

The vascular endothelium, the major interface of the vessel wall between blood and all tissues, must constantly adapt to fluctuations in environmental cues. Cellular metabolism is one mechanism that may contribute to rapid dynamic changes in cell phenotype. For example, glycolysis – one of the major forms of cellular metabolism that converts glucose to pyruvate–is required for immune cell activation, tumor cell proliferation and survival, and the phenotypic switch of endothelial cells (ECs) between quiescent and angiogenic states.1–8 ECs are exposed to a variety of stimuli such as turbulent/disturbed shear stress and hypoxia under pathophysiological states. To maintain homeostatic control of ECs, gene expression in ECs is subjected to highly tight regulation at multiple levels including transcriptional and epigenetic control. Exposure of vascular ECs to athero-protective laminar shear stress promotes anti-inflammatory, anti-thrombotic, and anti-oxidative properties largely through inducing the expression of a cassette of transcriptional regulators including Kruppel-like factor-2 (KLF2).9 Biomechanical stimuli also contribute to a resting quiescent state in ECs.10 However, the underlying mechanisms by which biomechanical stimuli such as laminar shear stress regulate cellular metabolism, including glycolysis and mitochondrial content, to maintain this resting metabolic state in ECs remains poorly understood.
In this issue of Arteriosclerosis, Thrombosis, and Vascular Biology, Doddaballapur et al.11 present elegant studies addressing the role of laminar shear stress in cellular metabolism of ECs. They report that laminar shear stress reduced EC glycolysis by regulating the expression of KLF2 and phosphofructokinase-2/fructose-2,6-bisphosphatase-3 (PFKFB3), an effect that maintained the quiescent metabolic state of ECs and inhibited angiogenesis (Figure). The authors demonstrated that laminar shear stress reduced glucose uptake in ECs in a KLF2-dependent manner as supported by siRNA-mediated knockdown studies of KLF2 demonstrating complete abrogation of laminar shear stress-induced reduction of glucose uptake. Overexpression of exogenous KLF2 reduced glucose uptake, lactate production, glycolysis, ATP levels, mitochondrial content, and basal mitochondrial respiration in ECs. Gain- and loss-of-function studies of PFKFB3 demonstrated that KLF2 reduced glycolysis at least partially depending on its repressive effect on PFKFB3 expression. To determine how KLF2 inhibits PFKFB3 expression, the authors scanned the PFKFB3 promoter region for potential KLF2 binding sites, and found KLF2 binds to the PFKFB3 promoter at ~14 bp upstream to the transcription start site, which mediated the repressive effect of KLF2. Transducing KLF2 into ECs reduced EC sprouting and network formation, a phenotype that is partially rescued by exogenously expressed PFKFB3. These effects appear to be independent of KLF2’s known regulatory effects on eNOS expression and nitric oxide (NO) production or 5′ adenosine monophosphate-activated protein kinase alpha 1 (AMPK-α1) expression. Furthermore, KLF2 does not increase EC senescence or apoptosis in vitro as quantified by β-galactosidase activity, p21 expression, cell cycle profiling, and caspase 3/7 and annexin staining. This study not only adds a new layer of complexity to the growing list of protective functions exerted by the master regulator KLF2, but also raises several provocative questions. While KLF2 may contribute to maintaining EC metabolic quiescence under laminar flow via PFKFB3 repression, is loss of KLF2 expression, whether by turbulent/disturbed shear stress (ie. as observed at vessel branch points) or by biochemical stimuli (ie. cytokines), necessary to increase EC metabolism in vivo? Under more stringent conditions of hyperglycemia, hyperlipidemia, and/or hypertension, is this EC resting metabolic state lost in a KLF2-dependent manner? Does KLF2-mediated regulation of PFKFB3 occur in the microvasculature where shear stress forces may not be as dominant as found in the macrovasculature? Given KLF2’s anti-angiogenic effects in ECs, would increased KLF2 expression confer deleterious effects in response to ischemic conditions? Conversely, will inhibition of KLF2 or overexpression of PFKFB3 expression rescue impaired angiogenesis found in relevant cardiovascular disease states such as myocardial or limb ischemia, or diabetic wound healing? Collectively, this study paves the way for new directions with considerable scientific interest.
Figure. The regulation of endothelial cellular metabolism by shear stress through Kruppel-like factor 2 (KLF2) and phosphofructokinase-2/fructose-2,6-bisphosphatase-3 (PFKFB3).
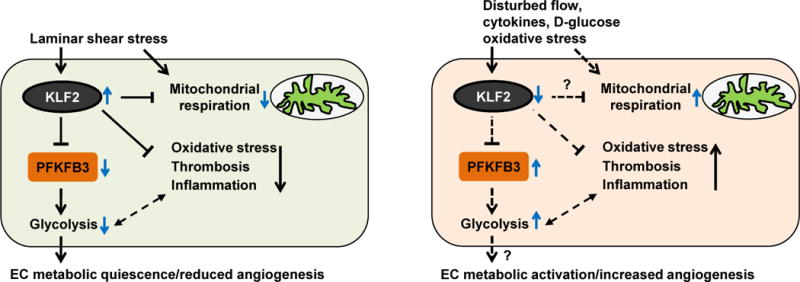
Laminar shear stress reduces endothelial cell (EC) glycolysis by repressing the expression of PFKFB3 in a KLF2-dependent manner, an effect that maintains the quiescent metabolic state of ECs and inhibits angiogenesis. Both KLF2 and laminar shear stress are involved in the regulation of mitochondrial respiration. The effect of laminar shear stress on mitochondrial respiration may depend on the induction of KLF2 or additional unknown regulators. KLF2 also confers EC anti-inflammatory, anti-thrombotic, and anti-oxidative properties. In regions of arteries where ECs are exposed to disturbed flow, or in response to biochemical stimuli such as cytokines, high glucose, and oxidative stress, KLF2 expression is reduced, which contributes to EC activation. Loss of the repressive function of KLF2 on PFKFB3 may induce PFKFB3 expression and increase glycolysis, an effect that may switch quiescent ECs to metabolically active ECs and induce angiogenesis.
In this study, both laminar shear stress and exogenously expressed KLF2 reduced mitochondrial content. KLF2 overexpression also reduced both mitochondrial respiration (decrease in oxygen consumption) and glycolysis (decrease in lactate production) (Figure). While both KLF2 and laminar flow reduced mitochondrial metabolism, the authors state that the laminar flow-dependent decrease was independent of KLF2, suggesting that additional regulators may be involved. Interestingly, PFKFB3 knockdown reduced glycolysis but not cellular oxygen consumption, suggesting that PFKFB3 did not regulate mitochondrial respiration and other metabolic molecules may be involved. Reactive oxygen species, which often contribute to endothelial dysfunction, is considered a major culprit of altered mitochondrial respiration.12–14 Because there was not a compensatory increase in oxidation and mitochondrial respiration when glycolysis was compromised after KLF2 overexpression or PFKFB3 deficiency, this suggests that both KLF2 and PFKFB3 could be targeted to control EC cellular metabolism without inducing oxidative stress. Surprisingly, while KLF2 overexpression in ECs increased eNOS expression and activity, KLF2-derived nitric oxide (NO) did not alter mitochondrial respiration as assessed using the nitric oxide inhibitor L-NAME. It remains unclear why ECs would not take advantage of the known inhibitory effect of NO on mitochondrial respiration to contribute to the KLF2-mediated inhibition of mitochondrial activity? It is possible that under different conditions or use of L-NAME in more than a single time point that effects of KLF2-derived NO on mitochondrial respiration might become apparent. Nonetheless, it raises the question of the dispensability of eNOS in this process – for example, will siRNA-mediated knockdown of eNOS have adverse effects on glycolysis in response to laminar flow or KLF2 overexpression?
Atherosclerotic lesions preferentially develop at arterial branch points, bifurcations, and the lesser curvature of the aorta where the vascular endothelium is exposed to disturbed flow.15–17 In these regions, KLF2 expression is low in endothelium of swine.18 It would be informative to examine whether PFKFB3 expression is inversely increased in these athero-susceptible regions, an effect that may reflect a switch in glycolytic state. In ECs, KLF2 confers anti-inflammatory effects in part by attenuating NF-κB signaling and attendant expression of proadhesive and prothrombotic genes. KLF2 has been implicated in maintaining quiescence of several leukocyte subsets.19 Indeed, myeloid-specific KLF2 protects mice against the development of atherosclerosis by virtue of preventing the activation and adhesion properties of macrophages and neutrophils to ECs.20 Given that aerobic glycolysis is also required for immune responses,1, 2, 21 these observations raise the potential causal link between altered glycolysis and inflammatory responses at predisposing regions of the vasculature that may progress to atherosclerotic lesion formation.
Mechanistically, the authors identified PFKFB3, hexokinase (HK), and phosphofructokinase-1 (PFK-1), as KLF2–repressed glycolytic genes. In contrast to PFKFB3, HK and PFK did not appear to be required for endothelial glycolysis in their studies. PFKFB3, identified over a decade ago22, 23 as a hypoxia-inducible gene, is stimulated through HIF-1alpha interaction with the consensus hypoxia response element in its promoter region.24 PFKFB3 generates fructose-2,6-bisphosphate, a key allosteric activator of PFK-1, itself a key rate-limiting enzyme in glycolysis that converts fructose-6-phosphate (F6P) to frustose-1,6,-phosphate. PFKFB3 is the highest expressed of these isoenzymes in ECs and exhibits >700-fold higher kinase activity compared to its phosphatase activity.3 Accumulating recent studies demonstrate PFKFB3 as a potential therapeutic target for pathological angiogenesis as well as other disease states. PFKFB3 promotes vessel formation, whereas PFKFB3 blockade inhibits pathological angiogenesis.3, 4 However, reduced PFKFB3 expression may have antithetical effects on inflammation in a range of cell types. For example, PFKFB3 inhibition attenuates T cell activation in vitro and suppresses T cell-dependent immunity in vivo5. Similarly, in human monocytes, PFKFB3 is rapidly induced by proinflammatory stimuli such as LPS.25 PFKFB3 expression is also induced in skeletal muscle of C57BL/6 mice by high-fat diet.26 However, PFKFB3 expression was decreased in epididymal fat from diabetic db/db mice.27 Intriguingly, knockdown of PFKFB3 in 3T3-L1 adipocytes reduced glucose incorporation into lipid, but increased the production of reactive oxygen species, which is associated with the activation of inflammatory signaling pathways.28 Moreover, PFKFB3 deficiency in mice exacerbated adipose tissue inflammatory response and systemic insulin resistance.28 In contrast, PFKFB3 overexpression in adipose tissue protects mice against diet-induced insulin resistance and inflammatory responses.29 As such, caution is required for considering PFKFB3 as a therapeutic target and additional studies are required to clarify the role of PFKFB3 in cardiovascular relevant cell types.
In summary, Doddaballapur et al.11 report a fascinating study linking laminar shear stress, KLF2, and endothelial cellular metabolism (Figure). These findings remind us how complicated the maintenance of EC quiescence can be in response to blood flow and shear forces. Given the role of a range of identified epigenetic regulators in response to laminar flow, including microRNAs,30, 31 it will be interesting to explore whether these factors impact KLF2-PFKFB3-dependent EC quiescence. Finally, whether the inhibitory effect on cellular metabolism mediated by the KLF2-PFKFB3 signaling axis is operative in other relevant cell types and in vivo in experimental models of vascular disease states in rodents and human tissues warrants further investigation.
Acknowledgments
Funding
This work was supported by funding from the National Institutes of Health (HL115141 and HL117994) to M.W.F.
Footnotes
Disclosures: None.
References
- 1.Chang CH, Curtis JD, Maggi LB, Jr, Faubert B, Villarino AV, O’Sullivan D, Huang SC, van der Windt GJ, Blagih J, Qiu J, Weber JD, Pearce EJ, Jones RG, Pearce EL. Posttranscriptional control of t cell effector function by aerobic glycolysis. Cell. 2013;153:1239–1251. doi: 10.1016/j.cell.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, Giamarellos-Bourboulis EJ, Martens JH, Rao NA, Aghajanirefah A, Manjeri GR, Li Y, Ifrim DC, Arts RJ, van der Meer BM, Deen PM, Logie C, O’Neill LA, Willems P, van de Veerdonk FL, van der Meer JW, Ng A, Joosten LA, Wijmenga C, Stunnenberg HG, Xavier RJ, Netea MG. Mtor- and hif-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014;345:1250684. doi: 10.1126/science.1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, Quaegebeur A, Ghesquiere B, Cauwenberghs S, Eelen G, Phng LK, Betz I, Tembuyser B, Brepoels K, Welti J, Geudens I, Segura I, Cruys B, Bifari F, Decimo I, Blanco R, Wyns S, Vangindertael J, Rocha S, Collins RT, Munck S, Daelemans D, Imamura H, Devlieger R, Rider M, Van Veldhoven PP, Schuit F, Bartrons R, Hofkens J, Fraisl P, Telang S, Deberardinis RJ, Schoonjans L, Vinckier S, Chesney J, Gerhardt H, Dewerchin M, Carmeliet P. Role of pfkfb3-driven glycolysis in vessel sprouting. Cell. 2013;154:651–663. doi: 10.1016/j.cell.2013.06.037. [DOI] [PubMed] [Google Scholar]
- 4.Schoors S, De Bock K, Cantelmo AR, Georgiadou M, Ghesquiere B, Cauwenberghs S, Kuchnio A, Wong BW, Quaegebeur A, Goveia J, Bifari F, Wang X, Blanco R, Tembuyser B, Cornelissen I, Bouche A, Vinckier S, Diaz-Moralli S, Gerhardt H, Telang S, Cascante M, Chesney J, Dewerchin M, Carmeliet P. Partial and transient reduction of glycolysis by pfkfb3 blockade reduces pathological angiogenesis. Cell metabolism. 2014;19:37–48. doi: 10.1016/j.cmet.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 5.Telang S, Clem BF, Klarer AC, Clem AL, Trent JO, Bucala R, Chesney J. Small molecule inhibition of 6-phosphofructo-2-kinase suppresses t cell activation. Journal of translational medicine. 2012;10:95. doi: 10.1186/1479-5876-10-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 7.Porporato PE, Dhup S, Dadhich RK, Copetti T, Sonveaux P. Anticancer targets in the glycolytic metabolism of tumors: A comprehensive review. Frontiers in pharmacology. 2011;2:49. doi: 10.3389/fphar.2011.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akhmedov AT, Rybin V, Marin-Garcia J. Mitochondrial oxidative metabolism and uncoupling proteins in the failing heart. Heart failure reviews. 2014 doi: 10.1007/s10741-014-9457-4. [DOI] [PubMed] [Google Scholar]
- 9.Nayak L, Lin Z, Jain MK. “Go with the flow”: How kruppel-like factor 2 regulates the vasoprotective effects of shear stress. Antioxidants & redox signaling. 2011;15:1449–1461. doi: 10.1089/ars.2010.3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Traub O, Berk BC. Laminar shear stress: Mechanisms by which endothelial cells transduce an atheroprotective force. Arteriosclerosis, thrombosis, and vascular biology. 1998;18:677–685. doi: 10.1161/01.atv.18.5.677. [DOI] [PubMed] [Google Scholar]
- 11.Doddaballapur A, Michalik KM, Manavski Y, Lucas T, Houtkooper RH, You X, Chen W, Zeiher AM, Potente M, Dimmeler S, Boon RA. Laminar shear stress inhibits endothelial cell metabolism via kruppel-like factor 2-mediated repression of 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase-3. Arteriosclerosis, thrombosis, and vascular biology. 2014 doi: 10.1161/ATVBAHA.114.304277. [DOI] [PubMed] [Google Scholar]
- 12.Turrens JF. Mitochondrial formation of reactive oxygen species. The Journal of physiology. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koopman WJ, Nijtmans LG, Dieteren CE, Roestenberg P, Valsecchi F, Smeitink JA, Willems PH. Mammalian mitochondrial complex i: Biogenesis, regulation, and reactive oxygen species generation. Antioxidants & redox signaling. 2010;12:1431–1470. doi: 10.1089/ars.2009.2743. [DOI] [PubMed] [Google Scholar]
- 14.Bratic A, Larsson NG. The role of mitochondria in aging. The Journal of clinical investigation. 2013;123:951–957. doi: 10.1172/JCI64125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davies PF, Polacek DC, Handen JS, Helmke BP, DePaola N. A spatial approach to transcriptional profiling: Mechanotransduction and the focal origin of atherosclerosis. Trends in biotechnology. 1999;17:347–351. doi: 10.1016/s0167-7799(99)01348-7. [DOI] [PubMed] [Google Scholar]
- 16.Chiu JJ, Chien S. Effects of disturbed flow on vascular endothelium: Pathophysiological basis and clinical perspectives. Physiological reviews. 2011;91:327–387. doi: 10.1152/physrev.00047.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davies PF, Civelek M, Fang Y, Fleming I. The atherosusceptible endothelium: Endothelial phenotypes in complex haemodynamic shear stress regions in vivo. Cardiovascular research. 2013;99:315–327. doi: 10.1093/cvr/cvt101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fang Y, Davies PF. Site-specific microrna-92a regulation of kruppel-like factors 4 and 2 in atherosusceptible endothelium. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:979–987. doi: 10.1161/ATVBAHA.111.244053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao Z, Sun X, Icli B, Wara AK, Feinberg MW. Role of kruppel-like factors in leukocyte development, function, and disease. Blood. 2010;116:4404–4414. doi: 10.1182/blood-2010-05-285353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lingrel JB, Pilcher-Roberts R, Basford JE, Manoharan P, Neumann J, Konaniah ES, Srinivasan R, Bogdanov VY, Hui DY. Myeloid-specific kruppel-like factor 2 inactivation increases macrophage and neutrophil adhesion and promotes atherosclerosis. Circulation research. 2012;110:1294–1302. doi: 10.1161/CIRCRESAHA.112.267310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cortese M, Sinclair C, Pulendran B. Translating glycolytic metabolism to innate immunity in dendritic cells. Cell metabolism. 2014;19:737–739. doi: 10.1016/j.cmet.2014.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamilton JA, Callaghan MJ, Sutherland RL, Watts CK. Identification of prg1, a novel progestin-responsive gene with sequence homology to 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase. Mol Endocrinol. 1997;11:490–502. doi: 10.1210/mend.11.4.9909. [DOI] [PubMed] [Google Scholar]
- 23.Nicholl J, Hamilton JA, Sutherland GR, Sutherland RL, Watts CK. The third human isoform of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (pfkfb3) map position 10p14-p15. Chromosome research : an international journal on the molecular, supramolecular and evolutionary aspects of chromosome biology. 1997;5:150. doi: 10.1023/a:1018482511456. [DOI] [PubMed] [Google Scholar]
- 24.Obach M, Navarro-Sabate A, Caro J, Kong X, Duran J, Gomez M, Perales JC, Ventura F, Rosa JL, Bartrons R. 6-phosphofructo-2-kinase (pfkfb3) gene promoter contains hypoxia-inducible factor-1 binding sites necessary for transactivation in response to hypoxia. The Journal of biological chemistry. 2004;279:53562–53570. doi: 10.1074/jbc.M406096200. [DOI] [PubMed] [Google Scholar]
- 25.Chesney J, Mitchell R, Benigni F, Bacher M, Spiegel L, Al-Abed Y, Han JH, Metz C, Bucala R. An inducible gene product for 6-phosphofructo-2-kinase with an au-rich instability element: Role in tumor cell glycolysis and the warburg effect. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:3047–3052. doi: 10.1073/pnas.96.6.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sparks LM, Xie H, Koza RA, Mynatt R, Bray GA, Smith SR. High-fat/low-carbohydrate diets regulate glucose metabolism via a long-term transcriptional loop. Metabolism: clinical and experimental. 2006;55:1457–1463. doi: 10.1016/j.metabol.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 27.Atsumi T, Nishio T, Niwa H, Takeuchi J, Bando H, Shimizu C, Yoshioka N, Bucala R, Koike T. Expression of inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase/pfkfb3 isoforms in adipocytes and their potential role in glycolytic regulation. Diabetes. 2005;54:3349–3357. doi: 10.2337/diabetes.54.12.3349. [DOI] [PubMed] [Google Scholar]
- 28.Huo Y, Guo X, Li H, Wang H, Zhang W, Wang Y, Zhou H, Gao Z, Telang S, Chesney J, Chen YE, Ye J, Chapkin RS, Wu C. Disruption of inducible 6-phosphofructo-2-kinase ameliorates diet-induced adiposity but exacerbates systemic insulin resistance and adipose tissue inflammatory response. The Journal of biological chemistry. 2010;285:3713–3721. doi: 10.1074/jbc.M109.058446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huo Y, Guo X, Li H, Xu H, Halim V, Zhang W, Wang H, Fan YY, Ong KT, Woo SL, Chapkin RS, Mashek DG, Chen Y, Dong H, Lu F, Wei L, Wu C. Targeted overexpression of inducible 6-phosphofructo-2-kinase in adipose tissue increases fat deposition but protects against diet-induced insulin resistance and inflammatory responses. The Journal of biological chemistry. 2012;287:21492–21500. doi: 10.1074/jbc.M112.370379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun X, Belkin N, Feinberg MW. Endothelial micrornas and atherosclerosis. Current atherosclerosis reports. 2013;15:372. doi: 10.1007/s11883-013-0372-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marin T, Gongol B, Chen Z, Woo B, Subramaniam S, Chien S, Shyy JY. Mechanosensitive micrornas-role in endothelial responses to shear stress and redox state. Free radical biology & medicine. 2013;64:61–68. doi: 10.1016/j.freeradbiomed.2013.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]