

Significance
The chemokine receptor axis plays a critical role in numerous physiological and pathological processes, yet the structural basis of receptor interaction with chemokines is poorly understood. Although the community agrees on the existence of two distinct epitopes for recognition of receptors by chemokines, conflicting evidence from structural and mutagenesis studies suggested several possibilities for receptor:chemokine complex stoichiometry. We use a combination of computational, functional, and biophysical approaches to show that despite its dimeric nature, chemokine receptor CXCR4 interacts with its chemokine ligand, CXCL12, in a 1:1 stoichiometry. This result is also likely relevant for other receptor:chemokine pairs. Structural modeling informed by restraints derived from cysteine trapping experiments enabled determination of the receptor:chemokine complex geometry at a medium resolution level.
Keywords: chemokine receptor, GPCR dimerization, molecular docking, functional complementation, cysteine trapping
Abstract
Chemokines and their receptors regulate cell migration during development, immune system function, and in inflammatory diseases, making them important therapeutic targets. Nevertheless, the structural basis of receptor:chemokine interaction is poorly understood. Adding to the complexity of the problem is the persistently dimeric behavior of receptors observed in cell-based studies, which in combination with structural and mutagenesis data, suggest several possibilities for receptor:chemokine complex stoichiometry. In this study, a combination of computational, functional, and biophysical approaches was used to elucidate the stoichiometry and geometry of the interaction between the CXC-type chemokine receptor 4 (CXCR4) and its ligand CXCL12. First, relevance and feasibility of a 2:1 stoichiometry hypothesis was probed using functional complementation experiments with multiple pairs of complementary nonfunctional CXCR4 mutants. Next, the importance of dimers of WT CXCR4 was explored using the strategy of dimer dilution, where WT receptor dimerization is disrupted by increasing expression of nonfunctional CXCR4 mutants. The results of these experiments were supportive of a 1:1 stoichiometry, although the latter could not simultaneously reconcile existing structural and mutagenesis data. To resolve the contradiction, cysteine trapping experiments were used to derive residue proximity constraints that enabled construction of a validated 1:1 receptor:chemokine model, consistent with the paradigmatic two-site hypothesis of receptor activation. The observation of a 1:1 stoichiometry is in line with accumulating evidence supporting monomers as minimal functional units of G protein-coupled receptors, and suggests transmission of conformational changes across the dimer interface as the most probable mechanism of altered signaling by receptor heterodimers.
The chemokine receptor CXCR4 regulates cell migration during many developmental processes (1, 2). Along with CCR5, it serves as one of the principal coreceptors for HIV entry into leukocytes (3), and is one of the most important chemokine receptors involved in cancer metastasis (4). Stromal-cell derived factor 1 (SDF-1 or CXCL12) was its only known ligand until recently, when CXCR4 was also shown to bind CXCL14 (5) and extracellular ubiquitin (6). Although structures of CXCR4 (7) and CCR5 (8) have been solved with synthetic antagonists, the structural basis for the interaction of CXCR4 (or any other chemokine receptor) with their natural ligands has yet to be determined. Numerous mutagenesis and NMR studies indicate that receptor:chemokine interactions involve two distinct sites (9–12), which has led to a two-site hypothesis of receptor activation (13). The so-called chemokine recognition site 1 (CRS1) (14) includes the N terminus of the receptor interacting with the globular core of the chemokine, whereas chemokine recognition site 2 (CRS2), located within the transmembrane (TM) domain pocket of the receptor, accommodates the flexible N terminus of the chemokine. Mutations in CRS1 typically reduce the binding affinity of the chemokine, whereas CRS2 is critical not only for binding but also for chemokine-induced activation (9, 10, 12, 15–20). Similarly, mutations to the core domain of the chemokine generally affect receptor-binding affinity, but truncations or modifications of as little as one amino acid in the N-terminal “signaling” domain frequently alter both ligand binding and pharmacology.
The two-site model has been envisioned in the context of a monomeric receptor. However, like many other G protein-coupled receptors (GPCRs) (21), CXCR4 has been shown to dimerize in cell membranes. Evidence supporting CXCR4 dimerization includes immunoprecipitation (22), bioluminescence and fluorescence resonance energy transfer [BRET (23) and FRET (24), respectively], fluorescence and luminescence complementation assays (25), and bivalent ligands (26). Dimerization of a WT CXCR4 with a C-terminally truncated mutant causing the “warts, hypogammaglobulinemia, infections and myelokathexis” (WHIM) syndrome has been implicated in its resistance to desensitization and enhanced signaling in heterozygous WHIM patients (27). CXCR4 has also been shown to heterodimerize with other chemokine receptors and with GPCRs outside the chemokine family (28–32), with consequences including transinhibition of ligand binding (28) and changes in G protein and β-arrestin coupling (30, 33, 34). These observations establish the dimeric nature of CXCR4; however, the functional role of CXCR4 dimers has yet to be elucidated.
In agreement with its persistently dimeric behavior, CXCR4 formed structurally similar parallel dimers in five crystal structures (7), despite being solved in different space groups and with different synthetic ligands. The cell-based and structure-based observations of CXCR4 dimers raised the key question as to whether CXCL12 binds to a single receptor subunit or to both subunits of the dimer, in a manner consistent with the two-site model. Several possible stoichiometries of the complex were suggested (7, 35, 36); among them, a 1:1 receptor:chemokine stoichiometry, a 2:1 stoichiometry with one chemokine molecule simultaneously binding to both subunits of a CXCR4 dimer, and a 2:2 stoichiometry with a chemokine dimer binding to the CXCR4 dimer. With respect to the latter, although CXCL12 dimers bind and act as partial agonists of CXCR4 (37), full agonist signaling requires a monomeric chemokine (37, 38). Consequently, the distinction between a 1:1 and a 2:1 receptor:chemokine stoichiometry is the most relevant question, and constituted the focus of the present study.
Our initial molecular modeling efforts encompassed the available structural information in the form of (i) the NMR structure of a cross-linked CXCL12 dimer in complex with an N-terminal peptide of CXCR4 (residues M1-K38) (39), and (ii) the X-ray structures of full-length CXCR4 (7). The former structure contains components of the CRS1 interaction (Fig. 1A), whereas the latter contains the receptor side of the CRS2 interaction. Although the crystallization constructs used in the CXCR4 X-ray study contained the intact N terminus of the receptor, only residues P27–S319 could be detected in the electron density; thus, the overlap between the NMR and X-ray structures was limited to residues P27–K38. Modeling demonstrated that a 2:1 receptor:chemokine model with decoupled CRS1 and CRS2 best accommodated the structural and mutagenesis data. In this model, the globular core of the chemokine interacts with the CRS1 of one receptor subunit and the N-terminal residues of the chemokine reach into CRS2 of its dimeric partner (Fig. 1B). In addition to being spatially consistent, this model provides a direct explanation for the negative cooperativity in chemokine binding that is frequently observed with receptor heterodimers (28, 40), and with the notion that CXCL12 triggers CXCR4 dimerization (41), stabilizes preformed dimers (42), or induces conformational changes within the dimers (23). The model is also consistent with the original two-site hypothesis of receptor activation. On the other hand, a 1:1 model, in which CXCL12 interacts with CRS1 and CRS2 of the same receptor subunit, required significant deviations from the CRS1 component of the CXCR4:CXCL12 interaction suggested by the NMR structure (39) to orient the CXCL12 N-terminal signaling domain toward the receptor binding pocket (Fig. 1 C and D).
Fig. 1.
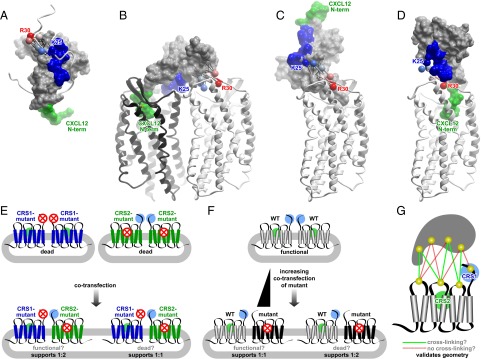
Molecular models and experimental designs used in the present study. (A) NMR structure of CXCL12 (skin mesh) in complex with the N terminus of CXCR4 (residues M1–K38, ribbon) (39). Chemokine N terminus (green) and N-loop (blue) correspond to the expected interactions in CRS2 and CRS1, respectively. Receptor residues K25–R30 are shown as spheres, labeled, and colored in order from blue to red. CRS1 residue proximities observed in the NMR structure and maintained throughout the docking simulations include the interaction of CXCR4 K25 (blue sphere) with CXCL12 S16, while the subsequent receptor residues up to R30 (red sphere) are directed away from the chemokine N-loop (blue surface) toward the chemokine C-terminal helix; these proximities are shown as thin black lines. (B) A hybrid 2:1 model of the receptor:chemokine interaction accommodates both NMR proximity restraints (black lines) and the mutagenesis data. (C) A hydrid 1:1 model that accommodates NMR proximity restraints (black lines) is inconsistent with mutagenesis and with the two-site interaction hypothesis, because the N terminus of the chemokine invariably points away from the receptor CRS2. (D) A 1:1 model consistent with the two-site interaction hypothesis contradicts NMR proximity restraints, as receptor residues K25–R30 are directed along the chemokine N-loop toward its N terminus. (E–G) Conceptual designs of the functional complementation (E), dimer dilution (F), and cysteine trapping (G) experiments used in this study to probe the receptor:chemokine stoichiometry and geometry hypotheses.
Three strategies were devised to elucidate the stoichiometry of the receptor:chemokine interaction. The first approach was based on functional complementation and designed to specifically probe the relevance of the 2:1 hypothesis. Functional complementation provides one of the strongest arguments for the existence and physiological role of GPCR dimers. In this type of experiment, different aspects of receptor function are restored by coexpression of two mutants of the receptor in question, each of which is incapable of producing the functional response when expressed alone (Fig. 1E). Functional rescue through dimerization has been demonstrated for several GPCRs. For example, domain swapping of histamine H1 receptor dimers reconstituted functional receptors from nonfunctional mutant components (43), and a related mechanism led to reconstitution of functional muscarinic and adrenergic receptors from receptor chimeras (44). Similarly, the binding site in the angiotensin II receptor was successfully reconstituted (45), and the function of the luteinizing hormone receptor was rescued by coexpression of two nonfunctional mutants (46). In the present study, the functional complementation strategy was used to probe the possibility of simultaneous interaction of CXCL12 with two CXCR4 monomers in the dimer.
Another strategy for exploring the role of dimers in general, and the stoichiometry of GPCR interactions with ligands and effectors in particular, is based on dimer dilution. In this approach, functional responses or binding events that are dependent on GPCR dimers are reduced or completely ablated by introducing increasing amounts of a mutant that is capable of dimerizing with the WT receptor but incapable of mediating the functional or binding response (Fig. 1F). The mutant receptors compete with WT receptors for dimer formation and lead to an increase in the surface density of WT/mutant dimers, with a simultaneous decrease in WT/WT dimers (47). In contrast, if dimers are unnecessary for the functional response or binding event, one should see no change with increasing concentration of mutant; thus, this approach distinguishes 1:1 vs. 2:1 interactions. There are important caveats associated with this strategy, as increasing expression of mutant receptors may interfere not only with formation of WT/WT dimers, but also with the expression of WT receptors because of expression competition. In this study, a modified dimer dilution strategy that addressed these problems was designed.
The above strategies are based on functional readouts and provide indirect evidence in favor of, or against, the different stoichiometries. We therefore complemented them by cysteine trapping studies, where pairs of cysteine mutations are introduced at different positions in the ligand and in the receptor, and spontaneous formation of disulfide bonds is monitored. These studies provide direct spatial proximity restraints that can be combined with modeling to determine the stoichiometry and geometry of the receptor:chemokine complex (Fig. 1G).
The results of all three complementary experimental strategies were supportive of a 1:1 and not a 2:1 receptor:chemokine stoichiometry. These results also informed further molecular modeling efforts, which led to construction of an experimentally validated model of the CXCR4:CXCL12 complex. The model elucidates key features of the receptor:chemokine interaction and may facilitate further structure-function studies to understand the molecular basis for CXCR4:CXCL12 signaling.
Results
Structural Constraints Are Incompatible with Mutagenesis in the Context of a 1:1 CXCR4:CXCL12 Model.
Using molecular modeling and chemical field-guided docking (48), we attempted reconstruction of the hybrid structure of the CXCR4:CXCL12 complex by simultaneously satisfying restraints from the X-ray structure of the CXCR4 TM domain (7) and the NMR structure of the CXCL12 dimer in complex with a CXCR4 N-terminal peptide (39) (Fig. 1A). The former set of restraints included the relative positioning of the CXCR4 TM helices, as well as a disulfide bond between the N-terminal cysteine (C28) of the receptor and its extracellular loop 3. The latter set involved harmonic distance restraints (thin black lines in Fig. 1 A–C) imposed between the Cβ atoms in the least uncertain portion of the receptor N terminus in the NMR structure (residues K25–R30, shown as spheres in Fig. 1 A–D and colored in order from blue to red) and the Cβ atoms of proximal chemokine residues F14–S16 (N-loop), I51–K56 (the loop connecting β3 and the C-terminal helix), and I58–E60 (C-terminal helix). As observed in the NMR structure, these restraints included the interaction of the receptor residue K25 (blue sphere in Fig. 1 A–D) with residue S16 in the chemokine N-loop (blue patch in Fig. 1 A–D), whereas the subsequent receptor residues up to R30 (red sphere in Fig. 1 A–D) were directed away from the N-loop and toward the chemokine C-terminal helix (Fig. 1A).
Docking simulations were carried out by explicit conformational sampling of CXCL12 and the N-terminal residues K25–R30 of CXCR4 in internal coordinates (49), with the remaining parts of the receptor represented by the potential grid maps (50). These simulations sought to optimize electrostatic, van der Waals, hydrogen bonding, and surface interactions within and between the two molecules while simultaneously satisfying the constraints from the existing structures. Despite allowing full flexibility in the N-terminal parts of both molecules, the simulations invariably resulted in models that were inconsistent with existing mutagenesis studies (9, 16, 18–20, 51–54) when undertaken in the context of the 1:1 stoichiometry hypothesis. Specifically, the N-terminal signaling domain of the chemokine was forced out of the receptor CRS2, separating the critical interacting residues by as much as 50 Å (Fig. 1C). In contrast, models built to test the 2:1 stoichiometry hypothesis appeared spatially compatible with the mutagenesis, as the N terminus of the chemokine could be freely directed into the CRS2 of one CXCR4 dimer partner when the core domain was bound to CRS1 of the other (Fig. 1B). Although coarse-grained and approximate, this modeling exercise raised the question of what the actual interaction stoichiometry is.
Design and Testing of a CXCR4-Free Cell Line for Functional Complementation Experiments.
For the purpose of testing loss-of-function CXCR4 mutant pairs in the functional complementation experiments, it was essential to use a cell line devoid of endogenous CXCR4 expression. However, we discovered that many cells commonly used in chemokine functional assays endogenously express CXCR4 (Fig. S1 A–E and Table S1) and mobilize calcium in response to CXCL12 (Fig. S1 F–I and Table S1). Among the few immortalized cell lines that did not express endogenous CXCR4, only Chinese hamster ovary (CHO) cells displayed both a robust transfection efficiency and signaling response using Ca2+ mobilization as a readout (Fig. S1 E and I). The latter was significantly improved when the cells were stably transfected with human Gα15 protein (55) (Fig. S1 J–P), which resulted in creation of a CHO-Gα15 cell line.
Design, Surface Expression, and Function of CXCR4 Point Mutants in CHO-Gα15.
CXCR4 mutants defective in chemokine binding and signaling were designed taking into account earlier mutagenesis studies (9, 16, 18–20, 51–54) and the residue contacts in the structural models of the complex. To disrupt CXCR4:CXCL12 interactions in CRS1, two mutants were generated: one with alanine substitutions in the positions of the three sulfotyrosines known to affect ligand binding (56) and signaling (57) (Y7A/Y12A/Y21A, further referred to as YYY), and another where I4 and I6 were also mutated to alanine (37) (I4A/I6A/Y7A/Y12A/Y21A, further referred to as IIYYY). To disrupt interactions in CRS2, the following mutations were introduced one at a time: D97N, D171A, D187A, and E288A (Fig. 2A). Mutants were cloned into multiple receptor constructs as described in SI Text.
Fig. 2.

CXCR4 mutants used in this study. (A) Location of mutated residues in the CXCR4 structure. Side view along the membrane plane and top view across the membrane plane from the extracellular side are shown. (B) Surface expression of mutants in HA-tagged and T7-tagged constructs when transiently expressed in CHO-Gα15 cells as determined by flow cytometry analysis of anti-HA and anti-T7 antibody staining. Data are presented as percent of WT receptor expression and represents the average and SD of relative geometric mean fluorescence intensity in at least two independent experiments. (C) Mutant functionality measured as the ability of CHO-Gα15 cells transiently transfected with the mutants to mobilize intracellular Ca2+ in response to stimulation with varying concentrations of CXCL12. Data are presented as percent maximal response elicited by the WT receptor and represents the average and SD of all replicates from at least two independent experiments.
It was expected that CRS1 mutations would impact CXCL12 binding affinity without affecting the maximal signaling capacity of the receptor, whereas CRS2 mutations would mainly disrupt signaling. Some of the CRS2 mutants have been shown to bind CXCL12 with affinities similar to the WT receptor; specifically, E288A and D187A bind CXCL12 with IC50 values of 4.4 nM and 4.7 nM, respectively, compared with 2.2 nM for WT CXCR4 in a radioligand competition binding assay (19). Similarly, Wong et al. reported Kd values of 47.6 nM and 44.1 nM for D97N and E288A, respectively, compared with 35.8 nM for WT CXCR4 in competition binding assays (20).
Using flow cytometry experiments described in SI Text, we found that all mutants were expressed similarly to WT when transiently transfected in CHO-Gα15 cells (Fig. 2B). The CRS1 YYY and IIYYY mutants were able to elicit a full Ca2+ mobilization response at high CXCL12 concentrations but had an ∼10-fold lower EC50 than WT CXCR4. Two of the CRS2 mutations, D171A and D187A, were significantly impaired in both EC50 and maximal Ca2+ responses, whereas the remaining two, D97N and E288A, were completely signaling-dead (Fig. 2C). These mutants therefore seemed viable candidates for use in the functional complementation and dimer dilution experiments.
CXCR4 Mutants Dimerize with Each Other and with WT CXCR4.
An important control for the functional complementation and dimer dilution experiments is that the mutants retain the ability to form dimers. BRET YFP titration experiments, which are commonly used to assess GPCR dimerization (58, 59), confirmed that none of the above mutations affected the oligomerization propensity of the receptor (Fig. 3). In these experiments, receptors C-terminally tagged with Renilla luciferase (Rluc) are expressed at a constant level, whereas expression of YFP-tagged receptors is increased. This results in a hyperbolic increase in BRETnet values if the interaction is specific; in contrast, a low linearly increasing signal is indicative of a nonspecific random interaction (60). The fluorescence/luminescence ratio at which the BRETnet value is half maximal (BRET50) is an indicator of affinity, whereas the BRETmax value depends on the conformation of the interacting receptors as well as the distance between the YFP and Rluc molecules in the complex (58). Mutant/mutant and mutation/WT combinations used in functional complementation and dimer dilution experiments are shown in Fig. 3 A and B, respectively. The BRET50 values for the different combinations indicate that none of the mutations adversely affected receptor dimerization capacity.
Fig. 3.

CXCR4 mutants retain ability to dimerize with each other (A) and with WT receptor (B and C). In A and B, BRET saturation experiments were performed in HEK293T cells as described in Materials and Methods. The resulting BRETnet ratio is plotted against the fluorescence/luminescence ratio. The BRET pair WT CXCR4-Rluc with CXCR4-YFP was used as a positive control and WT CXCR4-Rluc and GBR2-YFP was used as a negative control for nonspecific BRET. Data are from three independent experiments. In C, HA-tagged mutant receptors were pulled down with Flag-tagged WT receptors using anti-Flag affinity resin when the receptors were coexpressed in HEK293 cells, but not when lysates of two cell populations independently expressing the receptors were mixed (control). The amount of coimmunoprecipitated mutant receptor correlated with the levels of transfection (maximum vs. 50% max HA-tagged mutant).
Dimerization of mutant and WT receptors was also confirmed using coimmunoprecipitation (Fig. 3C). Following precipitation with anti-Flag affinity resin, bands indicating the presence of HA-tagged receptors were found in all samples coexpressing Flag-tagged WT and HA-tagged WT or mutant receptors, with the intensity of the bands correlating with the amount of transfected HA-tagged receptor. No coimmunoprecipitation was observed in the control sample where lysates of cells independently expressing the two types of receptors were mechanically mixed.
Mutant Functional Complementation Experiments Do Not Support 2:1 Model.
Functional complementation experiments were designed to specifically test the 2:1 model of the CXCR4:CXCL12 interaction shown in Fig. 1B. In the model, the globular core of CXCL12 interacts with CRS1 of one CXCR4 subunit in a dimer, and the N terminus of CXCL12 interacts with CRS2 of the other subunit. Therefore, these experiments were designed such that a binding-deficient CRS1 mutant was coexpressed with a nonfunctional CRS2 mutant. If the 2:1 hypothesis is correct, one would expect that coexpression would partially rescue the Ca2+ mobilization response to CXCL12 stimulation (Fig. 1E). However, no rescue response was observed with any of the CRS1/CRS2 mutant combinations tested (four representative combinations are shown in Fig. 4 A–D). As a control experiment, mutant coexpression was monitored by costaining the cells with antibodies conjugated to different fluorophores and specific to the two tags on the mutants; such coexpression was found efficient in all cases (Fig. 4 E–H). These data therefore suggest that the 2:1 receptor:chemokine hypothesis is incorrect, and that the CXCR4:CXCL12 stoichiometry is more likely 1:1.
Fig. 4.

The absence of functional rescue when coexpressing two complementary mutants of CXCR4 in CHO-Gα15 cells. (A–D) CHO-Gα15 cells were transfected with CRS1 mutants, CRS2 mutants, or cotransfected with both and their Ca2+ mobilization measured in response to varying concentrations of CXCL12. For all of the mutant pairs tested, the Ca2+ mobilization response of cotransfected cells did not exceed that of cells transfected with each of the mutants individually. In each experiment, cells transfected with WT CXCR4 were also tested as a positive control. Four representative mutant pairs are shown. Averages and SDs of all replicates in 2–12 independent experiments are shown. (E–H) Mutant coexpression in CHO-Gα15 cells was monitored via flow cytometry by costaining cotransfected cells with PE-conjugated anti-HA antibody and APC-conjugated anti-T7 antibody. Two-dimensional contour plots show that in all cases, the mutants were efficiently coexpressed.
Dimer Dilution Experiments Support the 1:1 Model.
To confirm that the stoichiometry of the CXCR4:CXCL12 complex is 1:1, dimer dilution experiments were performed in which nonfunctional CXCR4 mutant expression was systematically increased by transient transfection into HEK293 cells that stably express WT CXCR4, effectively “diluting” WT/WT CXCR4 homodimers with nonfunctional mutants (Fig. 1F). Empty vector complementation was used so that all cells were transfected with the same amount of DNA. The Ca2+ mobilization response of these cells was then tested after addition of 20 nM or a saturating concentration (100 nM) of CXCL12. If the 1:1 stoichiometry hypothesis is correct, then increasing amounts of mutant receptor to dilute WT CXCR4 homodimers should not reduce the signaling response. Indeed, there was no significant change in Ca2+ mobilization after diluting with either CRS1 or CRS2 mutants (Fig. 5 A–D). Importantly, we established that increasing expression of mutant receptors did not alter WT CXCR4 expression (which may have artificially resulted in decreased signaling), and moreover that the amount of mutant receptor exceeded that of WT CXCR4 by a factor sufficient to effectively dilute WT/WT dimers (Fig. 5 E–H). By costaining the cell population with antibodies conjugated to different fluorophores and directed against distinct tags on WT and mutant receptors, it was possible to ensure that the expression of WT CXCR4 was constant whereas the mutant CXCR4 constructs systematically increased, and that both constructs were coexpressed in the vast majority of cells (Fig. 5 I–L). Taken together, these data strongly support the 1:1 stoichiometry of interaction of monomeric CXCL12 with CXCR4.
Fig. 5.
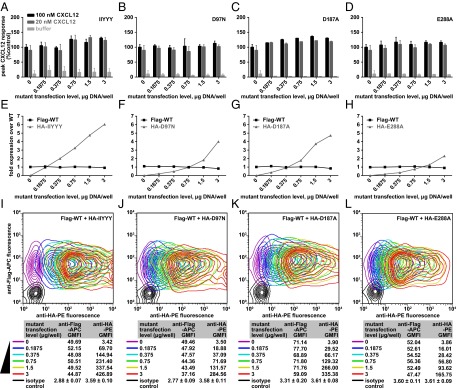
Diluting WT-WT dimers by increasing transfection of loss-of-function mutants does not lead to a decrease in signaling. (A–D) Peak fluorescence values from Ca2+ mobilization experiments in which CXCR4 HEK293 tetracycline-inducible cells transfected with the indicated amounts of CRS1 and CRS2 mutants were stimulated with the indicated CXCL12 concentrations. Data for four representative mutants are shown along with averages and SDs of replicates in two to four independent experiments. (E–H) WT and mutant receptor expression levels were monitored by flow cytometry; in all cases, the WT expression was constant and the transfected mutant expression exceeded it two- to sixfold. WT and mutant receptors N-terminally tagged with Flag and HA tags, respectively, were codetected on the cell surface with APC-conjugated anti-Flag antibody and PE-conjugated anti-HA antibody. To normalize geometric mean fluorescence intensity between the two antibodies, a series of samples coexpressing Flag-tagged and HA-tagged WT receptor was costained with these antibodies and also (independently) with anti-CXCR4 antibody (data now shown). (I–L) Coexpression of the two constructs was monitored by flow cytometry.
Cysteine Trapping Experiments Support the 1:1 Stoichiometry and Guide 1:1 Model Development.
To obtain validation of the 1:1 CXCR4:CXCL12 complex stoichiometry and insight into the structure of the complex, cysteine trapping experiments were developed. In these experiments, individual residues of CXCL12 and CXCR4 were mutated to cysteine, and the mutant pairs were coexpressed in Spodoptera frugiperda (Sf9) insect cells, purified by a His-tag on the receptor, and analyzed by SDS/PAGE and Western blotting for the presence of copurified chemokine. This approach assumes that when coexpressed, mutant receptors and chemokines bind in near-native geometry; and if this geometry brings the artificially introduced cysteines in proximity to one another, a disulfide bond spontaneously forms, resulting in an irreversible complex. As shown in Fig. 6A, cysteine trapping confirmed the proximity of K25 in CXCR4 with S16 in CXCL12, consistent with their relative orientation in the NMR structure (39) (Fig. 6D). However, multiple other residue pairs that were proximal in the NMR structure did not cross-link. Negative results were obtained for the following CXCR4:CXCL12 pairs: I6/N30, T8/L29, S9/L29, Y12/T31, P27/Q59, and F29/Q59 (Fig. 6 A and D). Systematic exploration of proximities in the CXCL12 region E15-V18 to CXCR4 K25 confirmed the initial finding of K25/S16; the nearby residues showed less or no cross-linking, providing evidence of specificity (Fig. 6B).
Fig. 6.
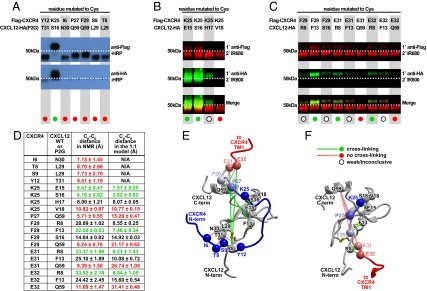
Cysteine trapping experiment with CXCR4 and CXCL12 coexpressed in insect Sf9 cells. (A–C) Nonreducing Western blot analysis of extracts from Sf9 cells coexpressing single Cys mutants of Flag-tagged CXCR4 with single Cys mutants of HA-tagged CXCL12 or its antagonist version, CXCL12(P2G) (15). Molecular weight shift and positive HA-tag staining in the purified material (green circles) indicates spontaneously formed disulfide bond and suggests spatial proximity of the two cysteine residues in the complex, whereas the absence of a chemokine band (red circles) is indicative of spatially distant position of the probed residues. Open circles indicate weak/inconclusive cross-linking. (A) Coexpression samples were probed with HRP-conjugated anti-FLAG and anti-HA antibodies (Top and Bottom, respectively). Flag-CXCR4(K25C) efficiently cross-linked with CXCL12-HA(P2G-S16C) as evidenced by the molecular weight shift and by positive staining with both anti-FLAG and anti-HA antibodies; other probed mutant pairs did not cross-link. (B and C) LI-COR IRDye conjugated secondary antibodies were used to differentially identify Flag-CXCR4 and CXCL12-HA on a single blot. (B) Specificity of the cross-linking reaction in the vicinity of CXCR4 K25 and CXCL12 S16. Flag-CXCR4(K25C) forms strong complexes with CXCL12-HA(S16C) and (E15C), a much weaker complex with CXCL12-HA(H17C) and no complex with CXCL12-HA(V18C). (C) Validation of residue proximities observed in the second-generation 1:1 model of the CXCR4:CXCL12 complex. Flag-CXCR4(F29C) forms a medium strength complex with CXCL12-HA(F13C); Flag-CXCR4(E31C) and (E32C) both form weak complexes with CXCL12-HA(R8C) and (F13C), but not at all with CXCL12-HA(Q59C). (D) Cβ-Cβ distances observed between the probed CXCR4:CXCL12 residue pairs in the NMR structure (39) and second-generation 1:1 complex models. Averages and SDs were calculated using the 20 structures of the NMR ensemble (PDB ID code 2K05) or two top-scoring model conformations. (E and F) Positive and negative cross-links mapped onto 3D structures of CXCR4:CXCL12 complex in the context of the NMR structure (39) (E) or a second-generation 1:1 complex model in Fig. 1D (F). Chemokine orientation is identical between E and F.
Based on this finding, we constructed second-generation 1:1 complex models by molecular docking, as described above, with one modification involving the introduction of a single disulfide bond between CXCR4 K25C and CXCL12 S16C instead of the NMR proximity restraints (Figs. 1D and 6F). The obtained models still featured the interaction of receptor residue K25 with S16 in the chemokine N-loop; however, the subsequent receptor residues up to R30 were directed along the N-loop, toward the chemokine N terminus, and away from the chemokine C-terminal helix. To functionally validate this prediction, we attempted cysteine trapping of residue pairs that were distant in the NMR structure but proximal in the new models (Fig. 6D). This included pairs of F29/F13, E31/R8, and E32/R8, which all showed positive cross-linking (although less efficient than with K25/S16) (Fig. 6 C, D, and F). We therefore concluded that the interactions observed in the NMR structure between the CXCL12 dimer and the N-terminal peptide of CXCR4 are different from those in the context of the full-length receptor. Our data suggest that the approximate complex geometry CXCL12 with full-length CXCR4 may be better represented by the computational model shown in Figs. 1D and 6F.
Discussion
Evidence firmly establishing class A GPCR dimerization in general, and chemokine receptor dimerization in particular, has accumulated for over a decade (23, 28, 41, 61). However, for most GPCRs, the functional purpose of dimerization is unclear. In the case of chemokine receptors, despite numerous reports suggesting functional interactions between receptors in both homodimers and heterodimers, the structural causes and functional consequences of such interactions remain unknown. CXCL12 stimulation was shown to cause conformational changes in the CXCR4 homodimer (23). Furthermore, chemokines and small-molecule antagonists were shown to transinhibit other receptors within heterodimers of chemokine receptors (28, 62), and chemokine receptors with other GPCRs (63, 64). The first report of a chemokine receptor crystal structure also showed that CXCR4 formed dimers (7), and moreover suggested that based on existing data, a 2:1 receptor:chemokine stoichiometry was a viable alternative to 1:1. Interesting examples in which a single GPCR agonist binds and activates a receptor homodimer with 2:1 receptor:ligand stoichiometry have been reported (46, 65). If confirmed for chemokine receptors, 2:1 stoichiometry could explain the observed dimer-mediated alteration of ligand binding and responses. On the other hand, definitively demonstrating a 1:1 stoichiometry between chemokines and their receptors would provide the impetus for investigating other, more complicated explanations for these phenomena.
Of the two possibilities, only the 2:1 stoichiometry was consistent with the NMR-based CXCL12 structure, the CXCR4 crystal structure, and the prevailing two-site model of receptor activation. Functional complementation experiments were designed to validate the 2:1 model, but the lack of functional rescue strongly suggested that such a model is incorrect and that the stoichiometry is more likely 1:1. Furthermore, dimer dilution experiments also supported the 1:1 interaction stoichiometry. Of note, although studying the functional importance of dimerization by coexpressing WT and nonfunctional mutant receptors is not new (27, 47, 64, 66, 67), care must be taken to ensure that the expression level of the WT receptor stays constant with increasing expression of mutant, and that the expression levels of the mutants are sufficient to effectively dilute WT/WT dimers. In the present study, such precautions were taken such that we could confidently conclude that dimerization of WT CXCR4 with CRS1 or CRS2 mutants does not alter its functional response, consistent with the 1:1 stoichiometry.
Finally, we used cysteine trapping experiments to elucidate the geometry of the 1:1 CXCR4:CXCL12 complex. These experiments confirmed that in the context of the full-length receptor, the pairwise residue proximity is different from that observed in the NMR structure (39) and is more consistent with a 1:1 model generated by computational docking with a single disulfide restraint. Similar contradictions are also present in other computationally generated 1:1 models of chemokine:receptor complexes (68, 69). One possible explanation for the inconsistency is that the NMR structure contains a dimer of CXCL12 in complex with two copies of the N-terminal CXCR4 peptide, rather than a monomer of CXCL12 complexed to a single receptor peptide. In fact, the CXCL12 dimer is known to be a partial agonist of CXCR4, and the NMR complex may be more representative of this alternative signaling complex (39). Alternatively, in the context of the full-length receptor, there may be some sort of rearrangement after docking of the CXCL12 globular domain to the CXCR4 N terminus, and engagement of the CXCL12 N terminus within the CXCR4 transmembrane binding pocket for activation.
The 1:1 stoichiometry and complex geometry suggested by our study answers a fundamental question regarding CXCR4:CXCL12, and likely most receptor:chemokine complexes. The results add to the accumulating data regarding the role of receptor monomers and not dimers in interactions with the various components of signaling complexes. Studies of rhodopsin (70), β2-adrenergic (71), neurotensin (72), and µ-opiod (73) receptors demonstrate that monomeric GPCRs are fully capable of activating G proteins. Monomeric rhodopsin can be phosphorylated by rhodopsin kinases and binds to visual arrestin in native disk membranes (74); and the structures of the β2-adrenergic receptor in complex with a heterotrimeric G protein (75) and β-arrestin1 (76) also suggest a 1:1 interaction. Along with the present work, these studies support receptor monomers as fully competent signaling units.
On the other hand, our results do not dismiss previously observed dimer-mediated phenomena, such as the conformational changes within CXCR4 homodimers upon CXCL12 stimulation (23), and ligand binding transinhibition within CXCR4 heterodimers with CCR2 and CCR5 (28, 62). Our data suggest that these phenomena must have other explanations than simultaneous binding of the chemokine to two protomers in the dimer. For example, they may originate from a conformational change that occurs upon 1:1 receptor:chemokine binding that is transmitted across the receptor dimer.
Similarly, our evidence adds to the interpretation of emerging data on regulation of CXCR4:CXCL12 signaling by the atypical chemokine receptor ACKR3 (CXCR7). CXCR4 and CXCR7 not only share CXCL12 as a chemokine ligand, but also heterodimerize in live cells. Such heterodimerization has been reported to negatively regulate CXCL12-mediated G protein signaling (77); for example, dilution of CXCR4:CXCR4 homodimers with CXCR4:CXCR7 heterodimers reduces G protein-mediated cellular responses to CXCL12 (30). Alteration of functional responses of one receptor by selective ligands of another has been reported as well (78–81). Our results suggest that these cross-talk phenomena occur because of propagation of conformational changes across the heterodimer interface and not because of in trans binding of CXCL12 to both receptors in the dimer. It is also possible that a heterodimer presents a new intracellular interface that supports altered signaling responses.
Finally, our data do not exclude the possibility that receptor oligomerization contributes to other processes, such as regulation of pharmacology and trafficking of homo- versus hetero-oligomers (82, 83). It may also contribute to the coordination and efficiency of sequential steps in the GPCR lifecycle.
Materials and Methods
Molecular Modeling.
Initial 1:1 and 2:1 receptor:chemokine models were generated by chemical field-guided molecular docking (48) of CXCL12 into the binding pocket of the CXCR4 monomer (for 1:1 model) or dimer (for 2:1 model) using the CXCR4 structures PDB ID codes 3OE0 and 3ODU (7). For the 2:1 models, CXCR4 dimers were derived from both chains in PDB ID code 3ODU or from chain A and its similarly oriented crystallographic neighbor in PDB ID code 3OE0. Chemical fields were generated from the structures of the cocrystallized ligands (IT1t and CVX15) as described in refs. 48 and 84 and attenuated for ligand atoms that are not in direct contact with the receptor. The ensemble of initial conformations of CXCL12 was generated from all available X-ray and NMR structures in the PDB; in cases where N-terminal residues of CXCL12 were missing from electron density, they were constructed ab initio. The receptor pocket was represented with potential grid maps as described in ref. 50, with the N-terminal residues K25–R30 and the side chains of residues E179–D182, I185, D187, F189, and D193 excluded from the calculations because of the uncertainty of their positions. A full-atom peptide representing CXCR4 residues K25–R30 was generated ab initio. For generation of models compatible with the NMR structure of the CXCL12 complex with the CXCR4 N terminus (39), this peptide was restrained to the Cβ atoms of proximal CXCL12 residues in the structure (F14–S16, I51–K56, and I58–E60) using soft harmonic restraints with target distances specified as observed in the structure. For second-generation NMR-independent models, a restraint was introduced in the form of the experimentally validated disulfide bond between CXCR4 K25C and CXCL12 S16C. The C-terminal part of the peptide was restrained to the positions of CXCR4 residues C28–R30 in the crystal structure, which are in turn tethered by a disulfide bond from C28 to C274 in the extracellular loop 3. Multiple orientations of CXCL12 were generated from each starting conformation by systematically flipping it along its principal axes. The system was then extensively sampled with a Biased Probability Monte Carlo search as implemented in the Internal Coordinate Mechanics software (49). During sampling, the backbone of chemokine residues P10–N67 was kept fixed except for switching between the multiple preselected conformations described above, and the side chains of these residues were sampled explicitly. Both backbone and side chains of the CXCR4 N-terminal peptide (residues K25–R30) and the chemokine N terminus (residues K1–C9) were sampled explicitly.
Ca2+ Mobilization Assay in CHO-Gα15 and HEK293 CXCR4 Tet-On Cells for Functional Complementation and Dimer Dilution Experiments, Respectively.
Calcium mobilization assays were carried out using the FLIPR Calcium 4 Assay Kit (Molecular Devices). Cells were cultured and transfected with relevant CXCR4 WT and mutant constructs as described in SI Text. For functional complementation experiments, CHO-Gα15 cells were lifted from dishes 6–8 h after transfection using sterile PBS containing 5 mM EDTA, plated at a density of 9 × 104 cells/well in poly-d-lysine–coated 96-well black/clear-bottom plates (Becton Dickinson Labware), cultured for another 16–20 h, and then tested using an adherent cell assay format. For dimer dilution experiments in HEK293 cells, cell culture media was replaced with fresh DMEM containing 10% (vol/vol) FBS 6–8 h after transfection, cells were cultured for another 16–20 h, and then tested in a detached cell assay format.
For adherent CHO-Gα15 cell assay, cell culture media in the 96-well plates was replaced with 112.5 µL per well of Ca2+ mobilization assay buffer consisting of 1× HBSS (Gibco), 20 mM Hepes, 0.1% BSA, and 4 mM probenecid. For detached HEK293 cell assay, cells were lifted from the culture dishes using PBS containing 5 mM EDTA, washed, resuspended in Ca2+ mobilization assay buffer consisting of 1× HBSS, 20 mM Hepes, and 0.1% BSA, and aliquoted into poly-d-lysine–coated 96-well black/clear-bottom plates at a density of 1.5 × 105 cells per well in 112.5 µL buffer. For both assay formats, 112.5 µL of the calcium indicator was added to the plate and mixed by gentle pipetting; detached cells were evenly settled at the bottom of the wells by centrifuging the plates at 250×g for 3 min. Following 75-min incubation of the plates at 37 °C with 5% CO2, ligand stimulation and response recordings were carried out using a FlexStation 3 plate reader (Molecular Devices). For functional complementation experiments, dose–response curves were generated with CXCL12 concentrations extending beyond the point of signal saturation. For dimer dilution experiments, CXCL12 concentrations of 0 nM, 20 nM, and 100 nM were tested. Triplicate measurements were made for each concentration, and each experiment was performed at least twice on different days. All responses were expressed as percent maximal response elicited by WT in the same experiment. The reported values are averages with SD of all replicates from all experiments for each concentration of CXCL12. Data were analyzed in Prism 6 (GraphPad Software) using a sigmoidal dose–response curve with variable slope as a model.
BRET and Co-Immunoprecipitation Experiments to Assess CXCR4 Mutant Dimerization Propensity.
BRET experiments were conducted with CXCR4-Renilla luciferase (CXCR4-Rluc) and CXCR4-YFP constructs possessing the same mutations as those used in the functional complementation and dimer dilution experiments. The BRET assay as applied to chemokine receptor dimerization was described previously (23, 59). Briefly, HEK293T cells were transfected with constant amounts of CXCR4-Rluc constructs and increasing amounts of CXCR4-YFP constructs, keeping the total amount of DNA transfected into each sample constant by empty vector complementation. After 48 h, cells were lifted from the culture plates with PBS containing 0.1% d-glucose, cell concentrations were normalized, and 105 cells were plated into each well of a 96-well white clear-bottom tissue-culture assay plate (BD Falcon). Fluorescence readings (excitation: 485 nm, emission: 538 nm) were recorded using a SpectraMax M5 fluorescent plate reader (Molecular Devices). Coelenterazine was then added to a final concentration of 5-µM, white backing tape (Perkin-Elmer) applied to the bottom of the assay plate, and both unfiltered and filtered luminescence (emission: 460 nm, 535 nm) readings were recorded with a VICTOR X Light 2030 luminometer (Perkin-Elmer). BRET ratios were calculated by dividing the luminescence signal at 535 nm by the signal at 460 nm. BRETnet values were calculated by subtracting the BRET ratio of Rluc-only transfected cells from all BRET ratios. BRETnet data were graphed as a function of increasing fluorescence/luminescence ratio. Resultant BRET YFP titration data were fit to a one site binding (hyperbolic) model in Prism (GraphPad Software).
For coimmunoprecipitation experiments, Flag-CXCR4-Tet-On and HEK293T cells were plated in a six-well plate and transiently transfected the next day with increasing amounts of HA-tagged WT and mutant CXCR4. Transfection media was replaced 6 h later with fresh culture media. Next, 16–18 h later, the culture media was removed, cells were rinsed with PBS, and lysed directly on the plate using 400 µL of cold lysis buffer [20 mM Tris⋅HCl, pH 7.5, 200 mM NaCl, 10% (vol/vol) glycerol, 1% DDM, 1× Protease Inhibitor mixture (Sigma)]. Lysates were transferred to 1.5-mL tubes, incubated with rocking at 4 °C for 1 h, and cleared by centrifugation for 20 min at 20,000 × g at 4 °C. Cleared lysates were incubated with anti-Flag affinity resin (Sigma) at 4 °C for 2 h with rocking, after which resin was washed four times with fresh lysis buffer, bound proteins were eluted with 150 ng/µL of 3× Flag peptide (Sigma), and analyzed by Western blot using high-affinity HRP-conjugated rat anti-HA antibody (3F10; Roche).
Analysis of Disulfide-Trapped Complexes Between Cysteine Mutants of Flag-CXCR4 and CXCL12-HA.
For the cysteine-trapping experiments, Flag-CXCR4 and CXCL12-HA cysteine mutant proteins were coexpressed in Sf9 insect cells and purified as described in SI Text. Purified protein samples were analyzed by nonreducing 10% SDS/PAGE gel where molecular weight shift and the relative band intensity were used as indicators of presence and relative abundance, respectively, of the irreversibly trapped complex. Western blotting was used to confirm the nature of Flag-CXCR4 and CXCL12-HA bands. For that process, ∼5 µL of purified sample was run on a 10% SDS/PAGE and transferred to a nitrocelluolose membrane. The membrane was blocked in TBS-T with 5% (wt/vol) milk for 1 h at room temperature. Primary staining was performed using 5 µL of mouse anti-Flag M2 primary antibody (Sigma) and 5 µL rat anti-HA 3F10 primary antibody (Roche) for the receptor and chemokine, respectively, in 10 mL of fresh TBS-T with 5% (wt/vol) milk for 1 h at room temperature. Secondary staining was done with 1 µL of IRDye 680 conjugated donkey anti-mouse IgG and IRDye 800 conjugated goat anti-rat IgG (LI-COR Biosciences) in 10 mL of TBS-T with 5% (wt/vol) BSA for 1 h at room temperature. Following incubation, the membrane was washed three times with 10 mL of fresh TBS-T for 10 min, transferred into 1× sterile PBS, and imaged using the Odyssey IR imaging system (LI-COR Bioscience).
Supplementary Material
Acknowledgments
We thank B. Volkman (Medical College of Wisconsin) for suggesting the CRS1 mutations; T. Gilliland, M. Gustavsson, Y. Zheng (University of California, San Diego), S. Katritch, and R. Stevens (The Scripps Research Institute) for valuable discussions; M. Bouvier (University of Montreal) for sharing the initial bioluminescence resonance energy transfer cloning vectors; and J. Morseman (Columbia Biosciences) for generously providing test samples of fluorescent antibodies. The work was partially supported by NIH Grants R01 GM071872 and U54 GM094618 (to R.A.), U01 GM094612 (to R.A. and T.M.H.), R01 AI37113, R01 GM081763, and R21 AI101687 (to T.M.H.); Cellular and Molecular Pharmacology Training Grant T32 GM007752 (to B.S.S.); Molecular Biophysics Training Grant T32 GM008326 (to B.S.S.); and 2012 Post Doctoral Fellowship in Pharmacology/Toxicology from the Pharmaceutical Research and Manufacturers of America (PhRMA) Foundation (to L.G.H.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1417037111/-/DCSupplemental.
References
- 1.Ma Q, et al. Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc Natl Acad Sci USA. 1998;95(16):9448–9453. doi: 10.1073/pnas.95.16.9448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zou Y-R, Kottmann AH, Kuroda M, Taniuchi I, Littman DR. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature. 1998;393(6685):595–599. doi: 10.1038/31269. [DOI] [PubMed] [Google Scholar]
- 3.Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: Functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. 1996;272(5263):872–877. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- 4.Zlotnik A, Burkhardt AM, Homey B. Homeostatic chemokine receptors and organ-specific metastasis. Nat Rev Immunol. 2011;11(9):597–606. doi: 10.1038/nri3049. [DOI] [PubMed] [Google Scholar]
- 5.Tanegashima K, et al. CXCL14 is a natural inhibitor of the CXCL12-CXCR4 signaling axis. FEBS Lett. 2013;587(12):1731–1735. doi: 10.1016/j.febslet.2013.04.046. [DOI] [PubMed] [Google Scholar]
- 6.Saini V, Marchese A, Majetschak M. CXC chemokine receptor 4 is a cell surface receptor for extracellular ubiquitin. J Biol Chem. 2010;285(20):15566–15576. doi: 10.1074/jbc.M110.103408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu B, et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330(6007):1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tan Q, et al. Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science. 2013;341(6152):1387–1390. doi: 10.1126/science.1241475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brelot A, Heveker N, Montes M, Alizon M. Identification of residues of CXCR4 critical for human immunodeficiency virus coreceptor and chemokine receptor activities. J Biol Chem. 2000;275(31):23736–23744. doi: 10.1074/jbc.M000776200. [DOI] [PubMed] [Google Scholar]
- 10.Zhou H, Tai HH. Expression and functional characterization of mutant human CXCR4 in insect cells: Role of cysteinyl and negatively charged residues in ligand binding. Arch Biochem Biophys. 2000;373(1):211–217. doi: 10.1006/abbi.1999.1555. [DOI] [PubMed] [Google Scholar]
- 11.Kofuku Y, et al. Structural basis of the interaction between chemokine stromal cell-derived factor-1/CXCL12 and its G-protein-coupled receptor CXCR4. J Biol Chem. 2009;284(50):35240–35250. doi: 10.1074/jbc.M109.024851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saini V, et al. The CXC chemokine receptor 4 ligands ubiquitin and stromal cell-derived factor-1α function through distinct receptor interactions. J Biol Chem. 2011;286(38):33466–33477. doi: 10.1074/jbc.M111.233742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gupta SK, Pillarisetti K, Thomas RA, Aiyar N. Pharmacological evidence for complex and multiple site interaction of CXCR4 with SDF-1alpha: Implications for development of selective CXCR4 antagonists. Immunol Lett. 2001;78(1):29–34. doi: 10.1016/s0165-2478(01)00228-0. [DOI] [PubMed] [Google Scholar]
- 14.Scholten DJ, et al. Pharmacological modulation of chemokine receptor function. Br J Pharmacol. 2012;165(6):1617–1643. doi: 10.1111/j.1476-5381.2011.01551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crump MP, et al. Solution structure and basis for functional activity of stromal cell-derived factor-1; Dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J. 1997;16(23):6996–7007. doi: 10.1093/emboj/16.23.6996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gerlach LO, Skerlj RT, Bridger GJ, Schwartz TW. Molecular interactions of cyclam and bicyclam non-peptide antagonists with the CXCR4 chemokine receptor. J Biol Chem. 2001;276(17):14153–14160. doi: 10.1074/jbc.M010429200. [DOI] [PubMed] [Google Scholar]
- 17.Rosenkilde MM, et al. Molecular mechanism of AMD3100 antagonism in the CXCR4 receptor: Transfer of binding site to the CXCR3 receptor. J Biol Chem. 2004;279(4):3033–3041. doi: 10.1074/jbc.M309546200. [DOI] [PubMed] [Google Scholar]
- 18.Choi W-T, et al. Unique ligand binding sites on CXCR4 probed by a chemical biology approach: Implications for the design of selective human immunodeficiency virus type 1 inhibitors. J Virol. 2005;79(24):15398–15404. doi: 10.1128/JVI.79.24.15398-15404.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tian S, et al. Distinct functional sites for human immunodeficiency virus type 1 and stromal cell-derived factor 1alpha on CXCR4 transmembrane helical domains. J Virol. 2005;79(20):12667–12673. doi: 10.1128/JVI.79.20.12667-12673.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong RSY, et al. Comparison of the potential multiple binding modes of bicyclam, monocylam, and noncyclam small-molecule CXC chemokine receptor 4 inhibitors. Mol Pharmacol. 2008;74(6):1485–1495. doi: 10.1124/mol.108.049775. [DOI] [PubMed] [Google Scholar]
- 21.Milligan G. The prevalence, maintenance, and relevance of G protein-coupled receptor oligomerization. Mol Pharmacol. 2013;84(1):158–169. doi: 10.1124/mol.113.084780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Babcock GJ, Farzan M, Sodroski J. Ligand-independent dimerization of CXCR4, a principal HIV-1 coreceptor. J Biol Chem. 2003;278(5):3378–3385. doi: 10.1074/jbc.M210140200. [DOI] [PubMed] [Google Scholar]
- 23.Percherancier Y, et al. Bioluminescence resonance energy transfer reveals ligand-induced conformational changes in CXCR4 homo- and heterodimers. J Biol Chem. 2005;280(11):9895–9903. doi: 10.1074/jbc.M411151200. [DOI] [PubMed] [Google Scholar]
- 24.Toth PT, Ren D, Miller RJ. Regulation of CXCR4 receptor dimerization by the chemokine SDF-1alpha and the HIV-1 coat protein gp120: Afluorescence resonance energy transfer (FRET) study. J Pharmacol Exp Ther. 2004;310(1):8–17. doi: 10.1124/jpet.103.064956. [DOI] [PubMed] [Google Scholar]
- 25.Luker KE, Gupta M, Luker GD. Imaging chemokine receptor dimerization with firefly luciferase complementation. FASEB J. 2009;23(3):823–834. doi: 10.1096/fj.08-116749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi W-T, et al. A novel synthetic bivalent ligand to probe chemokine receptor CXCR4 dimerization and inhibit HIV-1 entry. Biochemistry. 2012;51(36):7078–7086. doi: 10.1021/bi2016712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lagane B, et al. CXCR4 dimerization and beta-arrestin–mediated signaling account for the enhanced chemotaxis to CXCL12 in WHIM syndrome. Blood. 2008;112(1):34–44. doi: 10.1182/blood-2007-07-102103. [DOI] [PubMed] [Google Scholar]
- 28.Sohy D, Parmentier M, Springael J-Y. Allosteric transinhibition by specific antagonists in CCR2/CXCR4 heterodimers. J Biol Chem. 2007;282(41):30062–30069. doi: 10.1074/jbc.M705302200. [DOI] [PubMed] [Google Scholar]
- 29.Isik N, Hereld D, Jin T. Fluorescence resonance energy transfer imaging reveals that chemokine-binding modulates heterodimers of CXCR4 and CCR5 receptors. PLoS ONE. 2008;3(10):e3424. doi: 10.1371/journal.pone.0003424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Décaillot FM, et al. CXCR7/CXCR4 heterodimer constitutively recruits beta-arrestin to enhance cell migration. J Biol Chem. 2011;286(37):32188–32197. doi: 10.1074/jbc.M111.277038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kramp BK, Sarabi A, Koenen RR, Weber C. Heterophilic chemokine receptor interactions in chemokine signaling and biology. Exp Cell Res. 2011;317(5):655–663. doi: 10.1016/j.yexcr.2010.11.014. [DOI] [PubMed] [Google Scholar]
- 32.Salanga CL, O’Hayre M, Handel T. Modulation of chemokine receptor activity through dimerization and crosstalk. Cell Mol Life Sci. 2009;66(8):1370–1386. doi: 10.1007/s00018-008-8666-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Contento RL, et al. CXCR4-CCR5: A couple modulating T cell functions. Proc Natl Acad Sci USA. 2008;105(29):10101–10106. doi: 10.1073/pnas.0804286105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mellado M, et al. Chemokine receptor homo- or heterodimerization activates distinct signaling pathways. EMBO J. 2001;20(10):2497–2507. doi: 10.1093/emboj/20.10.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szpakowska M, et al. Function, diversity and therapeutic potential of the N-terminal domain of human chemokine receptors. Biochem Pharmacol. 2012;84(10):1366–1380. doi: 10.1016/j.bcp.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 36.Kufareva I, Abagyan R, Handel TM. Role of 3D structures in understanding, predicting, and designing molecular interactions in the chemokine receptor family. In: Tschammer N, editor. Chemokines, Top Med Chem. Heidelberg: Springer; 2014. pp. 1–45. [Google Scholar]
- 37.Drury LJ, et al. Monomeric and dimeric CXCL12 inhibit metastasis through distinct CXCR4 interactions and signaling pathways. Proc Natl Acad Sci USA. 2011;108(43):17655–17660. doi: 10.1073/pnas.1101133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rajarathnam K, et al. Neutrophil activation by monomeric interleukin-8. Science. 1994;264(5155):90–92. doi: 10.1126/science.8140420. [DOI] [PubMed] [Google Scholar]
- 39.Veldkamp CT, et al. Structural basis of CXCR4 sulfotyrosine recognition by the chemokine SDF-1/CXCL12. Sci Signal. 2008;1(37):ra4. doi: 10.1126/scisignal.1160755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.El-Asmar L, et al. Evidence for negative binding cooperativity within CCR5-CCR2b heterodimers. Mol Pharmacol. 2005;67(2):460–469. doi: 10.1124/mol.104.003624. [DOI] [PubMed] [Google Scholar]
- 41.Vila-Coro AJ, et al. The chemokine SDF-1alpha triggers CXCR4 receptor dimerization and activates the JAK/STAT pathway. FASEB J. 1999;13(13):1699–1710. [PubMed] [Google Scholar]
- 42.Steel E, Murray VL, Liu AP. Multiplex detection of homo- and heterodimerization of G protein-coupled receptors by proximity biotinylation. PLoS ONE. 2014;9(4):e93646. doi: 10.1371/journal.pone.0093646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bakker RA, et al. Domain swapping in the human histamine H1 receptor. J Pharmacol Exp Ther. 2004;311(1):131–138. doi: 10.1124/jpet.104.067041. [DOI] [PubMed] [Google Scholar]
- 44.Maggio R, Vogel Z, Wess J. Coexpression studies with mutant muscarinic/adrenergic receptors provide evidence for intermolecular “cross-talk” between G-protein-linked receptors. Proc Natl Acad Sci USA. 1993;90(7):3103–3107. doi: 10.1073/pnas.90.7.3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Monnot C, et al. Polar residues in the transmembrane domains of the type 1 angiotensin II receptor are required for binding and coupling. Reconstitution of the binding site by co-expression of two deficient mutants. J Biol Chem. 1996;271(3):1507–1513. doi: 10.1074/jbc.271.3.1507. [DOI] [PubMed] [Google Scholar]
- 46.Rivero-Müller A, et al. Rescue of defective G protein-coupled receptor function in vivo by intermolecular cooperation. Proc Natl Acad Sci USA. 2010;107(5):2319–2324. doi: 10.1073/pnas.0906695106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Trettel F, et al. Ligand-independent CXCR2 dimerization. J Biol Chem. 2003;278(42):40980–40988. doi: 10.1074/jbc.M306815200. [DOI] [PubMed] [Google Scholar]
- 48.Kufareva I, Chen Y-C, Ilatovskiy AV, Abagyan R. Compound activity prediction using models of binding pockets or ligand properties in 3D. Curr Top Med Chem. 2012;12(17):1869–1882. doi: 10.2174/156802612804547335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abagyan R, Totrov M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J Mol Biol. 1994;235(3):983–1002. doi: 10.1006/jmbi.1994.1052. [DOI] [PubMed] [Google Scholar]
- 50.Fernández-Recio J, Totrov M, Abagyan R. Soft protein-protein docking in internal coordinates. Protein Sci. 2002;11(2):280–291. doi: 10.1110/ps.19202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Våbenø J, Nikiforovich GV, Marshall GR. Insight into the binding mode for cyclopentapeptide antagonists of the CXCR4 receptor. Chem Biol Drug Des. 2006;67(5):346–354. doi: 10.1111/j.1747-0285.2006.00387.x. [DOI] [PubMed] [Google Scholar]
- 52.Hatse S, et al. Mutations at the CXCR4 interaction sites for AMD3100 influence anti-CXCR4 antibody binding and HIV-1 entry. FEBS Lett. 2003;546(2-3):300–306. doi: 10.1016/s0014-5793(03)00609-4. [DOI] [PubMed] [Google Scholar]
- 53.Hatse S, et al. Mutation of Asp(171) and Asp(262) of the chemokine receptor CXCR4 impairs its coreceptor function for human immunodeficiency virus-1 entry and abrogates the antagonistic activity of AMD3100. Mol Pharmacol. 2001;60(1):164–173. doi: 10.1124/mol.60.1.164. [DOI] [PubMed] [Google Scholar]
- 54.Zhou N, et al. Structural and functional characterization of human CXCR4 as a chemokine receptor and HIV-1 co-receptor by mutagenesis and molecular modeling studies. J Biol Chem. 2001;276(46):42826–42833. doi: 10.1074/jbc.M106582200. [DOI] [PubMed] [Google Scholar]
- 55.Offermanns S, Simon MI. G alpha 15 and G alpha 16 couple a wide variety of receptors to phospholipase C. J Biol Chem. 1995;270(25):15175–15180. doi: 10.1074/jbc.270.25.15175. [DOI] [PubMed] [Google Scholar]
- 56.Farzan M, et al. The role of post-translational modifications of the CXCR4 amino terminus in stromal-derived factor 1 α association and HIV-1 entry. J Biol Chem. 2002;277(33):29484–29489. doi: 10.1074/jbc.M203361200. [DOI] [PubMed] [Google Scholar]
- 57.Ziarek JJ, et al. Sulfopeptide probes of the CXCR4/CXCL12 interface reveal oligomer-specific contacts and chemokine allostery. ACS Chem Biol. 2013;8(9):1955–1963. doi: 10.1021/cb400274z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hamdan FF, Percherancier Y, Breton B, Bouvier M. Monitoring protein-protein interactions in living cells by bioluminescence resonance energy transfer (BRET) Curr Protoc Neurosci. 2006 doi: 10.1002/0471142301.ns0523s34. Chapter 5:Unit 5.23. [DOI] [PubMed] [Google Scholar]
- 59.Kufareva I, et al. A novel approach to quantify G-protein-coupled receptor dimerization equilibrium using bioluminescence resonance energy transfer. In: Cardona AE, Ubogu EE, editors. Chemokines: Methods and Protocols, Methods Mol Biol. Vol 1013. New York: Springer; 2013. pp. 93–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.James JR, Oliveira MI, Carmo AM, Iaboni A, Davis SJ. A rigorous experimental framework for detecting protein oligomerization using bioluminescence resonance energy transfer. Nat Methods. 2006;3(12):1001–1006. doi: 10.1038/nmeth978. [DOI] [PubMed] [Google Scholar]
- 61.Liang Y, et al. Organization of the G protein-coupled receptors rhodopsin and opsin in native membranes. J Biol Chem. 2003;278(24):21655–21662. doi: 10.1074/jbc.M302536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sohy D, et al. Hetero-oligomerization of CCR2, CCR5, and CXCR4 and the protean effects of “selective” antagonists. J Biol Chem. 2009;284(45):31270–31279. doi: 10.1074/jbc.M109.054809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.May LT, Bridge LJ, Stoddart LA, Briddon SJ, Hill SJ. Allosteric interactions across native adenosine-A3 receptor homodimers: Quantification using single-cell ligand-binding kinetics. FASEB J. 2011;25(10):3465–3476. doi: 10.1096/fj.11-186296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Szalai B, et al. Allosteric interactions within the AT₁ angiotensin receptor homodimer: Role of the conserved DRY motif. Biochem Pharmacol. 2012;84(4):477–485. doi: 10.1016/j.bcp.2012.04.014. [DOI] [PubMed] [Google Scholar]
- 65.Brock C, et al. Activation of a dimeric metabotropic glutamate receptor by intersubunit rearrangement. J Biol Chem. 2007;282(45):33000–33008. doi: 10.1074/jbc.M702542200. [DOI] [PubMed] [Google Scholar]
- 66.Hernandez PA, et al. Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat Genet. 2003;34(1):70–74. doi: 10.1038/ng1149. [DOI] [PubMed] [Google Scholar]
- 67.Benkirane M, Jin DY, Chun RF, Koup RA, Jeang KT. Mechanism of transdominant inhibition of CCR5-mediated HIV-1 infection by ccr5delta32. J Biol Chem. 1997;272(49):30603–30606. doi: 10.1074/jbc.272.49.30603. [DOI] [PubMed] [Google Scholar]
- 68.Xu L, Li Y, Sun H, Li D, Hou T. Structural basis of the interactions between CXCR4 and CXCL12/SDF-1 revealed by theoretical approaches. Mol Biosyst. 2013;9(8):2107–2117. doi: 10.1039/c3mb70120d. [DOI] [PubMed] [Google Scholar]
- 69.Liou J-W, et al. In silico analysis reveals sequential interactions and protein conformational changes during the binding of chemokine CXCL-8 to its receptor CXCR1. PLoS ONE. 2014;9(4):e94178. doi: 10.1371/journal.pone.0094178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Whorton MR, et al. Efficient coupling of transducin to monomeric rhodopsin in a phospholipid bilayer. J Biol Chem. 2008;283(7):4387–4394. doi: 10.1074/jbc.M703346200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Whorton MR, et al. A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc Natl Acad Sci USA. 2007;104(18):7682–7687. doi: 10.1073/pnas.0611448104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Inagaki S, et al. Modulation of the interaction between neurotensin receptor NTS1 and Gq protein by lipid. J Mol Biol. 2012;417(1-2):95–111. doi: 10.1016/j.jmb.2012.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kuszak AJ, et al. Purification and functional reconstitution of monomeric μ-opioid receptors: Allosteric modulation of agonist binding by Gi2. J Biol Chem. 2009;284(39):26732–26741. doi: 10.1074/jbc.M109.026922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bayburt TH, et al. Monomeric rhodopsin is sufficient for normal rhodopsin kinase (GRK1) phosphorylation and arrestin-1 binding. J Biol Chem. 2011;286(2):1420–1428. doi: 10.1074/jbc.M110.151043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rasmussen SGF, et al. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature. 2011;477(7366):549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shukla AK, et al. Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature. 2014;512(7513):218–222. doi: 10.1038/nature13430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Levoye A, Balabanian K, Baleux F, Bachelerie F, Lagane B. CXCR7 heterodimerizes with CXCR4 and regulates CXCL12-mediated G protein signaling. Blood. 2009;113(24):6085–6093. doi: 10.1182/blood-2008-12-196618. [DOI] [PubMed] [Google Scholar]
- 78.Kalatskaya I, et al. AMD3100 is a CXCR7 ligand with allosteric agonist properties. Mol Pharmacol. 2009;75(5):1240–1247. doi: 10.1124/mol.108.053389. [DOI] [PubMed] [Google Scholar]
- 79.Gravel S, et al. The peptidomimetic CXCR4 antagonist TC14012 recruits beta-arrestin to CXCR7: Roles of receptor domains. J Biol Chem. 2010;285(49):37939–37943. doi: 10.1074/jbc.C110.147470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zabel BA, et al. Elucidation of CXCR7-mediated signaling events and inhibition of CXCR4-mediated tumor cell transendothelial migration by CXCR7 ligands. J Immunol. 2009;183(5):3204–3211. doi: 10.4049/jimmunol.0900269. [DOI] [PubMed] [Google Scholar]
- 81.Zabel BA, Lewén S, Berahovich RD, Jaén JC, Schall TJ. The novel chemokine receptor CXCR7 regulates trans-endothelial migration of cancer cells. Mol Cancer. 2011;10(1):73. doi: 10.1186/1476-4598-10-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lin H, Trejo J. Transactivation of the PAR1-PAR2 heterodimer by thrombin elicits β-arrestin-mediated endosomal signaling. J Biol Chem. 2013;288(16):11203–11215. doi: 10.1074/jbc.M112.439950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stephens B, Handel TM. Chemokine receptor oligomerization and allostery. Prog in Mol Biol Transl Sci. 2013;115:375–420. doi: 10.1016/B978-0-12-394587-7.00009-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Totrov M. Atomic property fields: Generalized 3D pharmacophoric potential for automated ligand superposition, pharmacophore elucidation and 3D QSAR. Chem Biol Drug Des. 2008;71(1):15–27. doi: 10.1111/j.1747-0285.2007.00605.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.