

Survival of tissue engineered constructs after implantation depends on proper vascularization. The differentiation of endothelial cells into mature microvasculature requires dynamic interactions between cells, scaffold, and growth factors, which are difficult to recapitulate in artificial systems. Previously, we reported photocrosslinked poly(ethylene glycol) diacrylate (PEGDA) hydrogels displaying collagen mimetic peptides (CMPs), dubbed PEGDA-CMP, that can be further conjugated with bioactive molecules via CMP-CMP triple helix hybridization. Here, we report that a bifunctional peptide featuring pro-angiogenic domain mimicking vascular endothelial growth factor (VEGF), and a collagen mimetic domain that can fold into a triple helix conformation can hybridize with CMP side chains of the PEGDA-CMP hydrogel, which results in presentation of insoluble VEGF-like signals to endothelial cells. Presentation of VEGF-like signals on the surface of micropatterned scaffolds in this way transforms cells from a quiescent state to elongated and aligned phenotype suggesting that this system could be used to engineer organized microvasculature. We also show that the pro-angiogenic peptide, when applied topically in combination with modified dextran/PEGDA hydrogels, can enhance neovascularization of burn wounds in mice demonstrating the potential clinical use of CMP-mediated matrix-bound bioactive molecules for dermal injuries.
1. Introduction
Tissue engineering is one of the most promising approaches to help patients regenerate tissues and organs damaged by injury, weakened by disease, or compromised by congenital defects. Yet, thus far, successful tissue engineering has been limited to thin constructs, such as skin and cartilage.[1–3] Unlike thin tissues which can support growth by diffusion of nutrients, engineering thick complex organs (e.g. hearts and kidneys) requires functional vasculatures that can provide oxygen and nutrients to sustain cell viability.[4, 5] As a result, there is a great importance placed on the microvascularization of tissue engineering constructs.
The blood vessels are created by two major endothelial cell transformations. Vasculogenesis entails the morphogenesis of endothelial cells (ECs) into new blood vessels, whereas angiogenesis is the sprouting of new capillaries from existing blood vessels.[6, 7] Both of these processes are regulated by dynamic interactions between ECs, their surrounding matrix, and growth factors.[8, 9] One of the most extensively studied growth factors involved in vascular morphogenesis is the vascular endothelial growth factor (VEGF).
In humans, VEGF is found in three major isoforms, distinguished from one another by their propensity to bind to the extracellular matrix (ECM) (e.g. heparin).[10] The VEGF121 isoform exists exclusively as a soluble factor, while isoforms VEGF165 and VEGF189 are able to associate with the ECM via heparin binding domains. These heparin-binding domains are derived from basic residues encoded by exons 6 and 7 of the VEGF gene.[11] VEGF189 features additional basic residues that have strong affinity to heparin sulfate, rendering VEGF189 the isoform with the highest matrix affinity. Variation in binding affinity affects VEGF's diffusion characteristics through the ECM, and it can also elicit difference in cell-signaling responses. For example, endocytosis of growth factors and degradation of internalized ligand-receptor complexes, the nature of which are strong determinants of signaling pathways and the resulting angiogenic behavior of ECs, are believed to be quite different between matrix-bound factors and soluble factors.[12–14] For instance, matrix-bound factors may remain outside the cell and can repeatedly induce phosphorylation of tyrosine kinase. Although not completely understood, the VEGF isoforms seem to provide the basis for generating VEGF distribution and gradients in the ECM that can direct optimal angiogenesis during complex tissue development.
Therapeutic approaches for treating patients with reduced vascular perfusion and improving viability of tissue transplants often focus on delivery of angiogenic growth factors such as VEGF.[15–18] However, the use of protein growth factors for therapeutic purposes comes with challenges. Soluble growth factors are quickly degraded by enzymes in the body or undergo other physical and chemical transformations that shorten their biological half-life.[19] Studies using intravenously injected VEGF showed that the protein has a half-life of less than one hour,[20] and clinical trials of recombinant VEGF administered through bolus injection did not significantly enhance angiogenesis for patients with ischemic condition.[21]
Several research groups have used conjugation methods such as carbodiimide chemistry[22, 23] or crosslinkers and binding tags [24, 25] to link VEGF to a scaffold to prolong its bioavailability. However, the crosslinking reaction itself mitigates the bioactivity of VEGF due to non-specific conjugation at critical bioactive regions within the protein. Furthermore, reaction conditions such as pH, temperature, and presence of organic solvents may denature the protein and compromise its cell-signaling capability.[19] Therefore, it appears advantageous to use short peptides that mimic the growth factor in place of the entire natural protein.[26]
A promising VEGF mimetic peptide (15 amino acid peptide) was recently developed by Pedone and coworkers, which reproduces the VEGF-VEGF receptor binding interface domain of the ~42 kDa dimeric VEGF protein.[27] This mimetic peptide, referred to as QK, is based on the native VEGF sequence from Phe-17 to Tyr-25, and was modified to stabilize the alpha helical conformation of the binding domain critical for cell signaling. Pedone and coworkers demonstrated that the soluble QK competitively binds to VEGF receptors and induces angiogenic activation of ECs. Because of its simple structure, QK can be incorporated into various peptide delivery vehicles more readily than the entire VEGF protein. Researchers have adopted the QK peptide for immobilization onto inorganic substrates[28] and for incorporation into peptide amphiphiles that assemble into fibers[29] as well as for photocrosslinking into synthetic hydrogel scaffolds.[26] Furthermore, our lab has used the QK sequence in concert with collagen mimetic peptide (CMP) domains to develop a bifunctional QK-CMP peptide that binds to collagen scaffolds and at the same time activates early signs of angiogenic response in human umbilical vein endothelial cells (HUVECs).[30]
CMPs are short, synthetic peptides that mimic collagen's characteristic triple helical structure, made possible by the X-Y-glycine repeat sequence with high proline and hydroxyproline contents.[31–33] The self-assembly (homo-trimerization) of CMP strands is thermally reversible, with well defined Tm. When conjugated to multi-arm polymers, the CMP can induce triple helix mediated physical crosslinks that lead to the formation of stable hydrogels.[34, 35] Furthermore, CMPs covalently conjugated to hydrogels can hybridize with CMP derivatives with bioactive domains, allowing inert synthetic scaffolds to be modified with cellular cues.[36]
Aside from biochemical signals, cells require structural support from the viscoelastic ECM for growth and differentiation. This is often recapitulated by the use of viscoelastic hydrogels composed of synthetic polymers.[37] Among many polymers used for cell substrates, poly(ethylene glycol) (PEG) is interesting in that it is a hydrophilic polymer inert to cell interaction. Therefore, PEG-based hydrogels can be used as a blank slate that accentuates cellular responses to the scaffold-incorporated bioactive factors. In particular, photocrosslinked PEG diacrylate (PEGDA) hydrogels have seen wide use in various tissue engineering applications because gel formation can be triggered simply by exposure to UV light.[38–40] The PEGDA-based scaffolds have been used to incorporate VEGF proteins[26, 41, 42] and furthermore, the light mediated crosslinking enabled photopatterning of cell signals onto PEGDA scaffolds to control local cell behaviors.[43, 44] Our previously developed PEGDA-CMP platform demonstrated that CMPs conjugated to the PEGDA gel could hybridize with cell-adhesive CMP derivatives by triple helical association which led to spatial control of cell adhesion.[36]
In this work, we present a new type of bifunctional CMP-QK peptide that can bind to PEGDA-CMP scaffolds and exert its angiogenic activity to ECs (see Table of Content figure or Supporting Scheme in Supporting Information). In addition, we demonstrate fabrication of micrometer scale photopatterned substrates that, when modified with CMP-QK, can guide ECs to form long and thin aggregates that resemble early angiogenic response. We also show that the CMP-QK, when applied topically in combination with biodegradable dextran/PEGDA hydrogels, can enhance neovascularization of burn wounds in mice, which is, to our knowledge, the first in vivo therapeutic use of scaffold-bound collagen mimetic peptides.
2. Results and Discussion
2.1. Design and Characterization of CMP-QK
In our approach to combine the CMP-mediated VEGF signal delivery[30] with our previously developed PEGDA-CMP platform[36], we developed a new type of bifunctional peptide with sequence (POG)8-G3-KLTWQELYQLKYKGI (CMP-QK). The peptide was designed to bind to synthetic PEGDA-CMP scaffolds via triple helical assembly and act as a matrix-bound angiogenic factor. The CMP-QK peptide is comprised of i) a CMP domain consisting of eight Pro-Hyp-Gly, tri-amino acid repeats that can fold into triple helix at physiological temperature, ii) the VEGF-mimetic QK domain, and iii) a flexible three-glycine spacer that separates the CMP and VEGF-mimetic QK sequences enabling each domain to concurrently assume their secondary protein structure. Unlike the QK-CMP peptide reported previously by our group, the peptide was designed with QK at the C-terminus such that after the triple helical hybridization with PEGDA-CMP hydrogel, the bioactive QK domain is positioned away from the polymer backbone for facile interaction with VEGF receptors on ECs.
Circular dichroism (CD) studies of CMP-QK showed hybrid CD signals that indicated co-existence of CMP triple helix and QK alpha helix conformations (Figure 1). An ellipticity maximum at 225 nm which is characteristic of triple helix[45] was not observed in the hybrid CD trace as the ellipticity trace displayed continuous decrease from 260 nm to the characteristic alpha helix minimum at 222 nm (Figure 1A).[27] However, unlike the QK peptide with double minima (Figure 1B), the CMP-QK showed a single ellipticity minimum at 204 nm similar to that of the triple helical CMPs. The CD melting trace acquired at 225 nm (Figure 1C) showed a slow rise in ellipticity preceding the melting transition at 50 °C, which was also seen for QK-CMP in our prior work.[30] The initial increase in CD signal is due to the temperature dependency of QK's alpha helical signal, which gets stronger with rising temperature. When the sequence of QK domain was scrambled, the peptide appeared to lose its alpha helical structure, and the rise in ellipticity at 225 nm prior to the melting transition disappeared (Supporting Information, Figure S1). The derivative (dθ/dT) of CMP-QK's CD melting curve (Figure 1D) showed a clearly defined minimum at 50 °C, corresponding to the melting temperature of the peptide's triple helical structure. The results indicate that the CMP domain of CMP-QK forms a stable triple helix at physiological temperature, and that the triple helical folding propensity of CMP-QK will allow it to hybridize with other CMP derivatives including those conjugated to PEGDA hydrogels.
Figure 1.
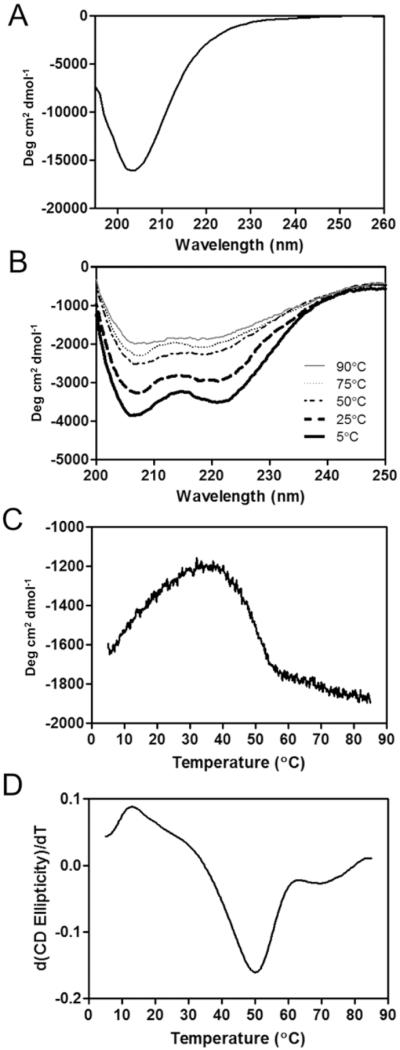
(A) CD spectrum of CMP-QK. (B) CD signals of QK peptide at different temperatures. (C) CD melting curve of CMP-QK acquired at 225 nm. (D) First derivative of the CD melting curve used to determine the Tm (50 °C) of CMP-QK. CD ellipticity is presented as mean residue ellipticity (degree cm2 dmol−1).
2.2. Development of PEGDA/CMP/RGD Photocrosslinked Scaffolds
Since the PEDGA-based hydrogel is generally inert to cell attachment, we first sought to create and test bioactive PEGDA-based hydrogel scaffolds that can support early angiogenic activity of ECs, one that is highly adhesive to cells and can present matrix-bound VEGF-mimetic signals. We wanted to explore this before synthesizing an entirely new degradable scaffold that can support formation of mature microvasculature. We created PEGDA hydrogels displaying cell adhesive RGD peptide[38] and CMP by copolymerizing PEGDA with two types of photocrosslinkable PEG conjugates, acrylate (Acrl)-PEG-RGD and Acrl-PEG-CMP (Supporting Information, Figure S2). Together, these components make up a cell-adhesive scaffold (PEGDA/CMP/RGD) that could be readily modified with other bioactive CMP species for diverse cell culture applications. Prior to UV photopolymerization of the hydrogel, the precursor solution was heated to 80 °C to melt the CMP triple helices into single strands to maximize the number of single strands in the hydrogel.[36]
2.3. Binding and Release Study of CMP-QK
To assess CMP-QK's capacity to hybridize to PEGDA/CMP/RGD scaffolds, we synthesized a fluorescently labeled version of the peptide CF-CMP-QK by adding a three-glycine spacer to the N-terminus of the peptide, followed by conjugation of carboxyfluorescein (CF). CF-CMP-QK solution was heated to 80 °C to melt the peptides into single strands, and the hot solution was added directly to a preformed PEGDA/CMP/RGD hydrogel. After incubating the sample overnight at 37 °C to allow for triple helical association, unbound peptides were removed by washing with blank buffer before measuring the fluorescence of the hydrogel.
The initial binding and cumulative release profiles of CF-CMP-QK from the PEGDA/CMP/RGD gels are shown in Figure 2, as well as those associated with two control gels- PEGDA/RGD gel (which lacks Acrl-PEG-CMP) and PEGDA-only gel (which lacks Acrl-PEG-CMP and Acrl-PEG-RGD). With an initial CF-CMP-QK binding of 5.4 ± 0.8 nmoles, the PEGDA/CMP/RGD gel supported significantly more peptide binding compared to the control gels (Student's t test, p < 0.01). Furthermore, release profiles indicated slower release of CF-CMP-QK from the PEGDA/CMP/RGD gel compared to control gels, with 64% of initially bound peptides remaining on the scaffold after 7 days versus 44% and 31% for PEGDA/RGD and PEGDA gels, respectively. Without the photocrosslinked Acrl-PEG-CMP, the gel lacks a binding partner for the CF-CMP-QK, and low initial binding and fast release are observed. The binding study also showed that PEGDA/RGD gels exhibit slightly stronger CF-CMP-QK binding compared to PEGDA gels. This is possibly due to weak charge-charge interactions between the QK domain and the RGDS groups. Nevertheless, the results clearly indicate that CMP-QK binding to the hydrogel is mediated primarily by association with CMPs of the hydrogel.[36]
Figure 2.

Initial binding (day 0) and release profiles (at 37 °C) of CF-CMP-QK from PEGDA/CMP/RGD scaffolds and control gels lacking CMP binding partners. Hot CF-CMP-QK (80 °C) solution was added to the gels and incubated at 37 °C overnight to allow for complete CMP triple helix hybridization. The gels were washed to remove unbound peptides, and incubated in PBS at 37 °C. The coverage solution was replaced with fresh deionized water immediately prior to each fluorescence measurement. Data reported as mean ± SD.
The binding study indicated that the CMP domain of CMP-QK can hybridize with CMPs in the hydrogel with a density of 3.5 nmoles cm−2. This density level is orders of magnitude higher than those achieved by conjugating VEGF protein to 2D surfaces, which is in the fmol cm−2 to pmol cm−2 range.[25, 46–48] The CMP-QK density demonstrated in this work falls in line with similar work by others such as immobilization of QK to PEGDA (2.6 nmol cm−2)[26] and collagen (5.5 nmol cm−2)[30] substrates. Therefore, we expected that adequate triple helix assembly between CMP-QK and PEGDA/CMP/RGD scaffold would create angiogenic QK signal with sufficient density to induce morphological changes in ECs.
When compared to covalent conjugation of QK, the triple helix mediated non-covalent binding of CMP-QK offers many advantages. For example, in CMP-QK approach, the QK signal is orthogonal to the photocrosslinked RGD peptide so that the two signaling molecules can be tuned independently. The bioactive CMP triple helix assembly strategy also has potential for displaying numerous types of bioactive signals in a combinatorial manner to investigate cell-scaffold interactions.”
2.4. Morphogenic Activation of Endothelial Cells
We tested CMP-QK's ability to mimic the angiogenic activity of matrix-bound VEGF isoforms, in particular, the ability to induce capillary network formation of ECs. PEGDA/CMP/RGD scaffolds were prepared by the same methods as those used in the CMP-QK binding studies. Hot CMP-QK solution (80 °C) was added to the preformed hydrogel and incubated overnight, allowing the CMP domain to form triple helical hybridizations with CMPs in the hydrogel. After washing off unbound peptides from the gel, 750 μL of HUVECs (110,000 cells mL−1) in EBM-2 media were seeded onto the scaffolds. The EBM-2 media devoid of soluble VEGF was used so that any VEGF-mediated signal could be attributed solely to CMP-QK activity. As a negative control, cells were also seeded on PEGDA/CMP/RGD scaffolds treated with blank hot PBS solution. As seen in Figure 3, HUVECs seeded on CMP-QK modified PEGDA/CMP/RGD scaffolds displayed migratory behavior that led to the formation of capillary network-like morphology. In comparison, HUVECs seeded on scaffolds without CMP-QK modification exhibited mostly cobblestone morphology.
Figure 3.

(A) Comparative phase contrast micrographs of HUVECs seeded onto PEGDA/CMP/RGD gels treated with CMP-QK or PBS (control) at 8 h and 24 h after seeding. Cells on CMP-QK modified scaffolds (left panels) extended filopodial projections and assumed migratory phenotype whereas cells on control scaffolds were mostly cobblestone in morphology (right panels). (B) Capillary network-like organization of cells on CMP-QK modified scaffolds. Examples of filopodial extensions are marked by arrowheads.
The HUVEC morphology was further analyzed using quantitative image analysis tools.[49, 50] We used CellProfiler to convert phalloidin and DAPI stained cell images (Figure 4A) into quantitative cell morphology data. Specifically, we compared eccentricity and compactness of the cell shape between CMP-QK modified and PBS modified samples. Figure 4B shows that the HUVECs seeded on CMP-QK modified scaffolds assumed a significantly more elongated morphology compared to those on PBS modified scaffolds, as deemed by higher compactness and eccentricity values (Student's t test, p < 0.001). To validate that this cell shape analysis can be applied to assess angiogenic behavior, we also conducted image analysis on positive and negative controls by seeding ECs on Geltrex basement membrane matrix and on tissue culture polystyrene (TCPS), respectively. Geltrex consists of diverse ECM components such as laminin, collagen IV, and heparin sulfate proteoglycans, as well as growth factors that cooperatively induce angiogenic differentiation of ECs. As expected, HUVECs seeded on Geltrex scaffolds exhibited higher compactness and eccentricity values than cells seeded on TCPS. While Geltrex substrates seemed to elicit more branching and formation of denser networks than CMP-QK modified PEGDA/CMP/RGD, both gels displayed similar values of cell compactness and eccentricity. These findings suggest that CMP-QK treated PEGDA/CMP/RGD scaffolds induce elongated EC morphology indicative of angiogenic activation.
Figure 4.

(A) Fluorescence micrographs of HUVECs cultured on PEGDA/CMP/RGD scaffolds modified with CMP-QK or PBS, as well as those cultured on Geltrex and TCPS substrate as positive and negative controls, respectively. Cells were stained with DAPI (stains nucleus in blue) and phalloidin (stains actin in green). (B) Results of quantitative image analysis of cell morphology showing the cell compactness and eccentricity values for different scaffold conditions (Student's t test: * p < 0.001). Data reported as mean ± SEM.
Matrix-bound VEGF signaling is known to cause EC polarization, formation of filopodial extensions containing thin processes of actin stress fibers, and enhanced migration.[13] In particular, this type of EC morphogenesis appears especially pronounced in genetically altered animal models that only produce the heparin-binding isoforms of VEGF. These animals are reported to feature ECs that first extend multidirectional filopodia and later develop into unusually thin capillary networks with high branching, consistent with a migratory EC phenotype.[51, 52] In drastic contrast, animal models that only express soluble VEGF isoforms produce thick capillaries with less branching. Although more thorough mechanistic study of the angiogenic pathway is needed, the filopodial extensions and migratory phenotype of HUVECs seeded on CMP-QK modified scaffolds seem to suggest that the hydrogel triggers matrix-bound VEGF-like signals to ECs.
In order to confirm that CMP-QK induced HUVEC morphogenesis occurred through a VEGF-like signaling pathway and not by non-specific alteration of cell-scaffold interactions, we conducted cell culture experiments on control scaffolds treated with CMP conjugated to scrambled QK sequence (CMP-RandomQK). RandomQK peptide had no alpha helical CD signature, but the CMP-RandomQK peptide was able to fold into triple helix with a Tm of 50 °C, similar to that of the CMP-QK, as evidenced by CD melting experiment (Supporting Information S1). HUVECs seeded onto scaffolds treated with CMP-RandomQK assumed cobblestone morphology with no indication of capillary network formation. Computerized image analysis showed high level of capillary-like tube coverage on scaffolds modified with CMP-QK; however, CMP-RandomQK treated scaffolds showed large node areas due to confluent cells (Figure 5). The lack of angiogenic response from CMP-RandomQK modified scaffolds strongly supports the notion that the HUVEC morphogenesis on CMP-QK treated PEGDA/CMP/RGD scaffolds is mediated by the QK signals. When human dermal neonatal fibroblasts were seeded, there were no distinct morphological differences between cells seeded onto CMP-QK or PBS treated scaffolds (Supporting Information, Figure S3). These findings further suggest that the CMP-QK mediated HUVEC stimulation arises from EC specific signaling pathway and not from alteration of cell-scaffold interactions through changes in scaffold stiffness or hydrophobicity.
Figure 5.

Computer-assisted image analysis (MetaMorph) of tube-like organization of endothelial cells that were seeded on PEGDA/CMP/RGD scaffolds modified with CMP-QK or CMP-RandomQK. Cells on CMP-QK modified scaffolds show greater tube coverage whereas CMP-RandomQK samples show confluent cells mostly characterized as nodes. The sequence of CMP-RandomQK is (POG)8-G3- KLTLEYQLKWYQKGI. Data reported as mean ± SD.
To confirm that the network-like morphology of HUVECs seeded on CMP-QK modified scaffolds was induced by VEGF mediated signaling pathways, we investigated ERK1/2 phosphorylation levels of ECs.[27, 53, 54] We examined ECs seeded on PEGDA/CMP/RGD scaffolds treated with either the CMP-QK, or blank PBS, as well as those cultured on the same scaffolds with media containing soluble VEGF. The p-ERK1/2 to ERK1/2 expression ratio in cells seeded on CMP-QK modified scaffolds was similar to that on VEGF-supplemented samples, but higher than control scaffolds treated with blank PBS (Figure 6). These findings suggest that scaffold-bound CMP-QK induces cell signaling pathways similar to that of VEGF.
Figure 6.
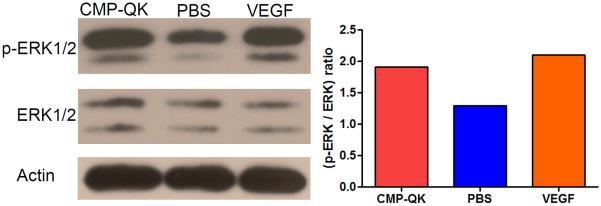
Expression and phosphorylation of ERK1/2 in HUVECs, in response to CMP-QK activation in PEGDA/CMP/RGD hydrogel. ERK1/2 Western Blot showed an increased phosphorylation to expression ratio when cells were exposed to scaffold bound CMP-QK or soluble VEGF as compared to the same scaffold treated with PBS.
2.5. CMP-QK Signaling with Photopatterned PEGDA Hydrogel
The photopolymerized PEGDA hydrogel system featuring CMP is a unique platform to combine spatial cell confinement with growth factor immobilization. In the past, geometric control over cell adhesion has been used to induce EC differentiation. For example, glass slides micropatterned with collagen can spatially confine EC adhesion to narrow lanes and induce ECs to exhibit elongated morphology with cytoskeleton alignment as well as alter the composition of ECM deposited by the cells.[55, 56] Microcontact printing of fibronectin strips resulted in guidance of cell adhesion and alignment of ECs, ultimately leading to lumen formation upon transfer to a fibrin gel.[50] Collagen-filled 3D microchannels were also used to spatially confine EC to induce tubulogenesis.[57] We were able to create narrow cell adhesive lanes by photopolymerizing a patterned layer of CMP/RGD on top of preformed PEGDA hydrogel. A precursor solution containing Acrl-PEG-CMP, Acrl-PEG-RGD, and photoinitiator was applied to the surface of a preformed PEGDA gel and exposed to UV light through a transparency mask with 100 or 200 μm lines. This process yielded scaffolds featuring micropatterned lanes containing both cell-adhesive RGD and CMP. The patterned scaffolds could be subsequently modified using 80 °C CF-labeled CMP solution with high spatial fidelity as evidenced by fluorescence microscopy (Figure 7A). HUVECs seeded on CMP-QK modified patterned scaffolds showed very narrow cell organization while exhibiting elongated cell shape and cytoskeleton alignment (Figure 7B and 7C); in contrast, cells on PBS treated scaffolds exhibited cobblestone morphology and a more proliferative phenotype. The morphology of ECs on CMP-QK modified scaffolds more closely resembled the migratory phenotype of ECs seen in animal models expressing only matrix-bound VEGF188 murine isoforms.[11]
Figure 7.
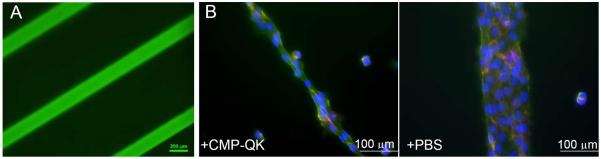
Fluorescence micrographs of PEGDA scaffolds micropatterned with CMP/RGD via surface photopolymerization and further treated with CF-CMP (A), and HUVECs seeded on the micropatterned hydrogels further treated with CMP-QK or blank PBS (B). Width of the line patterns are 200 µm in figure A and 100 μm in B. Cells on CMP-QK modified scaffolds displayed a polarized morphology aligned in the direction of long axis of the micropatterns, while cells on PBS scaffolds exhibited cobblestone morphology. Cells were stained with DAPI (stains nucleus in blue), phalloidin (stains actin in green), and CD31 (stains EC intercellular junctions in red).
We then tested if CMP-QK modification of synthetic scaffolds via triple helix hybridization could be used to locally control EC morphogenesis within a micropatterned lane of cells. After the RGD and CMP photopatterning, the hydrogels were partially dipped into a hot solution of CMP-QK (1 mM), allowing the peptide to diffuse into part of the gel and hybridize with the CMP. These gels were then transferred to a 24-well plate, rinsed with PBS to remove unbound CMP-QK, and subsequently seeded with HUVECs. Figure 8 shows that cells adhering to the micropatterned regions further modified by submersion in CMP-QK (with high CMP-QK signal) exhibited one to two cell layers within the pattern, with elongated and aligned cellular morphology, whereas those in the unsubmerged regions of the micropattern (no CMP-QK signal) appeared cobblestone and undifferentiated. The transition in cell morphology seemed to have occurred gradually along the photopatterned lane, possibly due to the spatial gradient of bioactive CMPs created through the gel.[36] These results demonstrate the novelty of our system, incorporation of both the cell adhesive cues to confine cell attachment and the spatial control of scaffold-bound QK signals to elicit local angiogenic activation of EC. This capability highlights the potential of using CMP hybridization to display VEGF-like signals in patterned scaffolds for directed angiogenesis. We are currently developing MMP-cleavable PEGDA scaffolds functionalized with CMP-QK, which could be used to study the tubulogenesis of ECs encapsulated in 3D scaffolds.
Figure 8.

Fluorescence micrographs of HUVECs seeded on PEGDA scaffolds micropatterned with CMP/RGD and further modified with CMP-QK solution by partial submersion. The spatial variation in the level of angiogenic QK signal induced spatially distinct cell morphology; elongated morphology at submerged regions (high CMP-QK signal) undifferentiated cobblestone morphology at unsubmerged regions (low CMP-QK signal). Cells were stained with DAPI (blue), phalloidin (green), and CD31 (red).
2.6. Neovascularization of Burn Wounds Promoted by CMP-QK Containing Degradable Scaffolds
Vascular function in defective tissues can be improved by promoting ingrowth of vasculatures from autologous blood vessels.[58] In deep burn wounds, vascular ingrowth is particularly critical for transporting of nutrients, oxygen, and immune cells to repair damaged tissues.[59, 60] Therefore, researchers have been working on developing biodegradable hydrogels that can promote tissue ingrowth and neovascularization.[61–63]
Gerecht and coworkers recently developed a dextran (Dex-AE) and PEGDA copolymer system that crosslinks upon UV exposure.[61] Dextran's reactive hydroxyl groups were first modified with allyl isocyanate to incorporate photoreactive vinyl groups, and with ethylamine to enhance hydrogel integration with host tissue. In third-degree burn wound mouse models, the implanted Dex-AE/PEGDA hydrogels allowed efficient infiltration of inflammatory cells that facilitated scaffold biodegradation.[63] This, in turn, promoted high levels of vascular cell infiltration and neovascularization at the burn site.
We thought that combining Dex-AE/PEGDA hydrogels with CMP-QK would further enhance the vascularization of burn wounds, which could be achieved by conjugating CMP-QK to the Dex-AE/PEGDA hydrogel using our hybridization strategy. Our approach was to first copolymerize Acrl-PEG-CMP with Dex-AE/PEGDA monomers, followed by hybridization with CMP-QK. We prepared three monomer mixture solutions consisting of 8 wt% Dex-AE and 2 wt% PEGDA, and 0, 0.5, or 1 wt% Acrl-PEG-CMP. This mixture solution was heated to 80 °C to ensure melting of the CMPs and photopolymerized by UV exposure (6.0 J cm−2). The resulting gels were implanted on third-degree burn wounds of mice 48 h after injury following current clinical practice. Histology of the tissue sections harvested at 7 and 21 days after treatment suggested that CMP-containing gels did not cause any evident necrosis or tissue toxicity, and the tissue exhibited abundant granulation tissues with macrophage and neutrophil activity levels similar to those of control gels. By 21 days, the gels in all samples were degraded and entirely replaced by regenerated dermal tissue featuring epidermal layer and growth of new hair follicles (Supporting Information, Figure S4).
Encouraged by the biocompatibility of the Dex-AE/PEGDA/Acrl-PEG-CMP system, we conducted preliminary in vivo test incorporating the QK signal into this hydrogel through three different approaches: 1) triple helix immobilization between CMP-QK and CMP of the hydrogel, 2) direct covalent conjugation into the hydrogel via photocrosslinking of Acrl-PEG-QK, and 3) encapsulation of QK as a soluble factor. For the first condition, we created Dex-AE/PEGDA gel precursor solution containing 0.5 wt% Acrl-PEG-CMP and 333 μM CMP-QK. This precursor solution was heated to 80 °C to melt the CMP-QK and Acrl-PEG-CMP into single strands and then photocrosslinked into a gel at room temperature, concurrently allowing CMP-QK to hybridize with crosslinked Acrl-PEG-CMP. The second test construct consisted of gels featuring covalently conjugated QK signal that were created by copolymerizing a precursor solution containing Dex-AE/PEGDA and 333 μM Acrl-PEG-QK. For the third condition, soluble QK (333 μM) was simply added into the Dex-AE/PEGDA precursor solution and encapsulated within the photocrosslinked gel. We also tested a blank control Dex-AE/PEGDA gel that did not contain any QK. After implanting these gels on burn wounds of mice, the tissues were harvested after 7 days and analyzed for ingrowth of vascular networks. Vascular networks stabilized by smooth muscle cells were identified by staining for alpha smooth muscle actin (α-SMA).
Figure 9 shows the histology of tissue sections and quantification of blood vessels for the different gel types tested. Although the results are from small sample size (two mice per each gel type), the gels clearly exhibited difference in the extent of neovascularization despite having equivalent molar amounts of QK signal. CMP-QK moieties bound to the scaffold via triple helical association with Acrl-PEG-CMP promoted the highest amount of neovascularization, nearly twice that of blank Dex-AE/PEGDA. When QK is photocrosslinked into the hydrogel via the copolymerization of Acrl-PEG-QK, there was only marginal enhancement in vascular development, whereas addition of soluble QK had no improvement when compared to the blank Dex-AE/PEGDA hydrogel. This could be due to the soluble QK quickly diffusing away from the wound site or fast proteolytic degradation.[19] In accordance with previously reported hydrogel matrices that contain covalently conjugated VEGF or QK,[26, 41, 64] our findings indicate that the immobilized QK provide signals that direct endothelial cell migration and vascular ingrowth. Interestingly, our results suggest that presenting angiogenic signals as a combination of matrix-bound and slowly-releasing forms as in the case of CMP-QK, is ideal for neovascularization, similar to the way VEGFs are presented in the ECM in natural tissues. We are currently conducting thorough investigations into the rates of matrix degradation, QK release, and neovascularization, in order to gain deeper insights into the benefits of QK-presenting gels in vivo.
Figure 9.

Dex-AE/PEGDA gels with different angiogenic QK moieties were implanted on burn wounds of mice, and the healing tissues were harvested after 7 days. (A) H&E-stained histological sections were stained for alpha smooth muscle actin to identify vascular networks (vessel examples marked by arrows; W, wound area; G, hydrogel scaffold). (B) Quantification of blood vessel density at wound sites treated with gels featuring different QK moieties showed that QK immobilization into the gel by CMP hybridization enhances neovascularization. Data reported as mean ± SD.
3. Conclusions
While the use of soluble angiogenic growth factors in tissue engineering studies has demonstrated promising results, recapitulating the matrix-bound growth factors found in vivo requires the development of new angiogenic factors that can bind to cell scaffolds. We designed a bifunctional CMP-QK peptide engineered to mediate VEGF-like signaling through its QK domain and at the same time hybridize with CMP-presenting scaffolds via triple helical assembly. ECs seeded on PEGDA/CMP/RGD scaffolds treated with this immobilized CMP-QK exhibited angiogenic behaviors including a migratory phenotype and the extension of multiple filopodial protrusions that led to the formation of a capillary network-like organization. Furthermore, the PEGDA-based system can be photopatterned with bioactive factors to spatially confine EC adhesion. On micropatterned lanes of 100–200 μm widths, ECs formed thin aggregates that are aligned in the direction of the long axis of the micropattern, suggesting the CMP-QK hybridization system may have potential for spatially directed angiogenesis. The CMP-QK peptide's angiogenic functionality also applies to in vivo settings as seen by its ability to promote neovascularization of burn wounds in a mouse model. To our knowledge, this is the first in vivo study that investigates the therapeutic benefits of scaffold-immobilized bioactive CMPs.
4. Experimental
Materials
Peptide synthesis reagents including Fmoc-amino acids, 2-(1H- benzotriazole-1-yl)-1,1,3,3-tetra-methyluronium hexafluoro-phosphate (HBTU), N,N-diisopropylethylamine (DIPEA), N-methylpyrrolidone (NMP), and trifluoroacetic acid (TFA) were obtained from Advanced ChemTech (Louisville, KY). Rink-type Tentagel R RAM peptide synthesis resin was obtained from Peptides International (Louisville, KY). Other peptide synthesis reagents including piperidine, carboxyfluorescein (CF), acrylic acid, triisopropylsilane (TIS), and 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone (Irgacure 2959) were purchased from Sigma-Aldrich (St Louis, MO). PyBOP and PyAOP were obtained from EMD Chemicals (Philadelphia, PA). N-Fmoc-amido-dPEG4-acid (dPEG) and poly(ethylene glycol) diacrylate (PEGDA, 3400 Da) were obtained from Advanced ChemTech and Glycosan BioSystems (Salt Lake City, UT), respectively. Succinimidyl valerate (SVA) activated PEG acrylate (acrylate-PEG3400-SVA) was purchased from Laysan Bio (Arab, Alabama). Dialysis membrane cassettes (3.5 kDa, MWCO) were obtained from Thermo Fischer Scientific (Rockford, IL). Round glass cover slips (15 mm diameter) were purchased from Electron Microscopy Sciences (Hatfield, PA), and 3-(trimethoxysilyl)propyl methacrylate was purchased from Sigma-Aldrich. HUVECs and EGM-2 endothelial cell media were purchased from Lonza (Walkersville, MD), and fibroblasts (human dermal, neonatal) were obtained from American Type Culture Collection (Manassas, VA). DMEM/F12 + GlutaMAX media and Geltrex basement membrane matrix were obtained from Life Technologies (Carlsbad, CA). For cell fixing and staining, 10% neutral buffered formalin was purchased from Sigma-Aldrich, tris-buffered saline (TBS) was obtained from Bio-Rad Laboratories (Hercules, CA), and Alexa-Fluor 488 phalloidin and 4,6-diamidino-2-phenylindole (DAPI) were obtained from Life Technologies and Roche Applied Science (Indianapolis, IN), respectively. Anti-human CD31 primary antibody (JC70A) was purchased from Dako (Glostrup, Denmark), and Alexa-Fluor 594 conjugated goat anti-mouse secondary antibody was obtained from Jackson ImmunoResearch Laboratories (West Grove, PA).
Peptide Synthesis
Peptides were manually synthesized on Tentagel resin using conventional Fmoc mediated chemistry. Coupling reactions were achieved using 4.5-fold molar excess of amino acids activated by HBTU in the presence of DIPEA. Fmoc protection groups were removed by treatment with piperidine. Acrl-PEG-CMP peptides were synthesized as described previously [36]. Briefly, the CMP was reacted on resin with the Fmoc-dPEG spacer (4-fold molar excess) activated by PyBOP, followed by deprotection and reaction of the dPEG's terminal amine with acrylic acid (5-fold molar excess) activated by PyBOP. Fluorescently labeled CMPs were synthesized by reacting peptide on resin with CF (6-fold molar excess) activated by PyAOP. Peptide conjugates were cleaved from the resin by treatment with a cleavage cocktail (95% TFA, 2.5% TIS, 2.5% deionized water) for at least 3 h. The cleaved peptide solution was filtered and added dropwise to cold ethyl ether, and kept at −20 °C overnight to precipitate the crude peptides. The precipitates were then pelleted by centrifugation and dried under vacuum.
Crude peptides were purified using reverse-phase high performance liquid chromatography (HPLC) on a Varian Polaris 210 series liquid chromatograph (Agilent Technologies, Santa Clara, CA) equipped with a semiprep Vydac reverse-phase C18 column. The mobile phase consisted of gradient mixtures of deionized water and acetonitrile with 0.1% TFA, which was pumped through the column at a flow rate of 4 mL min−1. Peptides were detected by UV absorption at 220 nm for Acrl-PEG-CMP and RGDS or at 275 nm for CMP-QK peptides. The elutions containing target peptides were collected and lyophilized. The purity of peptides was determined by matrix-assisted laser desorption/ionization time-of-flight (MALDI-ToF) mass spectrometry (Voyager DE-STR, Applied Biosystems, Foster City, CA) (Supporting Information, Figure S5).
Synthesis of acrylate-PEG-RGD
Acrylate (Acrl)-PEG-RGD was synthesized by conjugating RGDS to acrylate-PEG3400-SVA. Acrylate-PEG3400-SVA and 3-fold molar excess RGDS were each dissolved separately in 300 μL of 50 mM NaHCO3 buffer. RGDS solution was immediately added dropwise to the acrylate-PEG3400-SVA solution. The reaction mixture was stirred for 24 h at room temperature and dialyzed in the dark for 2 days using a 3.5 kDa MWCO membrane to remove excess RGDS peptide from the target Acrl-PEG-RGD product. HPLC chromatograms and MALDI mass spectra confirmed that the major product of this reaction was Acrl-PEG-RGD with negligible unreacted acrylate-PEG3400-SVA (Supporting Information, Figure S6).
Circular Dichroism Study
Prior to circular dichroism (CD) measurements, CMP-QK, QK, and CMP-RandomQK (100 μM) solutions were incubated at 4 °C overnight to ensure full conformational folding. The CD spectrum was measured using a JASCO 710 spectrometer with JASCO PTC-348 WI temperature controller (Easton, MD). CD thermal melting curves were generated by heating the sample from 5 °C to 85 °C at a rate of 1 °C min−1 while tracking the CD ellipticity at 225 nm. The melting temperature of the peptide (Tm) was designated as the minimum of the first derivative of ellipticity versus temperature curve.
PEGDA/CMP/RGD Hydrogel Synthesis
Hydrogel precursor solutions were prepared by combining 45 μL of 20 wt% PEGDA and 0.1 wt% Irgacure photoinitiator in PBS, 11.25 μL of 20 mM Acrl-PEG-CMP in PBS, and 33.75 μL of 10 mM Acrl-PEG-RGD in PBS to create a final composition of 10 wt% PEGDA, 0.05 wt% Irgacure, 2.5 mM Acrl-PEG-CMP, and 3.75 mM Acrl-PEG-RGD. This PEGDA/CMP/RGD gel precursor was heated to 80 °C for 30 min and then 85 μL was deposited onto a glass cover slip that was previously functionalized with methacrylate groups, which was achieved by swirling NaOH-etched cover slips in 2% solution of 3-(trimethoxysilyl)propyl methacrylate diluted in toluene for 30 min, followed by curing the cover slips in a 70 °C oven for 1 h [65]. An untreated cover slip was placed on top of the precursor solution to form a glass-solution-glass sandwich. The samples were then exposed for 7 min to a 365 nm UV light at ~10 mW cm−2 intensity (Omnicure S1000, Lumen Dynamics, Mississauga, Canada). After gel formation, the top cover slip was peeled away, and the gel bound to the methacrylated cover slip was transferred to a 24-well plate for further experiments.
CMP-QK Binding and Release Study
PEGDA/CMP/RGD gel and control samples PEGDA/RGD gel and PEGDA-only gel were prepared as described above. These gels were stored at 37 °C in a 24-well plate and covered with 500 μL deionized water. Next, 750 μM CF-CMP-QK solution was heated to 80 °C for 30 min to melt triple helices into single strands. After removing the coverage solution from the gels, 500 μL of hot CF-CMP-QK solution was added to the gels, which were then incubated at 37 °C overnight to allow for peptide-scaffold triple helical hybridization. On the following day, gels were washed with room temperature water (1 mL × 10) to remove unbound CF-CMP-QK. The fluorescence of the CF-labeled peptides bound to the gels was measured (excitation 489 nm, emission 533 nm) using a Spectramax plate reader (Molecular Devices, Sunnyvale, CA). The gels were incubated at 37 °C during subsequent days, and the coverage solution was replaced with fresh water immediately prior to each successive fluorescence reading. The fluorescence readings were converted into moles of peptide using a standard curve generated from fluorescence values of a series of solutions with known peptide concentration.
Cell Seeding on CMP-QK modified PEGDA/CMP/RGD Scaffolds
Gels were photocrosslinked as described above inside a cell culture hood under sterile condition, and transferred to a 24-well plate. Subsequently, 500 μL of CMP-QK in PBS (750 μM), (or either blank PBS or CMP-RandomQK as a control) preheated to 80 °C was added to the well and incubated at 37 °C overnight. The CMP-QK solution was removed from the gel by pipetting, and unbound peptides were removed by washing with blank PBS (1 mL × 10). HUVECs (passages 3 to 6) were seeded on the gels at a concentration of 110,000 cells mL−1 in 750 μL of EBM-2 medium containing 2% fetal bovine serum (FBS). Twenty four hours after seeding, HUVECs were fixed with neutral buffered 10% formalin solution for 30 min and blocked with TBS supplemented with 0.25% Triton X-100 and 5% donkey serum. Cells were stained using PBS solution containing DAPI (1 μg mL−1) and phalloidin (0.165 μM). Fluorescent cell images were captured using a Nikon Eclipse TE2000-E (Nikon Instruments, Melville, NY). CellProfiler software was used to obtain quantitative cell morphology data to evaluate cell compactness and eccentricity [66]. Compactness was defined as the variance of the radial distance of the cell's pixels from the centroid divided by the cell area. The eccentricity is the ratio of the distance between the foci of the ellipse and its major axis length. Eccentricity of a cell was determined from a corresponding ellipse that has the same second-moments as the cell. Only cells above a minimum cell area equal to twice the size of the average cell nucleus were included in the calculation. This threshold excludes cells found at the nexus of capillary networks whose rounded morphology can skew the cell shape measurements for CMP-QK modified scaffolds and Geltrex samples. To quantify the QK domain's influence on capillary network formation, the angiogenesis assay from MetaMorph software was used to analyze at least 5 different image fields from independent experiments (n ≥ 3) for scaffolds modified with CMP-QK or CMP-RandomQK.
ERK1/2 Western Blot Assay
To prepare cells for the ERK1/2 assay, HUVECs were starved for 4 h in reduced serum (0.25% FBS) EBM-2 media. Cells were trypsinized and seeded onto PEGDA/CMP/RGD gels modified with CMP-QK (or with blank PBS for control) as described above. Each gel was seeded with 750 μL of cell suspension with 150,000 cells mL−1 in EBM-2 containing 2% FBS. For positive control, select gels were seeded with cells using media supplemented with soluble VEGF (100 ng mL−1). After 2 h of incubation at 37 °C, the cells on the scaffolds were lysed with lysis buffer (25 mM Tris-HCL, 0.5% Triton X-100, 200 mM NaCl, 2 mM EDTA, phosphatase and protease inhibitors; Roche Applied Science). Cell lysates from multiple gels were combined for each condition and centrifuged at 14,000×g for 15 min at 4 °C. The total protein concentration in the supernatant was measured using a Pierce BCA protein assay kit. The lysate was loaded into a NuPAGE 10% Bis-Tris gel (Life Technologies), and the proteins were separated by electrophoresis in MOPS buffer (200 V for 50 min). The proteins on the gel were transferred to a nitrocellulose membrane using the iBlot Dry Blotting System (Life Technologies). The membranes were blocked for 1 h at room temperature with 5% milk or 5% BSA and then stained overnight at 4 °C with specific antibodies for total ERK1/2 or Phospho-ERK1/2, respectively. The membranes were stained with anti-rabbit horseradish peroxidase conjugated secondary antibody for 100 min at room temperature. Amersham ECL detection reagent was used to reveal the protein bands on films, and these bands were quantified using ImageQuant TL (GE Healthcare Life Sciences, Buckinghamshire, UK).
Photopatterning of PEGDA Gel Surfaces
Gel precursor solution (85 μL) consisting of 10 wt% PEGDA and 0.1% Irgacure in PBS were photopolymerized by exposing a glass-PEGDA-glass sandwich construct to 365 nm UV light at ~10 mW cm−2 intensity for 2 min. After removing the top glass coverslip, an 80 °C PBS solution (40 μL) consisting of 5 mM Acrl-PEG-CMP, 5 mM Acrl-PEG-RGD, and 0.5 wt% Irgacure was applied to cover the surface of PEGDA gel. The gel was covered with a transparency mask with linear gaps of 100 or 200 μm in width that were 1 mm apart, and the sample was exposed to UV light for 7 min. The gels were then transferred to 24-well plate and rinsed with PBS three times to remove uncrosslinked compounds. For CMP modification, the gel was treated with 500 μL of CMP-QK in PBS (750 μM, 80 °C). After overnight incubation at 37 °C, the gels were washed with PBS (1 mL × 10), followed by seeding with HUVECs (85,000 cells mL−1) in 750 μL of EGM-2 media devoid of VEGF.
For spatially-controlled CMP-QK modification, photopatterned gels were partially dipped into 80 °C CMP-QK solution (1 mM in PBS) with photopatterned lines perpendicular to the surface of the solution. The gels were incubated at 37 °C for 2 h while partially submerged in the solution. Gels were transferred to a 24-well plate, washed to remove unbound peptides, and seeded with cells as described above.
Mouse Surgery Procedure
We followed the animal experimental procedures previously reported by Sun and coworkers, which were approved by The Johns Hopkins University Animal Care and Use Committee [63]. Mice were anesthetized with isoflurane before shaving and applying a depilatory (Nair, Chruch & Dweight Co, Inc). A 220 gauge aluminum rod was heated in a 100 °C water bath and applied to the posterior-dorsum of each mouse for 4 sec to generate a burn wound. Following existing clinical procedure, we performed burn wound excision 48 h after the burn injury. We excised full-thickness skin (8 mm diameter) from the burn site and applied the Dex-AE/PEGDA gels onto the wound before covering the site with DuoDerm dressing.
Preparation of (Dex-AE)/PEGDA hydrogels featuring CMP and QK Analogs
A dextran-allyl isocyanate-ethylamine (Dex-AE)/PEGDA gel containing CMP was created by photocrosslinking a gel precursor solution consisting of Dex-AE/PEGDA (10 wt% in 80/20 ratio), Acrl-PEG-CMP (0, 0.5, or 1 wt%), and Irgacure 2959 photoinitiator (0.1 wt%). For neovascularization studies, the Dex-AE/PEGDA precursor solution was supplemented with either QK (333 μM), Acrl-PEG-QK (333 μM), or 0.5 wt% Acrl-PEG-CMP with 333 μM CMP-QK. Gel precursor solutions were then heated to 80 °C and pipetted into a sterile mold (70 μL volume per well) before photopolymerization by 10 min of UV exposure (6.0 J cm−2). The resulting hydrogel disks (8 mm in diameter, ~2 mm in thickness) were removed from the molds and immersed in sterile PBS before application to the burn wounds within 24 h.
Histology
Gel constructs were explanted 7 or 21 days after implantation and fixed with formalin-free fixative (Accustain, Sigma-Aldrich) following manufacturer's guidelines. Samples were then dehydrated in ethanol, embedded in paraffin, and serially sectioned before staining with hematoxylin and eosin (H&E). Mature vascular networks were identified by staining for alpha smooth muscle actin antibody (Abcam plc). Vessels were counted from samples harvested from two mice yielding between two to four tissue sections for each gel condition. The area in mm2 of each tissue section was traced using ImageJ (National Institutes of Health, Bethesda) which was used to calculate the density of vessels at the wound site.
Supplementary Material
Acknowledgements
The authors thank Andrew Wong and Peter Searson for help with microscopy, and Jesse Placone, Sarvenaz Sarabipour and Kalina Hristova for help with immunohistochemistry. This work was supported by grants from NIAMS/NIH (R01AR060484) and DOD (W81XWH-12-1-0555) awarded to S.M.Y., by NSF IGERT fellowship awarded to T.R.C., and by HHMI Graduate Training Program (NBMed) and NDSEG (32 CFR 168a) fellowships awarded to P.J.S. Supporting Information is available online from Wiley InterScience or from the author.
References
- [1].Mansbridge J. J. Biomater. Sci. 2008;19:955. doi: 10.1163/156856208784909417. [DOI] [PubMed] [Google Scholar]
- [2].Temenoff JS, Mikos AG. Biomaterials. 2000;21:431. doi: 10.1016/s0142-9612(99)00213-6. [DOI] [PubMed] [Google Scholar]
- [3].Marler JJ, Upton J, Langer R, Vacanti JP. Adv. Drug Del. Rev. 1998;33:165. doi: 10.1016/s0169-409x(98)00025-8. [DOI] [PubMed] [Google Scholar]
- [4].Griffith LG, Naughton G. Science. 2002;295:1009. doi: 10.1126/science.1069210. [DOI] [PubMed] [Google Scholar]
- [5].Ko HC, Milthorpe BK, McFarland CD. Eur. Cells Mater. 2007;14:1. doi: 10.22203/ecm.v014a01. [DOI] [PubMed] [Google Scholar]
- [6].Flamme I, Frölich T, Risau W. J. Cell. Physiol. 1997;173:206. doi: 10.1002/(SICI)1097-4652(199711)173:2<206::AID-JCP22>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- [7].Demir R, Seval Y, Huppertz B. Acta Histochem. 2007;109:257. doi: 10.1016/j.acthis.2007.02.008. [DOI] [PubMed] [Google Scholar]
- [8].Schmidt A, Brixius K, Bloch W. Circ. Res. 2007;101:125. doi: 10.1161/CIRCRESAHA.107.148932. [DOI] [PubMed] [Google Scholar]
- [9].Francis ME, Uriel S, Brey EM. Tissue Eng Pt. B-Rev. 2008;14:19. doi: 10.1089/teb.2007.0115. [DOI] [PubMed] [Google Scholar]
- [10].Roskoski R. Crit. Rev. Oncol./Hematol. 2007;62:179. doi: 10.1016/j.critrevonc.2007.01.006. [DOI] [PubMed] [Google Scholar]
- [11].Ruhrberg C. BioEssays. 2003;25:1052. doi: 10.1002/bies.10351. [DOI] [PubMed] [Google Scholar]
- [12].Carmeliet P, Ng YS, Nuyens D, Theilmeier G, Brusselmans K, Cornelissen I, Ehler E, Kakkar VV, Stalmans I, Mattot V. Nat. Med. 1999;5:495. doi: 10.1038/8379. [DOI] [PubMed] [Google Scholar]
- [13].Ruhrberg C, Gerhardt H, Golding M, Watson R, Ioannidou S, Fujisawa H, Betsholtz C, Shima DT. Genes Dev. 2002;16:2684. doi: 10.1101/gad.242002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chen TT, Luque A, Lee S, Anderson SM, Segura T, Iruela-Arispe ML. J. Cell Biol. 2010;188:595. doi: 10.1083/jcb.200906044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Annex BH, Simons M. Cardiovasc. Res. 2005;65:649. doi: 10.1016/j.cardiores.2004.09.004. [DOI] [PubMed] [Google Scholar]
- [16].Morishita R, Nakamura S, Hayashi S, Taniyama Y, Moriguchi A, Nagano T, Taiji M, Noguchi H, Takeshita S, Matsumoto K. Hypertension. 1999;33:1379. doi: 10.1161/01.hyp.33.6.1379. [DOI] [PubMed] [Google Scholar]
- [17].Simons M, Ware JA. Nat. Rev. Drug Discovery. 2003;2:863. doi: 10.1038/nrd1226. [DOI] [PubMed] [Google Scholar]
- [18].Carmeliet P. Nat. Med. 2003;9:653. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- [19].Tayalia P, Mooney DJ. Adv. Mater. 2009;21:3269. doi: 10.1002/adma.200900241. [DOI] [PubMed] [Google Scholar]
- [20].Lazarous DF, Shou M, Scheinowitz M, Hodge E, Thirumurti V, Kitsiou AN, Stiber JA, Lobo AD, Hunsberger S, Guetta E. Circulation. 1996;94:1074. doi: 10.1161/01.cir.94.5.1074. [DOI] [PubMed] [Google Scholar]
- [21].Henry TD, Annex BH, McKendall GR, Azrin MA, Lopez JJ, Giordano FJ, Shah PK, Willerson JT, Benza RL, Berman DS. Circulation. 2003;107:1359. doi: 10.1161/01.cir.0000061911.47710.8a. [DOI] [PubMed] [Google Scholar]
- [22].Shen YH, Shoichet MS, Radisic M. Acta Biomater. 2008;4:477. doi: 10.1016/j.actbio.2007.12.011. [DOI] [PubMed] [Google Scholar]
- [23].Chiu LLY, Radisic M. Biomaterials. 2010;31:226. doi: 10.1016/j.biomaterials.2009.09.039. [DOI] [PubMed] [Google Scholar]
- [24].Zisch AH, Schenk U, Schense JC, Sakiyama-Elbert SE, Hubbell JA. J. Controlled Release. 2001;72:101. doi: 10.1016/s0168-3659(01)00266-8. [DOI] [PubMed] [Google Scholar]
- [25].Backer MV, Patel V, Jehning BT, Claffey KP, Backer JM. Biomaterials. 2006;27:5452. doi: 10.1016/j.biomaterials.2006.06.025. [DOI] [PubMed] [Google Scholar]
- [26].Leslie-Barbick JE, Saik JE, Gould DJ, Dickinson ME, West JL. Biomaterials. 2011;32:5782. doi: 10.1016/j.biomaterials.2011.04.060. [DOI] [PubMed] [Google Scholar]
- [27].D'Andrea LD, Iaccarino G, Fattorusso R, Sorriento D, Carannante C, Capasso D, Trimarco B, Pedone C. Proc. Natl. Acad. Sci. U. S. A. 2005;102:14215. doi: 10.1073/pnas.0505047102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lee JS, Wagoner Johnson AJ, Murphy WL. Adv. Mater. 2010;22:5494. doi: 10.1002/adma.201002970. [DOI] [PubMed] [Google Scholar]
- [29].Webber MJ, Tongers J, Newcomb CJ, Marquardt KT, Bauersachs J, Losordo DW, Stupp SI. Proc. Natl. Acad. Sci. U. S. A. 2011;108:13438. doi: 10.1073/pnas.1016546108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chan TR, Stahl PJ, Yu SM. Adv. Funct. Mater. 2011;21:4252. doi: 10.1002/adfm.201101163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Holmgren SK, Taylor KM, Bretscher LE, Raines RT. Nature. 1998;392:666. doi: 10.1038/33573. [DOI] [PubMed] [Google Scholar]
- [32].Shoulders MD, Raines RT. Annu. Rev. Biochem. 2009;78:929. doi: 10.1146/annurev.biochem.77.032207.120833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kotch F, Raines R. Proc. Natl. Acad. Sci. U. S. A. 2006;103:3028. doi: 10.1073/pnas.0508783103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Stahl PJ, Romano NH, Wirtz D, Yu SM. Biomacromolecules. 2010;11:2336. doi: 10.1021/bm100465q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Pérez CM, Panitch A, Chmielewski J. Macromol. Biosci. 2011;11:1426. doi: 10.1002/mabi.201100230. [DOI] [PubMed] [Google Scholar]
- [36].Stahl PJ, Yu SM. Soft Matter. 2012;8:10409. doi: 10.1039/C2SM25903F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zhu J. Biomaterials. 2010;31:4639. doi: 10.1016/j.biomaterials.2010.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hern DL, Hubbell JA. J. Biomed. Mater. Res. A. 1998;39:266. doi: 10.1002/(sici)1097-4636(199802)39:2<266::aid-jbm14>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- [39].Lee HJ, Yu C, Chansakul T, Hwang NS, Varghese S, Yu SM, Elisseeff JH. Tissue Eng., Part A. 2008;14:1843. doi: 10.1089/ten.tea.2007.0204. [DOI] [PubMed] [Google Scholar]
- [40].Mann BK, Schmedlen RH, West JL. Biomaterials. 2001;22:439. doi: 10.1016/s0142-9612(00)00196-4. [DOI] [PubMed] [Google Scholar]
- [41].Phelps EA, Landázuri N, Thulé PM, Taylor WR, García AJ. Proc. Natl. Acad. Sci. U. S. A. 2010;107:3323. doi: 10.1073/pnas.0905447107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Moon JJ, Saik JE, Poché RA, Leslie-Barbick JE, Lee S-H, Smith AA, Dickinson ME, West JL. Biomaterials. 2010;31:3840. doi: 10.1016/j.biomaterials.2010.01.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hahn MS, Taite LJ, Moon JJ, Rowland MC, Ruffino KA, West JL. Biomaterials. 2006;27:2519. doi: 10.1016/j.biomaterials.2005.11.045. [DOI] [PubMed] [Google Scholar]
- [44].Chan V, Zorlutuna P, Jeong JH, Kong H, Bashir R. Lab on a Chip. 2010;10:2062. doi: 10.1039/c004285d. [DOI] [PubMed] [Google Scholar]
- [45].Ackerman MS, Bhate M, Shenoy N, Beck K, Ramshaw JAM, Brodsky B. J. Biol. Chem. 1999;274:7668. doi: 10.1074/jbc.274.12.7668. [DOI] [PubMed] [Google Scholar]
- [46].Anderson SM, Chen TT, Iruela-Arispe ML, Segura T. Biomaterials. 2009;30:4618. doi: 10.1016/j.biomaterials.2009.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Müller S, Koenig G, Charpiot A, Debry C, Voegel J-C, Lavalle P, Vautier D. Adv. Funct. Mater. 2008;18:1767. [Google Scholar]
- [48].Wang AY, Leong S, Liang Y-C, Huang RCC, Chen CS, Yu SM. Biomacromolecules. 2008;9:2929. doi: 10.1021/bm800727z. [DOI] [PubMed] [Google Scholar]
- [49].Robinet A, Fahem A, Cauchard JH, Huet E, Vincent L, Lorimier S, Antonicelli F, Soria C, Crepin M, Hornebeck W. J. Cell Sci. 2005;118:343. doi: 10.1242/jcs.01613. [DOI] [PubMed] [Google Scholar]
- [50].Dickinson LE, Moura ME, Gerecht S. Soft Matter. 2010;6:5109. [Google Scholar]
- [51].Stalmans I, Ng Y-S, Rohan R, Fruttiger M, Bouche A, Yuce A, Fujisawa H, Hermans B, Shani M, Jansen S, Hicklin D, Anderson DJ, Gardiner T, Hammes H-P, Moons L, Dewerchin M, Collen D, Carmeliet P, D'Amore PA. J. Clin. Invest. 2002;109:327. doi: 10.1172/JCI14362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kusters B, de Waal RMW, Wesseling P, Verrijp K, Maass C, Heerschap A, Barentsz JO, Sweep F, Ruiter DJ, Leenders WPJ. Cancer Res. 2003;63:5408. [PubMed] [Google Scholar]
- [53].Berra E, Milanini J, Richard DE, Le Gall M, Vinals F, Gothie E, Roux D, Pages G, Pouyssegur J. Biochem. Pharmacol. 2000;60:1171. doi: 10.1016/s0006-2952(00)00423-8. [DOI] [PubMed] [Google Scholar]
- [54].Qi JH, Ebrahem Q, Moore N, Murphy G, Claesson-Welsh L, Bond M, Baker A, Anand-Apte B. Nat. Med. 2003;9:407. doi: 10.1038/nm846. [DOI] [PubMed] [Google Scholar]
- [55].Vartanian KB, Kirkpatrick SJ, Hanson SR, Hinds MT. Biochem. Biophys. Res. Commun. 2008;371:787. doi: 10.1016/j.bbrc.2008.04.167. [DOI] [PubMed] [Google Scholar]
- [56].Vartanian KB, Kirkpatrick SJ, McCarty OJT, Vu TQ, Hanson SR, Hinds MT. J. Biomed. Mater. Res., Part A. 2009;91A:528. doi: 10.1002/jbm.a.32225. [DOI] [PubMed] [Google Scholar]
- [57].Raghavan S, Nelson CM, Baranski JD, Lim E, Chen CS. Tissue Eng., Part A. 2010;16:2255. doi: 10.1089/ten.tea.2009.0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ravi S, Chaikof EL. Regener. Med. 2010;5:107. doi: 10.2217/rme.09.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Li J, Zhang Y-P, Kirsner RS. Microsc. Res. Tech. 2003;60:107. doi: 10.1002/jemt.10249. [DOI] [PubMed] [Google Scholar]
- [60].Tredget EE. J. Trauma Acute Care Surg. 2007;62:S69. [Google Scholar]
- [61].Sun G, Shen Y-I, Ho CC, Kusuma S, Gerecht S. J. Biomed. Mater. Res., Part A. 2010;93A:1080. doi: 10.1002/jbm.a.32604. [DOI] [PubMed] [Google Scholar]
- [62].Sun G, Shen Y-I, Kusuma S, Fox-Talbot K, Steenbergen CJ, Gerecht S. Biomaterials. 2011;32:95. doi: 10.1016/j.biomaterials.2010.08.091. [DOI] [PubMed] [Google Scholar]
- [63].Sun G, Zhang X, Shen Y-I, Sebastian R, Dickinson LE, Fox-Talbot K, Reinblatt M, Steenbergen C, Harmon JW, Gerecht S. Proc. Natl. Acad. Sci. U. S. A. 2011;108:20976. doi: 10.1073/pnas.1115973108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Zisch AH, Lutolf MP, Ehrbar M, Raeber GP, Rizzi SC, Davies N, Schmökel H, Bezuidenhout D, Djonov V, Zilla P, Hubbell JA. FASEB J. 2003;17:2260. doi: 10.1096/fj.02-1041fje. [DOI] [PubMed] [Google Scholar]
- [65].Brafman DA, Chien S, Willert K. Nat. Protoc. 2012;7:703. doi: 10.1038/nprot.2012.017. [DOI] [PubMed] [Google Scholar]
- [66].Carpenter A, Jones T, Lamprecht M, Clarke C, Kang I, Friman O, Guertin D, Chang J, Lindquist R, Moffat J, Golland P, Sabatini D. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.