

Abstract
Mismatch repair (MMR) malfunction causes the accumulation of mismatches in the genome leading to genomic instability and cancer. The inactivation of an MMR gene (MSH2, MSH6, MLH1, or PMS2) with an inherited mutation causes Lynch syndrome (LS), a dominant susceptibility to cancer. MMR gene variants of uncertain significance (VUS) may be pathogenic mutations, which cause LS, may result in moderately increased cancer risks, or may be harmless polymorphisms. Our study suggests that an inherited MMR VUS individually assessed as proficient may, however, in a pair with another MMR VUS found in the same colorectal cancer (CRC) patient have a concomitant contribution to the MMR deficiency. Here, eight pairs of MMR gene variants found in cancer patients were functionally analyzed in an in vitro MMR assay. Although the other pairs do not suggest a compound deficiency, the MSH2 VUS pair c.380A>G/c.982G>C (p.Asn127Ser/p.Ala328Pro), which nearly halves the repair capability of the wild-type MSH2 protein, is presumed to increase the cancer risk considerably. Moreover, two MSH6 variants, c.1304T>C (p.Leu435Pro) and c.1754T>C (p.Leu585Pro), were shown to be MMR deficient. The role of one of the most frequently reported MMR gene VUS, MSH2 c.380A>G (p.Asn127Ser), is especially interesting because its concomitant defect with another variant could finally explain its recurrent occurrence in CRC patients.
Keywords: functional analysis, Lynch syndrome, mismatch repair, MSH2, MSH6, VUS
Introduction
The discovery of an inherited deleterious mutation in a cancer patient permits predictive gene testing in the family and enables targeted cancer surveillance. An inherited defect in any one of the mismatch repair (MMR) genes MSH2 (MIM# 609309, Ref-Seq NM_000251.1), MSH6 (MIM# 600678, RefSeq NM_000179.2), MLH1 (MIM# 120436, RefSeq NM_000249.3), or PMS2 (MIM# 600259, RefSeq NM_000535) is associated with a susceptibility to colorectal cancer (CRC; MIM# 114500) known as Lynch syndrome (LS, previously referred to as hereditary nonpolyposis CRC syndrome; HNPCC; MIM# 120435). Although the inactivation of an MMR gene seems to cause a dominant susceptibility to cancer, a significant proportion of mutations are nontruncating and, due to the uncharacterized effect of these variations on the function of the polypeptide, they can be difficult to distinguish from harmless polymorphisms. Such alterations are often referred to as variants of uncertain significance (VUS) [Goldgar et al., 2008].
The wide variety of clinical phenotypes in CRC families further complicates the pathogenicity assessments and the LS diagnostics [Lynch et al., 2007]. Nontruncating MMR gene alterations often associate with atypical clinical phenotypes with later age of cancer onset and low or no microsatellite instability (MSI) in tumors. The prediction of their pathogenicity without functional analysis is difficult because different alterations in the same functional domain and even in the same codon can cause complete elimination of MMR function or little to no effect on protein function [Ellison et al., 2001; Raevaara et al., 2005]. The interpretation of their significance is further complicated by the fact that cancer patients may carry more than one VUS in either the same or different MMR genes. For instance, Liu et al. (2003) and Wu et al. (2001) have reported second variations affecting MLH3 in an MSH2 VUS carrier and MSH6 in an MLH3 VUS carrier, respectively. These MLH3 variations have since been shown to be functionally MMR proficient [Korhonen et al., 2008] whereas the MSH2 and MSH6 variations have similarly been shown to have a decreased MMR activity [Cyr and Heinen, 2008; Gammie et al., 2007]. Carriers of more than one individually MMR proficient MLH1 and MSH2 variations have also been reported [Gargiulo et al., 2009; Raevaara et al., 2005]. Interestingly, based on mutation screening and yeast-based assays it has also been speculated that the enhancer effect of low risk MMR gene variations contribute to the risk of CRC [Liu et al., 2003; Martinez and Kolodner, 2010]. Nevertheless, the functional analysis of these VUS individually only distinguishes the MMR-deficient variations, and does not account for the potential enhancer or compound effect between the two coexisting VUS.
Although two simultaneously inherited VUS are not expected to cause a constitutive MMR deficiency such as the inheritance of homozygous or compound heterozygous truncating mutations that result in a complete lack or greatly compromised protein function and hematological and brain malignancies at early age of onset [Felton et al., 2007], by compound contribution they may increase the cancer risk significantly. Unfortunately, the pathogenicity caused by two or more MMR gene variants in one carrier is thus far not possible to predict in silico and needs much more complicated and laborious functional studies.
Indeed, a recent functional study on a Saccharomyces cerevisiae-based system identified a number of weak alleles of MMR genes and MMR gene polymorphisms that are capable of interacting with other weak alleles of MMR genes to produce strong polygenic increases in mutation rates [Martinez and Kolodner, 2010]. Continuing this kind of analysis of possible compound contribution to MMR deficiency, in this study, we functionally analyzed 8 pairs of MMR gene VUS, altogether 14 different variants found as pairs in cancer patients.
Materials and Methods
Variations
This study consists of eight VUS pairs affecting MMR genes MSH2 (NM_000251.1) and/or MSH6 (NM_000179.2). Nucleotide numbering reflects cDNA numbering with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence, according to the guidelines (www.hgvs.org/mutnomen). The initiation codon is codon 1. Three VUS pairs are in the MSH2 gene: c.380A>G/c.982G>C (p.Asn127Ser/p.Ala328Pro) [Samowitz et al., 2001], c.613G>C/c. 1099G>A (p.Glu205Gln/p.Val367Ile) [Gargiulo et al., 2009], and c.965G>A/c.1461C>G (p.Gly322Asp/p.Asp487Glu) [Hampel et al., 2006]; two in the MSH6 gene: (c.1304T>C/c.2633T>C (p.Leu435Pro/p.Val878Ala) [Hampel et al., 2006] and c.1754T> C/c.2030G>C (p.Leu585Pro/p.Ser677Thr) [the present study]; and in three pairs there is one VUS in the MSH2 gene and one in the MSH6 gene in the same patient: c.2726A> T/c.2633T>C (p.Lys909Ile/p.Val878Ala) [the present study]; c.435T>G/c.4061T>A (p.Ile145Met/p.Leu1354Gln) [Kariola et al., 2003]; and c.435T>G/c.3284G>A (p.Ile145Met/p.Arg1095His) [Kariola et al., 2003]). Of these, the VUS pairs MSH2 c.2726A>T/c.2633T>C (p.Lys909Ile/p.Val878Ala) and MSH6 c.1754T>C/c.2030G>C (p.Leu585Pro/p.Ser677Thr) have not been reported before. Altogether 14 different variants were constructed and functionally analyzed separately and together with their pairs in an in vitro MMR assay. The alterations, in silico predictions of their pathogenicity, and tumor pathological data of the VUS carriers are presented in Table 1.
Table 1.
Data of Studied Variants of Uncertain Significance (VUS) and Carriers
| VUS pairs | Nucleotide changea | Protein alteration | In silicob
|
VUS carrier
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Tumor typec | Age of onset | MSId | IHCe
|
Reference | ||||||||
| P | S | M | MLH1 | MSH2 | MSH6 | |||||||
| MSH2/2 | c.380A>G | p.Asn127Ser | − | − | + | CRC | 65 | MSI-H | N/A | N/A | N/A | [Samowitz et al., 2001] |
| c.982G>C | p.Ala328Pro | − | + | − | ||||||||
| c.613G>C | p.Glu205Gln | + | + | + | PC | 59 | N/A | N/A | N/A | N/A | [Gargiulo et al., 2009] | |
| c.1099G>A | p.Val367Ile | + | + | + | ||||||||
| c.965G>A | p.Gly322Asp | + | + | + | EC | 57 | MSI-L | − | + | − (5%) | [Hampel et al., 2006] | |
| c.1461C>G | p.Asp487Glu | + | + | + | ||||||||
| MSH2/6 | c.435T>G | p.Ile145Met | + | + | − | CRC | 65/74 | MSI-H | + | + | + | [Kariola et al., 2003] |
| c.3284G>A | p.Arg1095His | − | + | N/A | ||||||||
| c.435T>G | p.Ile145Met | + | + | − | CRC | 53 | MSI-H | + | − | − | [Kariola et al., 2003] | |
| c.4061T>A | p.Leu1354Gln | − | + | N/A | ||||||||
| c.2726A>T | p.Lys909Ilef | − | − | + | CRC | 79 | MSI-H | − | + | + | [The present study] | |
| c.2633T>C | p.Val878Alaf | + | + | N/A | ||||||||
| MSH6/6 | c.1304T>C | p.Leu435Pro | − | − | N/A | EC | 59 | MSI-H | + | + | − | [Hampel et al., 2006] |
| c.2633T>C | p.Val878Ala | + | + | N/A | ||||||||
| c.1754T>C | p.Leu585Prof | − | − | N/A | CRC | 38 | MSI-H | + | + | − (eqv.) | [The present study] | |
| c.2030G>C | p.Ser677Thrf | + | + | N/A | ||||||||
Data of compound carriers.
Nucleotide numbering reflects cDNA numbering with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence, according to journal guidelines (www.hgvs.org/mutnomen). The initiation codon is codon 1. MSH2 (MIM# 609309, RefSeq NM_000251.1) and MSH6 (MIM# 600678, RefSeq NM_000179.2)
Pathogenicity predictions by P, Polyphen-2; S, SIFT; M, MAPP-MMR; +, neutral; −, deleterious.
CRC, colorectal cancer; EC, endometrial cancer; PC, pancreatic cancer.
Microsatellite instability: MSI-H, ≥3 markers indicating MSI; MSI-L, ≤2 markers indicating MSI.
Immunohistochemistry: +, present; −, absent; N/A, data not available; Eqv., equivocal.
MSI and IHC analysis for novel variant carriers performed as described previously by Hampel et al. (2005) and variant data can be found from www.insight-group.org.
In Silico Analysis by Multiple Sequence Alignment Algorithms
The functional effects of the individual variations were predicted with three different in silico alignment analyses. These computational analyses identify conserved areas of a gene through multiple sequence alignment analyses across numerous species, and thereafter, deduce possible functional defects caused by the variation. Because of their high sensitivity and specificity [Tavtigian et al., 2008] the in silico prediction algorithms chosen to analyze the possible effects of the individual variations in this study included sorting intolerant from tolerant (SIFT) [Ng and Henikoff, 2001] (http://sift.jcvi.org/), the multivariate analysis of protein polymorphism (MAPP-MMR) [Chao et al., 2008; Stone and Sidow, 2005] (http://mendel.standford.edu/SidowLab/), and polymorphism phenotyping (PolyPhen-2 [version 2.1.0; HumDiv]) [Adzhubei et al., 2010] (http://genetics.bwh.harvard.edu/pph/) as the MAPP-MMR algorithm is not compatible for MSH6 VUS predictions.
Cell Lines and Nuclear Extracts
Cancer cell lines HeLa and LoVo were cultured according to providers’ instructions (American Type Culture Collection, Manassas, VA, USA). MMR proficient HeLa serves as a positive assay control whereas MMR-deficient LoVo, which lacks MSH2, MSH3, and MSH6 proteins [Drummond et al., 1997; Cannavo et al., 2005] is used as a negative control.
Nuclear proteins from HeLa and LoVo cells were extracted as previously described [Lahue et al., 1989; Holmes et al., 1990; Kantelinen et al., 2010]. In brief, approximately 2–10 × 108 cells were collected and treated with 30–40 ml of cold isotonic buffer (20 mM Hepes pH 7.5, 5 mM KCl, 1.5 mM MgCl, 250 mM sucrose, 0.2 mM PMSF, 1x Complete EDTA-free protease inhibitor mixture (Roche, Mannheim, Germany), 0.25 μg/ml aprotinin, 0.7 μg/ml pepstatin, 0.5 μg/ml leupeptin, 1mM DTT). The cells were resuspended in cold hypotonic buffer (isotonic buffer without sucrose) followed by immediate pelleting. Approximately 1 ml per 1–2 × 108 cells of hypotonic buffer was used to disrupt the cell membranes with a syringe and a narrow gauge needle. The nuclei were collected by centrifugation and suspended in cold extraction buffer (25 mM Hepes pH 7.5, 10% sucrose, 1 mM PMSF, 0.5 mM DTT, 1 μg/ml leupeptin) and NaCl up to 155 mM by rotation in + 4°C for 1 hr. The supernatant was dialyzed for 2 hr against cold dialysis buffer (25 mM Hepes pH 7.5, 50 mM KCl, 0.1 mM EDTA pH 8, 10% sucrose, 1 mM PMSF, 2 mM DTT, 1 μg/ml leupeptin) and collected after further centrifugation.
Heteroduplex Preparation
Heteroduplex DNA molecule is a circular 3193 bp long molecule with a single strand nick 369 bp upstream from the site of the loop (5′IDL1). Site-directed mutagenesis was carried out according to manufacturer’s instructions (QuikChange Site-directed mutagenesis, Stratagene, La Jolla, CA, USA) to create the 1 nucleotide (delA) deletion to the positive pGEM IDL40 plasmid strand at the BglII restriction site. Single-stranded (ss) DNA was prepared by infecting pGEM IDL40 transformed XL1-blue bacteria cells with the M13K07 bacteriophage (Amersham Biosciences, Piscataway, NJ, USA), which replicates the antisense strand. ssDNA was extracted from the bacteriophages and used in excess to reanneal with the linearized plasmid DNA creating heteroduplex molecules. Plasmid-safe DNAse (Epicentre, Madison, WI, USA) and BND cellulose (Sigma–Aldrich, St. Louis, MO, USA) treatments were carried out to purify the final product. 5′IDL1 heteroduplex was used as a substrate in the in vitro MMR assay.
Site-Directed Mutagenesis
The MSH2 and MSH6 missense variations were constructed with a PCR-based site-directed mutagenesis kit according to manufacturer’s instructions (QuikChange Lightning® Site-directed mutagenesis Kit, Stratagene, La Jolla, CA, USA) to the MSH2 and MSH6 cDNA cloned into a pFastBac1 vector (Invitrogen, Carlsbad, CA, USA). Each VUS was constructed individually and with its pair into the same molecule (MSH2 or MSH6 cDNA) or into different molecules (MSH2 and MSH6 cDNAs). Specific nucleotide and amino acid changes for each of the variations assayed can be found in Table 1. Prior to protein production, the mutated MSH2 and MSH6 cDNA constructs were sequenced (ABIPrism 3100 Genetic Analyzer; Applied Biosystems, Foster City, CA, USA). The primer sequences and PCR parameters are available on request.
Production of Heterodimer Protein Complexes
The MutSα heterodimer contains MSH2 and MSH6 proteins that form a functional recognition unit for the mispaired nucleotides during MMR [Jiricny, 2006]. To produce these proteins, coinfection yielding such heterodimer protein complexes was used since both in vivo study in mice [de Wind et al., 1999] and in vitro studies in human cell [Chang et al., 2000; Marra et al., 1998] have shown that the MSH6 protein is unstable without its cognate partner MSH2. Proteins were produced in Spodoptera frugiperda Sf(9) insect cells as described earlier [Kariola et al., 2002; Ollila et al., 2006]. The produced heterodimer protein complexes included either one VUS (Fig. 1A) or VUS pair (Fig. 1B) in one of the partners (MSH2 or MSH6) together with its wild-type (WT) partner (MSH6 or MSH2, respectively) or one VUS in both partners (MSH2 and MSH6) (Fig. 1C).
Figure 1.
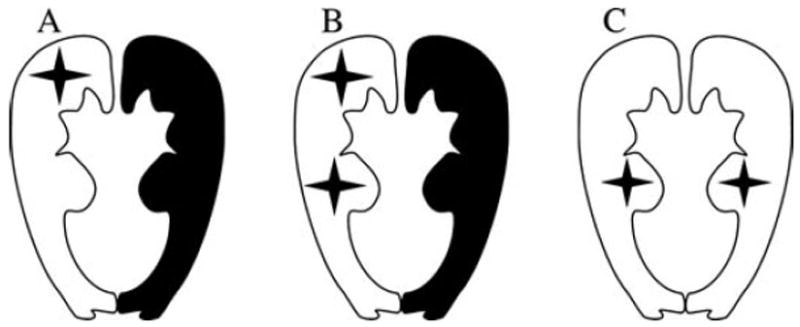
The MutSα heterodimer protein alternatives including either (A) one VUS or (B) a pair of VUS in one of the partners (MSH2 or MSH6) together with its WT partner (MSH6 or MSH2, respectively), or (C) one VUS in both partners (MSH2 and MSH6).
The amounts of produced proteins were estimated with a western blot analysis. The proteins were blotted onto nitrocellulose membranes (Hypond, PVDF, Amersham Pharmacia biotech, Uppsala, Sweden) and visualized with anti-MSH2 (Calbiochem, San Diego, CA, USA, MSH2- Ab1, NA-26) (0.2 μg/ml) and anti-MSH6 (BD Transduction Laboratories, Lexington, KY, USA, clone 44) (0.02 μg/ml) monoclonal antibodies.
The In Vitro MMR Assay
The repair efficiencies of the recombinant proteins were analysed by the in vitro MMR assay [Kantelinen et al., 2010; Nyström-Lahti et al., 2002] where the repair reactions were standardized to include 75 μg of MMR-deficient LoVo nuclear extract (NE) or an equal amount of MMR proficient HeLa extract with an excess amount of the heteroduplex DNA substrate (5′IDL1) set to 100 ng. The functionality of the WT and variant proteins was assayed by complementing LoVo NE with Sf9 total extracts (TE) including comparable amounts of MutSα heterodimer (when stable) in each sample. The amount of variant protein was determined by western blot analysis by adjusting the amount of its WT heterodimerization partner to be equal to that in the MutSα WT complex.
Because the clinical data of the families did not reveal the mode of inheritance, the study was designed to analyze two different possibilities, where the MSH2 or MSH6 VUS pairs are inherited either in a same gene allele (from the same parent) or in different alleles (from different parents). MOCK, which contains heteroduplex DNA with no added NE or recombinant protein, and MMR-deficient LoVo NE serve as negative controls and MMR proficient HeLa NE as well as LoVo NE complemented with MutSα WT serve as positive controls in the MMR assay. After analysis the substrates were linearized with Eco31I restriction enzyme. As the repair reaction fills the single nucleotide loop structure on the substrate, the BglII restriction site is recreated, and hence the repair efficiency (R%) can be measured by the efficiency of the double digestion. R% is measured using GeneTools 3.08 (SynGene, Cambridge, England) as the average of three independent assays. The relative R% was calculated in respect to the WT control as in previous work by Drost et al (2010). Statistical t-test analysis (one tailed, homoscedastic students t-test) was carried out to evaluate the significance of the R% differences between MutSα WT and the recombinant proteins.
Results
Clinical and Tumor Pathological Data of the VUS Carriers
As seen in Table 1, clinical data such as the tumor type, the age of onset, the MSI status, and the immunohistochemical (IHC) analysis of the tumors may represent the clinical phenotype caused by the coexistence of the two VUS, whereas the in silico predictions assess each VUS individually. Excluding the carrier of MSH6 p.Leu585Pro/p.Ser677Thr, all other compound VUS carriers have a relatively late age of cancer onset compared to that typically associated with LS. However, the tumor types such as CRC and endometrial cancer affecting most (7/8) of the carriers as well as the high MSI status seen in 6/8 cases belong to the typical LS tumor spectrum. Altogether, the families of 4 VUS pair carriers (MSH2/MSH2: p.Glu205Gln/p.Val367Ile; MSH2/MSH6: p.Lys909Ile/p.Val878Ala; MSH6/MSH6: p.Leu435Pro/p.Val878Ala and p.Leu585Pro/p.Ser677Thr fulfill the LS criteria, families of 2 carriers (MSH2/MSH6: p.Ile145Met/p.Leu1354Gln and p.Ile145Met/p.Arg1095His) do not fulfill, and of two families (MSH2/MSH2: p.Asn127Ser/p.Ala328Pro and p.Gly322Asp/p.Asp487Glu), the data is not available.
MMR Repair Deficiency Caused by the VUS Pairs
The amount of variant protein to be used in the in vitro MMR assay was determined by western blot analysis by adjusting the amount of its WT heterodimerization partner in MutSα to be equal to that in the MutSα WT complex (data not shown). Protein variants were analysed separately and together with the other VUS found in the same patient. Three types of pairs were tested—MSH2/MSH2, MSH2/MSH6, and MSH6/MSH6—with results shown in Figure 2.
Figure 2.
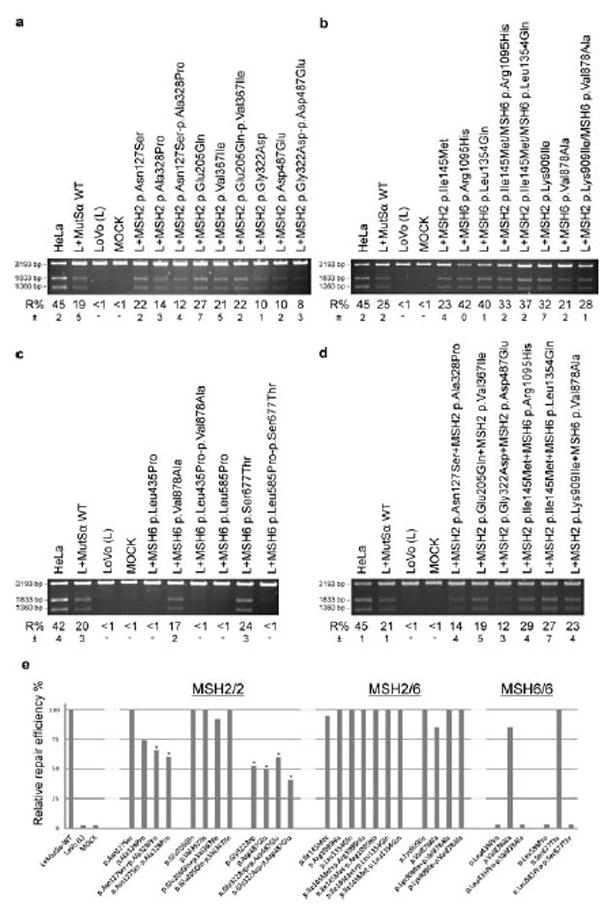
The in vitro MMR efficiency of the recombinant MutSα protein variants. Repair efficiency (R%) is measured as a percentage of the repaired double digested plasmid and calculated as the average of three independent assays, the standard deviation of which (±) is also marked. MMR proficient HeLa NE is used as a positive assay control, NE-free MOCK and uncomplemented MMR-deficient LoVo NE serve as negative controls, and LoVo NE complemented by MutSα WT serve as a positive control. A: MSH2 variations constructed into the same molecule (marked with −) and MutSα produced with MSH6 WT. B: MSH2/MSH6 variations produced into the same MutSα complexes. C: MSH6 variations constructed into the same molecule (−) and MutSα produced with MSH2 WT. D: MSH2 pairs or MSH6 pairs constructed into separate molecules (marked with +), MutSα produced with their cognate WT partners and assayed as compounds (1:1), mimicking the situation that the variations are inherited from different parents. E: The relative R% calculated in respect to the WT control. The R% of WT and other proficient MutSα complexes is marked as 100. Statistically significant (one tailed students t-test, n = 3) decrease in R% is marked with a star.
By comparing the relative repair efficiencies (Fig. 2E), of the MSH2/MSH2 VUS pairs p.Asn127Ser/p.Ala328Pro and p.Gly322Asp/p.Asp487Glu show significantly decreased repair efficiency when compared to that of the MutSα WT complex. When individually assayed, p.Asn127Ser was able to correct the mismatches as WT (22%, STD ±2% and 19%, STD ±5%, respectively) (P = 0.25), whereas the repair efficiency of p.Ala328Pro seems to be decreased (14%, STD ±3%), although not significantly (P = 0.11) (Fig. 2A, E). A statistically significant decrease in repair efficiency was seen in experiments, when the effect of these VUS pairs were tested in the same molecule, MSH2 p.Asn127Ser-p.Ala328Pro (12%, STD ±4%) and WT (19%, STD ±5%) (P = 0.04) (Fig. 2A, E) or in different molecules, MSH2 p.Asn127Ser+p.Ala328Pro (14%, STD ±4%) and WT (21%, STD ±1%) (P = 0.02) (Fig. 2D, E), while keeping the total amount of complementing recombinant protein at the level of MutSα WT. The decrease in repair efficiencies of MSH2 p.Gly322Asp and p.Asp487Glu proteins was significant when tested individually (10%, STD ±1% and 10%, STD ± 2%, respectively) (P = 0.02), as a pair in the same molecule MSH2 p.Gly322Asp-p.Asp487Glu (8%, STD ±3%) (P = 0.02) (Fig. 2A, E) or in different molecules MSH2 p.Gly322Asp+p.Asp487Glu (12%, STD ±3%) (P = 0.004) (Fig. 2D, E).
The pair MSH2/MSH2 p.Glu205Gln/p.Val367Ile and none of the variants in MSH2/MSH6 pairs including the novel pair p.Lys909Ile/p.Val878Ala show a significant decrease in the repair capability or any evidence of compound contribution to MMR deficiency (Fig. 2B, D, E). However, both MSH6/MSH6 pairs p.Leu435Pro/p.Val878Ala and p.Leu585Pro/p.Ser677Thr include one previously functionally uncharacterized VUS, p.Leu435Pro and p.Leu585Pro, respectively, which seem to be totally deficient in the in vitro MMR assay (Fig. 2C, E). Consequently, the repair capabilities of the pairs MSH6 p.Leu435Pro-p.Val878Ala and MSH6 p.Leu585Pro-p.Ser677Thr, including both VUS in the same molecule are also totally deficient (Fig. 2C, E) and the western blot analysis further revealed their instability in the total nuclear protein extract thus confirming the pathogenicity (Fig. 3).
Figure 3.
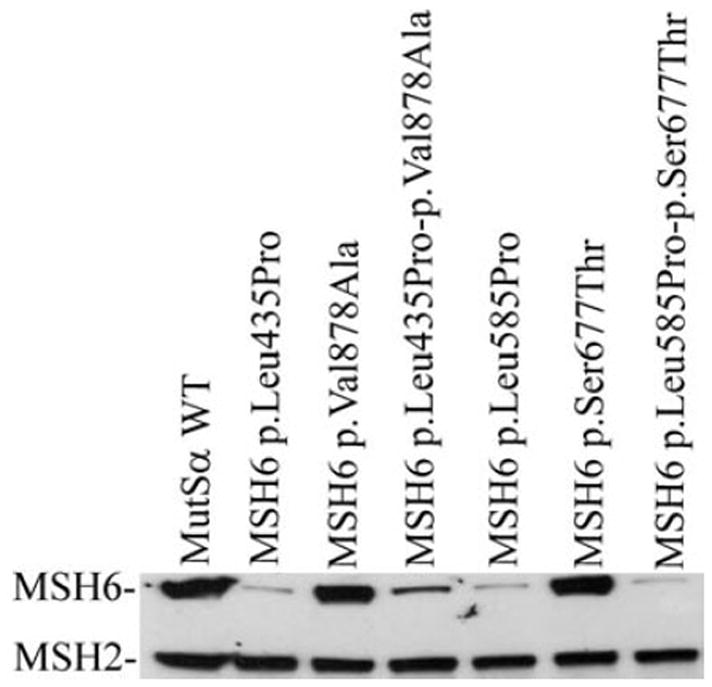
Western blot analysis of total protein extracts from Sf(9) cells coinfected with baculovirus constructs expressing MSH2 wild-type protein (MSH2 WT) with either MSH6 WT or with MSH6 variant(s) and showing instability of proteins MSH6 p.Leu435Pro and p.Leu585Pro, while MSH2 level stays the same as in MutSα WT.
Discussion
The functional analysis of MMR gene VUS as the VUS pairs allows the assessment of the potential compound effect caused by two VUS in one carrier. In the present study, of the eight analyzed VUS pairs, MSH2 p.Asn127Ser/p.Ala328Pro and MSH2 p.Gly322Asp/p.Asp487Glu show significantly decreased repair efficiency. In both pairs, one of the partners is a rare variation, whereas the other (p.Asn127Ser and p.Gly322Asp) is among the most frequently reported VUS in CRC [Hampel et al., 2006; Ollila et al., 2008]. Of these pairs, especially p.Asn127Ser/p.Ala328Pro suggests a concomitant contribution to the MMR deficiency. Although when individually assayed with the optimal amount of Sf9 total extract MSH2 p.Asn127Ser indicates proficiency, by halving its amount while maintaining the total recombinant MutSα amount in the assay at the same level as that of MutSα WT, MSH2 p.Asn127Ser cannot complement the deficiency caused by MSH2 p.Ala328Pro. Instead, their concomitant presence in the assay either in the same or different heterodimers slightly increases the MMR deficiency. According to our previous experiments, when MMR activity of MMR-deficient extract is complemented with different amounts of WT extract, its optimal amount can be reduced at least by a factor of 10 without notable reduction in the repair efficiency [Raevaara et al., 2003].
Thus, rather than haplo-insufficiency, the reason for the concomitant deficiency of MSH2 p.Asn127Ser/p.Ala328Pro is a functional defect in both. Although, MSH2 c.380A>G (p.Asn127Ser) is generally assessed as a nonpathogenic variation based on several different functional studies and healthy phenotype in many mutation carriers, it is among the most frequently reported VUS in CRC. Thus, all the previous data together with the present study suggest an extremely subtle MMR defect, which may not dominantly predispose to cancer but together with another inherited MMR gene variation like with a truncating mutation in MSH2 (c.1264G>T, Glu422X) as was reported by Tanyi et al. [Tanyi et al. 2006, 2008], the MSH2 c.380A>G (p.Asn127Ser) seems to increase the cancer risk, as in that study by decreasing the age of cancer onset into the early thirties.
Unfortunately, the original report of the CRC patient carrying the MSH2 variations c.380A>G and c.982G>C [Samowitz et al., 2001] does not tell the mutation carrier status of other cancer patients in the family to reveal if the latter variation could already alone predispose to cancer. The amino acid change in VUS MSH2 c.982G>C is predicted to be deleterious by Polyphen and MAPP-MMR alignment analyses and neutral by SIFT, while VUS MSH2 c.380A>G is predicted deleterious by Polyphen and SIFT, but not MAPP-MMR. The MutSα complex is an oval structure with allosteric communication between the DNA and ATP binding sites (Fig. 4). The substitutions p.Ala328Pro and p.Asn127Ser map to a helix in the lever domain and to a loop connecting domains 1 and 2, respectively. The distance between the substituted amino acids is fairly long (24 Å), and therefore their nonadditive effect is due to indirect interactions. Crystal structures with multiple substrates and normal mode analysis suggest that conserved domain motion is important for allostery [Mukherjee et al., 2009; Warren et al., 2007]. More particularly, domains 3 and 5 move together as a unit while domain 1 moves a lot during the reaction cycle. A kinked helix caused by the p.Ala328Pro mutation could affect the allosteric communication between the DNA and ATP binding domains, as the p.Ala328Pro mutation was already alone shown to impair activity. The substitution p.Asn127Ser alone in the hinge did not impair activity, so we may assume that all rotation states of domain 1 during the reaction cycle remain accessible. However, it is not impossible that a different transition path may be favoured in the presence of p.Asn127Ser. The nonadditive impairment seen in the double mutant would then be explained by blockage of also the alternative transition path between rotation states for domain 1 in the presence of also p.Ala328Pro. However, as long as the knowledge about the interactions and function of MutSα heterodimer molecules (one or more) with other players in the repair complex is under debate, this kind of concomitant contribution cannot be verified by biochemical experiments and rather serves as an example of how tricky the interpretation of the pathogenicity can be.
Figure 4.
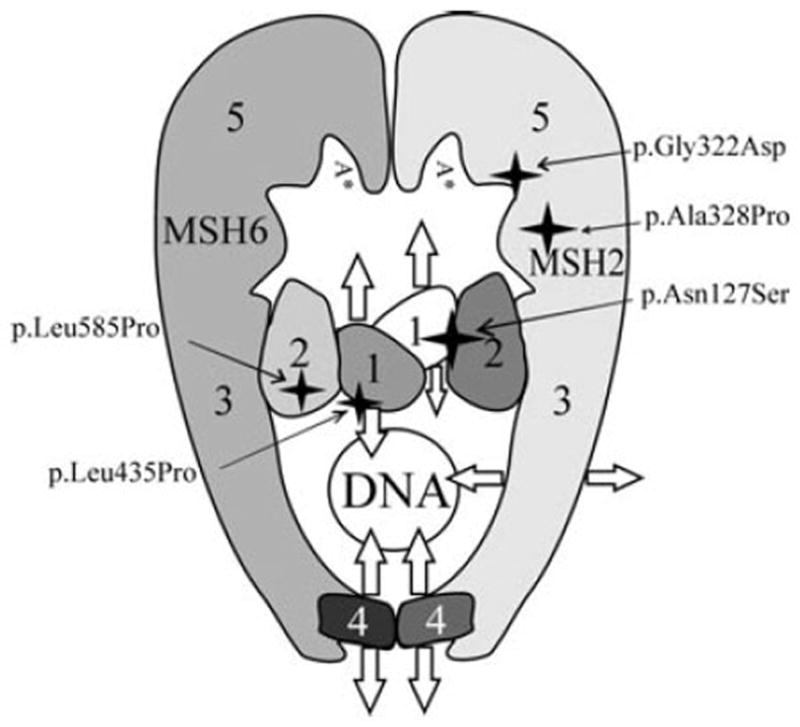
The three-dimensional structure of MutSα (adapted from [Warren et al., 2007] with permission) and locations of the mutations, MSH2: p.Asn127Ser, p.Gly322Asp, p.Ala328Pro and MSH6: p.Leu435Pro, p.Leu585Pro. MSH2 and MSH6 have similar domain architecture, consisting of a mismatch binding domain (1), a connector domain (2), a lever domain (3), a clamp domain (4), and an ATPase domain (5). Arrows indicate domain movements during the reaction cycle. Domains with the same shade move as rigid units.
The other MSH2/MSH2 variation pair, p.Gly322Asp/p.Asp487Glu, which shows significantly decreased repair efficiency, differs from the previous pair in that both variants also show decreased repair capability independently. MSH2 p.Asp487Glu has not been studied functionally before and, although the majority of the published data discusses p.Gly322Asp as a neutral polymorphism [Ollila et al., 2008; Martinez and Kolodner, 2010], it has also been hypothesized to be a low penetrance allele, supported by the functional analyses conducted with yeast assays [Drotschmann et al., 1999; Ellison et al., 2001]. Moreover, p.Gly322 is a conserved amino acid suggesting its importance. Irrespective of the recent study, where enhancer screens with the yeast homolog msh2 p.Gly317Asp did not yield enhancer mutations [Martinez and Kolodner, 2010] and our previous study, where the purified p.Gly322Asp variant did not show MMR deficiency [Ollila et al., 2008], here the total protein extract with over expressed MSH2 p.Gly322Asp individually and as a pair with p.Asp487Glu show a statistically significant decrease in repair efficiency. It is possible that the purification process has at least partly excluded structurally damaged heterodimers suggesting that the in vitro MMR assay performed with total extract is more reliable. Overall, it seems that results vary a lot dependent on the assay and assay path used for pathogenicity assessments. A recent and quite comprehensive study of the effect of MSH2 p.Gly322Asp on MMR in mouse embryonic stem cells (ESC) demonstrated that when expressed from its endogenous locus it behaved like WT MSH2 [Wielders et al., 2011]. The critical difference between that and the present study is that the functionality of MSH2 variant is assessed in undifferentiated stem cells or in human cell extracts originated from differentiated cancer cells, respectively. It is still unexplained why in Lynch syndrome a constitutional heterozygous MMR gene mutation predisposes to cancers only in some specific tissues or how a human embryo carrying a homozygous MMR gene mutation can succeed through all replications and recombinations occurring in several cell divisions and not lead to serious consequences until the first or second decade of life. Our results suggest that the in vitro MMR assay performed in human-based system and thus detecting defective protein function in its own environment and repair machinery may reveal problems not detectable in all other assay models.
Because the clinical data of the families did not reveal the type of inheritance, to analyze and compare these different possibilities in the present study, the MSH2/MSH2 and MSH6/MSH6 VUS pairs were constructed into the same molecule (allele) and different molecules. Although the locations of the two VUS did not significantly affect their repair efficiency, the repair efficiencies of MSH2 p.Asn127Ser/p.Ala328Pro and MSH2 p.Gly322Asp/p.Asp487Glu were lowest when constructed into the same allele suggesting stronger impairment on MSH2, MutSα, and its function in a repair complex.
Albeit, the other studied VUS pairs do not show a compound contribution to MMR deficiency, which as such is an important information to the families, two MSH6 variations p.Leu585Pro and p.Leu435Pro are, for the first time, shown to be functionally MMR-deficient in vitro. These MSH6 variations are also shown to be unstable when expressed in Sf(9) cells. Our results are compatible with results from IHC staining showing lack of MSH6 but presence of MSH2 and MLH1 in the VUS carriers’ tumors. Hence, the assessment of these two VUS as pathogenic mutations is based on several observations: the lack of the MSH6 protein in the tumor tissue, instability of the mutated protein in Sf(9) expression and MMR deficiency in the in vitro MMR assay as well as deficiency supported by the in silico analyses. Furthermore, MSH6 p.Leu435Pro has previously been shown to skip exon 4 already indicating its pathogenicity [Hampel et al., 2007]. Therefore, there are no additive effects seen when these two mutations are paired with other VUS in MSH6 since they sufficiently disrupt MMR function on their own.
When the ultimate aim in clinical work is to obtain a classification of the MMR VUS based on probability of being pathogenic as was proposed by using five probability classes from definitely pathogenic to not pathogenic or of no clinical significance [Plon et al., 2008], even small differences in repair capability such as is seen between MSH2 p.Ala328Pro alone and together with p.Asn127Ser become important. Although the validation assays and their cut-offs for decision making have not yet been determined and the critical level of needed repair capability in vivo depends on the circumstances, the concomitant defect of MSH2 p.Asn127Ser/p.Ala328Pro, which nearly halves the repair capability of MutSα WT, is most probably a cause of pathogenicity in the carrier. Especially interesting is the role of MSH2 c.380A>G, whose effect leading to even slightly increased cancer risk could finally explain discrepancy of its repeatedly analyzed proficiency albeit the frequent occurrence in CRC patients. As, epidemiologically, it might be of more significance to identify low-risk variations with high prevalence than high-risk rare variations in a population, it could be worth assessing the overall significance of MSH2 c.380A>G in CRC risk according to Bayes’ theorem.
Acknowledgments
Contract grant sponsor: Sigrid Juselius Foundation; European Research Council (2008-AdG-232635); Finnish Cancer Organizations; The Biocentrum Helsinki Organization; The Research Foundation of the University of Helsinki; and The Helsinki Graduate Program in Biotechnology and Molecular Biology.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavo E, Marra G, Sabates-Bellver J, Menigatti M, Lipkin SM, Fischer F, Cejka P, Jiricny J. Expression of the MutL homologue hMLH3 in human cells and its role in DNA mismatch repair. Cancer Res. 2005;65:10759–10766. doi: 10.1158/0008-5472.CAN-05-2528. [DOI] [PubMed] [Google Scholar]
- Chang DK, Ricciardiello L, Goel A, Chang CL, Boland CR. Steady-state regulation of the human DNA mismatch repair system. J Biol Chem. 2000;275:18424–18431. doi: 10.1074/jbc.M001140200. [DOI] [PubMed] [Google Scholar]
- Chao EC, Velasquez JL, Witherspoon MS, Rozek LS, Peel D, Ng P, Gruber SB, Watson P, Rennert G, Anton-Culver H, Lynch H, Lipkin SM. Accurate classification of MLH1/MSH2 missense variants with multivariate analysis of protein polymorphisms-mismatch repair (MAPP-MMR) Hum Mutat. 2008;29:852–860. doi: 10.1002/humu.20735. [DOI] [PubMed] [Google Scholar]
- Cyr JL, Heinen CD. Hereditary cancer-associated missense mutations in hMSH6 uncouple ATP hydrolysis from DNA mismatch binding. J Biol Chem. 2008;283:31641–31648. doi: 10.1074/jbc.M806018200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wind N, Dekker M, Claij N, Jansen L, van Klink Y, Radman M, Riggins G, van der Valk M, van’t Wout K, te Riele H. HNPCC-like cancer predisposition in mice through simultaneous loss of Msh3 and Msh6 mismatch-repair protein functions. Nat Genet. 1999;23:359–362. doi: 10.1038/15544. [DOI] [PubMed] [Google Scholar]
- Drost M, Zonneveld JB, van Dijk L, Morreau H, Tops CM, Vasen HF, Wijnen JT, de Wind N. A cell-free assay for the functional analysis of variants of the mismatch repair protein MLH1. Hum Mutat. 2010;31:247–253. doi: 10.1002/humu.21180. [DOI] [PubMed] [Google Scholar]
- Drotschmann K, Clark AB, Kunkel TA. Mutator phenotypes of common polymorphisms and missense mutations in MSH2. Curr Biol. 1999;9:907–910. doi: 10.1016/s0960-9822(99)80396-0. [DOI] [PubMed] [Google Scholar]
- Drummond JT, Genschel J, Wolf E, Modrich P. DHFR/MSH3 amplification in methotrexate-resistant cells alters the hMutSalpha/hMutSbeta ratio and reduces the efficiency of base-base mismatch repair. Proc Natl Acad Sci USA. 1997;94:10144–10149. doi: 10.1073/pnas.94.19.10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison AR, Lofing J, Bitter GA. Functional analysis of human MLH1 and MSH2 missense variants and hybrid human-yeast MLH1 proteins in Saccharomyces cerevisiae. Hum Mol Genet. 2001;10:1889–1900. doi: 10.1093/hmg/10.18.1889. [DOI] [PubMed] [Google Scholar]
- Felton KE, Gilchrist DM, Andrew SE. Constitutive deficiency in DNA mismatch repair. Clin Genet. 2007;71:483–498. doi: 10.1111/j.1399-0004.2007.00803.x. [DOI] [PubMed] [Google Scholar]
- Gammie AE, Erdeniz N, Beaver J, Devlin B, Nanji A, Rose MD. Functional characterization of pathogenic human MSH2 missense mutations in Saccharomyces cerevisiae. Genetics. 2007;177:707–721. doi: 10.1534/genetics.107.071084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gargiulo S, Torrini M, Ollila S, Nasti S, Pastorino L, Cusano R, Bonelli L, Battistuzzi L, Mastracci L, Bruno W, Savarino V, Sciallero S, et al. Germline MLH1 and MSH2 mutations in italian pancreatic cancer patients with suspected lynch syndrome. Fam Cancer. 2009;8:547–553. doi: 10.1007/s10689-009-9285-1. [DOI] [PubMed] [Google Scholar]
- Goldgar DE, Easton DF, Byrnes GB, Spurdle AB, Iversen ES, Greenblatt MS IARC Unclassified Genetic Variants Working Group. Genetic evidence and integration of various data sources for classifying uncertain variants into a single model. Hum Mutat. 2008;29:1265–1272. doi: 10.1002/humu.20897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, Nakagawa H, Sotamaa K, Prior TW, Westman J, Panescu J, Fix D, et al. Screening for the lynch syndrome (hereditary nonpolyposis colorectal cancer) N Engl J Med. 2005;352:1851–1860. doi: 10.1056/NEJMoa043146. [DOI] [PubMed] [Google Scholar]
- Hampel H, Frankel W, Panescu J, Lockman J, Sotamaa K, Fix D, Comeras I, La Jeunesse J, Nakagawa H, Westman JA, Prior TW, Clendenning M, et al. Screening for lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res. 2006;66:7810–7817. doi: 10.1158/0008-5472.CAN-06-1114. [DOI] [PubMed] [Google Scholar]
- Hampel H, Panescu J, Lockman J, Sotamaa K, Fix D, Comeras I, LaJeunesse J, Nakagawa H, Westman JA, Prior TW, Clendenning M, de la Chapelle A, et al. Comment on: screening for lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res. 2007;67:9603. doi: 10.1158/0008-5472.CAN-07-2308. [DOI] [PubMed] [Google Scholar]
- Holmes J, Jr, Clark S, Modrich P. Strand-specific mismatch correction in nuclear extracts of human and drosophila melanogaster cell lines. Proc Natl Acad Sci USA. 1990;87:5837–5841. doi: 10.1073/pnas.87.15.5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol. 2006;7:335–346. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- Kantelinen J, Kansikas M, Korhonen MK, Ollila S, Heinimann K, Kariola R, Nyström M. MutSbeta exceeds MutSalpha in dinucleotide loop repair. Br J Cancer. 2010;102:1068–1073. doi: 10.1038/sj.bjc.6605531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariola R, Otway R, Lönnqvist KE, Raevaara TE, Macrae F, Vos YJ, Kohonen-Corish M, Hofstra RM, Nyström-Lahti M. Two mismatch repair gene mutations found in a colon cancer patient—which one is pathogenic? Hum Genet. 2003;112:105–109. doi: 10.1007/s00439-002-0866-4. [DOI] [PubMed] [Google Scholar]
- Kariola R, Raevaara TE, Lönnqvist KE, Nyström-Lahti M. Functional analysis of MSH6 mutations linked to kindreds with putative hereditary non-polyposis colorectal cancer syndrome. Hum Mol Genet. 2002;11:1303–1310. doi: 10.1093/hmg/11.11.1303. [DOI] [PubMed] [Google Scholar]
- Korhonen MK, Vuorenmaa E, Nyström M. The first functional study of MLH3 mutations found in cancer patients. Genes Chromosomes Cancer. 2008;47:803–809. doi: 10.1002/gcc.20581. [DOI] [PubMed] [Google Scholar]
- Lahue RS, Au KG, Modrich P. DNA mismatch correction in a defined system. Science. 1989;245:160–164. doi: 10.1126/science.2665076. [DOI] [PubMed] [Google Scholar]
- Liu HX, Zhou XL, Liu T, Werelius B, Lindmark G, Dahl N, Lindblom A. The role of hMLH3 in familial colorectal cancer. Cancer Res. 2003;63:1894–1899. [PubMed] [Google Scholar]
- Lynch HT, Boland CR, Rodriguez-Bigas MA, Amos C, Lynch JF, Lynch PM. Who should be sent for genetic testing in hereditary colorectal cancer syndromes? J Clin Oncol. 2007;25:3534–3542. doi: 10.1200/JCO.2006.10.3119. [DOI] [PubMed] [Google Scholar]
- Marra G, Iaccarino I, Lettieri T, Roscilli G, Delmastro P, Jiricny J. Mismatch repair deficiency associated with overexpression of the MSH3 gene. Proc Natl Acad Sci USA. 1998;95:8568–8573. doi: 10.1073/pnas.95.15.8568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez SL, Kolodner RD. Functional analysis of human mismatch repair gene mutations identifies weak alleles and polymorphisms capable of polygenic interactions. Proc Natl Acad Sci USA. 2010;107:5070–5075. doi: 10.1073/pnas.1000798107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Law SM, Feig M. Deciphering the mismatch recognition cycle in MutS and MSH2-MSH6 using normal-mode analysis. Biophys J. 2009;96:1707–1720. doi: 10.1016/j.bpj.2008.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyström-Lahti M, Perrera C, Räschle M, Panyushkina-Seiler E, Marra G, Curci A, Quaresima B, Costanzo F, D’Urso M, Venuta S, Jiricny J. Functional analysis of MLH1 mutations linked to hereditary nonpolyposis colon cancer. Genes Chromosomes Cancer. 2002;33:160–167. [PubMed] [Google Scholar]
- Ollila S, Dermadi Bebek D, Greenblatt M, Nyström M. Uncertain pathogenicity of MSH2 variants N127S and G322D challenges their classification. Int J Cancer. 2008;123:720–724. doi: 10.1002/ijc.23573. [DOI] [PubMed] [Google Scholar]
- Ollila S, Sarantaus L, Kariola R, Chan P, Hampel H, Holinski-Feder E, Macrae F, Kohonen-Corish M, Gerdes AM, Peltomäki P, Mangold E, de la Chapelle A, et al. Pathogenicity of MSH2 missense mutations is typically associated with impaired repair capability of the mutated protein. Gastroenterology. 2006;131:1408–1417. doi: 10.1053/j.gastro.2006.08.044. [DOI] [PubMed] [Google Scholar]
- Plon SE, Eccles DM, Easton D, Foulkes WD, Genuardi M, Greenblatt MS, Hoger-vorst FB, Hoogerbrugge N, Spurdle AB, Tavtigian SV IARC Unclassified Genetic Variants Working Group. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat. 2008;29:1282–1291. doi: 10.1002/humu.20880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raevaara TE, Korhonen MK, Lohi H, Hampel H, Lynch E, Lonnqvist KE, Holinski-Feder E, Sutter C, McKinnon W, Duraisamy S, Gerdes AM, Peltomäki P, et al. Functional significance and clinical phenotype of nontruncating mismatch repair variants of MLH1. Gastroenterology. 2005;129:537–549. doi: 10.1016/j.gastro.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Raevaara TE, Vaccaro C, Abdel-Rahman WM, Mocetti E, Bala S, Lonnqvist KE, Kariola R, Lynch HT, Peltomäki P, Nyström-Lahti M. Pathogenicity of the hereditary colorectal cancer mutation hMLH1 del616 linked to shortage of the functional protein. Gastroenterology. 2003;125:501–509. doi: 10.1016/s0016-5085(03)00905-3. [DOI] [PubMed] [Google Scholar]
- Samowitz WS, Curtin K, Lin HH, Robertson MA, Schaffer D, Nichols M, Gruenthal K, Leppert MF, Slattery ML. The colon cancer burden of genetically defined hereditary nonpolyposis colon cancer. Gastroenterology. 2001;121:830–838. doi: 10.1053/gast.2001.27996. [DOI] [PubMed] [Google Scholar]
- Stone EA, Sidow A. Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity. Genome Res. 2005;15:978–986. doi: 10.1101/gr.3804205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanyi M, Olasz J, Kamory E, Csuka O, Tanyi JL, Ress Z, Damjanovich L. Difficulties in recognizing families with hereditary non-polyposis colorectal carcinoma. presentation of 4 families with proven mutation. Eur J Surg Oncol. 2008;34:1322–1327. doi: 10.1016/j.ejso.2008.01.006. [DOI] [PubMed] [Google Scholar]
- Tanyi M, Olasz J, Lukacs G, Csuka O, Toth L, Szentirmay Z, Ress Z, Barta Z, Tanyi JL, Damjanovich L. Pedigree and genetic analysis of a novel mutation carrier patient suffering from hereditary nonpolyposis colorectal cancer. World J Gastroenterol. 2006;12:1192–1197. doi: 10.3748/wjg.v12.i8.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavtigian SV, Greenblatt MS, Lesueur F, Byrnes GB IARC Unclassified Genetic Variants Working Group. In silico analysis of missense substitutions using sequence-alignment based methods. Hum Mutat. 2008;29:1327–1336. doi: 10.1002/humu.20892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren JJ, Pohlhaus TJ, Changela A, Iyer RR, Modrich PL, Beese LS. Structure of the human MutSalpha DNA lesion recognition complex. Mol Cell. 2007;26:579–592. doi: 10.1016/j.molcel.2007.04.018. [DOI] [PubMed] [Google Scholar]
- Wielders EA, Dekker RJ, Holt I, Morris GE, te Riele H. Characterization of MSH2 variants by endogenous gene modification in mouse embryonic stem cells. Hum Mutat. 2011;32:389–396. doi: 10.1002/humu.21448. [DOI] [PubMed] [Google Scholar]
- Wu Y, Berends MJ, Sijmons RH, Mensink RG, Verlind E, Kooi KA, van der Sluis T, Kempinga C, van dDer Zee AG, Hollema H, Buys CH, Kleibeuker JH, et al. A role for MLH3 in hereditary nonpolyposis colorectal cancer. Nat Genet. 2001;29:137–138. doi: 10.1038/ng1001-137. [DOI] [PubMed] [Google Scholar]