cohort was that, in contrast to the truncating CHD7 mutations observed typically in patients with CHARGE, patients with IGD harbored predominantly missense variants (n = 30 of 41; 73%), with a smaller number of patients harboring predicted splice-variants (n = 11 of 41; 27%). All patients harboring CHD7 RSVs were heterozygotes except for two individuals who each displayed two RSVs; however, no parental DNAs were available to determine their phase. All four patients who fulfilled the Verloes criteria for full CHARGE syndrome (17) harbored a single heterozygous CHD7 RSV (one nonsense, one frameshift, and two missense variants) (Table S3). All RSVs identified in IGD patients and in full CHARGE patients are shown in Fig. 1.
Fig. 1.

CHD7 protein domains and positions of RSVs identified in CHARGE syndrome and IGD. Both CHARGE- and IGD-associated variants in CHD7 were equally dispersed across its 37 exons without mutational hot spots. Most mutations identified in CHARGE syndrome are nonsense or frameshift, whereas the vast majority of RSVs found in IGD are missense variants. RSVs identified in family members with delayed puberty, anosmia, and unaffected carriers were depicted along with RSVs seen in probands. BRK, Brahma and Kismet domain; CD, chromodomain; DEXHc, DEAD-like helicase superfamily including an ATP-binding domain; NLS, nuclear localization signal.
IGD and CHARGE Patients Are Enriched for Functionally Deleterious RSVs.
Computational analysis of the functional impact of the identified CHD7 variants was performed using multiple software programs (SI Materials and Methods). Splice sequence analysis using three prediction programs predicted that only one putative splice variant would affect splice efficiency/utilization in two or more programs (Table S4). Modeling of the missense alleles produced significant discordance in the various predictions algorithms (Table S5). Given the known ambiguity of these prediction programs (18), we assessed the functionality of the IGD-associated RSVs using an in vivo zebrafish model. We selected a representative spectrum of 20 different nonsynonymous missense RSVs for extensive functional analyses based on their population frequency (Table S6) and associated human phenotypes (Tables S1–S3). The RSVs chosen for these analyses fell into three groups: (i) Group A (control alleles; n = 3): RSVs seen in IGD subjects but also seen with ≥0.5% minor allele frequency (MAF) in the National Heart, Lung, and Blood Institute (NHLBI) exome database (p.S103T, p.M340V, p.L2984F) (18); (ii) Group B (alleles associated with IGD without full CHARGE; n = 12): RSVs (MAF < 0.5%) consisting of six alleles found in IGD subjects lacking any CHARGE features (p.Y1616C, p.G1982E, p. I2064V, p.I2232V, p.T2532M, p.Q2621E), three from IGD patients without any CHARGE features but who carried additional heterozygous mutations in other genes previously identified to be associated with IGD (p.G1845R, p.E1897K, p.A2789V), and three from patients with IGD who had minor CHARGE features (p.P940L, p.F1362L, p.R2065G); and (iii) Group C (alleles associated with full CHARGE; n = 5): RSVs (MAF < 0.5%) from CHARGE syndrome patients identified either in this study (p.V1021G, p.A1289V) or reported previously in the CHD7 database (p.I1028V, p.D1596G, p.D1812H) (www.chd7.org/molgenis.do) (6).
To test the consequences of these missense mutations on protein function, we injected a previously reported splice-blocking morpholino (MO) against chd7 (chd7-MO) (19) into one- to four-cell–stage zebrafish embryos. Consistent with previous reports (19), injected embryos displayed abnormal otoliths at 3 days postfertilization (dpf), one of the many CHARGE phenotypes. We grouped the resulting embryos into four objective classes: normal, small (posterior otolith same size or smaller to anterior otolith), fused/three, or absent otoliths. Each of the 20 CHD7 missense RSVs was tested for either its relative ability to rescue the morphant phenotype or its ability to phenocopy MO-induced otolith defects when overexpressed (under a dominant paradigm). For in vivo rescue, the chd7-MO (6 ng) and each mutant CHD7 missense mRNA (5 pg) were injected and each experiment was scored blind to injection mixture and performed in triplicate. All morphant phenotypes could be rescued by coinjection of WT human CHD7 mRNA (Fig. 2A, Inset F), whereas WT overexpression in the absence of MO produced no appreciable phenotypes (Fig. 2C). Because the mutant CHD7 S103T control allele was able to rescue the morphant phenoptype indistinguishable from WT human CHD7 mRNA, all of the rescue experiments for mutant CHD7 alleles were statistically compared against the S103T allele (Fig. 2B). Results for all mutant alleles on both assays are presented except for the Q2621E allele for which the rescue experiment failed and is not shown.
Fig. 2.

Functional assessment of CHD7 RSVs in zebrafish assay. (A) The otic vesicle of control-MO–injected zebrafish contains two otoliths (Inset A). Larva treated with an MO against chd7 show normal (Inset B), three/fused (Inset C), or small (Inset D) or absent (Inset E) otoliths, whereas coinjection of WT human CHD7 rescues the morphant phenotype showing normal two otoliths (Inset F). (B) In vivo rescue assay: Coinjection of human CHD7 mRNA encoding mutant S103T allele along with chd7 MO results in significant rescue of morphant phenotype indistinguishable from WT rescue. Coinjection of chd7 MO with human CHD7 mRNAs encoding the two other control alleles (p.M340V, p.L2984F) from group A (see main text) shows rescue similar to S103T allele. Five IGD-associated CHD7 alleles (p.F1362L, p.G1845R, G1982E, p.I2064V, and p.I2232V) from group B (see main text) were partial LOF alleles (hypomorphs). Of the CHARGE-associated CHD7 alleles (group C, see main text), one was complete LOF (null) (p.I1028V) and two were hypomorphic (p.D1596G and p.D1812H). The remaining CHD7 alleles were benign on the rescue assay. (C) Overexpression of human mutant CHD7 mRNAs in zebrafish: overexpression of the human WT CHD7 mRNA did not induce any appreciable phenotypes. Four IGD-associated CHD7 alleles (p.P940L, p.E1897K, p.T2532M, and p.Q2621E) from group B (see main text) and two CHARGE-associated CHD7 alleles (p. A1289V and p.V1021G) from group C (see main text) showed a dominant effect in the overexpression assay. The remaining CHD7 alleles were benign on the overexpression assay. ***P < 0.0005, **P < 0.005, *P < 0.05; NS, not significant.
All three CHD7 RSVs from group A (control alleles) were able to rescue morphant phenotypes in a manner indistinguishable from WT human CHD7 and did not show any dominant activity upon overexpression (Fig. 2 B and C). These data support the population frequency-derived prediction that group A alleles, with a MAF > 0.5%, are likely benign and bolstered confidence on the specificity of the assay. Among the 12 CHD7 alleles found in IGD patients without full CHARGE (group B), five alleles (p.F1362L, p.G1845R, G1982E, p.I2064V, and p.I2232V) showed partial loss of function (LOF) in that the frequency of abnormal otolith phenotypes was significantly better than MO alone yet significantly worse than controls (P < 0.05 for each comparison, scored blind and in triplicate), indicating they were likely hypomorphic alleles (Fig. 2B). Four alleles (p.P940L, p.E1897K, p.T2532M, and p.Q2621E) showed a dominant effect in the overexpression assay (i.e., each induced the absence of otoliths in a small but significant number of embryos) (P < 0.05) (Fig. 2C). Three alleles (p.Y1616C, p.R2065G, and p.A2789V) tested benign in both assays (Fig. 2 B and C). Thus, 75% (9 of 12) of IGD-associated (group B) missense RSVs tested in the functional assays were either hypomorphic or dominant, indicating an enrichment of functionally deleterious variants in this group compared with the controls in group A. Of the five alleles associated with the full CHARGE syndrome in group C, one was null (p.I1028V), two were hypomorphic (p.D1596G and p.D1812H), and two (p. A1289V and p.V1021G) were dominant (Fig. 2 B and C).
Finally, integration of 3D structural modeling of CHD7 using an automated homology model by SWISS-MODEL (swissmodel.expasy.org/) (20) or the FoldX protein design algorithm (21) predicted that four variants (p.P940L, p.V1021G, p.A1289V, p.G1982E) disrupted the 3D protein structure of CHD7 (Fig. 3) and all of these four variants were also determined to be deleterious RSVs from the zebrafish experiments. These variants were located in the chromo, SNF2, or helicase domains, all known to play important roles in chromatin remodeling (22).
Fig. 3.
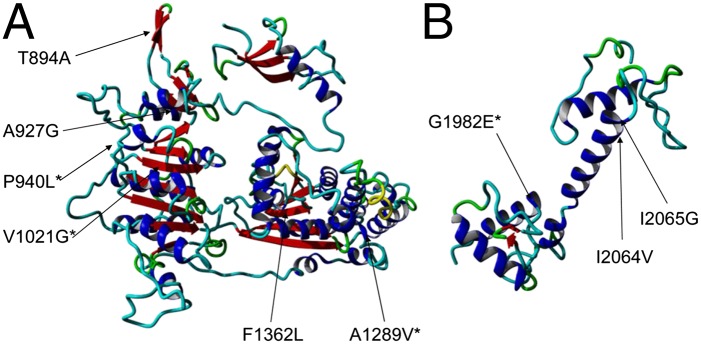
Structural modeling of the CHD7 chromo- and helicase domains (amino acids 799–1511) (A) and SANT domain (amino acids 1964–2115) (B) showing four variants (p.P940L, p.V1021G, p.A1289V, and p.G1982E) that were predicted to be detrimental to protein stability by FoldX algorithm (asterisk).
Deleterious CHD7 Missense Variants Cause both KS and nIGD Without Full CHARGE.
A total of 10 IGD subjects with both phenotypic forms (n = 6, KS; n = 4, nIGD) harbored pathogenic CHD7 missense mutations (Table 1). Only two of these patients had additional features of CHARGE but none fulfilled the Verloes criteria for full typical/atypical CHARGE syndrome (Table 2). However, when taking into consideration the presence of any CHARGE feature in patients harboring these deleterious CHD7 variants (Tables 1 and 2), patients with minor/full CHARGE were associated significantly with the KS phenotype (P = 0.0165), suggesting that the KS form of IGD is the primary allelic variant of CHARGE syndrome rather than its normosmic counterpart. However, the occurrence of deleterious CHD7 RSVs in the normosmic patients suggests that CHD7’s effect may not be limited to GnRH migration alone. In addition, one nIGD subject (Pedigree #35) (Fig. 1 and Table 1) had an adult-onset presentation of IGD, further confirming a neuroendocrine role for CHD7 in the pathogenesis of IGD. The clinical characteristics of patients with benign CHD7 alleles are also shown in Table S7. The clinical characteristics of patients with CHD7 alleles that were not tested in zebrafish assay are shown in Table S8.
Table 1.
Clinical characteristics of IGD patients without full CHARGE harboring pathogenic CHD7 mutations by zebrafish model
| Pedigree No. | Sex | Origin | De novo | Diagnosis | Olfaction | Nucleotide change | Amino acid change | Exon/Intron | Mutations in other genes |
| 11* | M | Caucasian | NA | KS | Self-reported anosmia | 2819C > T | P940L | 10 | None |
| 12 | M | Caucasian | NA | nIGD | Self-reported normal | 2819C > T | P940L | 10 | None |
| 18* | M | Caucasian | No | KS | UPSIT hyposmia | 4084T > C | F1362L | 19 | None |
| 24 | F | Caucasian | No | KS | UPSIT anosmia | 5533G > A | G1845R | 26 | FGFR1: 710G > A (G237D) |
| 25 | M | Asian | NA | nIGD | Self-reported normal | 5689G > A | E1897K | 29 | GNRHR: 836G > A (C279Y) homozygote |
| 28 | M | Caucasian | NA | KS | Self-reported anosmia | 5945G > A | G1982E | 30 | None |
| 32 | M | African American | NA | KS | UPSIT hyposmia | 6190A > G | I2064V | 31 | None |
| 35 | M | Asian | NA | nIGD, adult onset | UPSIT normal | 6694A > G | I2232V | 31 | None |
| 38 | F | African American | NA | nIGD | Self-reported normal | 7595C > T | T2532M | 34 | None |
| 44 | M | Caucasian | NA | KS | Self-reported anosmia | 8405G > A/7861C > G | G2802E/Q2621E | 38/36 | None |
F, female; M, male; NA, not assessed; UPSIT, University of Pennsylvania Smell Identification Test.
Probands with additional CHARGE features but not fulfilling Verloes CHARGE syndrome criteria.
Table 2.
Additional CHARGE features present in IGD patients with CHD7 RSVs that were pathogenic in zebrafish model
| Pedigree no. | Diagnosis | Mutation in CHD7 | Second hits | C | H | A | RG | RD | G | HL | E | CLP | FA | Verloes criteria for full CHARGE |
| 11 | KS | P940L | None | — | — | — | — | — | + | + | — | — | — | No |
| 18 | KS | F1362L | None | — | — | — | NA | + | + | + | — | — | — | No |
A, atresia of choanae; C, coloboma; CLP, cleft lip/plate; E, external ear defect; FA, facial asymmetry; G, genital defects; H, heart defects; HL, hearing loss; RG, retarded growth; RD, retarded development.
Probands with additional CHARGE features but not fulfilling Verloes CHARGE syndrome criteria.
Segregation Analysis Suggests Complex Inheritance Patterns.
For functionally deleterious CHD7 variants in the IGD pedigrees where inheritance information was available, none were de novo nor was there any apparent parent of origin bias (Fig. 4 and Table 1). These findings contrast previously reported CHARGE-causing variants that are typically de novo and show a preference for paternal inheritance (23). Segregation analysis also showed that the discovered CHD7 alleles are not sufficient to explain the observed phenotypes (Fig. 4). In two families, the pathogenic CHD7 variants were found in IGD patients who also carried a known pathogenic mutation in a second IGD gene (FGFR1 in pedigree #24 and GNRHR in pedigree #25), raising the possibility that some CHD7 alleles might interact genetically with other IGD mutations and thus contribute to the reported oligogenic nature of IGD (24) (Fig. 4 and Table 1). However, interpreting such genetic architecture based on a small number of candidate epistatic events must be done with caution because segregation analysis of the CHD7 alleles was incomplete in these oligogenic pedigrees (Fig. 4). However, in both of these oligogenic pedigrees, the presence of another IGD-associated genetic variant did not appear to heighten the occurrence of any CHARGE-related phenotypes; that is, these patients exhibited IGD only. This observation suggests that these other IGD-causing genes probably lack direct functional synergy with CHD7 in the developmental pathways involved in the non-GnRH organ systems involved in CHARGE syndrome.
Fig. 4.

Family pedigrees of probands with pathogenic CHD7 mutations. Pedigrees #11, #12, #18, #28, #33, #35, #38, and #44 harbored CHD7 mutations alone, and pedigrees #24 and #25 harbored CHD7 mutations oligogenic with FGFR1 and GNRHR genes, respectively. Among familial cases, pedigree #24 and pedigree #25 display incomplete penetrance for CHD7 mutations and pedigree #18 displays variable expressivity of GnRH deficiency/anosmia phenotypes for the CHD7 mutation. Probands are identified by arrows. “+” indicates WT allele.
Discussion
In this study, a large, well-phenotyped cohort of IGD patients, with both KS and nIGD that were assembled without preselection for or against known mutations in known IGD genes, were analyzed to assess the contribution of CHD7 to IGD. In contrast to truncating CHD7 mutations occurring in typical CHARGE syndrome, ∼5.2% of IGD patients without full CHARGE features harbored heterozygous, nonsynonymous missense or splice-altering variants in CHD7. The observed prevalence and the predominantly missense nature of the CHD7 variants is in accordance with recent reports (8, 16). However, to our knowledge for the first time, the functional impact of a representative subset of the missense variants in CHD7 was assessed in a zebrafish assay and compared with functional effects of low-frequency missense RSVs represented in the public Exome Sequencing Project (ESP) database. Whereas all variants from the ESP database tested benign, ∼75% of the tested IGD-associated CHD7 RSVs were pathogenic, acting either as clear hypomorphs that were unable to rescue the CHD7-MO phenotype or as dominant alleles when overexpressed. On phenotypic analysis, most IGD-patients with deleterious missense CHD7 alleles did not fulfill full CHARGE criteria, thus providing functional molecular validation that the nonsyndromic form of IGD is indeed an allelic variant of CHARGE syndrome. Segregation analysis of deleterious CHD7 variants shows incomplete penetrance and variable expressivity, suggesting that these deleterious variants contribute to the IGD phenotype but by themselves may be insufficient to cause it.
KS and nIGD represent the two distinct phenotypic forms of IGD. The KS phenotype results from the neurodevelopmental failure of GnRH neuronal migration during development and nIGD results from the neuroendocrine failure of GnRH secretion/action within the hypothalamus (12, 13). Whereas most genes exclusively cause either the KS or nIGD forms of the disease, a subset of genes (e.g., FGF8/FGFR1, PROK2/PROKR2) are associated with both KS and nIGD, indicating either (i) two potentially different pathophysiologic sites of actions on the ontogeny of GnRH neurons or (ii) the potential for dissociated effects on the ontogeny of GnRH neurons and olfactory axons, respectively (12, 13). Although the initial report by Kim et al. (8) implicated CHD7 in both KS and nIGD, subsequent investigators have failed to identify any predicted pathogenic CHD7 RSVs in nIGD (9, 16). However, some nIGD subjects did harbor rare CHD7 alleles but these were deemed to be benign based on prediction programs, which are recognized to be prone to ambiguity (16). The present study was able to address these issues by performing functional studies and by examining the phenotypes of patients with pathogenic CHD7 alleles, this study provides key insights into the role of CHD7 in GnRH neuronal ontogeny. Functionally deleterious CHD7 mutations were found in both KS and nIGD subjects, from which it can be inferred that CHD7 likely has both distinct developmental as well as neuroendocrine roles. Moreover, four subjects with pathogenic CHD7 alleles also had micropenis or cryptorchidism, implying an embryonic defect in GnRH development and secretion. These results also indicate that future genetic evaluations of all nonsyndromic KS and nIGD should be screened for CHD7 for sequence variants.
To date, only a few cases of parent-to-child transmission have been reported in CHARGE pedigrees with CHD7 mutation (23, 25). In addition, over 80% of CHARGE-causing CHD7 mutations are null alleles (5, 6). In our IGD patients, however, not only were CHD7 variants inherited (where possible to assess), but most IGD-associated CHD7 variants were either hypomorphs or had a modest dominant effect. Thus, it appears that it is the nature and severity of these variants, rather than their location within the CHD7 gene/protein that is critical for its selective perturbation on developing GnRH neurons producing IGD rather than their multiorgan involvement in CHARGE.
The present study has some limitations. The phenotypic details of additional CHARGE features in IGD patients, as well as phenotypes of parents, were partly ascertained by retrospective review of medical records, physician referral letters, and questionnaire data. Thus, it is possible that a prospective evaluation might uncover additional phenotypic features of CHARGE in these IGD patients. However, because the majority of our patients have had detailed physician reviews either via in-house or through referrals from multiple specialists, major CHARGE features are unlikely to have been missed. Typical of most sequencing studies, deep-intronic and regulatory-regions, deletions, and rearrangements were not evaluated. However, in previous studies of CHD7 mutations, no such deletions have been found (6, 26). While IGD subjects underwent Sanger sequencing, control CHD7 alleles were obtained from whole-exome seqencing data from the NHLBI ESP Exome Variant Server and, hence, potentially could be subject to platform-specific biases in variant ascertainment. However, reassuringly, limited validation of ESP singleton variants has been reported to be reliable (27) and hence biases, if any, are unlikely to be significant. Given the complexity of design and conduct of CHD7 functional assays, only a subset of alleles were examined. However, multiple alleles tested consistently as hypomorphs in the rescue experiments, validating the observed findings. Similarly, some alleles displayed reproducible altered dominant phenotype upon overexpression whereas WT allele overexpression did not produce any altered phenotype. Moreover, all alleles predicted as destabilizing on automated homology modeling were found to be deleterious in zebrafish, further strengthening the observations. Two alternative splice products of the CHD7 gene are known (28, 29) but the effect the RSVs identified in this study on these splice products were not examined in the present study. Finally, functional assessment of CHD7 alleles was ascertained using a surrogate otolith phenotype in zebrafish rather than GnRH or olfactory neuronal phenotypes. However, this assay is an outstanding proxy for CHD7 function (19) and, hence, the functional deficits observed are unlikely to represent false-positive results. In contrast, there is a possibility that the alleles that tested benign in the otolith assays may actually be detrimental to protein function below the dynamic range of the assay, representing false-negative observations. In addition, four patients with documented hypomorphic/dominant alleles in this study also displayed hearing loss, thus confirming the biological relevance of the examined zebrafish phenotype (Tables 1 and 2 and Table S3).
The question of where CHD7 fits into the developmental biology of the GnRH neurons remains a mystery. Mice with complete Chd7 deficiency mirror the observed human phenotypes (delayed puberty, genital hypoplasia, hypogonadotropism) and display neurodevelopmental defects, including reduced GnRH neurons in the hypothalamus and reduced expression of FGFR1 in the olfactory placode (30, 31). Recent work also suggests that CHD7 regulates genes involved in neural crest guidance (32), and there is increasing evidence that KS may also result from impaired neural crest guidance (33). Taken together, these observations suggest that CHD7 plays an important neurodevelopmental role in the formation of the hypothalamic-pituitary unit (31). That rare pathogenic CHD7 variants are associated with nIGD suggests that in addition to its neurodevelopmental role, CHD7 may well also influence subsequent neuroendocrine development pathways of GnRH neurons. The striking predominance of missense CHD7 variants in IGD implies that this gene and its associated biological pathways are especially critical for the ontogeny of GnRH neurons. Hence, working out the function of CHD7 via comparison of missense vs. truncating mutations might offer a route to unraveling its unique biological role in this complex system.
Materials and Methods
Study Subjects and Phenotypic Information.
Included in this study were 783 consecutive patients with IGD (367 KS, 411 nIGD) from the Reproductive Endocrine Unit of Massachusetts General Hospital, who underwent genetic screening for 14 genes associated with IGD (see below). Diagnostic criteria for IGD and CHARGE syndrome criteria are provided in SI Materials and Methods. Phenotypic records from individual medical charts, patient questionnaires, and referring provider’s summary were reviewed to ascertain additional phenotypic information related to CHARGE syndrome, such as coloboma, choanal atresia, external ear defects, cardiovascular malformations, cleft lip/palate, and developmental delay. Study protocols were approved by the Massachusetts General Hospital/Partners Institutional Review Board and informed consent obtained from all participants.
Genetic Analyses.
Genomic DNA was isolated from peripheral blood lymphocytes. In all IGD subjects, the 37 coding exons of CHD7 (exons 2–38) and their intronic flanking regions were amplified by PCR and directly sequenced, as described previously (9). The GenBank accession number NM_017780.2 was used as reference sequence for CHD7. In addition, all coding exons and intronic flanking regions of the GNRH1 (Gonadotropin-releasing hormone 1), GNRHR (Gonadotropin-releasing hormone receptor), KISS1 (KiSS-1 metastasis-suppressor), KISS1R (KISS1 receptor), PROK2 (prokineticin 2), PROKR2 (prokineticin receptor 2), TAC3 (tachykinin 3), TACR3 (tachykinin receptor 3), FGF8 (fibroblast growth factor 8), FGFR1 (fibroblast growth factor receptor 1), KAL1 (kallmann syndrome 1 sequence), HS6ST1 (heparan sulfate 6-O-sulfotransferase 1), and NSMF [NMDA receptor synaptonuclear signaling and neuronal migration factor; previously called NELF (nasal embroyonic leutinizing hormone releasing hormone factor)] genes were amplified by PCR with specific primers and directly sequenced as described previously (24). Protein-altering variants in CHD7 with a MAF < 0.5% in the NHLBI ESP Exome Variant Server (evs.gs.washington.edu/EVS/) (34) and not reported to be benign polymorphisms in previously published literature (5, 6), or in the CHD7 database (www.CHD7.org) were considered as RSVs. In addition, splice-site altering variants in sequences within 8 bp of splice junctions (the consensus sequences are AG and GT for the donor and acceptor sites, respectively) with MAF < 0.5% in the NHLBI Exome Variant Server were also considered as RSVs. For the other 13 IGD genes screened, previously published pathogenic RSVs were included in the oligogenicity analysis.
Generation of Zebrafish chd7 Knockdown and Rescue Experiments.
Zebrafish chd7 knockdown was done using a splice-blocking MO (TTATTTTCTGGCACTAACCATGTCC), previously reported (19). One- to four-cell–stage zebrafish embryos were injected with 6 ng MO or 5 pg mRNA. Injected embryos were scored live at 3 dpf and classified according to the presence, amount, and size of otoliths. To confirm the specificity of the MO, rescue experiments were performed. Human full-length CHD7 cDNA was kindly provided by Joanna Wysocka (Stanford School of Medicine, Stanford, CA) and Peter C. Scacheri (Case Western Reserve University School of Medicine, Cleveland, OH). Full-length WT CHD7 was amplified and cloned into pCS2. To generate CHD7 mutations, primers were designed to introduce the specific change and the reaction was performed using QuikChange Site-Directed Mutagenesis kit (Stratagene). CHD7 WT and mutations were confirmed through Sanger sequencing, cloned into pCS2 vector, and transcribed in vitro with the SP6 Message Machine kit (Ambion). Next, 10 pg of mRNA was injected alone into zebrafish embryos. For rescue experiments, CHD7 WT mRNA (5 pg) was coinjected with chd7-MO (6 ng).
Statistical Analysis.
All zebrafish experiments were repeated at least three times and a Pearson’s χ2 test was used to determine significance between S103T allele rescue vs. mutant rescue and between WT vs. mutant CHD7 allele overexpression. The χ2 test was also used to compare the occurrence of KS vs. nIGD in the presence of pathogenic CHD7 RSVs in IGD patients. A P value less than 0.05 were considered as statistically significant.
Supplementary Material
Acknowledgments
This study was supported by the National Institutes of Health Grants U54 HD028138 (to W.F.C.) and P50 DK096415 (to N.K.). R.B. is supported by the National Institutes of Health Grant K23 HD077043. K.M.S. is supported by National Institutes of Health Grant K08 DC010419 and the Bertarelli Foundation. J.F.G. is supported by National Institutes of Health Grant P01 GM061354 (Developmental Genome Anatomy Project).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1417438111/-/DCSupplemental.
References
- 1.Vissers LE, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004;36(9):955–957. doi: 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- 2.Sanlaville D, Verloes A. CHARGE syndrome: An update. Eur J Hum Genet. 2007;15(4):389–399. doi: 10.1038/sj.ejhg.5201778. [DOI] [PubMed] [Google Scholar]
- 3.Blake KD, Prasad C. CHARGE syndrome. Orphanet J Rare Dis. 2006;1:34. doi: 10.1186/1750-1172-1-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergman JE, et al. CHD7 mutations and CHARGE syndrome: The clinical implications of an expanding phenotype. J Med Genet. 2011;48(5):334–342. doi: 10.1136/jmg.2010.087106. [DOI] [PubMed] [Google Scholar]
- 5.Bartels CF, Scacheri C, White L, Scacheri PC, Bale S. Mutations in the CHD7 gene: The experience of a commercial laboratory. Genet Test Mol Biomarkers. 2010;14(6):881–891. doi: 10.1089/gtmb.2010.0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Janssen N, et al. Mutation update on the CHD7 gene involved in CHARGE syndrome. Hum Mutat. 2012;33(8):1149–1160. doi: 10.1002/humu.22086. [DOI] [PubMed] [Google Scholar]
- 7.Wheeler PG, Quigley CA, Sadeghi-Nejad A, Weaver DD. Hypogonadism and CHARGE association. Am J Med Genet. 2000;94(3):228–231. doi: 10.1002/1096-8628(20000918)94:3<228::aid-ajmg8>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 8.Kim HG, et al. Mutations in CHD7, encoding a chromatin-remodeling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet. 2008;83(4):511–519. doi: 10.1016/j.ajhg.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jongmans MC, et al. CHD7 mutations in patients initially diagnosed with Kallmann syndrome—The clinical overlap with CHARGE syndrome. Clin Genet. 2009;75(1):65–71. doi: 10.1111/j.1399-0004.2008.01107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foppiani L, Maffè A, Forzano F. CHARGE syndrome as unusual cause of hypogonadism: Endocrine and molecular evaluation. Andrologia. 2010;42(5):326–330. doi: 10.1111/j.1439-0272.2009.00994.x. [DOI] [PubMed] [Google Scholar]
- 11.Pinto G, et al. CHARGE syndrome includes hypogonadotropic hypogonadism and abnormal olfactory bulb development. J Clin Endocrinol Metab. 2005;90(10):5621–5626. doi: 10.1210/jc.2004-2474. [DOI] [PubMed] [Google Scholar]
- 12.Crowley WF. The developmental biology of the GnRH neurons. Mol Cell Endocrinol. 2011;346(1-2):1–3. doi: 10.1016/j.mce.2011.06.023. [DOI] [PubMed] [Google Scholar]
- 13.Balasubramanian R, Crowley WF., Jr Isolated GnRH deficiency: A disease model serving as a unique prism into the systems biology of the GnRH neuronal network. Mol Cell Endocrinol. 2011;346(1-2):4–12. doi: 10.1016/j.mce.2011.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwanzel-Fukuda M, Pfaff DW. Origin of luteinizing hormone-releasing hormone neurons. Nature. 1989;338(6211):161–164. doi: 10.1038/338161a0. [DOI] [PubMed] [Google Scholar]
- 15.Bergman JE, et al. The results of CHD7 analysis in clinically well-characterized patients with Kallmann syndrome. J Clin Endocrinol Metab. 2012;97(5):E858–E862. doi: 10.1210/jc.2011-2652. [DOI] [PubMed] [Google Scholar]
- 16.Marcos S, et al. The prevalence of CHD7 missense versus truncating mutations is higher in patients with Kallmann syndrome than in typical CHARGE patients. J Clin Endocrinol Metab. 2014;99(10):E2138–E2143. doi: 10.1210/jc.2014-2110. [DOI] [PubMed] [Google Scholar]
- 17.Verloes A. Updated diagnostic criteria for CHARGE syndrome: A proposal. Am J Med Genet A. 2005;133A(3):306–308. doi: 10.1002/ajmg.a.30559. [DOI] [PubMed] [Google Scholar]
- 18.Thusberg J, Olatubosun A, Vihinen M. Performance of mutation pathogenicity prediction methods on missense variants. Hum Mutat. 2011;32(4):358–368. doi: 10.1002/humu.21445. [DOI] [PubMed] [Google Scholar]
- 19.Patten SA, et al. Role of Chd7 in zebrafish: A model for CHARGE syndrome. PLoS ONE. 2012;7(2):e31650. doi: 10.1371/journal.pone.0031650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bordoli L, et al. Protein structure homology modeling using SWISS-MODEL workspace. Nat Protoc. 2009;4(1):1–13. doi: 10.1038/nprot.2008.197. [DOI] [PubMed] [Google Scholar]
- 21.Van Durme J, et al. A graphical interface for the FoldX forcefield. Bioinformatics. 2011;27(12):1711–1712. doi: 10.1093/bioinformatics/btr254. [DOI] [PubMed] [Google Scholar]
- 22.Bergman JE, et al. A novel classification system to predict the pathogenic effects of CHD7 missense variants in CHARGE syndrome. Hum Mutat. 2012;33(8):1251–1260. doi: 10.1002/humu.22106. [DOI] [PubMed] [Google Scholar]
- 23.Pauli S, et al. CHD7 mutations causing CHARGE syndrome are predominantly of paternal origin. Clin Genet. 2012;81(3):234–239. doi: 10.1111/j.1399-0004.2011.01701.x. [DOI] [PubMed] [Google Scholar]
- 24.Sykiotis GP, et al. Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc Natl Acad Sci USA. 2010;107(34):15140–15144. doi: 10.1073/pnas.1009622107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Delahaye A, et al. Familial CHARGE syndrome because of CHD7 mutation: Clinical intra- and interfamilial variability. Clin Genet. 2007;72(2):112–121. doi: 10.1111/j.1399-0004.2007.00821.x. [DOI] [PubMed] [Google Scholar]
- 26.Vuorela P, et al. Molecular analysis of the CHD7 gene in CHARGE syndrome: Identification of 22 novel mutations and evidence for a low contribution of large CHD7 deletions. Genet Med. 2007;9(10):690–694. doi: 10.1097/gim.0b013e318156e68e. [DOI] [PubMed] [Google Scholar]
- 27.Tennessen JA, et al. Broad GO Seattle GO NHLBI Exome Sequencing Project Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337(6090):64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Colin C, Tobaruella FS, Correa RG, Sogayar MC, Demasi MA. Cloning and characterization of a novel alternatively spliced transcript of the human CHD7 putative helicase. BMC Res Notes. 2010;3:252. doi: 10.1186/1756-0500-3-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kita Y, Nishiyama M, Nakayama KI. Identification of CHD7S as a novel splicing variant of CHD7 with functions similar and antagonistic to those of the full-length CHD7L. Genes Cells. 2012;17(7):536–547. doi: 10.1111/j.1365-2443.2012.01606.x. [DOI] [PubMed] [Google Scholar]
- 30.Bergman JE, Bosman EA, van Ravenswaaij-Arts CM, Steel KP. Study of smell and reproductive organs in a mouse model for CHARGE syndrome. Eur J Hum Genet. 2010;18(2):171–177. doi: 10.1038/ejhg.2009.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Layman WS, Hurd EA, Martin DM. Reproductive dysfunction and decreased GnRH neurogenesis in a mouse model of CHARGE syndrome. Hum Mol Genet. 2011;20(16):3138–3150. doi: 10.1093/hmg/ddr216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schulz Y, et al. CHD7, the gene mutated in CHARGE syndrome, regulates genes involved in neural crest cell guidance. Hum Genet. 2014;133(8):997–1009. doi: 10.1007/s00439-014-1444-2. [DOI] [PubMed] [Google Scholar]
- 33.Pingault V, et al. Loss-of-function mutations in SOX10 cause Kallmann syndrome with deafness. Am J Hum Genet. 2013;92(5):707–724. doi: 10.1016/j.ajhg.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP) Seattle, WA. Available at evs.gs.washington.edu/EVS. Accessed November 26, 2013.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.