

Abstract
Background
Transposable elements (TEs, transposons) are mobile genetic DNA sequences. TEs can insert copies of themselves into new genomic locations and they have the capacity to multiply. Therefore, TEs have been crucial in the shaping of hosts’ current genomes. TEs can be utilized as genetic markers to study population genetic diversity. The rice stem borer Chilo suppressalis Walker is one of the most important insect pests of many subtropical and tropical paddy fields. This insect occurs in all the rice-growing areas in China. This research was carried out in order to find diversity between C. suppressalis field populations and detect the original settlement of C. suppressalis populations based on the piggyBac-like element (PLE). We also aim to provide insights into the evolution of PLEs in C. suppressalis and the phylogeography of C. suppressalis.
Results
Here we identify a new piggyBac-like element (PLE) in the rice stem borer Chilo suppressalis Walker, which is called CsuPLE1.1 (GenBank accession no. JX294476). CsuPLE1.1 is transcriptionally active. Additionally, the CsuPLE1.1 sequence varied slightly between field populations, with polymorphic indels (insertion/deletion) and hyper-variable regions including the identification of the 3′ region outside the open reading frame (ORF). CsuPLE1.1 insertion frequency varied between field populations. Sequences variation was found between CsuPLE1 copies and varied within and among field populations. Twenty-one different insertion sites for CsuPLE1 copies were identified with at least two insertion loci found in all populations.
Conclusions
Our results indicate that the initial invasion of CsuPLE1 into C. suppressalis occurred before C. suppressalis populations spread throughout China, and suggest that C. suppressalis populations have a common ancestor in China. Additionally, the lower reaches of the Yangtze River are probably the original settlement of C. suppressalis in China. Finally, the CsuPLE1 insertion site appears to be a candidate marker for phylogenetic research of C. suppressalis.
Electronic supplementary material
The online version of this article (doi:10.1186/s12867-014-0028-y) contains supplementary material, which is available to authorized users.
Keywords: Transposon, piggyBac, Molecular characterization, Evolution, Chilo suppressalis
Background
Transposable elements (TEs, transposons) are mobile genetic DNA sequences, and they are found in the genomes of nearly all eukaryotes [1,2]. TEs can insert copies of themselves into new genomic locations and they have the capacity to multiply. Therefore, TEs make up a significant portion of the eukaryotic genome and have driven genome evolution in many ways, including gene expression alterations, gene deletions and insertions, chromosome rearrangements and others [3-6]. TEs are divided into two major classes based on their transposition intermediate and distinct structural features [7]. Class I TEs, which are also called retrotransposons, use a “copy-and-paste” mechanism that involves an RNA intermediate. This intermediate is reverse transcribed before its reintegration into a new position. Class II TEs, which are also called DNA transposons, use a DNA-mediated mode of “cut-and-paste” transposition.
The piggyBac element, which is a class II transposon, was originally discovered in the TN-368 cell line of the cabbage looper moth Trichoplusia ni [8,9]. It transposes via a “cut-and-paste” mechanism, inserting exclusively at 5′-TTAA-3′ tetranucleotide target sites and excising with precision, leaving no footprint [10]. Transposons similar to the original functional piggyBac IFP2 called piggyBac-like elements (PLEs) have been found in diverse organisms, including fungi, plants, insects, crustaceans, urochordates, amphibians, fishes and mammals [1,11-15]. PLEs are highly divergent and can be classified into three main classes, namely by high sequence similarity to IFP2, moderate sequence similarity to IFP2 and very distantly related ancient elements [14].
The rice stem borer Chilo suppressalis Walker is one of the most important insect pests of many subtropical and tropical paddy fields in Asia, North Africa and southern Europe. This insect occurs in all the rice-growing areas in China, and it colonizes a wide range of hosts such as rice (Oryza sativa), water-oat (Zizania aquatica) and chufa (Eleocharis tuberosa) [16]. It is assumed that all C. suppressalis field populations in China have a common ancestor. However, there is no clear evidence of this. We want to know if C. suppressalis field populations have a common ancestor, and if so, where this common ancestor originated.
For this paper, we isolated a group of endogenous PLEs from the C. suppressalis genome, which were designated as CsuPLE1s. The CsuPLE1 copy with an intact open reading frame (ORF) was named CsuPLE1.1. The frequency of CsuPLE1.1 insertion at a specific locus in the C. suppressalis genome varied among populations. This study will contribute to our understanding of the distribution and characteristics of the piggyBac family. In addition, the analysis CsuPLE1s sequence variants identified in C. suppressalis from different field populations provides insights into the evolution of CsuPLE1s. Based on the insertion sites and sequence variations of CsuPLE1s, the phylogeography of C. suppressalis is discussed.
Results
Characterization of piggyBac-like element (PLE) in C. suppressalis
A full-length PLE from C. suppressalis was obtained and named CsuPLE1.1 (GenBank accession no. JX294476, Figure 1). This PLE is 2406 bp in length and contains all the characteristic structures of a PLE, including 13 bp inverted terminal repeats (ITRs), asymmetrically located 25 bp sub-terminal inverted repeats and a single open reading frame (ORF) encoding a transposase of 505 amino acids. The putative transposase contains all the aspartate residues of the “DDD” motif, which correspond to D268, D346, D447 and D450 in T. ni IFP2 transposase. As in other PLEs, the CsuPLE1.1 was inserted into typical tetranucleotide target-site TTAA duplications and flanked by a sequence (912 bp at 5′-end and 576 bp at 3′) that was not significantly homologous to any gene sequences in the GenBank. Notably, CsuPLE1.1 also has a putative CAAT site, and a TATA site exists at nt 392–395 and nt 478–481. There is also a polyadenylation signal site at nt 2188–2193, which is characteristic of an actively translated protein. Alignments showed that, among the known PLEs, the putative transposase of CsuPLE1.1 shared the highest similarity (53%) with HsaPGBD3 transposase (Figure 2), and belongs to a class that is moderately similar to IFP2.
Figure 1.

Nucleotide sequence, transcript and putative transposase of the CsuPLE1.1 . The nucleotide sequence in the light gray shadow is the CsuPLE1.1 transcript. The inverted terminal repeats (ITRs) are underlined and italicized. The sub-terminal inverted repeats are double-underlined and italicized. The 4 bp (TTAA) target site duplications (TSDs) are boxed and in bold. Putative CAAT, TATA and AATAAA polyadenylation signal site sequences are underlined and in bold. The primers used in flanking PCR are indicated by left and right arrows below the corresponding sequences. The “DDD-motif” amino acid residues in the putative transposase are indicated by open circles. The bipartite nuclear localization signal (NLS) is boxed and in bold.
Figure 2.
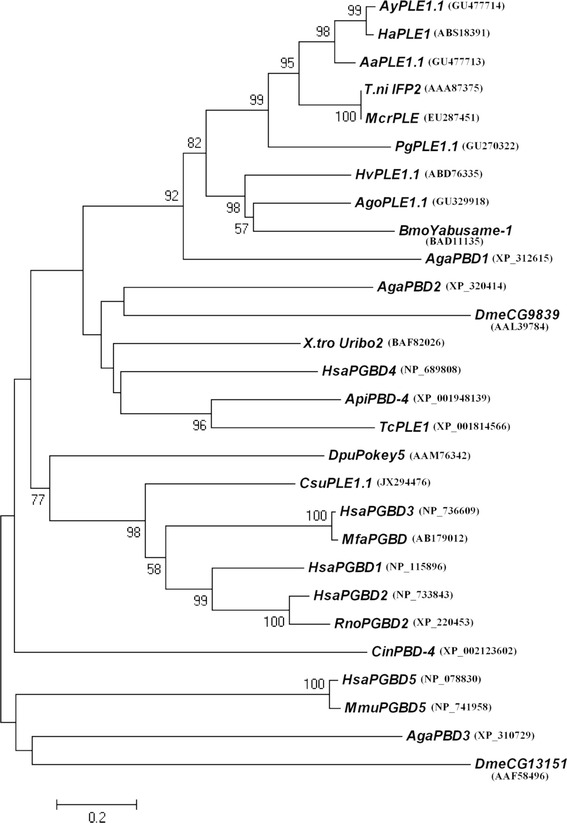
Phylogenetic relationships among piggyBac -like element transposase amino acid sequences. The tree was generated by the neighbor-joining method. The numbers at the nodes are the bootstrap values (>50%) for 5000 replications. The GenBank accession numbers are in brackets. Abbreviations: Aa, Argyrogramma agnata; Aga, Anopheles gambiae; Ago, Aphis gossypii; Api, Acyrthosiphon pisum; Ay, Agrotis ypsilon; Bmo, Bombyx mori; Cin, Ciona intestinalis; Csu, Chilo suppressalis; Dme, Drosophila melanogaster; Dpu, Daphnia pulicaria; Ha, Helicoverpa armigera; Hsa, Homo sapiens; Hv, Heliothis virescens; Mcr, Macdunnoughia crassisigna; Mfa, Macaca fascicularis; Mmu, Mus musculus; Pg, Pectinophora gossypiella; Rno, Rattus norvegicus; Tc, Tribolium castaneum; T.ni, Trichoplusia ni; X.tro, Xenopus tropicalis.
RACE amplification using a cDNA template revealed that CsuPLE1.1 was expressed as a 1748 bp transcript with a 111 bp 5′ untranslated region (UTR) and a 119 bp 3′ UTR containing a 24 bp poly (A) tail (Figure 1).
Insertion site, TSDs and ITR variations
The 5′ TE display showed that the insert sites of CsuPLE1s varied between populations (Additional file 1: Figure S1). Further sequencing results obtained 21 different insertion sites among 72 flanking sequences from 12 field populations (Table 1 and Additional file 2: Table S1). Two insertion sites were found in all populations (insertion sites 1 and 2). One insertion site was found in nine populations (insertion site 8). One insertion site was found in seven populations (insertion site 7). One insertion site was found in five populations (insertion site 3). Two insertion sites were found in four populations (insertion sites 4 and 14). One insertion site was found in three populations (insertion site 9) and the remaining 13 insertion sites were found in only one or two populations. Almost one half of all populations had a unique insert site, found only in that population (Table 1).
Table 1.
5′ insertion sites of CsuPLE1 in C. suppressalis genome
| 5′ Insertion site | Sampling locations | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GY | JJ | XY | SY | JZ | DY | LS | GZL | HX | TC | YJ | YX | |
| Site 1 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ |
| Site 2 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ |
| Site 3 | √ | √ | × | × | × | √ | √ | × | × | × | √ | × |
| Site 4 | √ | √ | × | × | × | × | × | × | √ | × | √ | × |
| Site 5 | √ | × | × | × | × | × | × | × | × | × | × | × |
| Site 6 | √ | × | × | × | × | × | × | × | × | × | × | × |
| Site 7 | √ | × | × | × | × | √ | √ | √ | √ | √ | × | √ |
| Site 8 | √ | √ | √ | √ | √ | √ | × | √ | √ | × | × | √ |
| Site 9 | × | √ | × | × | × | √ | √ | × | × | × | × | × |
| Site 10 | × | × | √ | × | × | × | × | × | × | × | × | × |
| Site 11 | × | × | × | √ | × | × | × | × | × | × | × | × |
| Site 12 | × | × | × | √ | × | × | × | × | × | × | × | × |
| Site 13 | × | × | × | √ | × | × | × | √ | × | × | × | × |
| Site 14 | × | × | × | × | √ | × | √ | × | × | √ | √ | × |
| Site 15 | × | × | × | × | × | √ | × | × | × | × | × | × |
| Site 16 | × | × | × | × | × | × | √ | × | × | × | × | × |
| Site 17 | × | × | × | × | × | × | √ | √ | × | × | × | × |
| Site 18 | × | × | × | × | × | × | × | √ | √ | × | × | × |
| Site 19 | × | × | × | × | × | × | × | × | √ | × | × | × |
| Site 20 | × | × | × | × | × | × | × | × | × | √ | × | × |
| Site 21 | × | × | × | × | × | × | × | × | × | × | √ | × |
The presence and absence of each insertion site within each population is indicated by a √ and × respectively.
Among these 21 different insertion sites, most of the insertions occurred at a TTAA target site, which is characteristic of the TTAA-specific family of piggyBac transposons. Only four target site duplications (TSDs) contained variations. In insertion site 5, the TSD was CTAT; In insertion site 9 and 16, the TSDs were ATAT; In insertion site 10, the TSD was CTAA. The ITR analysis showed that the 13 bp ITRs of CsuPLE1 were conserved in most individuals, with only two ITRs containing slight variations. In insertion site 14, there was a C-A variation in the ITRs; In insertion site 16, there was a G-A variation in the ITRs (Table 2).
Table 2.
The variations of 5′TSDs and 5′ITRs of CsuPLE1 s
| Insertion site | 5′ TSDs | 5′ ITRs |
|---|---|---|
| 1 | TTAA | 5′-CCCAGATTAGCCT |
| 2 | TTAA | 5′-CCCAGATTAGCCT |
| 3 | TTAA | 5′-CCCAGATTAGCCT |
| 4 | TTAA | 5′-CCCAGATTAGCCT |
| 5 | CTAT | 5′-CCCAGATTAGCCT |
| 6 | TTAA | 5′-CCCAGATTAGCCT |
| 7 | TTAA | 5′-CCCAGATTAGCCT |
| 8 | TTAA | 5′-CCCAGATTAGCCT |
| 9 | ATAT | 5′-CCCAGATTAGCCT |
| 10 | CTAA | 5′-CCCAGATTAGCCT |
| 11 | TTAA | 5′-CCCAGATTAGCCT |
| 12 | TTAA | 5′-CCCAGATTAGCCT |
| 13 | TTAA | 5′-CCCAGATTAGCCT |
| 14 | TTAA | 5′-ACCAGATTAGCCT |
| 15 | TTAA | 5′-CCCAGATTAGCCT |
| 16 | ATAT | 5′-CCCAAATTAGCCT |
| 17 | TTAA | 5′-CCCAGATTAGCCT |
| 18 | TTAA | 5′-CCCAGATTAGCCT |
| 19 | TTAA | 5′-CCCAGATTAGCCT |
| 20 | TTAA | 5′-CCCAGATTAGCCT |
| 21 | TTAA | 5′-CCCAGATTAGCCT |
The variant nucleotides are in italics.
CsuPLE1.1 insertion frequency and sequence variations
Flanking PCRs for testing the presence or absence of CsuPLE1.1 insertions were performed on 45 randomly collected individuals from the 21 populations. The frequencies of individuals with the insertion varied between populations (Table 3). From the 945 individuals tested, 384 were heterozygous for the CsuPLE1.1 insertion. All remaining individuals did not have a CsuPLE1.1 insertion.
Table 3.
The frequency of CsuPLE1.1 insertion in each field population
| Rank of CsuPLE1.1 insertion frequency | Sampling locations | Frequency of CsuPLE1.1 insertion (mean ± SEM) |
|---|---|---|
| 1 | YX | 0.8 ± 0.0387 |
| 2 | HX | 0.6667 ± 0.0771 |
| 3 | GZL | 0.578 ± 0.0588 |
| 3 | SY | 0.578 ± 0.089 |
| 5 | GY | 0.511 ± 0.022 |
| 5 | YZ | 0.511 ± 0.097 |
| 7 | TC | 0.489 ± 0.097 |
| 8 | QC | 0.4667 ± 0.0384 |
| 9 | JZ | 0.4443 ± 0.0588 |
| 10 | GX | 0.3557 ± 0.0802 |
| 10 | LH | 0.3557 ± 0.0588 |
| 12 | DY | 0.3333 ± 0.0771 |
| 12 | NC | 0.3333 ± 0.0667 |
| 12 | MH | 0.3333 ± 0.0384 |
| 15 | JJ | 0.311 ± 0.0588 |
| 16 | FN | 0.289 ± 0.0443 |
| 16 | YS | 0.2887 ± 0.0802 |
| 18 | YJ | 0.2667 ± 0.0667 |
| 19 | XY | 0.222 ± 0.0588 |
| 19 | GN | 0.222 ± 0.0588 |
| 21 | LS | 0.1777 ± 0.0447 |
A total of 84 copies of the CsuPLE1.1 insertion were cloned from 84 individuals across the 21 field populations. These sequences share high levels of similarity with the exception of the Flk-PLE1JZ4 copy, which has an approximately 130 bp sequence deletion. There were substitutions, deletions and/or insertions in each copy (Additional file 3: Table S2). Furthermore, in some CsuPLE1.1 copies, there were indels of three or more bases. We defined this position as special variation position (SVP). Six SVPs were identified (Figure 3). SVP1 from copy Flk-PLE1GZL4 has an 11 bp deletions at nt ~170. In SVP3, the copies Flk-PLE1NC2, Flk-PLE1NC3, Flk-PLE1NC4 and Flk-PLE1SY4 have 2 bp deletions and 7 bp insertions at nt ~630. In SVP4, the copies Flk-PLE1JZ3 and Flk-PLE1LS4 have 9 bp deletions at nt ~842. In SVP5, the copies Flk-PLE1GX3, Flk-PLE1YS3 and Flk-PLE1GN4 have 7 bp insertions at nt ~1889. In SVP6, the copies Flk-PLE1YJ2, Flk-PLE1YJ3, Flk-PLE1YJ4 and Flk-PLE1LH4 have 9 bp deletions at nt ~2210. Three bases, ACG, were found in only some PLE1 copies particularly at nt ~298 of SVP2.
Figure 3.
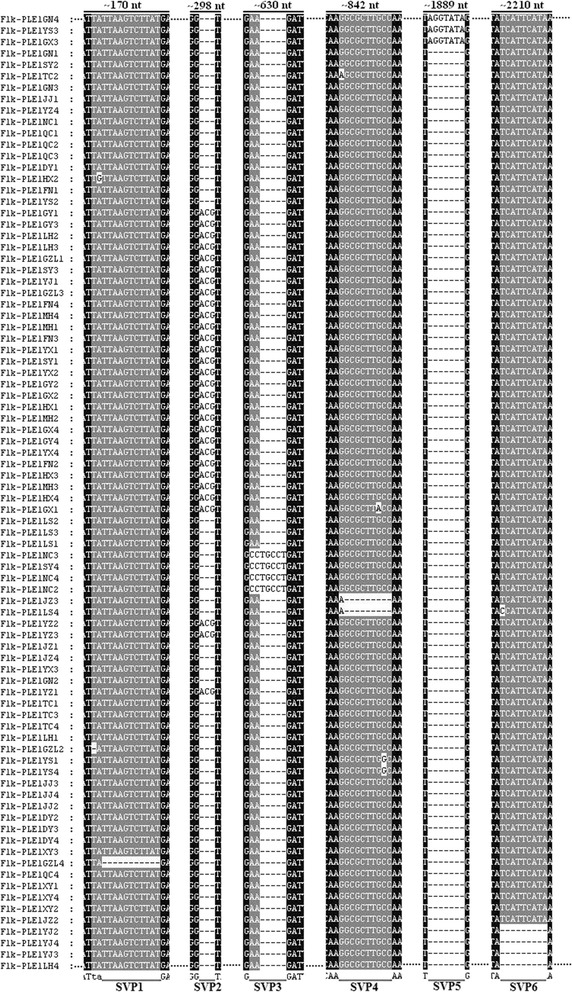
Six special variation positions (SVP). The location of each SVP in the sequence is indicated by Arabic numbers on the top. The omitted sequences are indicated by ellipses. The black short line indicates that there is no base. Flk-PLE1GN is from the Guangning (GN) population. Flk-PLE1YS is from the Yangshuo (YS) population. Flk-PLE1GX is from the Ganxian (GX) population, and so on.
Among the 84 sequences, 63 sequences have a putative intact ORF. These 63 sequences shared a high degree of sequence similarity. While, position nt ~2310, in the 3′ region outside of the ORF, showed the highest nucleotide variability between CsuPLE1.1 copies. This site, which we called the variation hotspot, contained a number of indels (Additional file 4: Figure S2).
The variation rate (Rv) of the CsuPLE1.1 copies differed inside and outside the ORF region. The variation rates inside the ORF region and in the PLE 5′ region outside the ORF were significantly lower than the PLE 3′ outside the ORF region (Table 4).
Table 4.
The variation rate in different areas of CsuPLE1.1
| Area | Length (average ± SEM) | No. of position variances* (average ± SEM) | Variation rate* (average ± SEM) |
|---|---|---|---|
| 5′ outside the ORF region | 613.2 ± 0.3 | 3.8 ± 0.4 | 0.0062 ± 0.0007 B |
| Inside the ORF region | 1518 ± 0 | 8 ± 0.8 | 0.0053 ± 0.0005 B |
| 3′ outside the ORF region | 278 ± 0.1 | 3.1 ± 0.3 | 0.0113 ± 0.0011 A |
*A certain position contains an/a insertion, deletion, transition or transversion was recorded as one variance position. For the variation rate, were significant variations (p < 0.01) between the 3′ outside the ORF region and inside the ORF region, and the 5′ outside the ORF region.
Phylogenetic tree of C. suppressalis field populations
A MP phylogenetic tree and a UPGMA phylogenetic tree were constructed independently. In the MP phylogenetic tree, there were many clusters and the phylogenetic relationship between each was ambiguous (Additional file 5: Figure S3). In the UPGMA tree, there were three clusters: JZ, GY and XY formed one clade; YJ, LS, JJ and DY formed a second clade; and TC, SY, HX, GZL and YX formed a third clade (Additional file 6: Figure S4).
Discussion
Since piggyBac is one of the most popular transposons used for transgenesis, searching for new active PLEs has attracted lots of attention. However, only a few active PLEs have been reported to date, including IFP2, Uribo2, McrPLE and AgoPLE1.1 [15,17-19]. Here we identified another potentially active PLE. This PLE has the intact structure of a piggyBac transposon, including TTAA insertion sites, 13 bp ITRs, 25 bp subterminal inverted repeats, and a single ORF encoding a transposase of 505 amino acids with a perfect “DDD-motif”. The transposase was also shown to be transcriptionally active with a 1748 bp transcript cloned from C. suppressalis.
There was a high degree of sequence similarity in CsuPLE1s from different field populations. All the tested populations shared two identical insertion sites, and each population also had their own unique insert sites. Since all inactive copies of TE will be fixed or lost in the population over time if they are neutral, insertion sites will become more homogeneous in populations over time [20,21]. Our results therefore suggest that a few CsuPLE1 copies in C. suppressalis may be functional and still moving. Investigators have previously hypothesized that if TEs with high sequence similarities could maintain their original structure in their hosts, then the invasion of the TE was a recent event [15,22,23]. The high sequence similarities between CsuPLE1 copies found in this study suggest that the invasion of CsuPLE1 was a recent event.
The transposition activities of intact transposons are often regulated or silenced at the transcriptional, translational, or transpositional level for the survival of transposons and their hosts [6,24-26]. As a result, there are many transposable elements with mutations or variations within their host organisms [27-29]. Sequence mutations may be randomly distributed in transposons. However our results indicate that the 3′ region outside the ORF in the CsuPLE1 transposons sequence had the highest variation rates. A variation hotspot was also found in this 3′ region. Generally, regions with high numbers of mutation are the result of complex cellular processes including (i) interactions between DNA and mutagens, (ii) repair of premutational lesions, (iii) local reduction in the fidelity of DNA polymerization, and (iv) expression and selection of a protein (RNA) molecule from which mutations have been detected [30,31]. This region is complex and deserves further study.
Of the 21 insertion sites found, two occurred in all field populations and five occurred in multiple field populations. Thus, these results imply that CsuPLE1 existed in C. suppressalis prior to the expansion of the insect host populations into new regions. Meng et al. stated that C. suppressalis had strong population structure with three genetic clusters, i.e. a central China (CC) clade, a northern plus northeastern China (NN) clade and a southwestern China (SW) clade [32]. In these three clades, C. suppressalis had arisen from separate refuges and experienced parallel evolution. However, our results suggest that Chinese C. suppressalis populations have a common ancestor. The research of retrotransposon Ty3/gypsy in C. suppressalis shown that one insertion site of Ty3/gypsy existed in all C. suppressalis filed populations [33]. This also supports our conclusion that C. suppressalis populations have a common ancestor in China.
The MP phylogenetic tree showed that many small clusters. This was due to the high sequence similarity. However, in the 84 CsuPLE1.1 copies, there were six SVPs. Based on our results and Meng et al.’s findings [32], we conclude that the C. suppressalis populations of SY, GX, YS and GN belong to the central China (CC) clade; and the C. suppressalis populations in JZ and LS belong to the southwestern China (SW) clade.
In the UPGMA phylogenetic tree, three clades were found. In the first clade, JZ and XY come from similar geographic areas. However, they are far from GY population. In the second clade, LS, JJ and DY come from the same geographic (the Sichuan Basin). YJ belongs to coastal areas, with a similar temperature, humidity and seasonal temperature difference to the Sichuan Basin. In the third clade, TC and HX is close to each other and have similar environmental conditions. However, SY, GZL and YX are far from each other, and have different environments. This result is not entirely consistent with Meng et al.’s result. This may be due to our small sample size or may be because our choice of method reveals a different phylogenetic relationship between C. suppressalis populations. Our research indicated that the insertion sites were a candidate marker for phylogenetic research of C. suppressalis.
Rice is the main host plant for C. suppressalis. Gene flow in C. suppressalis follows a similar pattern to the expansion of rice domestication in China [32]. It has been suggested that the lower reaches of the Yangtze River in China were the first rice farming region, although there are debates about the origin of rice [34-36]. Meng et al. found gene flow of C. suppressalis in CC and SW regions tends to move west. In the CC region this is from Ningbo towards regions such as Quzhou, Nangchang, and in the SW region, this is from Liuzhou towards regions such as Guiyang and Yaan. In the NN region gene flow moves northward from Wuhan or Zhumadian to Changchun [32]. These results together suggest that the lower reaches of the Yangtze River are probably the original settlement of C. suppressalis in China. Our results have shown that the CsuPLE1.1 insertion frequency was the highest in YX populations, located in the lower reaches of the Yangtze River. Moreover, the frequency of CsuPLE1.1 insertions decreases with increasing distance from YX. If the lower reaches of the Yangtze River are the original settlement of C. suppressalis in China, we proposed that the CsuPLE1.1 invasion event initially occurred at the lower reaches of the Yangtze River. As the transposition of transposons can help host organisms adapt, we suggest that the CsuPLE1.1 had more transpositional opportunities as C. suppressalis expanded into new areas and new environments. The CsuPLE1.1 insertion frequencies in Clade A (including GZL, GY, YZ and HX populations) were higher than in Clade B (including YJ, MH and LH populations) (Figure 4 and Table 3). This may be due to fewer C. suppressalis generations each year in Clade A (2 ~ 3 generations per year) compared to Clade B (3 ~ 4 generations per year) and therefore less opportunity for CsuPLE1.1 transposition in Clade A. Another reason for differences in insertion frequency could be differences in environmental stress. For example, the CsuPLE1.1 insertion frequency in FN population was lower than all other nearby populations (Figure 4 and Table 3). In FN, upland rice, which has lower nutrient levels than non-upland rice, was planted. Our previous research showed that the average individual body weight of C. suppressalis in FN was lighter than other field population [37]. Also, winters in FN are colder and drier than other nearby populations. C. suppressalis in FN therefore faces greater challenges to survive and such stress potentially provides more transpotition opportunity for the CsuPLE1.1 in this population.
Figure 4.
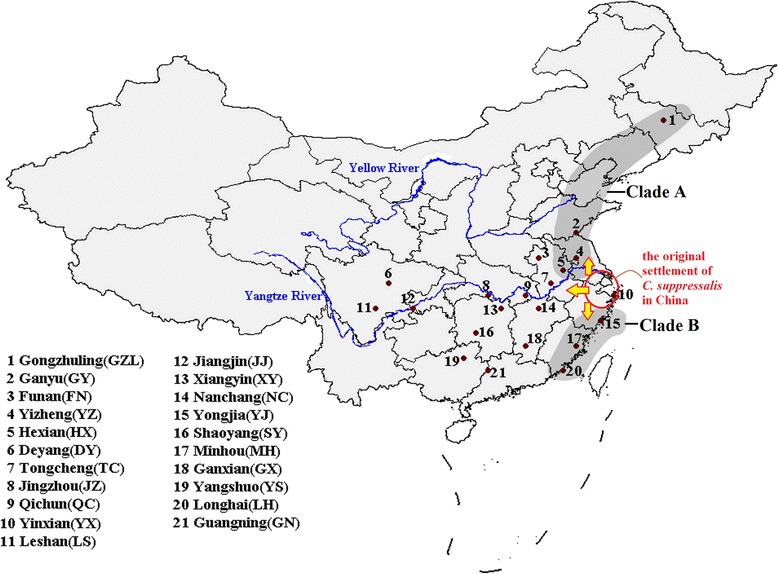
Sampling locations and potential migration directions of C. suppressalis . The letters in the brackets are the location abbreviations. The putative original settlement of C. suppressalis in China is indicated by red circle. The putative migration directions of C. suppressalis are indicated by yellow arrows.
Conclusions
C. suppressalis occurs in all rice-growing areas in China, and they are non-migratory insects. Based on our results, we suggest that C. suppressalis populations have a common ancestor in China. The initial invasion of CsuPLE1 in C. suppressalis occurred before C. suppressalis populations spread throughout China, and the invasion of CsuPLE1 transposons was a recent event. Additionally, the lower reaches of the Yangtze River are probably the original settlement of C. suppressalis in China. Moreover, the insertion sites of CsuPLE1s should be a candidate marker for the phylogenetic research of C. suppressalis.
Methods
Sample collection and DNA isolation
The C. suppressalis samples were collected from 21 paddy rice field locations in China (Figure 4 and Additional file 7: Table S3), and were kept at −80°C until DNA extraction. Forty-five individual samples were randomly picked from each field population, and genomic DNA (gDNA) was prepared using an AxyPrep DNA Extraction Kit (Axygen Biosciences, Hangzhou, China) by following the protocol provided by the manufacturer.
PCR amplification and sequence analysis
In order to obtain intact sequences of PLEs from C. suppressalis, the transcriptome of C. suppressalis was surveyed and a putative PLE fragment with the longest sequence (about 1700 bp) was found. Based on this sequence, one pair of specific primers was designed (SPF1: 5′-TGTATTCTACCTCCTCCTGTTG-3′; SPR1: 5′- AAAACACTTGACACACACTCCA-3′). Using these specific primers, a 1616 bp fragment of CsuPLE1 was amplified from Hexian (HX) individual samples by PCR. The purpose of this PCR was to verify the PLE fragment sequence originating from the transcriptome of C. suppressalis. The PCR was performed using LA Taq polymerase (TaKaRa Biotechnology, Dalian, China) with the following protocol: 95°C for 3 min; 94°C for 30 s, after which the annealing temperature of the reaction was decreased by 1°C for 30 s every cycle, from 60°C to 50°C, followed by 72°C for 1 min 40 s; 26 cycles of 94°C for 30 s, 53°C for 30 s, and 72°C for 1 min 40 s; and final elongation at 72°C for 10 min. The final PCR volume was 25 μl containing approximately 50 ng gDNA, 0.2 mM of each dNTP, 1.5 mM of Mg2+, 0.2 μM of each primer, 2.5 μl of 10 × LA PCR buffer (Mg2+ free) and 0.25 μl (5 U/μl) of LA Taq polymerase (TaKaRa).
Based on this 1616 bp fragment, two pairs of nested primers for inverse PCR were designed. Inverse PCR was then performed on HX individuals with these two pairs of nested primers to obtain the full-length CsuPLE1. The following two pairs of primers were used for the nested inverse PCR: external primer pair (IPS1: 5′-TTGGCTTTCGCAGAAGGGTA-3′ and IPA1: 5′-CGTTTTCTCCATGCGTAGGTA-3′); Internal primer pair (IPS2: 5′-CATGCTGATAGTCGTAAAGACCA-3′ and IPA2: 5′-ATCACTCACCATAATCTGCCCT-3′). The inverse PCR was performed using LA Taq polymerase (TaKaRa) with the following protocol: 95°C for 3 min; 94°C for 30 s, after which the annealing temperature of the reaction was decreased by 1°C for 30 s every cycle, from 63°C to 53°C, followed by 72°C for 4 min; 25 cycles of 94°C for 30 s, 53°C for 30 s, and 72°C for 4 min; and final elongation at 72°C for 10 min. The final PCR volume is 25 μl containing approximately 50 ng gDNA, 0.2 mM of each dNTP, 1.5 mM of Mg2+, 0.2 μM of each primer, 2.5 μl of 10 × LA PCR buffer (Mg2+ free) and 0.25 μl (5 U/μl) of LA Taq polymerase (TaKaRa).
All PCR products were purified with an AxyPrep DNA Gel Extraction Kit (Axygen) and directly cloned into the pGEM-T Easy vector (Promega, Madison, WI, USA), and three clones were sequenced by GenScript Biotechnology Co., Ltd. Nanjing, China. The sequencing results were compared with non-redundant databases in the NCBI server using BLASTX and TBLASTX (http://blast.ncbi.nlm.nih.gov/Blast.cgi). CLUSTAL X1.8 [38] was used to align the putative transposase sequence of CsuPLE1 to 27 PLEs with full-length transposases from other species sourced from Genbank. Phylogenetic analysis of the different PLE transposases was then performed on the aligned sequences in MEGA version 4 using the neighbor-joining method [39].
RNA extraction and first-strand cDNA synthesis for RT-PCR
Chilo suppressalis individuals from our laboratory strain were chosen at random for total RNA extraction. One forth-instar larvae was stored at −80°C for subsequent RNA extraction. Total RNA was isolated using a Promega SV Total RNA Isolation system (Promega) using the manufacturer’s protocol.
Approximately 1 μg of total RNA was used as a template for first-strand cDNA synthesis with the PrimeScript RT reagent kit (TaKaRa) in a 20 μl reaction. Reactions were conducted at 37°C for 15 min, followed by 85°C for 5 s, and stopped by cooling on ice for 5 min.
Determination of CsuPLE1 transcript by RACE
In order to determine the sequence of the intact transcript of CsuPLE1, 5′- and 3′-RACE was conducted using the SMART RACE cDNA Amplification Kit (Clontech, Mountain View, CA, USA) using the manufacturer’s protocol. The first-strand 5′-RACE-ready cDNA and 3′-RACE-ready cDNA was synthesized from 1 μg of total RNA using SMARTScribe Reverse Transcriptase (Clontech). The synthesized first-stand 5′-RACE-ready cDNA was used as a template to amplify the 5′ end of CsuPLE1 cDNA using the Universal Primer A Mix (UPM) and the Nested Universal Primer A (NUP) with the two CsuPLE1-specific reverse primers (5RACE01: 5′ CTCCAGGCAAATTCTTTTTGACAGCA-3′ and 5RACE02: 5′- TCAAACCACTCAATTGGGCGCTTATC-3′). The first round of PCR was performed using LA Taq polymerase (TaKaRa) with the following protocol: 95°C for 3 min; 94°C for 30 s, after which the annealing temperature of the reaction was decreased by 2°C for 30 s every cycle, from 65°C to 55°C, followed by 72°C for 1 min; 25 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 1 min; and final elongation at 72°C for 10 min. The primers used were UPM and 5RACE01. The final PCR volume was 25 μl containing approximately 50 ng cDNA, 0.2 mM of each dNTP, 1.5 mM of Mg2+, 0.2 μM of each primer, 2.5 μl of 10 × LA PCR buffer (Mg2+ free) and 0.25 μl (5 U/μl) of LA Taq polymerase (TaKaRa). The second round of PCR, also LA Taq polymerase (TaKaRa), was performed on 1 μl of the first round PCR product. The PCR conditions were: 95°C for 3 min; 30 cycles of 94°C for 30 s, 66°C for 30 s, and 72°C for 1 min; and final elongation at 72°C for 10 min. The primers used were NUP and 5RACE02. The final PCR volume was 25 μl and concentrations of all other reagents within the reaction were the same as the first round of PCR. Likewise, the synthesized first-stand 3′-RACE-ready cDNA was employed as a template to amplify the 3′ end of CsuPLE1 cDNA using UPM and NUP and the two CsuPLE1-specific forward primers (3RACE01: 5′-GAAATGGTACATGCCGTTGCTCACAC-3′ and 3RACE02: 5′-TCATCTTGGCTTTCGCAGAAGGGTAG-3′). The conditions of these two rounds of PCR were the same as those for the amplication of the 5′ end of CsuPLE1 cDNA, respectively. All the PCR products were subcloned into the pGEM-T Easy vector (Promega) and three clones were sequenced as described above.
Vectorette PCR for TE-display and UPGMA tree construction
Vectorette PCR was used to isolate the CsuPLE1’s flanking sequences and to examine insertion site diversity. Vectorette PCR was performed as previously described [40]. Two anchoring bubble linker oligonucleotides were designed to make the vectorette unit for ligation to the Hind III digested gDNA.
The vectorette unit was prepared as described in Ko et al. [40]. For this analysis, eight individuals were randomly selected from each of 12 field populations. Approximately 1 μg of gDNA was digested at 37°C for 6 h using Hind III (NEB, Ipswich, MA, USA) in a 20 μl reaction. The digested gDNA was ligated with the vectorette unit using T4 DNA ligase (NEB). Two rounds of nested PCR with two pairs of primers were then carried out using the following primers: VPCR1: 5′-CCCTTCTCGAATCGTAACCG-3′ (vectorette external primer), VPCR2: 5′-CGTAACCGTTCGGTCCTCTG-3′ (vectorette internal primer), VP5R1: 5′-TGCCATGTCTGCAACGCACT-3′ (5′ specific external primer), VP5R2: 5′-AGCTGACACGTTTCTTACTGC-3′ (5′ specific internal primer). The Vectorette PCR was performed in a 10 μl reaction volume containing approximately 25 ng gDNA, 0.2 mM of each dNTP, 1.5 mM of Mg2+, 0.2 μM of each primer, 2.5 μl of 10 × LA PCR buffer (Mg2+ free) and 0.1 μl (5 U/μl) of LA Taq polymerase (TaKaRa). These two rounds of Vectorette PCR amplification conditions were 3 min at 95°C for initial denaturation, then 94°C for 30 s, after which the annealing temperature of the reaction was decreased by 1°C for 1 min every cycle, from 65°C to 51°C, followed by 72°C for 2 min 30 s; then 23 cycles of 94°C for 30 s, 52°C for 1 min, and 72°C for 2 min 30 s; and final elongation at 72°C for 10 min. For the second PCR, 1 μl of 100-fold diluted first round PCR product was used as the template. All the PCR products were visualised on a 2% agarose gel with ethidium bromide (EB) staining. To obtain the flanking sequences of CsuPLE1 in C. suppressalis genome, the vectorette PCR products were cloned and sequenced as described above.
Based on the 5′ insertion sites of CsuPLE1, a 0 (no insertion) / 1 (with insertion) binary matrix was constructed. Genetic similarities (GS) (Additional file 8: Table S4) between pairs of field populations were measured as GS(ij) = 2a/(2a + b + c) where a is the number of co-existed insertion sites in both samples, b is the number of presence insertion sites in i but absent in j, and c is the number of presence insertion sites in j but absent in i [41,42]. Genetic similarities were used to construct a UPGMA phylogenetic tree in Phylip version 3.695 [43].
The CsuPLE1.1 insertion frequency, sequence variations and MP tree construction
Inverse PCR identified approximately 1480 bp of flanking sequence from the putative intact copy of CsuPLE1.1. The absence or presence of CsuPLE1.1 in the insertion site (corresponding to the 5′ insertion site 2 in Table 1) in individual C. suppressalis was examined by flanking PCR with the primer pairs Flk-F (5′-TAACTAAGGTTCGCTGATGAC-3′) and Flk-R (5′-GATGCGCCTATCTATTTCG-3′). These primers flank the insertion site. For this analysis, a total of 945 individuals were randomly selected from 21 field populations. For each field populations, 45 individuals were selected (three groups, 15 individuals in each group). The absence of CsuPLE1.1 at the insertion site was indicated by a 264 bp amplicon, and its presence was indicated by an approximately 2670 bp amplicon. The flanking PCR amplification conditions were as follows: initial denaturing at 94°C for 3 min, followed by 30 cycles of 94°C for 30 s, 55°C for 30 s, 72°C for 3 min, and a final extension at 72°C for 10 min. The PCR products were run on a 1.5% agarose gel with EB staining to detect the presence or absence of the CsuPLE1.1 insertion in each individual. The resulting PCR products were purified, cloned and sequenced as described above.
To compare nucleotide variation inside and outside ORF regions, the CsuPLE1.1 transposon sequence was divided into three parts: (i) the PLE 5′ outside the ORF region, (ii) inside the ORF region (from the initiator codon to the termination codon) and (iii) the PLE 3′ outside the ORF region. Four individuals containing the CsuPLE1.1 copy in each of the 21 field populations were randomly selected from those samples mentioned in this section above and the CsuPLE1.1 copy was purified, cloned and sequenced as described above. The four CsuPLE1.1 copies were aligned independently for each of the 21 field populations using CLUSTAL X1.8. All nucleotide variation, including indels and single nucleotide polymorphisms, were scored as a single variant. The number of variants (Nv) and the length of each part (L) were used to calculate the variation rate (Rv) where Rv = Nv/L.
To examine variation in CsuPLE1.1 sequences between field populations, all 84 CsuPLE1.1 copies (4 CsuPLE1.1 copies from 21 populations) were aligned using CLUSTAL X1.8, A phylogenetic tree was generated using Maximum Parsimony in MEGA 4.
Acknowledgements
We are grateful to Dr. Kostas D. Mathiopoulos for helpful suggestions for improving this manuscript. This work was supported by National Rice Industry Technology System Project grant Cars-001-25, National Key Technology R&D Program grant 2012BAD19B03, National Natural Science Foundation of China grant 31201505 and 31101435, and Jiangsu Agriculture Science and Technology Innovation Fund grant CX(13)3038.
Abbreviations
- TE
Transposable element
- PLE
piggyBac-like element
- ITRs
Inverted terminal repeats
- ORF
Open reading frame
- TSDs
Target site duplications
- NLS
Nuclear localization signal
- UPGMA
Unweighted pair group method analysis
Additional files
Gel analysis of the 5′ Vectorette PCR of CsuPLE1s in 21 populations.
5′ insertion sites and flanking sequences of CsuPLE1 in C. suppressalis.
The viorations in CsuPLE1.1 copies.
{kind=link}
The Variation hotspot in CsuPLE1.1 copies.
Phylogenetic tree constructed based on multiple sequence alignments of CsuPLE1.1 copies.
Phylogenetic tree constructed based on the 5′ insertion sites of CsuPLE1s.
Longitude and latitude of sampling locations.
The genetic similarities between pairs of field populations.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
G-HL and X-HL performed most of the experiments together. G-HL and J-CF conceived and designed the study together. G-HL, Z-CZ and B-SL collected the insect samples together. G-HL, Z-JH, H-FG and J-CF wrote the paper together. All authors analysed the data. All authors read and approved the final manuscript.
Contributor Information
Guang-Hua Luo, Email: luogh_cn@163.com.
Xiao-Huan Li, Email: xhli2012@163.com.
Zhao-Jun Han, Email: zjhan@njau.edu.cn.
Hui-Fang Guo, Email: guohf@jaas.ac.cn.
Qiong Yang, Email: mlyangqiong@163.com.
Min Wu, Email: wumin_au@163.com.
Zhi-Chun Zhang, Email: zczhangjs@163.com.
Bao-Sheng Liu, Email: liubsnj@126.com.
Lu Qian, Email: kinglune@126.com.
Ji-Chao Fang, Email: fangjc@jaas.ac.cn.
References
- 1.Sarkar A, Sim C, Hong YS, Hogan JR, Fraser MJ, Robertson HM, Collins FH. Molecular evolutionary analysis of the widespread piggyBac transposon family and related “domesticated” sequences. Mol Genet Genomics. 2003;270(2):173–180. doi: 10.1007/s00438-003-0909-0. [DOI] [PubMed] [Google Scholar]
- 2.Feschotte C, Pritham EJ. DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet. 2007;41:331–368. doi: 10.1146/annurev.genet.40.110405.090448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Langley CH, Montgomery E, Hudson R, Kaplan N, Charlesworth B. On the role of unequal exchange in the containment of transposable element copy number. Genet Res. 1988;52(3):223–235. doi: 10.1017/S0016672300027695. [DOI] [PubMed] [Google Scholar]
- 4.Kazazian HH., Jr Mobile elements: drivers of genome evolution. Science. 2004;303(5664):1626–1632. doi: 10.1126/science.1089670. [DOI] [PubMed] [Google Scholar]
- 5.Oliver KR, Greene WK. Transposable elements: powerful facilitators of evolution. Bioessays. 2009;31(7):703–714. doi: 10.1002/bies.200800219. [DOI] [PubMed] [Google Scholar]
- 6.Feschotte C. Transposable elements and the evolution of regulatory networks. Nat Rev Genet. 2008;9(5):397–405. doi: 10.1038/nrg2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Finnegan DJ. Transposable elements. Curr Opin Genet Dev. 1992;2(6):861–867. doi: 10.1016/S0959-437X(05)80108-X. [DOI] [PubMed] [Google Scholar]
- 8.Fraser MJ, Smith GE, Summers MD. Acquisition of host cell DNA sequences by baculoviruses: relationship between host DNA Insertions and FP mutants of autographa californica and galleria mellonella nuclear polyhedrosis viruses. J Virol. 1983;47(2):287–300. doi: 10.1128/jvi.47.2.287-300.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cary LC, Goebel M, Corsaro BG, Wang HG, Rosen E, Fraser MJ. Transposon mutagenesis of baculoviruses: analysis of Trichoplusia ni transposon IFP2 insertions within the FP-locus of nuclear polyhedrosis viruses. Virology. 1989;172(1):156–169. doi: 10.1016/0042-6822(89)90117-7. [DOI] [PubMed] [Google Scholar]
- 10.Fraser MJ, Ciszczon T, Elick T, Bauser C. Precise excision of TTAA-specific lepidopteran transposons piggyBac (IFP2) and tagalong (TFP3) from the baculovirus genome in cell lines from two species of Lepidoptera. Insect Mol Biol. 1996;5(2):141–151. doi: 10.1111/j.1365-2583.1996.tb00048.x. [DOI] [PubMed] [Google Scholar]
- 11.Zimowska GJ, Handler AM. Highly conserved piggyBac elements in noctuid species of Lepidoptera. Insect Biochem Mol Biol. 2006;36(5):421–428. doi: 10.1016/j.ibmb.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 12.Xu HF, Xia QY, Liu C, Cheng TC, Zhao P, Duan J, Zha XF, Liu SP. Identification and characterization of piggyBac-like elements in the genome of domesticated silkworm, Bombyx mori. Mol Genet Genomics. 2006;276(1):31–40. doi: 10.1007/s00438-006-0124-x. [DOI] [PubMed] [Google Scholar]
- 13.Wang J, Miller ED, Simmons GS, Miller TA, Tabashnik BE, Park Y. piggyBac-like elements in the pink bollworm, Pectinophora gossypiella. Insect Mol Biol. 2010;19(2):177–184. doi: 10.1111/j.1365-2583.2009.00964.x. [DOI] [PubMed] [Google Scholar]
- 14.Wu M, Sun Z, Luo G, Hu C, Zhang W, Han Z. Cloning and characterization of piggyBac-like elements in lepidopteran insects. Genetica. 2011;139(1):149–154. doi: 10.1007/s10709-010-9542-0. [DOI] [PubMed] [Google Scholar]
- 15.Luo GH, Wu M, Wang XF, Zhang W, Han ZJ. A new active piggyBac-like element in Aphis gossypii. Insect Sci. 2011;18(6):652–662. doi: 10.1111/j.1744-7917.2011.01406.x. [DOI] [Google Scholar]
- 16.Sheng C, Wang H, Sheng S, Gao L, Xuan W. Pest status and loss assessment of crop damage caused by the rice borers, Chilo suppressalis and Tryporyza incertulas in China. Entomol Knowl. 2003;40(4):289–294. [Google Scholar]
- 17.Ding S, Wu X, Li G, Han M, Zhuang Y, Xu T. Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell. 2005;122(3):473–483. doi: 10.1016/j.cell.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 18.Hikosaka A, Kobayashi T, Saito Y, Kawahara A. Evolution of the Xenopus piggyBac transposon family TxpB: domesticated and untamed strategies of transposon subfamilies. Mol Biol Evol. 2007;24(12):2648–2656. doi: 10.1093/molbev/msm191. [DOI] [PubMed] [Google Scholar]
- 19.Wu M, Sun ZC, Hu CL, Zhang GF, Han ZJ. An active piggyBac-like element in Macdunnoughia crassisigna. Insect Sci. 2008;15(6):521–528. doi: 10.1111/j.1744-7917.2008.00241.x. [DOI] [Google Scholar]
- 20.Deceliere G, Charles S, Biemont C. The dynamics of transposable elements in structured populations. Genetics. 2005;169(1):467–474. doi: 10.1534/genetics.104.032243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J, Ren X, Miller TA, Park Y. piggyBac-like elements in the tobacco budworm, Heliothis virescens (Fabricius) Insect Mol Biol. 2006;15(4):435–443. doi: 10.1111/j.1365-2583.2006.00653.x. [DOI] [PubMed] [Google Scholar]
- 22.Garcia Guerreiro MP, Fontdevila A. The evolutionary history of Drosophila buzzatii. XXXVI. Molecular structural analysis of Osvaldo retrotransposon insertions in colonizing populations unveils drift effects in founder events. Genetics. 2007;175(1):301–310. doi: 10.1534/genetics.106.064378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bui QT, Delauriere L, Casse N, Nicolas V, Laulier M, Chenais B. Molecular characterization and phylogenetic position of a new mariner-like element in the coastal crab, Pachygrapsus marmoratus. Gene. 2007;396(2):248–256. doi: 10.1016/j.gene.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 24.Lippman Z, May B, Yordan C, Singer T, Martienssen R. Distinct mechanisms determine transposon inheritance and methylation via small interfering RNA and histone modification. PLoS Biol. 2003;1(3):E67. doi: 10.1371/journal.pbio.0000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tran RK, Zilberman D, de Bustos C, Ditt RF, Henikoff JG, Lindroth AM, Delrow J, Boyle T, Kwong S, Bryson TD, Jacobsen SE, Henikoff S. Chromatin and siRNA pathways cooperate to maintain DNA methylation of small transposable elements in Arabidopsis. Genome Biol. 2005;6(11):R90. doi: 10.1186/gb-2005-6-11-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Castro JP, Carareto CM. Drosophila melanogaster P transposable elements: mechanisms of transposition and regulation. Genetica. 2004;121(2):107–118. doi: 10.1023/B:GENE.0000040382.48039.a2. [DOI] [PubMed] [Google Scholar]
- 27.Wang J, Du Y, Wang S, Brown SJ, Park Y. Large diversity of the piggyBac-like elements in the genome of Tribolium castaneum. Insect Biochem Mol Biol. 2008;38(4):490–498. doi: 10.1016/j.ibmb.2007.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Osanai-Futahashi M, Suetsugu Y, Mita K, Fujiwara H. Genome-wide screening and characterization of transposable elements and their distribution analysis in the silkworm, Bombyx mori. Insect Biochem Mol Biol. 2008;38(12):1046–1057. doi: 10.1016/j.ibmb.2008.05.012. [DOI] [PubMed] [Google Scholar]
- 29.Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA, Minx P, Reily AD, Courtney L, Kruchowski SS, Tomlinson C, Strong C, Delehaunty K, Fronick C, Courtney B, Rock SM, Belter E, Du F, Kim K, Abbott RM, Cotton M, Levy A, Marchetto P, Ochoa K, Jackson SM, Gillam B, et al. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326(5956):1112–1115. doi: 10.1126/science.1178534. [DOI] [PubMed] [Google Scholar]
- 30.Rogozin I, Kondrashov F, Glazko G. Use of mutation spectra analysis software. Hum Mutat. 2001;17(2):83–102. doi: 10.1002/1098-1004(200102)17:2<83::AID-HUMU1>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 31.Rogozin IB, Pavlov YI. Theoretical analysis of mutation hotspots and their DNA sequence context specificity. Mutat Res. 2003;544(1):65–85. doi: 10.1016/S1383-5742(03)00032-2. [DOI] [PubMed] [Google Scholar]
- 32.Meng XF, Shi M, Chen XX. Population genetic structure of Chilo suppressalis (Walker) (Lepidoptera: Crambidae): strong subdivision in China inferred from microsatellite markers and mtDNA gene sequences. Mol Ecol. 2008;17(12):2880–2897. doi: 10.1111/j.1365-294X.2008.03792.x. [DOI] [PubMed] [Google Scholar]
- 33.Li XH, Luo GH, Zhang ZC, Liu BS, Fang JC. Coling and characterization of Ty3/gypsy retrotransposon in Chilo suppressalis (Lepidoptera: Pyralidae) Chin J Rice Sci. 2014;28(3):314–321. [Google Scholar]
- 34.Wang W-M, Ding J-L, Shu J-W, Chen W. Exploration of early rice farming in China. Quat Int. 2010;227(1):22–28. doi: 10.1016/j.quaint.2010.06.007. [DOI] [Google Scholar]
- 35.Molina J, Sikora M, Garud N, Flowers JM, Rubinstein S, Reynolds A, Huang P, Jackson S, Schaal BA, Bustamante CD, Boyko AR, Purugganan MD. Molecular evidence for a single evolutionary origin of domesticated rice. Proc Natl Acad Sci U S A. 2011;108(20):8351–8356. doi: 10.1073/pnas.1104686108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crawford G. Early rice exploitation in the lower Yangzi valley: what are we missing? The Holocene. 2012;22(6):613–621. doi: 10.1177/0959683611424177. [DOI] [Google Scholar]
- 37.Luo GH, Zhang ZC, Han GJ, Han ZJ, Fang JC. Characteristics of overwintering populations of rice stem borers and mutation frequencies of resistance to triazophos. Chin J Rice Sci. 2012;26(4):481–486. [Google Scholar]
- 38.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25(24):4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24(8):1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 40.Ko WY, David RM, Akashi H. Molecular phylogeny of the Drosophila melanogaster species subgroup. J Mol Evol. 2003;57(5):562–573. doi: 10.1007/s00239-003-2510-x. [DOI] [PubMed] [Google Scholar]
- 41.Dice LR. Measures of the amount of ecologic association between species. Ecology. 1945;26(3):297–302. doi: 10.2307/1932409. [DOI] [Google Scholar]
- 42.Nei M, Li WH. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci U S A. 1979;76(10):5269–5273. doi: 10.1073/pnas.76.10.5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Felsenstein J. PHYLIP-phylogeny inference package (version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]