

Significance
Useful antimalarial drugs must be rapidly acting, highly efficacious, and have low potential for developing resistance. (+)-SJ733 targets a Plasmodium cation-transporting ATPase, ATP4. (+)-SJ733 cleared parasites in vivo as quickly as artesunate by specifically inducing eryptosis/senescence in infected, treated erythrocytes. Although in vitro selection of pfatp4 mutants with (+)-SJ733 proceeded with moderate frequency, during in vivo selection of pbatp4 mutants, resistance emerged slowly and produced marginally resistant mutants with poor fitness. In addition, (+)-SJ733 met all other criteria for a clinical candidate, including high oral bioavailability, a high safety margin, and transmission blocking activity. These results demonstrate that targeting ATP4 has great potential to deliver useful drugs for malaria eradication.
Keywords: malaria, PfATP4, drug discovery
Abstract
Drug discovery for malaria has been transformed in the last 5 years by the discovery of many new lead compounds identified by phenotypic screening. The process of developing these compounds as drug leads and studying the cellular responses they induce is revealing new targets that regulate key processes in the Plasmodium parasites that cause malaria. We disclose herein that the clinical candidate (+)-SJ733 acts upon one of these targets, ATP4. ATP4 is thought to be a cation-transporting ATPase responsible for maintaining low intracellular Na+ levels in the parasite. Treatment of parasitized erythrocytes with (+)-SJ733 in vitro caused a rapid perturbation of Na+ homeostasis in the parasite. This perturbation was followed by profound physical changes in the infected cells, including increased membrane rigidity and externalization of phosphatidylserine, consistent with eryptosis (erythrocyte suicide) or senescence. These changes are proposed to underpin the rapid (+)-SJ733-induced clearance of parasites seen in vivo. Plasmodium falciparum ATPase 4 (pfatp4) mutations that confer resistance to (+)-SJ733 carry a high fitness cost. The speed with which (+)-SJ733 kills parasites and the high fitness cost associated with resistance-conferring mutations appear to slow and suppress the selection of highly drug-resistant mutants in vivo. Together, our data suggest that inhibitors of PfATP4 have highly attractive features for fast-acting antimalarials to be used in the global eradication campaign.
In 2010, there were 220 million cases of malaria, leading to 660,000 deaths. More than 400,000 of those who died were African children (1). Recent successes in antimalarial drug discovery and development have led to renewed hope for global eradication of malaria (2, 3). Antimalarial drugs are a critical component of the eradication campaign, and single-dose regimens of cheap, safe, and efficacious drugs, which ideally also reduce transmission, are required (4). The discovery of agents that act rapidly against novel targets would greatly aid in the development of such drugs (5). Additionally, given the ready propensity with which the parasite develops resistance to new antimalarials (6), targets for which drug-resistance–conferring mutations cause a high fitness cost are of particular interest.
In a high-throughput screen to identify molecules that block the proliferation of Plasmodium falciparum in cocultures with human erythrocytes in vitro, we identified three high-priority series (7). Pharmacological and chemical optimization of one of these series, the dihydroisoquinolones (DHIQs), led to the selection of a clinical candidate, (+)-SJ733, for development as a fast clearance component (Target Candidate Profile 1; TCP1) of a single-exposure radical cure and prophylaxis (SERCaP) drug (4). Here, we present evidence that (+)-SJ733 acts on the recently recognized target PfATP4 (pfatp4; PFL0590c) (8) to give rapid clearance of the parasite in vivo by inducing eryptosis or senescence in parasite-infected erythrocytes. Mutation of pfatp4 to confer resistance to (+)-SJ733 carries a high fitness cost, and the development of resistance in vivo is slow and produces only low-level resistance.
Results and Discussion
(+)-SJ733 Is a Candidate for TCP1 of a SERCaP Drug.
SJ733 was highly potent in vitro against all tested strains of P. falciparum (EC50 range 10–60 nM; SI Appendix, Fig. S1A and Table S1), including those resistant to other antimalarials, and was equally potent against all stages of the erythrocytic life cycle (SI Appendix, Fig. S3E). Of the two enantiomers of SJ733, the (+)-enantiomer was significantly more potent. (+)-SJ733 was, however, 10-fold less potent against Plasmodium berghei and other rodent malarias ex vivo (SI Appendix, Fig. S1B). (+)-SJ733 bound to a single receptor site in P. falciparum-infected erythrocytes with equivalent affinity to its growth-inhibitory potency (kd = 50 nM; SI Appendix, Fig. S3F).
(+)-SJ733 was highly potent and efficacious against P. falciparum 3D70087/N9 in vivo when administered as four sequential daily oral doses in the NOD-scid IL2Rγnull mouse model (Fig. 1A) (9), with a 90% effective dose, (ED90 1.9 mg/kg) and exposure [area under the curve at ED90 (AUCED90), 1.5 μM⋅h] superior to artesunate (11.1 mg/kg; AUCED90 not determined), chloroquine (4.3 mg/kg; AUCED90 3.1 μM⋅h), and pyrimethamine (0.9 mg/kg; AUCED90 5. μM⋅h) in the same model. When treated with the ED90 dose, (+)-SJ733 concentrations in blood remained above the average in vitro EC90 for 6–10 h after each dose (SI Appendix, Fig. S1C). In line with ex vivo results, SJ733 was 10-fold less potent in vivo against P. berghei when administered as four sequential daily oral doses (racemate ED90 40 mg/kg; AUCED90 80 μM⋅h; SI Appendix, Fig. S1D), with (r)-SJ733 concentrations in blood remaining above the average ex vivo EC90 for 10–12 h after each dose (SI Appendix, Fig. S1E). (+)-SJ733 had robust in vivo pharmacokinetic behavior in all preclinical species, including mouse, rat, and dog, with excellent exposure (3–10 μM Cmax, 5–40 μM–h AUC, at 20–30 mg/kg doses) and reasonable clearance (Fig. 1B). The compound also had oral bioavailability of >65% in both rats (SI Appendix, Fig. S1F) and dogs (SI Appendix, Fig. S1G).
Fig. 1.
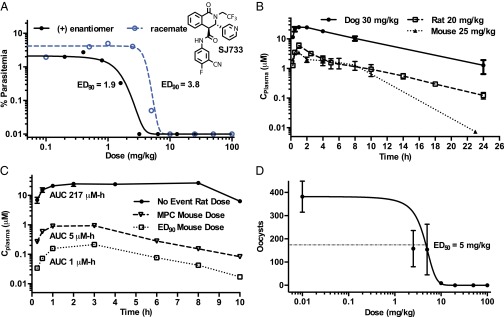
SJ733 is an efficacious and safe orally active drug candidate. (A) SJ733 is efficacious against P. falciparum 3D70087/N9 in the nonobese diabetic Scid interleukin-2 receptor γ chain null (NSG) mouse model with the (+)-enantiomer exhibiting excellent potency (1.9 mg/kg); similar to pyrimethamine (ED90 0.9 mg/kg) and superior to chloroquine (ED90 4.3 mg/kg) in this model. (+)-SJ733 achieves this efficacy from exposure (AUCED90 1.5 µM⋅h) similar to that of chloroquine (AUCED90 3.1 µM⋅h) and superior to that of pyrimethamine (AUCED90 5.2 µM⋅h) in the same model. (B) (+)-SJ733 exhibits excellent exposure after oral administration in mouse, rat, and dog. In rodents, (+)-SJ733 reaches peak plasma concentrations of ∼5 µM within 1 h after 20–25 mg/kg doses and >20 µM in dogs following a 30 mg/kg oral dose. (C) (+)-SJ733 exhibits no significant toxicology at doses up to 200 mg/kg in rats with an exposure (AUC) ∼43-fold higher than that required to produce the maximum parasitological response (fastest rate of killing) and 220-fold that required to produce the ED90 in the mouse, indicating the potential for an excellent therapeutic ratio. (D) (+)-SJ733 potently and efficaciously blocks transmission of P. berghei from infected mice to mosquitos when the mice are treated 1 h before feeding the mosquitos as measured by counting oocysts from dissected mosquitos after the sexual stage is allowed to mature.
(+)-SJ733 has not exhibited either significant safety liabilities at any dose in extensive profiling in vitro (see summary in SI Appendix) or significant safety or tolerability liabilities in either single- or repeat-dose studies at any dose tested in any preclinical species (no observed adverse effect level and maximum tolerated dose >240 mg/kg from 7-d repeat dosing study in rat; see summary in SI Appendix). Therefore, (+)-SJ733 is expected to have a safety margin of at least 43-fold [comparing AUCmax in the rat safety studies to AUC from the dose giving maximal parasitological response in mice (Fig. 1C)]. In addition, (+)-SJ733 blocked transmission of P. berghei from infected mice to mosquitos (ED50 5 mg/kg; Fig. 1D), at a dose lower than that efficacious against asexual blood stages. Finally, (+)-SJ733 was readily synthesized in good yield from commercially available precursors in five steps with a route that is expected to provide low cost of goods (SI Appendix, Fig. S1H). The only potential liability noted to date was a relatively high clearance in human microsomal models (SI Appendix). However, predicted efficacious human doses, allometrically derived from preclinical pharmacokinetics experiments, fall into the range of 3–6 mg/kg, and the projected half-life is long enough to allow (+)-SJ733 to be the fast acting component (TCP1) of a SERCaP drug (4). Thus, (+)-SJ733 is highly potent, efficacious against both asexual and sexual blood stages, safe, and highly orally bioavailable. (+)-SJ733 was selected for clinical development by Medicines for Malaria Venture in March 2013.
(+)-SJ733 Targets ATP4.
To gain insight into the mechanism of action of (+)-SJ733 and the potential for acquisition of resistance, we selected drug-resistant mutant strains of P. falciparum in vitro and P. berghei in vivo, using (+)-SJ733 and other DHIQ analogs, and characterized the alleles conferring resistance (6, 10). Mutant strains were generated in vitro by pressuring erythrocytic cocultures with (+)-SJ733 or other DHIQ analogs (SI Appendix, SI Materials and Methods) until apparently resistant parasites emerged. Resistance, confirmed by measuring the potency of (+)-SJ733, varied between twofold to ∼750-fold (SI Appendix, Table S1) and emerged with minimum inocula of between 107 and 109 parasites (3D7 strain). In most cases, resistance was ∼10-fold and emerged from 108 to 109 parasites (SI Appendix, Fig. S2A). The single mutant strain generated in vivo was produced by serial passage of parasites in mice with multiple rounds of intermittent high-dose treatment with (+)-SJ311, a close analog of (+)-SJ733 (SI Appendix, Fig. S2C), with weak (twofold) stable resistance emerging only after 4 weeks of treatment.
For six independently produced resistant P. falciparum strains, we used whole-genome sequencing to identify mutations (SI Appendix, Table S2). Genomic DNA was prepared for parental and mutant strains, and libraries were sequenced (SI Appendix). After quality filtering and alignment to the reference sequence, the average coverage ranged from >50-fold to >700-fold for the 14 nuclear chromosomes and >200-fold to >30,000-fold for the mitochondrial and plastid genomes. The presence of copy number variants (CNVs) was assessed, relative to the parental strains, by using the R-package CNV-Seq (11). Other than the variable sequences proximal to the telomeres, no significant (P < 0.001) deletions were detected relative to the drug-sensitive parental strains (SI Appendix, Table S3). Similarly, no amplified regions were detected for any strain. To detect genomic mutations we compared exonic sequences for both the parental and selected strains. Because the mutant strains were not necessarily clonal populations, we reasoned that true drivers of drug resistance would be gain-of-function mutations driven to purification as opposed to passenger mutations present at various frequencies. Therefore, we considered only nonsynonymous mutations that were present at 99% purity or higher, where purity was defined as the percent of the total reads confirming a mutation for a given position in the genome, assuming that mutations occurred in single-copy genes.
By using these criteria, only a single mutated gene, encoding the putative Na+-ATPase PfATP4 (PFL0590c), was shared among all six selections (SI Appendix, Table S2). In five of six selections, 100% of the mapped reads confirmed the detected mutation, all of which were confirmed by Sanger sequencing. In five of six strains, pfatp4 was the only mutated gene detected using these criteria. We also investigated whether expression of the mRNA for pfatp4, as measured by quantitative RT-PCR (qRT-PCR), was altered in wild-type or mutated strains, with or without treatment with (+)-SJ733. Relative to a housekeeping gene, we detected no significant change in expression (SI Appendix, Table S4).
Two additional independently derived resistant strains were generated by selection with SJ733 or another DHIQ. Sanger sequencing of the pfatp4 coding sequence identified nonsynonymous mutations in both cases (SI Appendix, Table S3). For the sole resistant P. berghei strain, described above, sequencing of the orthologous gene pbatp4 revealed a nonsynonymous mutation absent in parental parasites (SI Appendix, Table S5).
Finally, we examined whether cross-resistance existed for mutant strains of malaria previously selected with other putative PfATP4 targeted compounds, including the DHIQ family, the spiroindolones, and a series developed at GlaxoSmithKline (SI Appendix, Table S5). In all cases there was complete and bidirectional cross-resistance for all compounds and strains (SI Appendix, Table S1). The fold resistance exhibited by each strain tracked with the genotype and was consistent from compound to compound. This finding strongly suggests that the mutation(s) in pfatp4 are the sole determinant of resistance for each of the compounds.
Two DHIQ-resistant mutant strains (P. falciparum ATP4L350H and ATP4P966T) were tested in competition assays and were found to be substantially less fit than their parental strains in the absence of drug pressure (SI Appendix, Fig. S2B). The slow emergence of DHIQ resistance observed during our in vivo selection experiment is consistent with our finding of a fitness cost associated with DHIQ resistance.
Together, the complete association of resistance with point mutations in pfatp4, the absence of copy-number variations or expression-level differences for this gene, and the cross-resistance with spiroindolones all support the notion that mutation of PfATP4 is both necessary and sufficient for resistance to SJ733.
A homology model for PfATP4, built from crystal structures of the closest mammalian homolog (sarco/endoplasmic reticulum Ca2+-ATPase; SERCA), revealed that the majority of the resistance-conferring mutations clustered in a small portion of the predicted protein structure within the transmembrane, ion-transporting channel of the protein (Fig. 2 A and C). All other resistance-conferring mutations were predicted to perturb the protein structure near this cluster by distorting the orientation of helices adjoining the cluster. Unconstrained docking of the structure of (+)-SJ733 to this homology model revealed one predicted binding site, which is enveloped by the cluster of residues bearing resistance-conferring mutations (Fig. 2B).
Fig. 2.

SJ733 targets the PfATP4 protein. (A) All SJ733-resistant strains of malaria generated worldwide contain mutations in the pfatp4 gene that cluster in a single region of the protein. The mutations are illustrated with a homology model of the protein structure for PfATP4 (gold/blue ribbon), built from the available SERCA crystal structures, with the location of mutations causing either high-fold resistance (red), medium-fold resistance (yellow), or low-fold resistance (blue) shown as solid fill. The blue helices represent those in the transmembrane domain and are indicated in the gene block diagram in C. (B) Theoretical docking studies with (+)-SJ733 reproducibly generate a single pose that places (+)-SJ733 in contact with the two residues inducing the highest fold resistance and the one residue selected by in vivo passage with cycling drug exposure. This pose is illustrated by the docked structure of (+)-SJ733 placed into the putative binding site on the PfATP4 structure, with residues conferring resistance color-coded as in A. A loop with sequence variability among Plasmodium spp. that may lead to differing sensitivities to (+)-SJ733 is highlighted in magenta. (C) Block diagram of pfatp4 gene structure and positioning of mutations detected in strains resistant to (+)-SJ733. The selected mutations are color-coded to match those in A, with the location in the coding sequence indicated. The blue cylinders represent the helical portions of the protein shown as blue on the ribbon diagram in A. (D) SJ733 disrupted resting [Na+]i within asexual blood-stage P. falciparum as illustrated by dose–response experiments with the active (+) and inactive (−) enantiomers. Resting [Na+]i was measured >60 min after addition of the compound to sodium-binding benzofuran isophthalate-loaded, saponin-isolated parasites. Each data point represents the mean final [Na+]i averaged from at least three independent experiments (shown ± SD). The relative potencies of the two SJ733 isomers in the [Na+]i assays shown here, on the two different strains (the SJ733-resistant mutant ATP4L350H and the wild-type parent) matched those seen in parasite growth assays.
The three rodent malaria species (P. berghei, Plasmodium vinckei, and Plasmodium chabaudi) were all significantly less sensitive to (+)-SJ733 than was P. falciparum both ex vivo (SI Appendix, Fig. S1B) and in vivo (SI Appendix, Figs. S1D and S2D). The atp4 alleles from these species carry a short variant sequence (SI Appendix, Fig. S2E), which is predicted to encode a loop within the (+)-SJ733 binding site (purple ribbon, Fig. 2B). Replacing the pbatp4 gene with the pfatp4 gene fully conferred increased susceptibility to (+)-SJ733 ex vivo (SI Appendix, Fig. S1B) and partially (2-fold vs. 10-fold) restored sensitivity in vivo (SI Appendix, Figs. S1D and S2D), thus strongly suggesting that the sequence variations underpin the reduced susceptibility of the rodent malarias to (+)-SJ733. The lack of a full restoration of sensitivity in vivo may be due to differences between the humanized mouse model used for P. falciparum and the normal mouse model used for P. berghei, but is not driven by differing pharmacokinetics, because there were no significant exposure variations.
PfATP4 has been proposed to be a Na+-efflux ATPase that maintains a low Na+ concentration in the parasite cytosol ([Na+]i) (12). As with the spiroindolones (12), treatment of saponin-isolated trophozoites with (+)-SJ733 resulted in a rapid increase in [Na+]i from a resting level of 6 ± 1 mM to a maximal level of 110 ± 10 mM within ∼1.5 h of initiating treatment (Fig. 2D and SI Appendix, Fig. S2F). As is also seen for the spiroindolones (13), (+)-SJ733 caused a rapid increase in the transmembrane pH gradient (0.11 ± 0.03 pH units; mean ± SD n = 6) and reduced the extent of acidification of the parasite cytosol seen following the inhibition of the parasite’s plasma membrane V-type H+ ATPase by concanamycin A (SI Appendix, Fig. S2G).
The EC50 for the effect of (+)-SJ733 on [Na+]i in wild-type parasites is ∼200 nM, within fivefold of the growth inhibition potency in the same strain (SI Appendix, Fig. S1A) and well within the concentrations achieved in the blood of effectively treated animals (SI Appendix, Fig. S1C). The P. falciparum ATP4L350H strain, which is resistant to (+)-SJ733, showed a ∼50-fold decrease in sensitivity (relative to the parental strain) to the [Na+]i-disrupting effects of (+)-SJ733. Furthermore, the relative potency of the enantiomers of SJ733 in the [Na+]i assays for these strains was the same as in the proliferation assays (Fig. 2D and SI Appendix, Fig. S1A). The mutant strain had a significantly higher resting [Na+]i than the parent (15.8 ± 0.8 mM vs. 5.7 ± 0.5 mM; mean ± SEM from 9 or 10 independent experiments; P < 0.001 with unpaired t test), which may account for the decreased fitness of this strain. The data are consistent with the primary growth inhibition effects of (+)-SJ733 stemming from its ability to increase [Na+]i through inhibition of PfATP4.
Inhibition of Plasmodium ATP4 Leads to Rapid Clearance in Vivo Through Inducing Physical Changes in Infected, Treated Red Cells.
Treatment of P. falciparum-infected NOD-scid IL2Rγnull mice (9) with (+)-SJ733 caused rapid clearance of parasites, which were 80% depleted within the first 24 h and undetectable by 48 h (Fig. 3A). Notably, (+)-SJ733 cleared parasites faster than artesunate in the same model. The same pharmacodynamics were observed after treatment of P. falciparum-infected splenectomized NOD-scid IL2Rγnull mice (Fig. 3B) or normal mice infected with rodent malarias, with or without liposomal clodronate depletion of Kupffer cells (SI Appendix, Fig. S3A). As discussed above, although the rate of action remains the same, the potency of (+)-SJ733 varied by >10-fold between these models. Thus, the rate of clearance in vivo was independent of both malaria species and host immune status.
Fig. 3.

SJ773 causes rapid clearance in vivo that is not dependent upon innate immunity. (A) (+)-SJ733 causes rapid clearance of parasites in vivo, with speed equivalent to artesunate, the fastest-acting antimalarial drug, as measured by clonal dilution assays. When total parasites present in blood are measured, all parasites are cleared systemically within 48 h of initiation of therapy. Similar pharmacodynamics are seen in P. berghei-infected animals (SI Appendix). (B) The in vivo clearance rate of (+)-SJ733 is independent of the presence of a spleen. When total parasites in blood are measured, all parasites are cleared within 48 h of initiation of treatment in both splenectomized and nonsplenectomized animals. Similar experiments in clodronate-treated animals infected with P. berghei indicate that macrophages are not required for rapid pharmacodynamics (SI Appendix). (C) (+)-SJ733 arrests growth of parasites rapidly in vitro, reaching a maximal effect within 24 h as measured by proliferation of a luciferase-labeled 3D7 strain. (D) (+)-SJ733 kills parasites in vitro, but does so with modest speed, equivalent to pyrimethamine as measured by clonal dilution assays. When viable parasites are measured, 96 h of continuous exposure above the EC99 is required for maximal effect.
When infected erythrocytes were treated with (+)-SJ733 in vitro, growth was fully arrested within 24 h, as measured by using luciferase-labeled parasites (Fig. 3C), but fully killing parasites required 96 h (Fig. 3D), as measured by using clonal dilution methods (14). Given that a typical P. falciparum-infected mouse has ∼108 parasites, the same load used in the in vitro cidality assays, it appears that (+)-SJ733 acts at least fourfold faster in vivo than it does in vitro. These data suggest that compounds targeting ATP4 induce physical changes in the infected erythrocyte that allow recognition and clearance of treated, infected cells by the host.
The most obvious explanation for enhanced clearance in vivo is that (+)-SJ733 induces an erythrocyte phenotype leading to rapid clearance such as eryptosis (suicidal erythrocyte death) or senescence, which can be caused by a wide range of stresses (15, 16). Eryptotic and senescent erythrocytes share several key features: an increase in exposed phosphatidylserine (PS), increased membrane rigidity, more spherical shape, and decreased size (17, 18). The eryptotic/senescent phenotype has been correlated with increased clearance of erythrocytes, primarily through erythrophagocytosis (19–21). However, mechanical clearance in the spleen (22) and sinusoidal clearance, independent of Kupffer cells, in the liver (16) also both play a role and can compensate for a lack of erythrophagocytosis in macrophage-depleted animals. We simultaneously measured each of these variables for uninfected and infected erythrocytes in vitro using multimodal fluorescence activated cell sorting measuring forward/side scatter (size and shape), parasite infection (SYBR green binding), and PS exposure (Annexin V binding).
Plasmodium-infected erythrocytes (iRBCs) have been reported to possess a mild eryptotic/senescent phenotype (23) relative to uninfected erythrocytes (uRBCs). In our studies iRBCs were small relative to uRBCs (Fig. 4A, lower forward scatter in Top Left vs. Bottom Left) and possessed more exposed PS [color scale in Fig. 4A; uRBCs, green (<1,000 Annexin binding) and iRBCs, white to light pink (1,000–5,000 Annexin binding)]. Treatment of iRBCs with (+)-SJ733, or with another ATP4-targeted compound (the spiroindolone NITD-246), caused the majority of the cells to shift to the population with the most severe phenotype possessing a 10-fold increase in exposed PS relative to uRBCs [Fig. 4A; increased pink intensity (10,000 to >1000,000 Annexin binding, Middle Left and Middle Right)]. The potency of (+)-SJ733 in shifting cells from the mild to more severely eryptotic/senescent phenotype was almost identical to that observed for growth inhibition (EC50 30 nM; Fig. 4B). No other class of antimalarial drug has this profile. For example artesunate, another rapidly acting drug (Fig. 4A, Top Right) did not produce the more pronounced phenotype, regardless of dose. Importantly, this response is dependent upon the presence of both the parasite and the drug, because treatment of uninfected erythrocytes did not induce eryptosis (Fig. 4A, Bottom Right).
Fig. 4.

SJ733 arrests parasite development and induces eryptosis selectively in infected erythrocytes. (A) (+)-SJ733 causes a significant proportion of infected erythrocytes to shrink and expose PS. Uninfected erythrocytes, whether treated with (+)-SJ733 or not (Bottom Left and Bottom Right), remain normally sized, do not have exposed PS, and do not possess substantial DNA, as evidenced by examining the cell population for forward scatter, Annexin V binding, and SYBR green binding in FACS. Cells are shown as a population contour plot that compared scatter with SYBR green binding and are color-coded to indicate Annexin binding. After infection, magnetically purified trophozoite/schizont-infected erythrocytes can be detected in three populations (large, with little exposed PS; small, with moderate levels of exposed PS; and intermediate-sized, with high levels of exposed PS), with the majority being in the small- and large-sized populations. Treatment with (+)-SJ733 causes the population distribution to shift strongly toward the intermediate-sized population with high amounts of exposed PS—consistent with strong induction of eryptosis. This shift is not seen with either control drugs or DMSO treatment. (B) When the eryptosis induction effect is examined for (+)-SJ733 by using a concentration-response experiment, the maximal effect is seen at a concentration of ∼40 nM and the EC50 at 30 nM—congruent with the doses causing similar levels of response for proliferation inhibition and parasite Na+ levels. Artesunate has no such effect. (C) (+)-SJ733 causes a significant increase in rigidity of infected, treated erythrocytes. As previously reported, infection of erythrocytes (iRBC) causes a significant increase in their rigidity (P < 0.001; Jonckheere–Terpstra test). After treatment with (+)-SJ733, these infected erythrocytes become significantly more rigid, with the degree of rigidity peaking at ∼7 h after treatment. *P < 0.001 (Jonckheere–Terpstra test). There are no detectable effects on unparasitized erythrocytes at any time point. (D) (+)-SJ733 induces a rapid arrest of parasite motility inside infected erythrocytes that is maintained for the period of treatment as shown by time-lapse microscopy of the treated and untreated cells. The first row of images shows typical motility and growth of a ring-stage parasite over 18 h, which is in stark contrast to the treated parasite in the second row of images that immediately arrests both motility and growth. In some cases the parasite can be observed to lyse within the erythrocyte as shown in the third row. In additional cases the infected, treated erythrocyte itself will lyse after lysis of the parasite as shown in the fourth row. No changes are noted to erythrocyte morphology after treatment of uninfected erythrocytes.
The biconcave cell shape of RBCs causes a multimodal distribution of forward scatter scores, whereas changing RBC morphology to a more spherical shape characteristic of eryptosis/senescence leads to a more monomodal distribution (24) that can be captured by regression modeling of forward and side scatter data (25). The treatment of iRBCs with (+)-SJ733 (or NITD246) collapsed the multimodal distribution largely to a monomodal distribution (containing >85% of cells) of the iRBCs, which when modeled is similar to spherical cells (MI −0.7) and not to discoid cells (MI −0.9). This finding suggests that inhibition of ATP4 in the parasite leads to a spherical morphology, perhaps through parasite swelling. Individual iRBCs that are more spherical are also observed after treatment with (+)-SJ733 in video microscopy (Fig. 4D, comparing the first row to the following three). These cell shape changes were not observed for either uRBCs or untreated iRBCs.
The eryptotic/senescent phenotype also includes an increase in rigidity as measured by deformation when the cell is pushed through a capillary with fluid pressure. (+)-SJ733 treatment of iRBCs caused them to become significantly more rigid compared with either uRBCs or untreated iRBCs (Fig. 4C; P < 0.001; Jonckheere–Terpstra test). Again, there was no effect on the rigidity of uRBCs exposed to (+)-SJ733. This increase in rigidity was larger in magnitude than previously reported to take place after infection (26) and is congruent with the magnitude of loss of deformability that is associated with rapid clearance of eryptotic/senescent erythrocytes (17, 27, 28).
Finally, treatment of infected erythrocytes led to the arrest of parasite development (Fig. 4D, row 2, relative to row 1) and to changes in the morphology of both the parasite (swelling) and the erythrocyte (becoming pyknotic) as observed by serial single cell phase contrast microscopy. All of these changes reached maximal effect within 1 h of treatment and were maintained for the duration of treatment—congruent with the time scale on which [Na+]i increases. In many cases the swelling of the parasite was followed by parasite bursting (Fig. 4D, row 3) or by sequential bursting of the parasite and the infected, treated erythrocyte (Fig. 4D, row 4). These effects become evident after 7–10 h. In all cases, the effects on erythrocyte morphology and integrity were limited to treated, infected erythrocytes, with no changes observed in uninfected, treated erythrocytes.
Together, these data strongly suggest that (+)-SJ733 produces structural changes in infected erythrocytes consistent with eryptosis/senescence, while leaving uninfected erythrocytes unscathed. This effect is clearly not due to simple changes in erythrocyte ion content; similar (eryptotic) changes were not seen on treatment of uninfected erythrocytes with the Na+/K+ ATPase inhibitor ouabain under conditions shown previously to cause major perturbation of the Na+/K+ ratio in the erythrocyte cytosol (SI Appendix, Fig. S3G) (29). Our data suggest that eryptosis may be induced by mechanical changes caused by parasite swelling. Induction of the eryptosis/senescence phenotype leads to clearance by several documented mechanisms. Our results in animals with varying phagocyte and macrophage defects, as described above, suggest that all of these clearance mechanisms are used in a complementary way to drive the rapid clearance of parasites in vivo by (+)-SJ733.
The data presented here are consistent with the model outlined in Fig. 5. Treatment of parasitized erythrocytes with (+)-SJ733 blocks the normal function of PfATP4 and leads to a rapid increase of [Na+]i within the parasite, reaching a maximum by 1.5 h after treatment. This immediate response triggers other cellular responses, probably including the arrest of protein synthesis (8), and these are integrated at the level of both the parasite (manifested as a swollen, development-arrested phenotype) and the host erythrocyte (manifested as induction of an eryptotic/senescent phenotype). These morphological changes reach a maximum by 7–8 h after treatment. In vivo, these effects trigger rapid clearance of the infected, treated erythrocytes through host-mediated mechanisms used to clear aging red cells. Conversely, in vitro, without the physiological clearance mechanisms, the parasites go into a growth-arrested state, with maximal arrest being reached by 24 h after treatment. Parasites then die by a number of mechanisms, including spontaneous lysis, with the maximal effect ultimately reached within 96 h.
Fig. 5.

Induction of eryptosis by SJ733 triggers rapid clearance in vivo. When (+)-SJ733 is added to erythrocytes infected with Plasmodium spp., it prevents the action of the putative Na+ ATPase PfATP4 and causes a significant increase in cytosolic Na+ within the parasite, reaching maximal effect within 90 min after treatment. This process results in the immediate arrest of parasite motility and blockade of intracellular parasite replication that reaches maximal potency after 24 h of exposure. Simultaneously, infected, treated erythrocytes begin to enter eryptosis, as characterized by their shrinking and becoming more spherical, becoming significantly more rigid, and exposing PS on their plasma membrane. These effects maximize by 7 h after treatment. In vitro, the drug effects lead to complete arrest of replication within 24 h of treatment, followed by slow death, which is maximal by 96 h. In stark contrast to the in vitro setting, the induction of eryptosis leads to rapid clearance of the infected, treated erythrocytes in vivo.
In conclusion, these studies of (+)-SJ733 indicate that PfATP4 inhibitors, including both the DHIQs and the spiroindolones, possess multiple properties that make them ideal for development as fast-acting components of combination therapies for single dose cure of malaria. First, the host-mediated mechanism leads to extremely rapid clearance in vivo, thus obviating the need for extended coverage. Indeed, the spiroindolones have proven faster acting in the clinic than expected from preclinical modeling (30). Second, they have exquisite selectivity due to a host-mediated mechanism—only affecting infected and treated erythrocytes. Third, there is a high fitness cost associated with resistance-conferring mutations. This trait is potentially due to the observed higher resting [Na+]i causing cellular stress. The combination of this fitness cost and the extremely rapid action of the compounds make it very difficult for the parasite to acquire resistance in vivo despite a frequency of in vitro resistance acquisition that is in line with other targeted drugs. Therefore, we hypothesize that the acquisition of resistance during clinical use will occur at a very low frequency, on par with artemisinin-containing drugs. The rapid in vivo clearance produced by compounds that target ATP4 make it an attractive drug development target and the favorable properties of (+)-SJ733 encourage further development.
Materials and Methods
All procedures were carried out according to published methods. Details of the methods are included in SI Appendix. The detailed methods included in SI Appendix are the in vivo efficacy studies for blood stages in both the P. falciparum and P. berghei models; in vivo transmission blocking studies; in vivo and in vitro pharmacokinetics studies in all species; in vitro and ex vivo efficacy studies in P. falciparum and P. berghei; synthesis of (+)-SJ733; homology modeling and docking studies of (+)-SJ733 binding to PfATP4; in vitro and in vivo selection of mutant strains of P. falciparum and P. berghei resistant to (+)-SJ733; whole-genome and Sanger sequencing of mutant strains; quantitative PCR of mutant strains; production of transgenic parasites; measurement of cytosolic Na+ and pH; mutant fitness studies; time-lapse microscopy studies; erythrocyte rigidity studies; FACS analysis of erythrocytes; and measurement of speed of effect on P. falciparum. The SI Appendix includes additional data related to the generation, characterization of the chemical sensitivity, and genotype of P. falciparum strains resistant to (+)-SJ733; summaries of the in vivo efficacy and pharmacokinetics data; and the details of safety pharmacology and tolerability studies.
Supplementary Material
Acknowledgments
We thank Dr. Alexis LaCrue for mosquito dissections; Case McNamara, Elizabeth Winzeler, and Thierry Diagana for sharing mutant strains of P. falciparum selected by exposure to the spiroindolones and samples of the spiroindolones; the National Institute of Mental Health Psychoactive Drug Screening Program for profiling (+)-SJ733 in their assay panel; Deqing Pei for biostatistical analyses; Dr. Leonard D. Shultz and The Jackson Laboratory for providing access to nonobese diabetic SCID IL2Rγc null mice through their collaboration with GlaxoSmithKline Tres Cantos Medicines Development Campus; and the Australian and Spanish Red Cross Blood Service for the provision of blood. This work was supported by National Institute of Allergy and Infectious Diseases Contract HHSN2722011000221; NIH Grants AI090662 and AI075517; the Medicines for Malaria Venture; Australian National Health and Medical Research Council (NHMRC) Project Grant 1042272; NHMRC Overseas Biomedical Fellowships 585519 and 1072217; the Howard Hughes Medical Institute; The David and Lucille Packard Foundation; the American Lebanese Syrian Affiliated Charities; and St. Jude Children’s Research Hospital. We acknowledge the support of the High-Throughput Screening Center, the Biostatistics Department, and the Flow Cytometry and Cell Sorting Shared Resource at St. Jude Children’s Research Hospital.
Footnotes
Conflict of interest statement: M.B.J.-D., M.G.G., M.S.M., A.R.-A., L.S., F-J.G., S.F., and I.A.-B. are employees of GlaxoSmithKline and are engaged in commercial development of antimalarial drugs, although not this compound.
*This Direct Submission article had a prearranged editor.
Data deposition: The sequencing data have been deposited in the National Center for Biotechnology Information BioProject database, www.ncbi.nlm.nih.gov/bioproject (project ID PRJNA253899).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1414221111/-/DCSupplemental.
References
- 1.World Health Organization . World Malaria Report. WHO; Geneva: 2012. [Google Scholar]
- 2.Anthony MP, Burrows JN, Duparc S, Moehrle JJ, Wells TN. The global pipeline of new medicines for the control and elimination of malaria. Malar J. 2012;11:316. doi: 10.1186/1475-2875-11-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spangenberg T, et al. The open access malaria box: A drug discovery catalyst for neglected diseases. PLoS ONE. 2013;8(6):e62906. doi: 10.1371/journal.pone.0062906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burrows JN, van Huijsduijnen RH, Möhrle JJ, Oeuvray C, Wells TN. Designing the next generation of medicines for malaria control and eradication. Malar J. 2013;12:187. doi: 10.1186/1475-2875-12-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chatterjee AK, Yeung BK. Back to the future: Lessons learned in modern target-based and whole-cell lead optimization of antimalarials. Curr Top Med Chem. 2012;12(5):473–483. doi: 10.2174/156802612799362977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neafsey DE. Genome sequencing sheds light on emerging drug resistance in malaria parasites. Nat Genet. 2013;45(6):589–590. doi: 10.1038/ng.2648. [DOI] [PubMed] [Google Scholar]
- 7.Guiguemde WA, et al. Chemical genetics of Plasmodium falciparum. Nature. 2010;465(7296):311–315. doi: 10.1038/nature09099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rottmann M, et al. Spiroindolones, a potent compound class for the treatment of malaria. Science. 2010;329(5996):1175–1180. doi: 10.1126/science.1193225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiménez-Díaz MB, et al. Improved murine model of malaria using Plasmodium falciparum competent strains and non-myelodepleted NOD-scid IL2Rgammanull mice engrafted with human erythrocytes. Antimicrob Agents Chemother. 2009;53(10):4533–4536. doi: 10.1128/AAC.00519-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park DJ, et al. Sequence-based association and selection scans identify drug resistance loci in the Plasmodium falciparum malaria parasite. Proc Natl Acad Sci USA. 2012;109(32):13052–13057. doi: 10.1073/pnas.1210585109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xie C, Tammi MT. CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinformatics. 2009;10:80. doi: 10.1186/1471-2105-10-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spillman NJ, et al. Na+ regulation in the malaria parasite Plasmodium falciparum involves the cation ATPase PfATP4 and is a target of the spiroindolone antimalarials. Cell Host Microbe. 2013;13(2):227–237. doi: 10.1016/j.chom.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spillman NJ, Allen RJ, Kirk K. Na+ extrusion imposes an acid load on the intraerythrocytic malaria parasite. Mol Biochem Parasitol. 2013;189(1-2):1–4. doi: 10.1016/j.molbiopara.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 14.Rathod PK, Leffers NP, Young RD. Molecular targets of 5-fluoroorotate in the human malaria parasite, Plasmodium falciparum. Antimicrob Agents Chemother. 1992;36(4):704–711. doi: 10.1128/aac.36.4.704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lutz HU, Bogdanova A. Mechanisms tagging senescent red blood cells for clearance in healthy humans. Front Physiol. 2013;4:387. doi: 10.3389/fphys.2013.00387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee SJ, Park SY, Jung MY, Bae SM, Kim IS. Mechanism for phosphatidylserine-dependent erythrophagocytosis in mouse liver. Blood. 2011;117(19):5215–5223. doi: 10.1182/blood-2010-10-313239. [DOI] [PubMed] [Google Scholar]
- 17.Kwan JM, Guo Q, Kyluik-Price DL, Ma H, Scott MD. Microfluidic analysis of cellular deformability of normal and oxidatively damaged red blood cells. Am J Hematol. 2013;88(8):682–689. doi: 10.1002/ajh.23476. [DOI] [PubMed] [Google Scholar]
- 18.Mohandas N, Groner W. Cell membrane and volume changes during red cell development and aging. Ann N Y Acad Sci. 1989;554:217–224. doi: 10.1111/j.1749-6632.1989.tb22423.x. [DOI] [PubMed] [Google Scholar]
- 19.de Back DZ, Kostova EB, van Kraaij M, van den Berg TK, van Bruggen R. Of macrophages and red blood cells: A complex love story. Front Physiol. 2014;5:9. doi: 10.3389/fphys.2014.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwartz RS, et al. Increased adherence of sickled and phosphatidylserine-enriched human erythrocytes to cultured human peripheral blood monocytes. J Clin Invest. 1985;75(6):1965–1972. doi: 10.1172/JCI111913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schroit AJ, Madsen JW, Tanaka Y. In vivo recognition and clearance of red blood cells containing phosphatidylserine in their plasma membranes. J Biol Chem. 1985;260(8):5131–5138. [PubMed] [Google Scholar]
- 22.Klausner MA, et al. Contrasting splenic mechanisms in the blood clearance of red blood cells and colloidal particles. Blood. 1975;46(6):965–976. [PubMed] [Google Scholar]
- 23.Föller M, et al. Suicide for survival—death of infected erythrocytes as a host mechanism to survive malaria. Cell Physiol Biochem. 2009;24(3-4):133–140. doi: 10.1159/000233238. [DOI] [PubMed] [Google Scholar]
- 24.Piagnerelli M, et al. Assessment of erythrocyte shape by flow cytometry techniques. J Clin Pathol. 2007;60(5):549–554. doi: 10.1136/jcp.2006.037523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahlgrim C, Pottgiesser T, Sander T, Schumacher YO, Baumstark MW. Flow cytometric assessment of erythrocyte shape through analysis of FSC histograms: Use of kurtosis and implications for longitudinal evaluation. PLoS ONE. 2013;8(3):e59862. doi: 10.1371/journal.pone.0059862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo Q, Reiling SJ, Rohrbach P, Ma H. Microfluidic biomechanical assay for red blood cells parasitized by Plasmodium falciparum. Lab Chip. 2012;12(6):1143–1150. doi: 10.1039/c2lc20857a. [DOI] [PubMed] [Google Scholar]
- 27.Evans E, Leung A. Adhesivity and rigidity of erythrocyte membrane in relation to wheat germ agglutinin binding. J Cell Biol. 1984;98(4):1201–1208. doi: 10.1083/jcb.98.4.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Williams RJ, Shaw SK. The relationship between cell injury and osmotic volume reduction: II. Red cell lysis correlates with cell volume rather than intracellular salt concentration. Cryobiology. 1980;17(6):530–539. doi: 10.1016/0011-2240(80)90067-x. [DOI] [PubMed] [Google Scholar]
- 29.Lee P, Ye Z, Van Dyke K, Kirk RG. X-ray microanalysis of Plasmodium falciparum and infected red blood cells: Effects of qinghaosu and chloroquine on potassium, sodium, and phosphorus composition. Am J Trop Med Hyg. 1988;39(2):157–165. doi: 10.4269/ajtmh.1988.39.157. [DOI] [PubMed] [Google Scholar]
- 30.White NJ, et al. Spiroindolone KAE609 for falciparum and vivax malaria. N Engl J Med. 2014;371(5):403–410. doi: 10.1056/NEJMoa1315860. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.