

Abstract
Proinflammatory mediators trigger intensive postischemic inflammatory remodeling of the blood–brain barrier (BBB) including extensive brain endothelial cell surface and junctional complex changes. Junctional adhesion molecule-A (JAM-A) is a component of the brain endothelial junctional complex with dual roles: paracellular route occlusion and regulating leukocyte docking and migration. The current study examined the contribution of JAM-A to the regulation of leukocyte (neutrophils and monocytes/macrophages) infiltration and the postischemic inflammatory response in brain ischemia/reperfusion (I/R injury). Brain I/R injury was induced by transient middle cerebral artery occlusion (MCAO) for 30 min in mice followed by reperfusion for 0–5 days, during which time JAM-A antagonist peptide (JAM-Ap) was administered. The peptide, which inhibits JAM-A/leukocyte interaction by blocking the interaction of the C2 domain of JAM-A with LFA on neutrophils and monocytes/macrophages, attenuated I/R-induced neutrophil and monocyte infiltration into brain parenchyma. Consequently, mice treated with JAM-A peptide during reperfusion had reduced expression (~3-fold) of inflammatory mediators in the ischemic penumbra, reduced infarct size (94 ± 39 vs 211 ± 38 mm3) and significantly improved neurological score. BBB hyperpermeability was also reduced. Collectively, these results indicate that JAM-A has a prominent role in regulating leukocyte infiltration after brain I/R injury and could be a new target in limiting post-ischemic inflammation.
Keywords: Tight junctions, JAM-A, Stroke, Inflammation, Blood–brain barrier
Introduction
Inflammation contributes to secondary brain injury after ischemia. Inflammation initiation and progression involve a complex set of interactions between brain endothelial cells and leukocytes, predominantly neutrophils and monocytes/macrophages, allowing first neutrophils and then monocytes to adhere to endothelial cells, traverse the blood–brain barrier (BBB) and eventually migrate into the injured brain tissue (Becker, 2001; del Zoppo et al., 1991; Garcia et al., 1994; Jander et al., 2007). Infiltrating neutrophils and monocytes/macrophages further amplify the inflammatory reaction and exacerbate brain injury (Becker, 2001; Jander et al., 2007). Post-ischemic inflammation is considered a good target for potential therapy, with a relatively long duration (hours or several days) providing a longer treatment window (Becker, 2001; Garcia et al., 1994; Tuttolomondo et al., 2009).
Perturbations in BBB integrity, with subsequent increased vascular permeability and leukocyte infiltration, play a pivotal role in post-ischemic inflammation (Fernandez-Lopez et al., 2012; Liu et al., 2012; Yang and Rosenberg, 2011). During this process, most tight junction (TJ) complex components, particularly the transmembrane proteins occludin and claudin-5, undergo redistribution from the membrane cell surface (Dimitrijevic et al., 2006; Fernandez-Lopez et al., 2012; Jiao et al., 2011; Liu et al., 2012; Yang and Rosenberg, 2011). In this way, a paracellular route is opened and leukocyte transmigration enhanced. However, leukocyte migration into brain parenchyma also requires chemotactic signals and close interaction with the brain endothelial lateral surface, where the specific adhesion molecules (e.g. CD31, CD99 and junction adhesion molecules) control leukocyte movement (Mamdouh et al., 2009; Schenkel et al., 2002).
Junctional adhesion molecules (JAMs) are members of the immunoglobulin superfamily. In general, JAMs are expressed at cell-to-cell junctions in endothelia and epithelia and display different patterns of homotypic and heterotypic adhesion (Martin-Padura et al., 1998; Sobocki et al., 2006; Weber et al., 2007). JAMs also have an important role in regulating leukocyte/endothelial cell interactions controlling monocyte, neutrophil or lymphocyte recruitment in several models of inflammation (Nourshargh et al., 2006; Weber et al., 2007; Wojcikiewicz et al., 2009; Woodfin et al., 2009). For example, JAM-A and JAM-C are upregulated on atherosclerotic endothelium and in unstable plaques (Azari et al., 2010; Babinska et al., 2007; Cavusoglu et al., 2007; Manetti et al., 2013). JAM-A is also an important player in ischemia/reperfusion (I/R) injury in the heart, liver and kidney, particularly in post-ischemic inflammation and monocyte recruitment (Corada et al., 2005; Khandoga et al., 2005; Lakshmi et al., 2012; Nourshargh et al., 2006). On the one hand, JAM-C expression is more extensive in tissues with chronic inflammatory diseases, such as asthma, bronchitis, interstitial nephritis, autoimmune hepatitis and alcoholic cirrhosis; on the other hand, JAM-B expression is correlated with monocyte and lymphocyte recruitment (Johnson-Leger et al., 2002; Liang et al., 2002; Manetti et al., 2013; Weber et al., 2007). JAM expression and function may be regulated by a variety of inflammatory mediators, but there is limited evidence on this issue. Regarding the CNS, there are limited data on JAM expression. JAM-A has a role in perivascular neutrophil recruitment in cytokine-induced meningitis, with a blocking anti-JAM-A antibody significantly reducing leukocyte recruitment (Del Maschio et al., 1999). However, in bacteria-induced meningitis, the contribution of JAM-A is minor, indicating that JAM-A function and expression could depend on the type of inflammation, inducing factors and tissue (Lechner et al., 2000). There is still no compelling evidence about the role of JAM-A in brain I/R injury.
The present study addresses the role of JAM-A in regulating brain post-ischemic inflammation and defines whether JAM-A may be a useful target for anti-inflammatory therapy after stroke.
Methods
Mouse brain microvascular endothelial cells
Mouse brain microvascular endothelial cells (mBMECs) were prepared from 6 to 8 weeks old CD1 mice (Charles River, Portage, MI) using a modified protocol already described (Dimitrijevic et al., 2006; Stamatovic et al., 2009; Stamatovic et al., 2012). Briefly, microvessels isolated from cerebral cortex were digested in HBSS solution containing 1 μg/ml collagenase/dispase (Roche, Indianapolis, IN), 10 U/ml DNase I (Sigma-Aldrich, St Louis, MO) and 1 μg/ml Na-p-tosyl-l-lysine chloromethyl ketone (TLCK) for 20 min at 37 °C and purified/precipitated with CD31 coated magnetic beads (Dynabeads, Life Technologies, Grand Island, NY). These vessels were cultured in Dulbecco’s Modified Eagles medium (DMEM) supplemented with 10% inactivated fetal calf serum, 2.5 μg/ml heparin (Sigma-Aldrich), 20 mM HEPES, 2 mM glutamine, 1× antibiotic/antimycotic (all from Life Technologies), and endothelial cell growth supplement (BD Bioscience, San Jose, CA) and grown in 6 well plates coated with collagen type IV (BD Bioscience). This protocol typically produces primary endothelial cell cultures that are approximately 99% pure (as determined by immunocytochemistry with anti-PECAM-1 antibody; BD Bioscience). mBMEC in the first passage was used in all experiments.
In vitro OGD/reoxygenation injury
mBMECs were subjected to oxygen-glucose deprivation (OGD) injury. Injury was initiated by placing cultures in an anaerobic chamber (O2 level 0–5 ppm; Coy Laboratory, Great Lake MI) in the presence of OGD media (DMEM glucose free solution purged with an anaerobic gas mixture, 5% CO2/95%N2 and 10% H2, to remove residual oxygen) for 5 h at 37 °C. This was denoted as the ischemic condition. Then, cultures were removed from the chamber, medium exchanged with oxygenated DMEM, and cells placed in an incubator at 37 °C, with 5% CO2-containing normal oxygen (21% O2) to mimic reperfusion for 0 to 48 h. This was denoted as the reoxygenation condition. For every set of experiments, viability assays were performed (Lactate Dehydrogenase (LDH) Assay, Promega WI). Only cell cultures with cell viability of ~95% after OGD exposure, were used in experiments (Andjelkovic et al., 2003; Dimitrijevic et al., 2006).
Transient middle cerebral artery occlusion in mice
Experiments were performed on male 10–12 weeks old C57BL/6 mice (Jackson Laboratory, Bar Harbor, MA). Mice were anesthetized with ketamine and xylazine (100 and 10 mg/kg; i.p.). Body temperature was maintained at 37 ± 0.5 °C by heating blanket and heating lamp during the entire experimental procedure. Focal cerebral ischemia was induced by left middle cerebral artery occlusion (MCAO) using an intraluminal filament technique (Dimitrijevic et al., 2007). Briefly, the common carotid artery was exposed through a midline neck incision. Next, a 6-0-silicon suture was introduced into the external carotid artery and advanced into the internal carotid artery a distance of ~9 mm from the common carotid artery bifurcation according to animal weight. MCAO was confirmed by a Laser Doppler Flow(LDF) probe (Model BPM System, Vasomedics, St. Paul, MN) positioned 3mmposterior and 5mm lateral to bregma. After 30 min MCAO, mice were reperfused by suture withdrawal and allowed to awaken from anesthesia. Sham-operated animals underwent all procedures except the occlusion. Physiological parameters (pO2, pCO2, pH, blood glucose, cerebral blood flow rCBF) were monitored before, during and after MCAO (Table 1). Reperfusion periods were 1–10 days.
Table 1.
Physiological variables of mice before, during and 6 hrs after tMCAO.
| Variables | before n = 10 | 15 min MCAO n = 10 | 6 hr post –tMCAO n = 10 |
|---|---|---|---|
| ip physiological saline injection | |||
| pH | 7.37±0.02 | 7.33±0.01 | 7.35±0.02 |
| PCO2 | 33±0.6 | 34±1 | 35±1 |
| PO2 | 122±3 | 109±4 | 118±7 |
| Glucose mg/dL | 136±6 | 145±4 | 130±7 |
| rCBF (%) | 100±0 | 14±1 | 99±1 |
| JAM-Ap ip injection | |||
| pH | 7.36±0.03 | 7.32±0.012 | 7.36±0.018 |
| PCO2 | 32±2 | 34±2 | 36±1 |
| PO2 | 123±5 | 110±7 | 115±10 |
| Glucose g/dL | 135±3 | 142±6 | 128±9 |
| rCBF (%) | 100±0 | 13±2 | 100±1 |
During reperfusion, neurological deficit was evaluated with the following scoring scheme by a blinded investigator: 0, no deficits; 1, flexion of the torso and contralateral forelimb when lifted by the tail; 2, contralateral forelimb weakness upon application of pressure to the side of the body; 3, circling to the affected side; and 4, no spontaneous locomotor activity. The duration of reperfusion was chosen based on our evaluation of physiological parameters as well as the survival rate after transient MCAO.
Morphometric measurement of infarct volume
Animals were sacrificed 1, 3 and 5 days after MCAO, the brain removed and sliced. Slices were incubated in 2% 2,3,5-triphenyltetrazolium chloride (TTC; Sigma Aldrich) solution for 1 h at 37 °C. The area of infarction in each slice was determined by computerized image analysis system and infarct volume calculated by multiplying the distance between sections. In addition, to account for cerebral edema or infarct resolution, an indirect measurement of infarction was performed. Infarct volume was calculated as (contralateral hemisphere volume – (ipsilateral hemisphere volume – measured injury volume)) (16). Cresyl violet staining of 200 μm thick serial sections was also used to examine infarct size after 1, 3 and 5 days of reperfusion.
Peptide treatment and binding
JAM-A peptides (P1-ELVFDPLSASDTGEYSCEAR; P2-TVNIPSSATGNRAVLTCSE, P3-NGYGTPMTSNAVR; P4_-NPKSTRAFSNDDYVLNPTTG; P5-QDGSPPSEYTWFKDGIV-MPT) were synthesized by New England Peptide (Gardner, MA) using solid-phase synthesis by FMOC chemistry. Peptide purity was assessed by high-performance liquid chromatography, and molecular mass confirmed by mass spectrometry. Control scrambled peptide (cp), which contains the same amino acids as JAM-A peptide was also synthesized. The mice were randomly assigned to control peptide (cp) and JAM-A inhibitory peptide (JAM-Ap) groups. Male C57BL/6 mice were treated with peptide for 5 consecutive days during reperfusion, starting before and 6 h after MCAO. JAM-A peptide (1 μg) or control scrambled peptide (1 μg) was dissolved in sterile 0.9% NaCl and injected i.p. or iv.
To determine the distribution and binding, JAM-Ap and control scrambled peptide (cp) were labeled using the Lightning-Link® Rapid Fluorescein kit (Innova Bioscience) according tomanufacturer’s instruction. JAM-Ap and cp (1 μg) were injected iv or i.p. and 6 and 24 h later the brain and blood were collected. Brain slides were analyzed for fluorescent labeling in tissue and blood vessels using confocal microscopy. Blood samples were analyzed for JAM-Ap-FITC and control peptide-FITC (cp-FITC) using flow cytometry. The analysis also included the total number of neutrophils and monocytes in JAM-Ap and cp treated groups.
Saturation, competitive and specificity binding analyses
The binding affinity of JAM-Ap on brain endothelial cells was examined by 1) JAM-A saturation binding curves with labeled JAM-Ap-FITC, and 2) competition curves with unlabeled JAM-Ap competing for binding with JAM-Ap-FITC. mBMEC was plated on 96-well plates and for saturation binding experiments and determining EC50 values, cells were incubated with the labeled JAM-Ap-FITC at the range of concentrations of 0.1–1000 μg (final volume of 200 μl) in Dulbecco’s phosphate buffered saline (D-PBS, Life Science technology) at 4 °C for 30 min. For competitive binding assay, the cells were incubated with constant concentration of JAM-Ap-FITC with increased concentration of unlabeled JAM-Ap. After incubation, cells were washed 3 times and fixed with 4% paraformaldehyde for 15 min at room temperature. Fluorescence intensity was detected on a fluorescent reader (Infinity FL200). The fluorescence intensity values or percentage of inhibition was plotted against concentration of JAM-Ap-FITC and EC50 (saturation binding curves) or IC50 (inhibition binding curves) was determined from the graphs using GraphPad Prism software.
The specificity of JAM-A-FITC binding to JAM-A on brain endothelial cells was tested. We previously generated the JAM-A knockdown (JAMA-KD) cells by stable transfection of JAM-A shRNA in bEnd.3 cells (ATCC, Manassas, VA) with JAM-A shRNA incorporated in vector pGFP-V-RS (OriGene, Rockville, MD) (Stamatovic et al., 2012). These cells have a >C98% reduction in JAM-A mRNA and protein expression. Both bEnd.3 JAM-A-KD cells and mBMEC with JAM-A expression were plated in the 96 well plates and exposed to inflammatory condition either IL1β and TNF-α for 6 h or OGD/reoxygenation for 24 h in DMEM media. After that incubation media was removed and bEnd.3 JAM-A-KD cells or mBMECs were treated with JAM-Ap-FITC with or without the increasing concentration of ICAM-1 antibody (0.1–20 μg/ml, R&DSystem) in D-PBS for 30 min at 4 °C. Cells were then washed 3 times with D-PBS and fixed with 4% paraformaldehyde for 15 min at room temperature. The fluorescence intensity was detected on a fluorescent reader (Infinity FL200). The additional control groups included cells treated with cp-FITC under the same experimental condition.
Cell treatment
For in vitro experiments, JAM-A peptide was dissolved in culture media (DMEM) and applied to mBMEC in two experimental conditions at the beginning of reperfusion time and after 12 h reperfusion. Cell viability assay (LiveDeath assay, Life Technologies) indicated >99% viability after treatment with JAM-A peptide.
For other treatments, cells were exposed to either CCL2 (100 ng/ml), a cocktail of IL-1β/TNFα (10 ng/ml each) (all from PeproTech, Rocky Hill, NJ) or media only (negative control) for 0–120 min. A matrix metalloprotease inhibitor, GM6001 (100 ng/ml, EMP Millipore, Billerica, MA) or cycloheximide (5 μg/ml, Sigma Aldrich) was introduced at the beginning of reperfusion and cells exposed for 24 h. Control cells were exposed to assay media (DMEM) without inhibitors. Cell viability assays were performed to exclude possible toxic effects of inhibitors and only cells with 98% viability were used in experiments. The effects of treatment and inhibitor were evaluated by Western blot.
Adhesion and migration assay
Mouse blood (~1.5 ml) was collected by cardiac puncture from CD1 mice (Charles River Laboratory). Neutrophils were prepared as described previously (Yuan and Fleming, 1990). Briefly, blood was centrifuged to separate white blood cells from erythrocytes (white buffy coat), followed by separation of neutrophils on a Percoll density gradient. Formonocytes, we used thioglycollate-elicitedmØ from the peritoneal cavity as a surrogate mononuclear leukocyte source (Cohn, 1974). Neutrophil or monocyte purity was evaluated by Diff–Quik staining solution (Fisher Scientific, Pittsburgh, PA) and viability tested by trypan blue exclusion. Purity was >98% for neutrophils and >95% for monocytes/macrophages, with small portion (1–3%) of thrombocytes. Viability was >99%. Isolated monocytes/macrophages or neutrophils were labeled with Calcein-AM (4 μM; Life Technologies) for 30 min at 37 °C.
mBMEC was plated and grown to confluence on collagen type IV-coated filters in a Transwell dual chamber system. They were then exposed to OGD followed by reoxygenation. As a negative control, cells were exposed to assay media only. Calcein-AM labeled polymorphonuclear leukocytes (PMNs) and monocytes/macrophages (104) were then added to the upper Transwell chamber at 0, 6, 12, and 24 h of reoxygenation and left for 1 h at 37 °C. After medium removal and washing cells once, the total number of adherent cells on the mBMEC surface as well as the number of migrated cells on the bottom of the Transwells chambers were evaluated by florescent reader (Infinity FL200, Tecan Group Ltd, Männedorf, Switzerland). The number of cells was calculated from a standard curve.
To neutralize JAM-A activity, either JAM-A neutralizing antibody (10 μg/ml, R&D System, Minneapolis, MN) or antagonist peptide (1 μg/ml, New England Peptide, Gardner, MA) was added 1 h prior to the addition of PMN or monocytes and present during the adhesion/migration assay. A mismatch control peptide or isotypic control for anti-JAM-A antibodies, IgG2a (R&D System) was used as control groups. Cell adhesion was also blocked by neutralizing anti-ICAM-1 antibody (10 μg/ml, R&D System) in a control study. In addition, to confirm JAM-A involvement in leukocyte adhesion, assays were performed on JAM-A knockdown (KD) cells prepared after stable transfection of immortalized brain endothelial cells (bEnd.3) with JAM-A shRNA (pGFP-V-RS-JAMAshRNA). The selected concentration of antibodies (anti-JAM-A and -ICAM-1) was chosen based on a dose dependency curve for both antibodies. The concentration of 10 μg/ml was the lowest dose that could reduce the leukocyte adhesion and migration on brain endothelial cells by >50%.
In vitro permeability assay
The permeability of mBMEC monolayers was measured as described in Kazakoff et al. (1995) and modified in our laboratory (Dimitrijevic et al., 2006; Stamatovic et al., 2009; Stamatovic et al., 2012). The permeability coefficient (PC; cm/min) of monolayers was calculated for the tracer FITC-inulin or dextran-Texas red (1 μg/ml, Sigma-Aldrich) at different reperfusion time points with/without JAM-A peptide. FITC-inulin and dextran-Texas Red concentrations were evaluated on a fluorescent reader (Infinity FL200).
In vivo permeability assay
BBB integrity of mice was assessed by determining the transfer coefficient (Ki) for FITC-inulin (5 kDa) and dextran-Texas red (40 kDa) as described previously (Dimitrijevic et al., 2007). Briefly, dextran-Texas red and FITC-inulin were injected as bolus into a femoral vein 20 min prior to the end of the experiment. Serial arterial blood samples were taken from a femoral artery every 5 min from 0 to 20 min to determine the dextran-Texas red and FITC-inulin plasma profile. At the end of the experiment, mice were killed by decapitation. Brains were rapidly removed, cleaned of meninges and dissected into right and left hemispheres. These were weighed and homogenized in 50 mM Tris buffer solution (pH 7.4). The homogenates were centrifuged 3000 rpm for 30 min and the supernatant collected. Methanol was added to the collected supernatant (1:1) and the mixture centrifuged again under the same conditions. The fluorescence intensity of the supernatant and plasma samples was measured with a fluorescent reader (Tecam; emission 485 and excitation 540 nm) and concentration calculated using a standard curve. The Ki for dextran-Texas Red and FITC-inulin was determined using an equation developed by Ohno et al. (1980).
where Cbr is the concentration in brain tissue at the time of decapitation (ng/g), Cbl is the concentration in the last blood sample, Vo is the regional blood volume (ml/g), ∫ Cpl · dt is the integral of the arterial concentration over time t. Vo was determined in a second set of animals where mice were sacrificed 1 min after intravenous injection of Dextran-Texas red and FITC-Inulin. Assuming no extravasation during this short circulation time, Vo = Cbr / Cbl·. Vo, ∫ Cpl and Cbl were determined for sham control, JAM-Ap treated and non-treated mice and there were no significant inter-group differences.
Real-time PCR
Samples of mBMEC and isolated microvessels were collected at the end of experiments. In vivo, the brain area (penumbra) around the ischemic lesion was collected using a “pinch-out” method where the visible ischemic lesion was removed and the surrounding area (1mmfrom the lesion) sampled. The corresponding area in the contralateral hemisphere and sham-operated mice was collected as control (Dimitrijevic et al., 2007). For isolated microvessels, brain tissue was mechanically dissociated and homogenized in Dounce type of homogenizer. After washing with Hanks balanced solution, the myelin and erythrocytes were cleaned by 18% Dextran solution and Percol gradient, respectively. Total RNAwas prepared using TRIZOL (Life Technologies). Single strand cDNA from2 μg total RNA was synthesized using RT2 first strand kit and Real-Time PCR performed using SABioscience JAM-A primers (QIAGEN Inc., Valencia, CA).
Immunofluorescence
Samples were fixed in 4% paraformaldehyde and then preincubated in blocking solution containing 5% normal goat serum and 0.05% Tween in PBS. Samples were then incubated with primary antibodies, anti-JAM-A (R&D System), anti-claudin-5-FITC, occludin-FITC, ZO-1-FITC, Alexa Fluor 594 (Life Technologies), anti-CD31 (BD Bioscience), anti-myeloperoxidase antibody (Novus Biologicals) or anti-ED1 antibody (Abcam) overnight at 4 °C. Reactions were visualized by fluorescein-conjugated anti-mouse and/or anti-rabbit antibodies. All samples were viewed on a confocal laser scanning microscope (LSM 510 Zeiss, Germany).
Cell fractionation and Western blotting of tight junction proteins
Cell fractionation and Western blot analysis were performed on mBMEC exposed to OGD-reoxygenation condition in vitro, as well as isolated penumbral microvessels. Cell fractionation was performed using a ProteoExtract Subcellular Proteome Extraction kit (Calbiochem, La Jolla, CA). Membrane, cytosolic, cytoskeletal and nuclear fractions were separated. Fraction specificity was confirmed using anti-cytochrome P450 reductase (membrane fraction), anti-calpain (cytosolic fraction) and anti-vimentin (actin cytoskeletal fraction) antibodies. For “total cell lysate”, cells were washed in PBS, scraped and rinsed in 1 ml of lysis buffer (25 mM Tris–HCl pH 7.4 with 150 mM NaCl, 0.1% SDS, 1% Triton X-100 and 1% deoxycholate).
Western blotting was performed with the following antibodies: mouse anti-JAM-A (R&D System) anti-claudin-5, anti-occludin and anti-ZO-1 (Life Technologies). Immunoblots were exposed to secondary anti-mouse or rabbit-HRP conjugated antibody, visualized with a chemiluminescent HRP substrate kit and analyzed using Image J software (NIH, Bethesda, MD).
Flow cytometry
Mice were anesthetized and then perfused via the ascending aorta with saline containing 2 U/ml heparin to remove vascular blood, PMNs and monocytes/macrophages. Brains were rapidly isolated, meninges was removed and divided into ischemic and non-ischemic hemispheres and washed in ice-cold-PBS. Hemispheres were mechanically dissociated and sieved through a 40-μm pore size mesh to generate single-cell suspensions. Isolated cells were placed in media (RPMI 1640, 10% FCS), pelleted and resuspended in isotonic 30% Percoll (Amersham Pharmacia, Piscataway, NJ) and spun at 1300 × g for 30 min at 4 °C, to separate myelin debris. Cell viability was tested by trypan blue exclusion. Cells were further processed for flow cytometry using the following antibodies: anti-mouse leukocyte common antigen (LCA; CD45-FITC, BD Bioscience), anti-mouse Ly-6G (Ly-6G-PerCP-Cy5.5, BD Bioscience), anti-mouse CD11b (CD11b-PeCy7), and anti-mouse F4/80 (F4/80-FITC, eBioscience, San Diego, CA) and corresponding isotype control IgG2aκ. Data were analyzed with a FACSCalibur using CellQuest software (BD Immunocytometry Systems, Mountain View, CA).
Cytokine antibody array
A Mouse Cytokine Antibody Array 3 kit (RayBiotech Inc., Atlanta, GA) was used to simultaneously detect and semi-quantify 62 cytokines in samples collected under both in vitro and in vivo conditions. For tissues, samples were homogenized in 1.8 ml Tris buffer solution (TBS, pH 8.5) supplemented with Triton X-100 at a final concentration of 1% and stirred for 12 h at 4 °C. Samples were centrifuged 100,000 g for 60 min at 4 °C to remove cell debris. Supernatant was collected and protein levels evaluated by BCA protein assay (Thermo Fisher Scientific, Rockford IL) and adjusted to 2 μg/ml. The cytokine antibody array was performed according to the manufacturer’s instructions. Membranes were developed with the Pierce ECL substrate kit (Thermo Fisher Scientific) and underwent densitometric analysis using ImageJ software. The relative level of inflammatory cytokines was evaluated using software provided by the manufacturer. In addition, IL-1β, IL-1α, IL-6, IL-12, INF-γ, TNF-α, CCL5, CCL2, CCL3, CXCL12 and CCL11 protein levels were quantified by ELISA assay (QIAGEN Inc.).
Cell-based ELISA
mBMEC monolayers were exposed to OGD for 5 h followed by 0–48 h of reperfusion. Cells were then washed with PBS and incubated with goat anti-mouse JAM-A antibody in PBS/0.1% BSA, for 1 h at 4 °C with occasional shaking. They were then washed with PBS/0.1% BSA followed by incubation with HRP-conjugated secondary antibody for 30 min at room temperature. After washing with PBS/0.1% BSA buffer, cells were fixed in 4% paraformaldehyde for 20 min and washed with PBS. For detection, equal parts of substrate reagents hydrogen peroxide and 3,3′,5,5′-tetramethylbenzidine solution (Sigma-Aldrich) were added and incubated in the dark at room temperature for 15 min while shaking. The color reaction was stopped by the addition of 1 N HCL. Absorbance was measured at 450 nm using a TECAN microplate reader. To adjust for cell number, wells were washed with H2O and stained with 0.08% crystal violet in PBS for 5 min. The nuclear stain was solubilized with 33% acetic acid and optical density measured at 595 nm. To assess potential JAM-A internalization during exposure to anti-JAM-A antibody, mBMEC was treated with primary antibody, fixed with 4% paraformaldehyde, permeabilized with 0.05% Triton in PBS and exposed to HRP-conjugated secondary antibody followed by visualization with hydrogen peroxide and 3,3′,5,5′-tetramethylbenzidine solution. Samples were compared with those without fixation and permeabilization. Controls were untreated cells (Stamatovic et al., 2012).
Statistical analysis
Unpaired Student’s t-test and one-way analysis of variance (ANOVA), with Bonferroni post hoc tests were used to test group level differences, Chi-squared tests (neurological scores) and Kaplan–Meier survival curves were used (Prism analysis software). A probability value < 0.05 was regarded as statistically significant. EC50 and IC50 were determined by GraphPad Prism software using the nonlinear regression curve fits.
Results
Brain I/R injury and JAM-A relocalization
Ischemia/reperfusion injury elicits a series of events at the BBB which, in the early phase, cause BBB disruption and TJ complex degradation, while the later phase is associated with inflammatory processes and cytokine-induced BBB changes (Dimitrijevic et al., 2006; Fernandez-Lopez et al., 2012). Alterations in BBB permeability are manifested as modifications in TJ protein immunostaining in penumbral vessels. For most TJ proteins, such as claudin-5, occludin and ZO-1, there is a very specific fragmentation or loss of staining in vessels after MCAO compared to the corresponding area in sham-operated mice or contralateral hemisphere (Fig. 1A). In contrast, JAM-A showed a different pattern of expression, with intense staining in penumbral vessels (Fig. 1A).
Fig. 1.
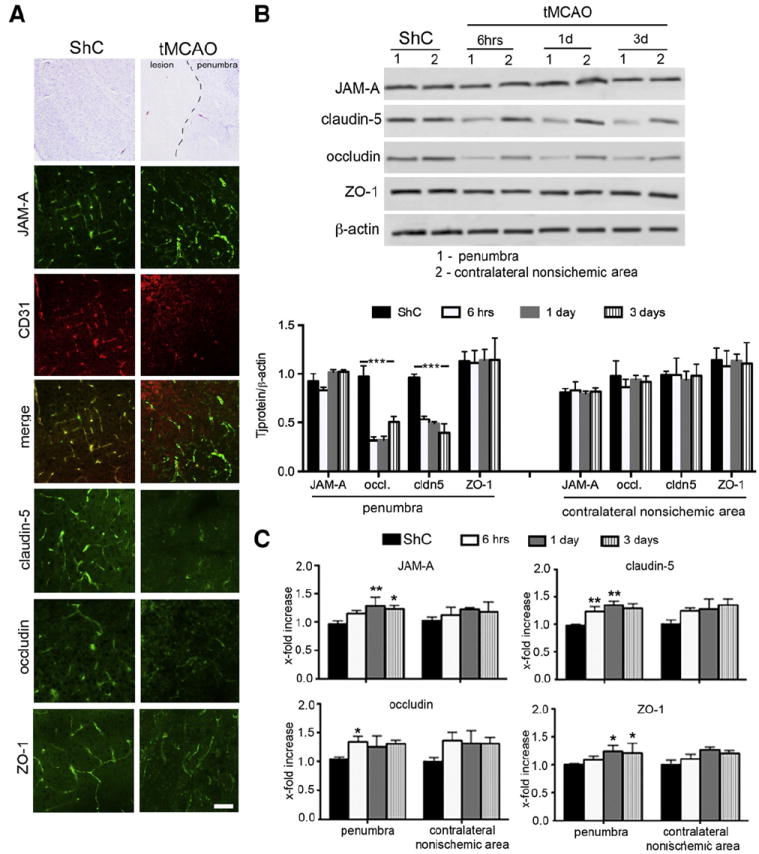
A) JAM-A expression was examined by immunohistochemistry in brains of mice subjected to transient middle cerebral artery occlusion (MCAO, 30 min occlusion) at 3 days of reperfusion or sham controls (ShC). JAM-A + staining was mostly present in CD31 + blood vessels, with continuous staining along vessels in both control and I/R brains. This was contrary to immunohistochemistry for other TJ proteins, claudin-5, occludin and ZO-1, which showed fragmented staining along vessels in the area surrounding lesion (penumbra) after MCAO. Scale bar 20 μm. B) Western blot semiquantitative densitometric analysis of TJ protein expression in isolated penumbral blood vessels after MCAO and corresponding area in the non-ischemic contralateral hemisphere and sham controls. Notice the decreased expression of occludin and claudin-5 while JAM-A showed slightly increased expression after MCAO. Graph data represent means ± SD, n=5; ***p < 0.001. Western blotting image is one of five independent experiments. C) Real time RT-PCR analysis of TJ protein mRNA expression in isolated penumbral blood vessels after MCAO compared to contralateral hemisphere and sham controls (ShC). There was sight elevation of JAM-A, but also claudin-5, occludin and ZO-1 expression compared to ShC, particularly at day 3 of reperfusion. Graph data represent means ± SD, n=5; **p < 0.01, *p < 0.05.
Analysis of isolated penumbral microvessels showed that this specific pattern of immunostaining is associated with significantly decreased claudin-5 and occludin protein expression (p < 0.001), but slightly increased expression of JAM-A during reperfusion (Fig. 1B). In contrast, mRNA of all three proteins showed a small increase during reperfusion compared with the corresponding area in the contralateral non-ischemic hemisphere (Fig. 1C).
In a previous study, we also found a “different behavior” of JAM-A compared to other TJ proteins in response to a chemokine, CCL2 (Stamatovic et al., 2012). In that study, during CCL2-induced redistribution of transmembrane TJ proteins, JAM-A became relocalized from the lateral to the apical membrane of brain endothelial cells and gained a new role as a leukocyte adhesion molecule (Stamatovic et al., 2012). Accordingly, we further examined JAM-A redistribution during OGD/reoxygenation injury in vitro and I/R injury in vivo. Under control conditions, JAM-A and other TJ proteins (e.g. claudin-5) are localized at the cell–cell border of mBMEC and it is characterized as continuous staining. Immunostaining and confocal 3D image analysis of mBMEC exposed to 5 h OGD followed by 30 min or 24 h reoxygenation showed JAM-A redistribution, mostly to the apical membrane, with intense punctate staining. In contrast, at 24 h, there was rare punctate staining for claudin-5 and not on the apical membrane (Fig. 2A). These alterations in TJ proteins were associated with decreased mBMEC monolayer TEER values. At 30 min, TEERs were 10 ± 2 Ω · cm2 vs 118 ± 9 Ω · cm2, ~85% drop from control values) and 61 ± 7 Ω · cm2 at 24 h (~52% drop from control).
Fig. 2.
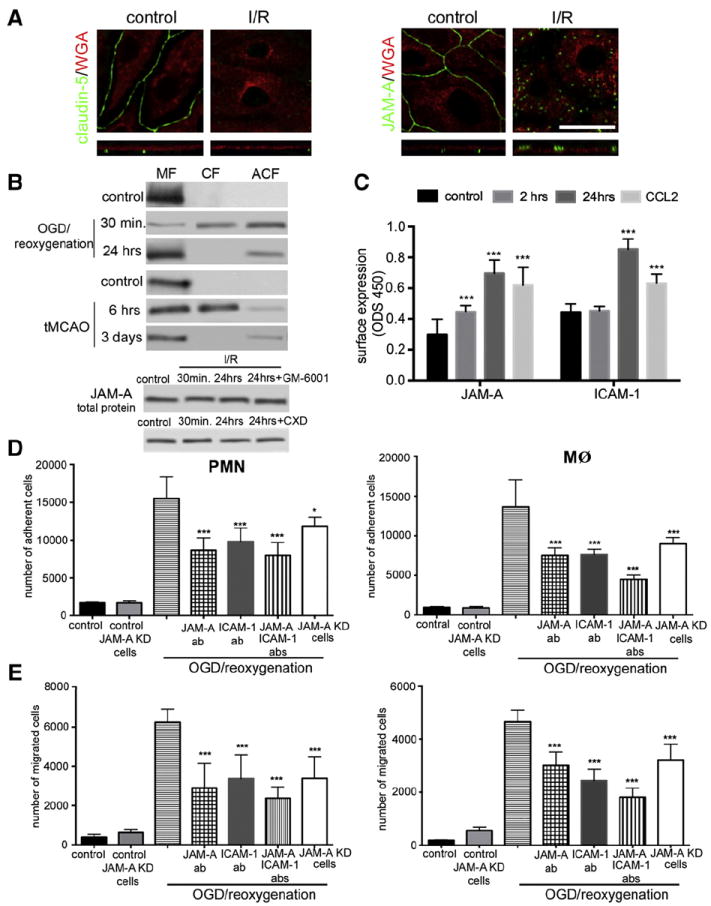
A) Immunostaining with either anti-JAM-A or claudin-5 antibodies supported by optical z-section and 3D-analysis of mBMEC monolayers exposed to 5 h OGD followed by 24 h reoxygenation. Cell membranes were labeled with Alexa496 conjugated WGA. Notice the localization of JAM-A on the apical side at 24 h of reoxygenation. Top and bottom columns show projections of x–y planes and z sections, respectively. Scale bar 50 μm. B) Cell fractionation and Western blot analysis of JAM-A in mBMEC and isolated penumbral blood vessels during reperfusion showed redistribution of JAM-A from membrane to cytosol fraction at early time points, 30 min (mBMEC) and 6 h (blood vessels), and return to membrane fraction 24 h later (in vitro) or 3 days (in vivo). MF, CF and ACF indicate membrane, cytosolic and actin cytoskeletal cell fractions, respectively. The total amount of JAM-A protein was not changed by in vitro I/R, even in the presence of a matrix metalloproteinase inhibitor (GM-6001, 100 ng/ml) or cycloheximide (5 μg/ml). Representative images of Western blot of three independent experiments. C) Cell based ELISA to examine JAM-A surface expression upon exposure to OGD (5 h) followed by reoxygenation for 2 and 24 h or exposure to CCL2 (100 ng/ml) for 2 h. After exposure, each sample was incubated with anti-JAM-A antibody followed by HRP conjugated secondary antibody and fixation. Notice the increased apical membrane JAM-A expression after OGD and CCL2 exposure. Similar experiments were performed to examine ICAM-1 surface expression during reoxygenation and CCL2 treatment. ICM-1 surface expression was also increased by these treatments. Data represent means ± SD for n = 3 independent experiments. ***p < 0.001 compared with control non-treated cells. D) and E) Freshly prepared PMN and macrophages were labeled with Calcein-AM and layered on top of mBMEC monolayers previously exposed to OGD for 5 h followed by 24 h reoxygenation. At the beginning of reperfusion cells were treated with neutralizing anti-JAMA antibody (JAM-A-Ab, 10 μg/ml) or anti-ICAM-1 antibody (ICAM-1Ab, 10 μg/ml) or a cocktail of these antibodies. As a control, we also used JAM-A knockdown (KD) cells, generated by stable transfection of bEnd.3 cells with JAM-A shRNA. Cells were incubated for 2 h and then the media with non-adherent cells were removed and the sample washed, fixed and fluorescence determined on a microplate reader. OGD/reoxygenation condition increased the number of adherent (D) and migrated (E) neutrophils (PMN) and macrophages (MØ) and that was blocked by treatment with JAM-A antibody or if JAM-A KD cells were used. Data represent averages ± SD for n= 5 independent experiments. ***Indicates p < 0.001 vs. OGD/reoxygenation alone.
Under control conditions, cell fractionation/Western blot analysis of mBMEC and isolated penumbral blood vessels also indicated that JAMA-A is found in the cell membrane fraction, but after 30 min (in vitro) or 6 h (in vivo) of reperfusion, it was found in the cytosolic (CF) and actin cytoskeletal (ACF) fractions. By 24 h (in vitro) and 3 days (in vivo) of reperfusion, JAM-A is predominantly localized in the membrane and actin cytoskeletal fractions (Fig. 2B). In vitro, the total amount of JAM-A protein was not changed by OGD/reoxygenation injury and adding an inhibitor of matrix metalloproteinase (GM-6001) as well as an inhibitor of protein synthesis, cycloheximide, which can modulate the TJ protein levels during ischemic injury (Yang et al., 2013), did not affect JAM-A expression (Fig. 2B). Thus, during OGD/reoxygenation injury, JAM-A has a redistribution pattern similar to that found in inflammatory conditions (treatment with CCL2 or lipopolysaccharide) (Stamatovic et al., 2012) which could, based on the observed time, be linked to the proinflammatory response at brain endothelial cells during OGD/reoxygenation injury.
The apical relocalization of JAM-A during OGD/reoxygenation in vitro was further established by cell-based ELISA assay (Fig. 2C). Exposure of mBMEC monolayers to either OGD/reoxygenation or CCL2 for 2 or 24 h caused an increase in cell surface (apical) expression of JAM-A (2 and 24 h; p < 0.001). A similar degree of surface accumulation was found for another adhesion molecule, ICAM-1, known to be mostly present on the apical surface of endothelial cells (Fig. 2C).
CCL2-induced JAM-A relocalization was associated with a new role for JAM-A as leukocyte adhesion molecule (Stamatovic et al., 2012). This was also tested in OGD/reoxygenation injury in vitro using monocytes/macrophages and neutrophils (PMN). mBMECs exposed to OGD and 24 h reperfusion (which causes high apical membrane JAM-A expression), showed increased adhesion and migration of both neutrophils and macrophages compared to control untreated cells (p < 0.001; Figs. 2D & E). To confirm that JAM-A contributed to this adhesion/migration, mBMECs were treated with a neutralizing anti-JAM-A antibody (BV11 clone), a neutralizing anti-ICAM antibody or a cocktail of both. Antibodies were added at the beginning of reperfusion for 24 h. All three experimental conditions significantly decreased neutrophil and macrophage adhesion (p < 0.001) and migration (p < 0.001) compared to mBMEC exposed to OGD/reoxygenation injury but treated with isotypic IgG2 (Figs. 2D & E). Inhibition was slightly amplified when a combination of anti-JAM-A and anti-ICAM antibodies was used (p < 0.001; Figs. 2D & E). The role of JAM-A was further assessed by depleting brain endothelial cells by transfection with JAMA shRNA (JAM-A KD cells). Depleting JAM-A significantly decreased neutrophil (p < 0.01) and macrophage adhesion (p < 0.001) and migration (p < 0.001), confirming that JAM-A plays an important role in leukocyte adhesion and migration during OGD/reoxygenation injury (Figs. 2D & E).
Targeting JAM-A/leukocyte interaction: JAM-A antagonist peptide
A potential site for interaction between endothelial JAM-A with PMN and monocytes/macrophages is the JAMA-A extracellular D2 domain, which may interact with leukocyte LFA (lymphocyte function associated antigen) (Severson and Parkos, 2009; Sobocki et al., 2006; Weber et al., 2007). Therefore, we designed and had synthesized by New England Peptide Inc., five different peptides (labeled P1, P2, P3, P4 and P5) that might serve as antagonists to the D2 extracellular domain. The ability of these peptides to inhibit leukocyte/endothelial JAM-A interaction was tested in adhesion assay using CCL2-pretreated mBMEC monolayers. These were overlaid with macrophages in the presence of increasing concentrations (1 to 1000 μg/ml) of JAM-A peptides. As shown in Supplemental Fig. 1A, only two peptides, P2 and P4, inhibited macrophage adhesion to activated brain endothelial cells. While P2 peptide showed an effect at high concentrations (100 and 1000 μg/ml, p < 0.001), P4 peptide had an inhibitory effect at concentrations between 1 and 1000 μg/ml (p < 0.001) compared with cells treated with CCL2 in the absence of any JAM-A peptide.
The JAM-A peptide P4 (now referred to as JAM-Ap) was further tested by examining binding specificity, selective antagonism JAM-Ap on brain endothelial cells, efficiency in blocking JAM-A in different inflammatory states (IL-1β/TNF-α treatment and OGD/reoxygenation injury) and comparing efficacy to JAM-A and ICAM-1 neutralizing antibodies. JAM-Ap binding to JAM-A was estimated by saturation binding curves and competitive binding assay in experiments with JAM-Ap-FITC (Figs. 3A and B). The EC50 value = 0.042 μg/ml (19 nM), while the IC50 was 0.055 μg/ml (25 nM) in experiments where JAM-Ap-FITC binding was inhibited by increasing unlabeled JAM-Ap concentrations (Figs. 3A and B).
Fig. 3.
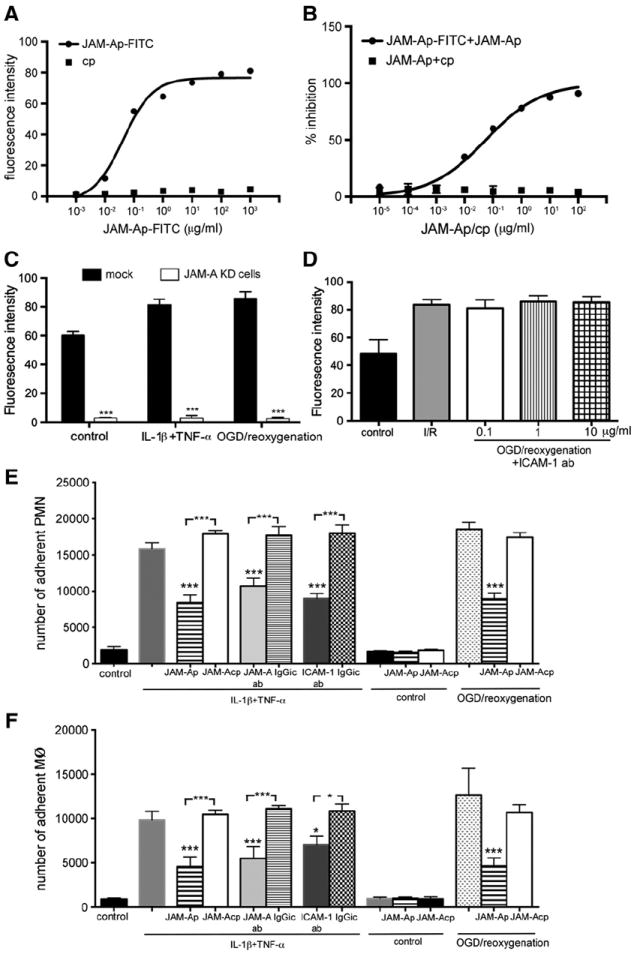
A) Saturation binding curve and B) dose-dependent inhibition curve for JAM-Ap-FITC and cp-FITC to brain endothelial cells. For dose-dependent inhibition JAM-Ap-FITC was challenged with increasing concentrations of either unlabeled JAM-Ap or cp. The data represent value from 3 independent experiments. C) and D) Specificity of JAM-Ap-FITC binding to brain endothelial cells was tested by knocking down JAM-A expression by stable transfection with JAM-A shRNA (C) and by treated cells with increased concentration of anti-ICAM-1 neutralizing antibody (0.1–10 μg/ml; D). In both experiments, cells were exposed to proinflammatory condition where JAMA-A occurs (cocktails of IL-1β and TNF-α or OGD/reoxygenation). Data represent values of mean ± SD. ***p < 0.001 for n=3 independent experiments. E) and F) Neutrophil (PMN) and monocyte/macrophage (MØ) adhesion on brain endothelial cells in the presence of JAM-A antagonist peptide (JAM-Ap) or control peptide (JAM-Acp). mBMEC was pretreated with a cocktail of IL1β and TNF-α (10 ng/ml) or exposed to OGD/reoxygenation injury in vitro, treated with antagonist JAM-A peptide (P4, 1 μg/ml) for 1 h and overlaid with isolated macrophages and PMN. Notice the significant reduction in the number of adherent cells with JAM-A peptide compared to control peptide. As controls, cells treated with IL1β and TNF-α were also treated with JAM-A or ICAM-1 blocking antibodies (JAM-A ab, ICAM-1 ab; 10 μg/ml) or isotypic control antibody (IgGic). Both JAM-A and ICAM-1 antibodies reduced PMN and MØ adherence. Data represent values of mean ± SD. ***p < 0.001 for n=5 independent experiments.
JAM-Ap-FITC binding was higher in proinflammatory exposed brain endothelial cells exposed to proinflammatory conditions (~65%, p < 0.01) compared with control, non-treated, cells. This increased binding was not the result of de novo synthesis of JAM-A as it was unaffected by inhibiting protein synthesis with cycloheximide (Fig. 2B). Rather, the increased JAM-Ap-FITC binding in proinflammatory conditions probably resulted from JAM-A relocalization to the apical membrane of the endothelial cells, with easier access by JAM-Ap-FITC compared to control resting conditions when JAM-A is mostly on the lateral membrane. In addition, JAM-Ap-FITC showed high specificity for JAM-A binding on brain endothelial cells. Cells without JAM-A (JAM-A KO cells) showed very low levels of JAM-Ap-FITC binding on the cell surface in proinflammatory conditions (<5% of the value of the JAM-A binding to JAM-A+/+ cells; Fig. 3C). The JAM-Ap “cross binding” to another adhesion molecule, ICAM-1, was tested in both the presence and absence of JAM-A in brain endothelial cells treated with increasing concentrations of anti-ICAM-A antibody. There was very low binding affinity of JAM-Ap-FITC for ICAM-1 (Fig. 3D).
JAM-Ap had a strong inhibitory effect on neutrophil and macrophage adhesion on mBMEC exposed to IL-1β and TNF-α or OGD/reoxygenation injury (p < 0.001; Figs. 3E & F), the magnitude of which was similar to neutralizing antibodies. This decreased adhesion with JAM-Ap was also associated with reduced neutrophil and monocyte migration (p < 0.0001; Figs. 1A & B supplemental data). We also tested the effect of JAM-Ap on mBMEC barrier function and it had no effect on barrier permeability to inulin (5 kDa) at different concentrations (0.1–1000 μg/ml) and times (0–72 h) (Supplemental Figs. 2A & B). In contrast, JAM-A antibody increased the permeability to inulin at concentration of 1–100 μg/ml in a time dependent manner (p < 0.001; Supplemental Figs. 2A & B).
Effect of targeting JAM-A on brain I/R injury
Given the ability of JAM-Ap to block leukocyte adhesion and migration during OGD/reoxygenation injury, we examined the effects of this peptide on leukocyte infiltration in vivo after I/R injury. Mice were divided into three groups: sham control, JAM-Ap and control scrambled peptide (cp). They were first exposed toMCAO for 30 min or “sham” operation (sham control) and were followed for 1–10 days after reperfusion. Mice receiving a peptide were injected 6 h after MCAO with reinjection every 24 h for 1–5 days. In preliminary experiments, we followed the distribution of ip or iv JAM-Ap-FITC administered under the same scheme as used with that of the unlabeled JAM-Ap for treatment propose. Confocal microscopy analysis of JAM-Ap-FITC distribution in the liver, lung and brain demonstrated high accumulation of JAM-Ap-FITC luminal on the luminal side of CD31 + brain microvessel. Comparing between ischemic and contralateral hemisphere, JAMAp-FITC was predominately present at luminal side of the microvessels (Fig. 4A). Furthermore, immunohistochemical analysis of JAM-Ap-FITC distribution in brain tissue (contralateral hemisphere, penumbra and ischemic tissue) demonstrated that JAM-Ap-FITC was not associated with CD45 + cells (mostly infiltrated leukocytes) and nonspecific trapped JAM-Ap-FITC in ischemic lesion was comparable with labeled control peptide (cp), at the level of threshold (Fig. 4A). We did not detect significant amount of JAM-Ap-FITC accumulation either in liver and lung microvessels or tissue after i.p. injection (data now showed).
Fig. 4.
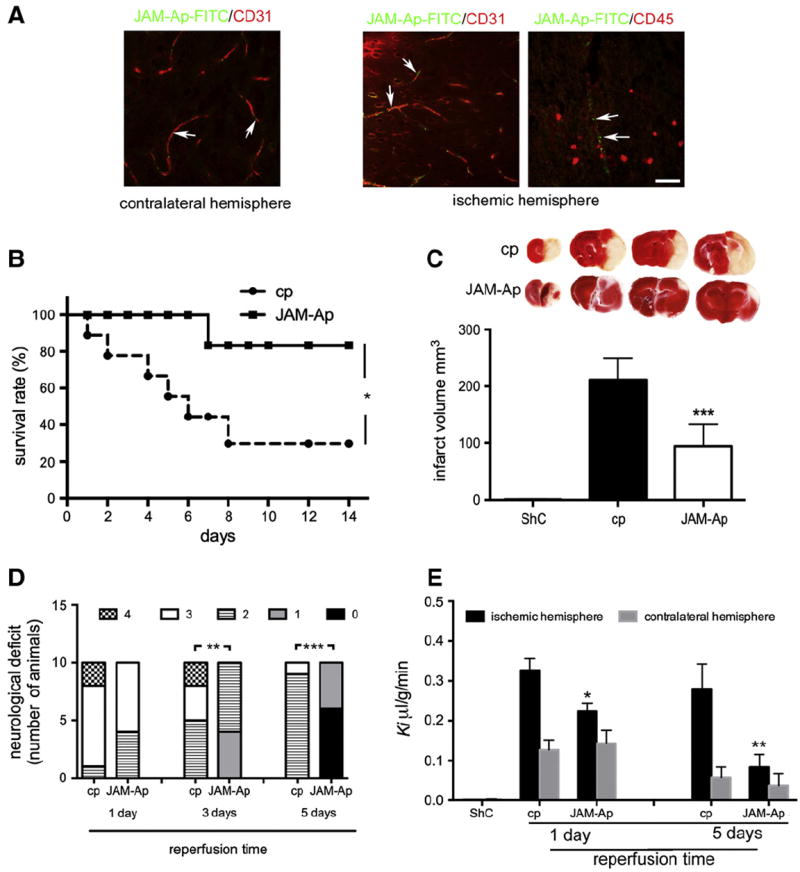
A) Representative images of JAM-Ap-FITC distribution in brain (ischemic and contralateral hemisphere) after tMCAO. The immunohistochemical labeling of brain blood vessels with anti-CD31 antibody and leukocytes with anti-CD45 antibody. Arrows indicated accumulated JAM-Ap-FITC at the blood vessels. Scale bar 50 μm. B) Kaplan–Meier curve of survival rate of mice treated with JAM-A peptide (JAM-Ap) and control peptide (cp). Survival rate was evaluated for 14 days after MCAO. JAM-Ap significantly improved survival after tMCAO. *p < 0.05 vs. cp (log-rank t-test; cp n = 5/15, JAM-Ap n = 12/15). C) Representative TTC-stained coronal brain sections from mice at day 5 after MCAO and were treated with JAM-A antagonist peptide (JAM-Ap) or control nonsense peptide (cp; 1 μg/kg, i.p.). Brain infarct size at day 5 of reperfusion in mice subjected to MCAO and treated with JAM-Ap or cp. Values are mean ± SD. **p < 0.01 (n = 10 each group). ShC = sham control. D) Summary of neurological scores at days 1, 3 and 5 after transient MCAO in mice treated with JAM-Ap or cp. No neurological deficit score is 0; maximal deficit score is 4. cp (n = 10) and JAM-Ap (n = 10), **p < 0.01 by Chi-squared test. E) Influx rate constant (Ki) for Dextran-Texas red (40 kDa) in ischemic and non-ischemic hemispheres at days 1 and 5 after transient MCAO. ShC = sham control. Values are means ± SD, n = 9; *p < 0.05; **p < 0.01. JAM-Ap mice treated with JAM-1 peptide (1 μg/kg, i.p.), cp-mice treated with control peptide.
JAM-Ap increased survival time, significantly reduced infarct size, improved neurological score and also had a protective effect on BBB permeability (Fig. 4). The survival rate in cp-treated mice 7 days after MCAO was only 30%, but it was 83% with JAM-Ap treatment (p < 0.05, Fig. 4B). JAM-Ap treated mice had smaller infarct volumes compared with cp treatment (e.g. 94 ± 29 vs. 211 ± 38 mm3 at day 5, p < 0.001; Fig. 4C). In addition, JAM-Ap treated mice had decreases in ischemic (but not non-ischemic) hemispheric volume compared to cp-treated mice at day 3 of reperfusion (118 ± 32 vs. 234 ± 37 mm3; p < 0.001). Because of potential confounding effects of edema and infarct resolution, in an indirect measure of infarct volume was also calculated and this also showed a protective effect of JAM-Ap (Fig. 4C).
Mice treated with JAM-Ap during reperfusion also had a better neurological score after MCAO (Fig. 4D). Thus, on a scale of 0–4, at day 1, 60% of the mice showed circling to the affected side (score 3) while 80% of the mice in the cp treated group had scores of 3 and 4 (no spontaneous locomotor activity). At days 3 and 5, 100% of JAM-Ap-treated mice had scores of 0 to 2 (~50% has score 1), while cp treated mice scored in neurological examination typically 2 to 3 (~50% at day 3, and 90% at day 5 had score 2, p < 0.01). Taking these results together, treatment with JAM-Ap had the greatest effect in the subacute phase (days 3 and 5) after transient MCAO.
JAM-Ap also reduced BBB hyperpermeability after MCAO, mostly for the higher molecular size tracer dextran (40 kDa). JAM-Ap treatment from 6 h after MCAO decreased dextran permeability significantly at day 1 (cp 0.33 ± 0.06 vs JAM-Ap 0.22 ± 0.08 μl/g/min, p < 0.05) and day 5 (cp 0.28 ± 0.06 vs JAM-Ap 0.083 ± 0.032 μl/g/min; p < 0.01) (Fig. 4E). On the one hand there was only a small tendency to reduce permeability to a small molecular weight tracer (FITC-inulin) that did not reach statistical significance (data not shown). On the other hand, in control mice, JAM-Ap had no effect on permeability, in contrast to JAM-A neutralizing antibody, which caused an increase in BBB permeability to inulin (Supplemental Fig. 2B).
Effect of targeting JAM-A on leukocyte recruitment and the inflammatory response
Brain I/R injury is closely associated with an inflammatory response. This develops in the first 6 h of reperfusion, with a peak in inflammation at 3 days after MCAO. One classic hallmark of I/R injury is leukocyte infiltration, first neutrophils and then monocytes. As JAM-Ap showed significant effects on leukocyte adhesion and migration, we tested the effect of JAM-Ap on leukocyte infiltration into brain after MCAO. Analyzing the total number of PMN and monocytes/macrophages in ischemic and non-ischemic hemispheres at days 1, 3 and 5 after MCAO in mice treated with control peptide (cp) and JAM-Ap showed significant differences in number of cells in the ischemic hemisphere (day 1: cp 70,935 ± 6023 vs. JAM-Ap 39,306 ± 1700. Day 3: cp 85,857 ± 7243 vs. JAM-Ap 30,516 ± 3700; p < 0.001).
Two populations of cells were prominent in the ischemic hemisphere at days 1 and 3 of reperfusion, neutrophils (Cd45highCD11b+Ly6G+ cells) and monocytes (CD45highCd11b+Ly6G− cells). JAM-Ap treatment significantly reduced neutrophils at days 1 and 3 (p < 0.01 and p < 0.001, respectively) compared with cp treatment, while a significant reduction in monocytes was found at days 3 and 5 after MCAO (p < 0.01; Figs. 5A, B &C). This reduced number of neutrophils (MPO + cells) and monocytes (ED1 + cells) was mostly present in and around the ischemic lesion as determined by immunohistochemistry (Fig. 5D). JAM-Ap treatment affected neutrophil and monocyte infiltration into the ischemic lesion rather than the total number of neutrophils and monocytes in blood. Fluorocytometric analysis of the blood for neutrophils and monocytes in sham control and mice with tMCAO treated with JAM-Ap and cp did not show any changes in total number of these cells (blood day 5 after tMCAO and beginning of peptide injection PMN-sham control −1 × 109/l, cp-1.02 109/l, JAM-Ap 0.99 × 109/l; MO-sham control 4 × 108; cp-3.89 × 108; and JAM-Ap-3.95 × 108).
Fig. 5.
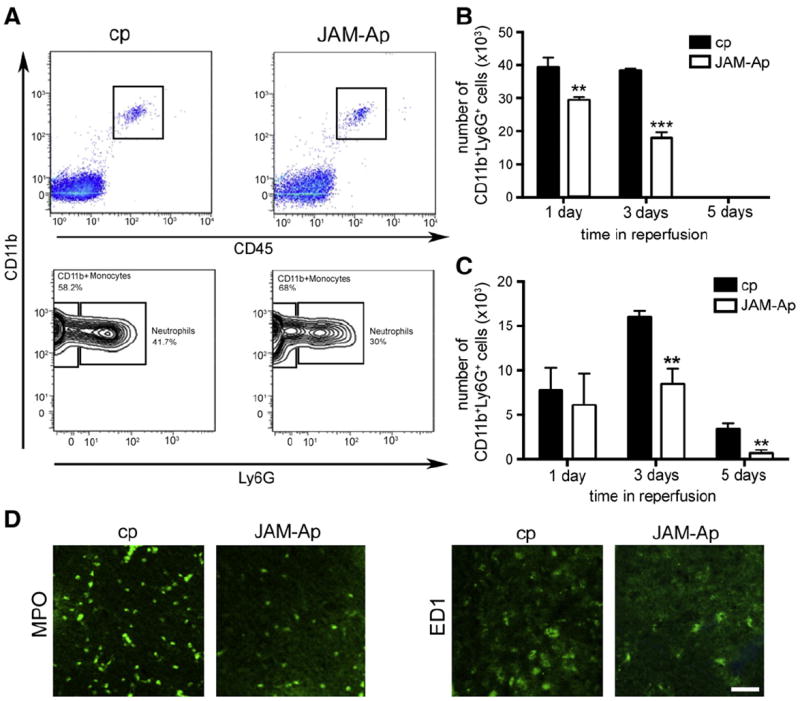
A) Representative flow cytometric analyses of infiltrating neutrophils (PMN) and monocytes/macrophages in brain in mice treated with JAM-A peptide (JAM-Ap) or control peptide (cp) at day 3 after transient MCAO. B) and C) Total number of infiltrated neutrophils (CD11b+Ly6G+ cells) and monocytes (CD11b+Ly6G− cells) at 1, 3 and 5 days of reperfusion. Note significant reduction in neutrophil infiltration into brain parenchyma at days 1 and 3 in mice treated with JAM-Ap compared to treatment with control peptide. JAM-Ap also reduced monocyte infiltration at days 3 and 5. Values are mean ± SD **p < 0.01, ***p < 0.001, n = 5 mice. D) Representative image of infiltrated neutrophils (MPO + cells) and monocytes (ED1 + cells) in ischemic hemisphere 3 days after tMCAO in mice treated with cp or JAM-Ap. Scale bar 50 μm.
Infiltrating PMN and monocytes/macrophages have an important role in the development of reperfusion-associated inflammation (Tuttolomondo et al., 2009). Thus, a reduction of leukocyte infiltration by JAM-Ap could also impact the magnitude of inflammatory response by modulating the expression of inflammatory mediators. The profile of proinflammatory mediator expression in ischemic penumbra at day 3 of reperfusion in JAM-Ap and cp-treated mice was analyzed by protein array. The analysis determined the fold increases in expression in MCAO mice treated with JAM-Ap or control peptide compared to sham-operated mice (Fig. 6A). In cp-treated mice, post-ischemic inflammation at day 3 of reperfusion was characterized by significant elevations (1.4–4-fold) in cytokines (IL-1β, IL-6, TNF-α, IL12p70, M-CSF, GM-CSF) and chemokines (CCL7, CCL9, CXCL13, CX3CL1 and CXCL5). However, JAM-Ap treated mice had lower level of proinflammatory cytokine/chemokine expression in ischemic hemisphere compared to cp-treated mice (Fig. 6A).
Fig. 6.
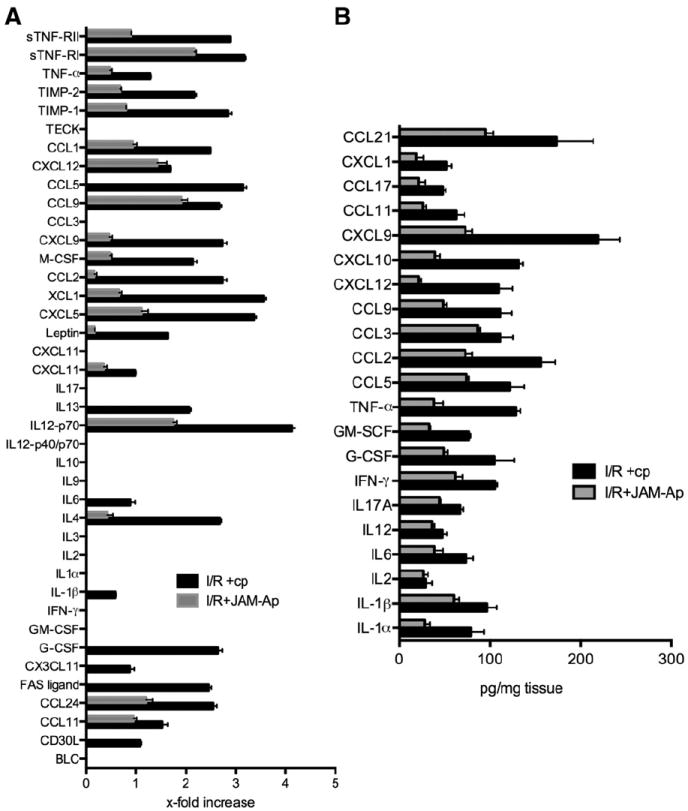
A) Protein array and B) ELISA assay for proinflammatory mediators present in brain tissue (penumbra) of mice exposed to MCAO followed by 3 days of reperfusion. Mice were treated with JAM-A peptide (JAM-Ap) or control peptide (cp) starting from 6 h after transient MCAO with further injections at 24 and 48 h. For the protein array (A), data represent the fold increase compared to sham-operated animals. For the ELISA assay (B), absolute data (pg/mg tissue) are reported. Both assays indicated a decrease in cytokine/chemokine expression with the JAM-A antagonist peptide. Values are means ± SD, n = 5 mice.
Quantification of proinflammatory cytokines in brain tissue by ELISA after MCAO further confirmed that JAM-Ap treated mice had significant lower levels compared to cp (i.e. IL-1β, TNF-α, IL-6, IL-17a, CCL2, CCL9; p < 0.001; Fig. 6B). Therefore, inhibition of JAM-A during reperfusion injury affects leukocyte infiltration into brain parenchyma, consequently modulating the post-ischemic inflammatory response, lesion size and neurological outcome.
Discussion
Ischemia/reperfusion injury triggers a complex process in brain endothelial cells resulting in intense inflammatory remodeling of the endothelial surface (Woodfin et al., 2009). At the apical (blood-facing) membrane, this remodeling includes significant upregulation of adhesion molecules, critical for leukocyte interaction and transmigration (Weber et al., 2007;Woodfin et al., 2009). At the lateral membrane, remodeling mostly affects transmembrane tight and adherens junction proteins, which alters adhesion and is associated with opening of the paracellular space for leukocyte migration (Dimitrijevic et al., 2006; Mamdouh et al., 2009; Stamatovic et al., 2009). Several recent studies have shown that JAM-A has dual roles, a TJ complex protein under physiological conditions and a leukocyte adhesion molecule during inflammation (Severson and Parkos, 2009; Weber et al., 2007; Wojcikiewicz et al., 2009;Woodfin et al., 2009). There is now accumulating evidence regarding beneficial effects of blocking JAM-A during inflammatory responses (Del Maschio et al., 1999; Khandoga et al., 2005; Lakshmi et al., 2012; Nourshargh et al., 2006). The present study aimed to establish the contribution of JAM-A to leukocyte infiltration and post-ischemic inflammation in brain I/R injury. We found that: a) during post-ischemic brain inflammation, JAM-A undergoes redistribution from the lateral to the apical membrane of the brain endothelial cell; and b) apical JAM-A acts as a leukocyte adhesion molecule, modulating neutrophil and monocyte adhesion and migration through the brain endothelial barrier; c) modulating JAM-A interaction with PMN and monocytes/macrophages significantly affects brain I/R injury (lesion size, neurological score) and inflammation (number of infiltrating PMN and monocytes/macrophages and brain cytokine/chemokine levels). This study also highlights a new approach to targeting JAM-A, a small peptide inhibitor designed to block interaction between the JAM-A D2 domain and LFA present on PMN and monocytes/macrophages. Implications of these findings are discussed below.
Reperfusion after ischemia elicits a marked inflammatory response at the BBB and adversely affects brain endothelial barrier function by disrupting intercellular junctions, affecting polarization of proteins, and altering protein folding in the endoplasmic reticulum (Fernandez-Lopez et al., 2012; Latour et al., 2004; Neumann-Haefelin et al., 2000). During post-ischemic reperfusion, there is a biphasic BBB ‘opening’, with an early phase occurring at reperfusion onset and a late phase occurring after 12–24 h (Fernandez-Lopez et al., 2012; Latour et al., 2004; Neumann-Haefelin et al., 2000; Yang and Rosenberg, 2011). Although the early phase of “opening” of BBB is closely associated with severe BBB breakdown caused by oxidative mediators and matrix metalloproteinases, the later “opening” is the result of ongoing inflammation (Liu et al., 2012; Yang and Rosenberg, 2011). Proinflammatory cytokines play a pivotal role in inducing BBB hyperpermeability, which can persist for up to 14 days, and be responsible for ischemic lesion progression (Jander et al., 2007; Neumann-Haefelin et al., 2000; Tuttolomondo et al., 2009). In both early and late BBB “opening”, there are alterations in TJ structure manifested as lost or fragmented staining of the transmembrane TJ proteins, occludin and claudin-5, although there are different underlying mechanisms for these TJ changes (Dimitrijevic et al., 2006; Dimitrijevic et al., 2007; Fernandez-Lopez et al., 2012; Jiao et al., 2011). A transmembrane TJ protein that does not follow the pattern of changes found for occludin and claudin-5 is JAM-A. As shown in our recent study, in the presence of inflammatory stimuli, JAM-A relocates from the endothelial lateral membrane via macropinocytosis and is delivered to the apical (luminal) membrane where it has a new role as adhesion molecule for PMN and monocytes/macrophages (Stamatovic et al., 2012). Brain I/R injury creates an inflammatory condition (burst of proinflammatory cytokines, chemokines) and it may not be surprising that JAM-A also shows apical accumulation (shown by cell surface ELISA) in this condition. The timing of JAM-A apical accumulation (24 h after I/R injury in vitro) correlates with peak expression of proinflammatory molecules such as CCL2, TNF-α, both of which can induce JAM-A relocalization (Dimitrijevic et al., 2006; Dimitrijevic et al., 2007; Jander et al., 2007). Additional evidence that JAM-A-dependent apical relocalization occurs is a significant increase in leukocyte adhesion/migration at the brain endothelium (data from adhesion assay) even though JAM-A total protein levels in endothelial cells remain constant during OGD/reoxygenation injury in vitro. JAM-A relocalization, rather than de novo synthesis during inflammatory remodeling, was additionally confirmed in using the protein synthesis inhibitor cycloheximide. A slight increase in total JAM-A levels during in vivo I/R could be due to infiltrating PMN and monocytes/macrophages in brain parenchyma during reperfusion, which also expressed JAM-A (Corada et al., 2005; Mamdouh et al., 2009; Nourshargh et al., 2006).
JAM-A relocalization and its presence on the apical side could be considered as part of inflammatory remodeling of the TJ complex in order to form a paracellular route and support leukocyte transmigration. JAM-A and other transmembrane TJ proteins, claudin-5 and occludin, are redistributed from the lateral membrane of brain endothelial cells, removing adhesive interactions between cells. This process is mostly associated with actual uptake/internalization of these TJ proteins without altering total protein levels. In recent studies by Utech et al. and Bruewer et al., proinflammatory (TNF-α and INF-γ) remodeling of epithelial barriers was described as ‘block internalization’ of JAM-A, occludin and claudin-1 via macropinocytosis (Bruewer et al., 2005; Utech et al., 2005). However, our recent studies dissected different mechanisms of claudin-5, occludin and JAM-A protein redistribution during inflammatory remodeling of brain endothelial cells (Stamatovic et al., 2012) Internalization via macropinocytosis was pinpointed as a pathway for JAM-A removal from the lateral membrane. By sorting through endosomal vesicles, from early to recycling, JAM-A is ultimately rapidly delivered to the apical membrane. The different pathways of TJ protein internalization (caveolin vs macropinocytosis) and their end-results, delivery on apical side vs residing in the recycling vesicles (claudin-5 and occludin) is probably related to the role of JAM-A in leukocyte–zendothelial cell interaction. JAM-A relocalization during inflammation via internalization (macropinocytosis) should be considered as establishing a new position of JAM-A for interaction with leukocytes and together with CD31 and CD99 in regulating the process of directing leukocyte migration through the paracellular space.
The role of JAM-A as leukocyte adhesion and transmigration molecule has been described in a variety of inflammatory conditions such as atherosclerosis, hepatic I/R injury, as well as chronic inflammatory diseases (i.e. asthma, bronchitis, interstitial nephritis, autoimmune hepatitis and alcoholic cirrhosis) (Babinska et al., 2007; Cavusoglu et al., 2007; Corada et al., 2005; Khandoga et al., 2005; Lakshmi et al., 2012; Manetti et al., 2013). Those results showed that JAM-A inhibition, either by knocking down JAM-A expression or by neutralizing JAM-A antibody, reduces leukocyte infiltration, particularly neutrophils (Azari et al., 2010; Babinska et al., 2007; Cera et al., 2009; Corada et al., 2005; Del Maschio et al., 1999; Lakshmi et al., 2012; Nourshargh et al., 2006). The current study shows a similar effect at the BBB, with JAM-A inhibition diminishing neutrophil and monocyte adhesion and migration in vitro and in vivo.
It is important to pinpoint that cocktails of JAM-A and ICAM neutralizing antibodies had an amplifying effect, indicating a potential collaborative role of these two molecules in regulating leukocyte adhesion on brain endothelial cells. One intriguing part of our findings is that inhibition of JAM-A in vivo had comparable effects to that in vitro. Early reports showed that JAM-A neutralizing antibody predominantly had effects in vitro or in certain inflammatory conditions (i.e. cytokine-induced meningitis vs. bacterial meningitis) (Del Maschio et al., 1999; Lechner et al., 2000) and greater effects were found in JAM-A deficient mice (Lakshmi et al., 2012; Nourshargh et al., 2006). Our data, however, showed that JAM-A inhibition using the peptide JAM-Ap in vivo had greater effects compared with data obtained with a neutralizing antibody. A potential reason for this outcome may lie in the approach of inhibiting JAM-A/PMN or JAM-A monocyte/macrophage interaction via small peptide designed for the JAM-A C2 domain. Most earlier published studies using neutralizing antibody against JAM-A tested the BV-11 clone, which targets the membrane-distal domain of JAM-A, important for dimerization, but not the C2 domain important for leukocyte/endothelial LFA/JAM-A interaction (Severson and Parkos, 2009; Severson et al., 2008; Weber et al., 2007; Wojcikiewicz et al., 2009). The BV-11 clone predominantly has indirect effects on leukocyte interaction by diminishing JAM-A dimerization, which reduces the opportunity of JAM-A/leukocyte interaction (Severson and Parkos, 2009; Severson et al., 2008;Wojcikiewicz et al., 2009). Although dimerization is important for JAM-A to function as a leukocyte adhesion molecule, some recent results from atomic force microscopy have shown that heterophilic JAM-A/LFA interaction had better binding affinity and stronger interaction than homophilic JAM-A/JAM-A interaction (dimerization interaction) (Wojcikiewicz et al., 2009). With the neutralizing antibody in vivo, ICAM-1 may compensate resulting in the absence of an overall beneficial effect (Severson and Parkos, 2009; Severson et al., 2008). We do not also exclude the potential of JAM-A to compensate in the case of ICAM-1 neutralization. These adhesion molecules are already indicated as having the ability to cause each other’s clustering, which could support the leukocyte recruitment. Thus, for potential anti-adhesion therapy, inhibition of ICAM-1 and JAM-A might represent the best strategy for reducing leukocyte recruitment in brain injury.
The important finding from this study is the efficacy of a small inhibitory peptide targeting the JAM-A C2 domain and JAM-A/LFA interaction in inhibiting leukocyte adhesion both in vitro and in vivo. The peptide not only affected leukocyte adhesion, it also affected transmigration as LFA/JAM-A interaction is essential for initiating PMN or monocyte diapedesis. This peptide approach to inhibit endothelial JAM-A function had a significant effect on leukocyte recruitment at the site of the ischemic lesion. Our results on barrier permeability, however, suggest another benefit of the JAM-Ap approach. JAM-Ap had no effect on normal barrier permeability whereas the neutralizing antibody increased permeability in vivo and in vitro, which may relate to the specificity of the JAM-Ap approach in targeting JAM-A/LFA interaction omitting potential antibody (IgG) effect on barrier function described in some earlier studies (Enlimomab Acute Stroke Trial, 2001; Frijns and Kappelle, 2002).
Reducing leukocyte recruitment in brain has an important role in tissue protection during reperfusion. PMN and monocytes/macrophages are a source of proinflammatory cytokines by which both cells can damage neurons and also trigger local glial cells to produce a variety of proinflammatory mediators and further amplify the inflammatory response (Jander et al., 2007; Tuttolomondo et al., 2009). As shown in our data, inflammatory cytokine expression/production was greatly reduced in when JAM-Ap was used to reduce leukocyte infiltration and this may contribute to improved neurological outcome and smaller lesion volume.
An important issue is that inhibiting JAM-A/LFA interaction also improved BBB integrity after I/R injury. It is important to highlight that we believe that this improvement is due to reduced leukocyte recruitment rather than a direct effect of JAM-Ap on homophilic interactions of JAM-A between endothelial cells. This hypothesis is based on the fact that PMN and monocytes/macrophages are source of the proinflammatory mediators and significantly contribute to BBB injury and progression on I/R injury. In addition, JAM-Ap had no effect on I/R-induced endothelial barrier disruption in vitro in the absence of other cell types.
It is also important to note that complete inhibition of inflammation may affect recovery after ischemic injury. Exclusion of PMN and monocytes/macrophages affects the magnitude of the immune response in parenchyma as they are important source of inflammatory cytokines and their interaction with local cells plays a significant role in lesion progression (Becker, 2001; Dimitrijevic et al., 2007; Tuttolomondo et al., 2009). However, controlling/stopping leukocyte recruitment in brain parenchyma does not affect the function of resident microglia/macrophages during the recovery process and JAM-Ap may shift the inflammatory response and recovery process towards resident sources. Therefore, JAM-A inhibition may display at several levels a protective role in brain I/R injury.
Conclusion
In conclusion, targeting JAM-A during brain I/R injury may benefit by decreasing leukocyte infiltration and reducing the inflammation-mediated brain damage. Targeting JAM-A was beneficial over a time frame of several hours to several days and was not selective for neutrophils or monocytes, suggesting a wide area for using JAM-A in treating brain I/R injury. This study provides new insights into how to develop new therapeutic strategies for stroke and for other neurological disorders involving inflammation.
Supplementary Material
Acknowledgments
This work was supported by the Public Health Service grants NS062853 from the National Institute of Neurological Disorders and Stroke (AVA), NS034709 (RFK) and GM29507 and GM61656 from the National Institute for General Medical Science (PAW).
The confocal microscopy work was performed in the Microscopy and Image-analysis Laboratory (MIL) at the University of Michigan, Department of Cell & Developmental Biology.
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.nbd.2014.03.010.
References
- Andjelkovic AV, et al. The protective effects of preconditioning on cerebral endothelial cells in vitro. J Cereb Blood Flow Metab. 2003;23:1348–1355. doi: 10.1097/01.WCB.0000091762.61714.FE. [DOI] [PubMed] [Google Scholar]
- Azari BM, et al. Silencing of the F11R gene reveals a role for F11R/JAM-A in the migration of inflamed vascular smooth muscle cells and in atherosclerosis. Atherosclerosis. 2010;212:197–205. doi: 10.1016/j.atherosclerosis.2010.05.014. [DOI] [PubMed] [Google Scholar]
- Babinska A, et al. The F11 receptor (F11R/JAM-A) in atherothrombosis: overexpression of F11R in atherosclerotic plaques. Thromb Haemost. 2007;97:272–281. [PubMed] [Google Scholar]
- Becker KJ. Targeting the central nervous system inflammatory response in ischemic stroke. Curr Opin Neurol. 2001;14:349–353. doi: 10.1097/00019052-200106000-00014. [DOI] [PubMed] [Google Scholar]
- Bruewer M, et al. Interferon-gamma induces internalization of epithelial tight junction proteins via a macropinocytosis-like process. FASEB J. 2005;19:923–933. doi: 10.1096/fj.04-3260com. [DOI] [PubMed] [Google Scholar]
- Cavusoglu E, et al. Association of plasma levels of F11 receptor/junctional adhesion molecule-A (F11R/JAM-A) with human atherosclerosis. J Am Coll Cardiol. 2007;50:1768–1776. doi: 10.1016/j.jacc.2007.05.051. [DOI] [PubMed] [Google Scholar]
- Cera MR, et al. JAM-A promotes neutrophil chemotaxis by controlling integrin internalization and recycling. J Cell Sci. 2009;122:268–277. doi: 10.1242/jcs.037127. [DOI] [PubMed] [Google Scholar]
- Cohn ZA. The isolation and cultivation of mononuclear phagocytes. Methods Enzymol. 1974;32:758–765. doi: 10.1016/0076-6879(74)32079-4. [DOI] [PubMed] [Google Scholar]
- Corada M, et al. Junctional adhesion molecule-A-deficient polymorphonuclear cells show reduced diapedesis in peritonitis and heart ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2005;102:10634–10639. doi: 10.1073/pnas.0500147102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Maschio A, et al. Leukocyte recruitment in the cerebrospinal fluid of mice with experimental meningitis is inhibited by an antibody to junctional adhesion molecule (JAM) J Exp Med. 1999;190:1351–1356. doi: 10.1084/jem.190.9.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Zoppo GJ, et al. Polymorphonuclear leukocytes occlude capillaries following middle cerebral artery occlusion and reperfusion in baboons. Stroke. 1991;22:1276–1283. doi: 10.1161/01.str.22.10.1276. [DOI] [PubMed] [Google Scholar]
- Dimitrijevic OB, et al. Effects of the chemokine CCL2 on blood–brain barrier permeability during ischemia-reperfusion injury. J Cereb Blood Flow Metab. 2006;26:797–810. doi: 10.1038/sj.jcbfm.9600229. [DOI] [PubMed] [Google Scholar]
- Dimitrijevic OB, et al. Absence of the chemokine receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice. Stroke. 2007;38:1345–1353. doi: 10.1161/01.STR.0000259709.16654.8f. [DOI] [PubMed] [Google Scholar]
- Enlimomab Acute Stroke Trial, I. Use of anti-ICAM-1 therapy in ischemic stroke: results of the Enlimomab Acute Stroke Trial. Neurology. 2001;57:1428–1434. doi: 10.1212/wnl.57.8.1428. [DOI] [PubMed] [Google Scholar]
- Fernandez-Lopez D, et al. Blood–brain barrier permeability is increased after acute adult stroke but not neonatal stroke in the rat. J Neurosci. 2012;32:9588–9600. doi: 10.1523/JNEUROSCI.5977-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frijns CJ, Kappelle LJ. Inflammatory cell adhesion molecules in ischemic cerebrovascular disease. Stroke. 2002;33:2115–2122. doi: 10.1161/01.str.0000021902.33129.69. [DOI] [PubMed] [Google Scholar]
- Garcia JH, et al. Influx of leukocytes and platelets in an evolving brain infarct (Wistar rat) Am J Pathol. 1994;144:188–199. [PMC free article] [PubMed] [Google Scholar]
- Jander S, et al. Imaging inflammation in acute brain ischemia. Stroke. 2007;38:642–645. doi: 10.1161/01.STR.0000250048.42916.ad. [DOI] [PubMed] [Google Scholar]
- Jiao H, et al. Specific role of tight junction proteins claudin-5, occludin, and ZO-1 of the blood–brain barrier in a focal cerebral ischemic insult. J Mol Neurosci. 2011;44:130–139. doi: 10.1007/s12031-011-9496-4. [DOI] [PubMed] [Google Scholar]
- Johnson-Leger CA, et al. Junctional adhesion molecule-2 (JAM-2) promotes lymphocyte transendothelial migration. Blood. 2002;100:2479–2486. doi: 10.1182/blood-2001-11-0098. [DOI] [PubMed] [Google Scholar]
- Kazakoff PW, et al. An in vitro model for endothelial permeability: assessment of monolayer integrity. In Vitro Cell Dev Biol Anim. 1995;31:846–852. doi: 10.1007/BF02634568. [DOI] [PubMed] [Google Scholar]
- Khandoga A, et al. Junctional adhesion molecule-A deficiency increases hepatic ischemia-reperfusion injury despite reduction of neutrophil transendothelial migration. Blood. 2005;106:725–733. doi: 10.1182/blood-2004-11-4416. [DOI] [PubMed] [Google Scholar]
- Lakshmi SP, et al. Effects of JAM-A deficiency or blocking antibodies on neutrophil migration and lung injury in a murine model of ALI. Am J Physiol Lung Cell Mol Physiol. 2012;303:L758–L766. doi: 10.1152/ajplung.00107.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latour LL, et al. Early blood–brain barrier disruption in human focal brain ischemia. Ann Neurol. 2004;56:468–477. doi: 10.1002/ana.20199. [DOI] [PubMed] [Google Scholar]
- Lechner F, et al. Antibodies to the junctional adhesion molecule cause disruption of endothelial cells and do not prevent leukocyte influx into the meninges after viral or bacterial infection. J Infect Dis. 2000;182:978–982. doi: 10.1086/315765. [DOI] [PubMed] [Google Scholar]
- Liang TW, et al. Vascular endothelial-junctional adhesion molecule (VE-JAM)/JAM 2 interacts with T, NK, and dendritic cells through JAM 3. J Immunol. 2002;168:1618–1626. doi: 10.4049/jimmunol.168.4.1618. [DOI] [PubMed] [Google Scholar]
- Liu J, et al. Matrix metalloproteinase-2-mediated occludin degradation and caveolin-1-mediated claudin-5 redistribution contribute to blood–brain barrier damage in early ischemic stroke stage. J Neurosci. 2012;32:3044–3057. doi: 10.1523/JNEUROSCI.6409-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamdouh Z, et al. Transcellular migration of leukocytes is mediated by the endothelial lateral border recycling compartment. J Exp Med. 2009;206:2795–2808. doi: 10.1084/jem.20082745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manetti M, et al. Differential expression of junctional adhesion molecules in different stages of systemic sclerosis. Arthritis Rheum. 2013;65:247–257. doi: 10.1002/art.37712. [DOI] [PubMed] [Google Scholar]
- Martin-Padura I, et al. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol. 1998;142:117–127. doi: 10.1083/jcb.142.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann-Haefelin T, et al. Serial MRI after transient focal cerebral ischemia in rats: dynamics of tissue injury, blood–brain barrier damage, and edema formation. Stroke. 2000;31:1965–1972. doi: 10.1161/01.str.31.8.1965. discussion 1972–3. [DOI] [PubMed] [Google Scholar]
- Nourshargh S, et al. The role of JAM-A and PECAM-1 in modulating leukocyte infiltration in inflamed and ischemic tissues. J Leukoc Biol. 2006;80:714–718. doi: 10.1189/jlb.1105645. [DOI] [PubMed] [Google Scholar]
- Ohno K, et al. Cerebrovascular integrity in protein-deprived rats. Brain Res Bull. 1980;5:251–255. doi: 10.1016/0361-9230(80)90166-5. [DOI] [PubMed] [Google Scholar]
- Schenkel AR, et al. CD99 plays amajor role in the migration ofmonocytes through endothelial junctions. Nat Immunol. 2002;3:143–150. doi: 10.1038/ni749. [DOI] [PubMed] [Google Scholar]
- Severson EA, Parkos CA. Structural determinants of Junctional Adhesion Molecule A (JAM-A) function and mechanisms of intracellular signaling. Curr Opin Cell Biol. 2009;21:701–707. doi: 10.1016/j.ceb.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severson EA, et al. Cis-dimerization mediates function of junctional adhesion molecule A. Mol Biol Cell. 2008;19:1862–1872. doi: 10.1091/mbc.E07-09-0869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobocki T, et al. Genomic structure, organization and promoter analysis of the human F11R/F11 receptor/junctional adhesion molecule-1/JAM-A. Gene. 2006;366:128–144. doi: 10.1016/j.gene.2005.08.025. [DOI] [PubMed] [Google Scholar]
- Stamatovic SM, et al. Caveolae-mediated internalization of occludin and claudin-5 during CCL2-induced tight junction remodeling in brain endothelial cells. J Biol Chem. 2009;284:19053–19066. doi: 10.1074/jbc.M109.000521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatovic SM, et al. Relocalization of junctional adhesion molecule A during inflammatory stimulation of brain endothelial cells. Mol Cell Biol. 2012;32:3414–3427. doi: 10.1128/MCB.06678-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuttolomondo A, et al. Inflammation as a therapeutic target in acute ischemic stroke treatment. Curr Top Med Chem. 2009;9:1240–1260. doi: 10.2174/156802609789869619. [DOI] [PubMed] [Google Scholar]
- Utech M, et al. Mechanism of IFN-gamma-induced endocytosis of tight junction proteins: myosin II-dependent vacuolarization of the apical plasma membrane. Mol Biol Cell. 2005;16:5040–5052. doi: 10.1091/mbc.E05-03-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber C, et al. The role of junctional adhesion molecules in vascular inflammation. Nat Rev Immunol. 2007;7:467–477. doi: 10.1038/nri2096. [DOI] [PubMed] [Google Scholar]
- Wojcikiewicz EP, et al. LFA-1 binding destabilizes the JAM-A homophilic interaction during leukocyte transmigration. Biophys J. 2009;96:285–293. doi: 10.1529/biophysj.108.135491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodfin A, et al. Endothelial cell activation leads to neutrophil transmigration as supported by the sequential roles of ICAM-2, JAM-A, and PECAM-1. Blood. 2009;113:6246–6257. doi: 10.1182/blood-2008-11-188375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Rosenberg GA. Blood–brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke. 2011;42:3323–3328. doi: 10.1161/STROKEAHA.110.608257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, et al. Early inhibition of MMP activity in ischemic rat brain promotes expression of tight junction proteins and angiogenesis during recovery. J Cereb Blood Flow Metab. 2013;33:1104–1114. doi: 10.1038/jcbfm.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Fleming BP. A method for isolation and fluorescent labeling of rat neutrophils for intravital microvascular studies. Microvasc Res. 1990;40:218–229. doi: 10.1016/0026-2862(90)90021-i. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.