

Abstract
The EphA4 receptor is highly expressed in the nervous system, and recent findings suggest that its signaling activity hinders neural repair and exacerbates certain neurodegenerative processes. EphA4 has also been implicated in cancer progression. Thus, EphA4 inhibitors represent potential therapeutic leads and useful research tools to elucidate the role of EphA4 in physiology and disease. Here, we report the structure of a cyclic peptide antagonist, APY, in complex with the EphA4 ligand-binding domain (LBD), which represents the first structure of a cyclic peptide bound to a receptor tyrosine kinase. The structure shows that the dodecameric APY efficiently occupies the ephrin ligand-binding pocket of EphA4 and promotes a “closed” conformation of the surrounding loops. Structure-guided relaxation of the strained APY β-turn and amidation of the C terminus to allow an additional intrapeptide hydrogen bond yielded APY-βAla8.am, an improved APY derivative that binds to EphA4 with nanomolar affinity. APY-βAla8.am potently inhibits ephrin-induced EphA4 activation in cells and EphA4-dependent neuronal growth cone collapse, while retaining high selectivity for EphA4. The two crystal structures of APY and APY-βAla8.am bound to EphA4, in conjunction with secondary phage display screens, highlighted peptide residues that are essential for EphA4 binding as well as residues that can be modified. Thus, the APY scaffold represents an exciting prototype, particularly since cyclic peptides have potentially favorable metabolic stability and are emerging as an important class of molecules for disruption of protein–protein interactions.
EphA4, a member of the Eph family of receptor tyrosine kinases, represents a very promising target for promoting neural repair and counteracting neurodegenerative processes.1,2 EphA4 signaling can be activated by all ephrin ligands, including the five GPI-linked ephrin-As and the three transmembrane ephrin-Bs. Ephrin binding stimulates EphA4 tyrosine kinase activity and downstream signaling, which in neurons leads to inhibition of axon growth and retraction of synaptic structures known as dendritic spines.3−5 In addition, EphA4 interaction with the ephrin-A3 ligand expressed in astrocytes stimulates “reverse” signals through the ephrin that limit the uptake of the extracellular neurotransmitter glutamate, thus modulating synaptic transmission.6,7 Dysregulation of these EphA4 activities can hinder regeneration in the injured nervous system as well as promote neurotoxicity and neurodegeneration. Indeed, EphA4 has been identified as a possible inhibitor of nerve regeneration after spinal cord injury3,8,9 and as a modifier gene that accelerates the progression of amyotrophic lateral sclerosis (ALS).10 Recent reports also suggest the possible involvement of EphA4 in the pathogenesis of other neurological disorders, including Alzheimer’s disease11,12 and stroke.13
Increasing evidence also implicates EphA4 in various types of cancer. For example, EphA4 downregulation studies have suggested a role for EphA4 in leukemia, prostate, pancreatic, and gastric cancer cell growth and in liver cancer metastasis.14−18 High EphA4 expression has also been correlated with shorter survival in breast and gastric cancer patients,16,19 although the opposite correlation was found in lung cancer patients.20 Finally, EphA4 can enhance the oncogenic effects of fibroblast growth factor receptor 1 in glioblastoma cells.21
Central to its signaling ability, EphA4 has a ligand-binding domain (LBD) at the N terminus of its extracellular region and a tyrosine kinase domain in its cytoplasmic region.22 Thus, the main strategies to inhibit ephrin-dependent EphA4 activities involve the use of either kinase inhibitors or antagonists that block ephrin binding to the LBD.23,24 Kinase inhibitors typically target multiple kinases due to the high conservation of the ATP binding pocket,25 explaining the difficulties in identifying kinase inhibitors selective for EphA4.26 In contrast, the ephrin-binding pocket of Eph receptors has unique features that can be exploited for more selective targeting with small molecules and peptides.24 Small molecules targeting subsets of Eph receptors, including EphA4, have been identified but are not very potent and some have problematic features.12,24,27 Peptide antagonists that selectively target EphA4 include three dodecapeptides identified by phage display, with the most potent being the linear KYL (KYLPYWPVLSSL).28,29 The potential of these peptides is highlighted by the successful use of KYL in studies from various groups,3,4,11,12,28,30−32 including the recent study implicating EphA4 in ALS pathogenesis.10 However, with a KD value of ∼1 μM,32 the linear KYL peptide does not appear ideally suited for therapeutic development. In addition, both a recent phage display screen of a cyclic nonapeptide library33 and an NMR-based screen for smaller EphA4 peptidomimetic antagonists29 failed to yield peptides more potent than KYL.
The difficulties in obtaining submicromolar EphA4 antagonists are likely due to the nature of the ephrin-binding pocket of EphA4, which is very broad (exceeding an estimated 900 Å2), lacks sufficient hot spot regions,34 and is highly dynamic.35−39 These features reduce the potential free energy gain for the binding of small molecules and flexible linear peptide ligands. In contrast to linear peptides, cyclic peptides are more structured in their unbound form, which can improve binding affinity and pharmacokinetic properties.40 Furthermore, cyclic peptides are able to better occupy a wide cavity such as the ephrin-binding pocket of EphA4 due to their circular conformation. We therefore sought to optimize the cyclic peptide APYCVYRGSWSC (APY) that we originally discovered along with KYL.28 The APY peptide has an intramolecular disulfide bond, is only marginally less potent toward EphA4 than KYL, and selectively inhibits EphA4 but not other Eph receptors.32 We solved the crystal structure of APY bound to the EphA4 LBD. This revealed the mechanism of APY-mediated antagonism and enabled rational design of an improved APY derivative. The crystal structure of this derivative reveals features contributing to its increased potency. We also performed secondary phage display screens to discriminate the importance of different peptide residues. The new more potent APY derivative can serve as a research tool and a promising therapeutic lead for targeting EphA4 in neurodegenerative diseases and cancer.
Results and Discussion
Crystal Structure of the APY Cyclic Peptide in Complex with EphA4 Reveals a Unique Binding Mode
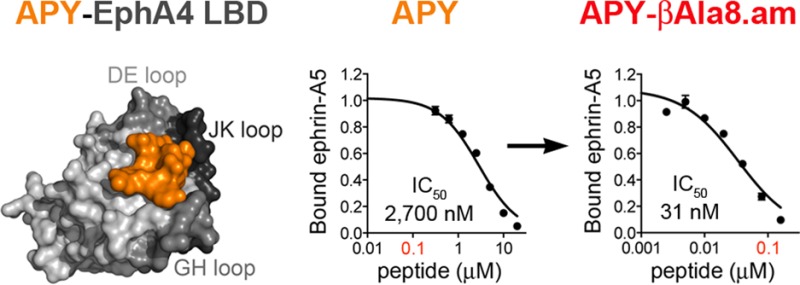
While the EphA4 LBD has been crystallized in its unbound form and in complex with ephrin ligands,35−39,41 complexes containing peptides and small molecules that target the ephrin-binding pocket of EphA4 have evaded structural evaluation.32 After extensive screening of crystallization conditions, we succeeded in generating crystals of the APY-EphA4 LBD complex and solved the crystal structure at a resolution of 2.4 Å (Supporting Information Table 1). The structure contains four APY–EphA4 complexes in the asymmetric unit and provides detailed information on the APY–EphA4 interaction. The APY peptide efficiently utilizes ∼70% (∼600 Å2) of the available surface within the ephrin-binding pocket (Figure 1A,B; Supporting Information Figure 1). Comparison of the structure of the EphA4 LBD bound to APY (Figure 1A,B) with structures not bound to a ligand (PDB entries 3CKH, 2WO1, 4BK4, and 4M4P) or bound to ephrins (Figure 1C,D,E,F)35−39,41 reveals that APY induces conformational changes in critical loops of EphA4. Upon binding, the peptide locks the DE and JK loops in highly structured antiparallel β-sheet conformations, which causes the ephrin-binding pocket to adopt a “closed” conformation (compare Figure 1B with Figure 1D,F), counteracting the reported extraordinary structural flexibility of the EphA4 LBD.35−38,42 This structural rearrangement suggests that the APY peptide not only functions as a competitive inhibitor that sterically precludes ephrin ligand binding to EphA4 but also promotes a distinct conformation of the EphA4 LBD that could inhibit the receptor preclustering interactions that have been proposed to facilitate subsequent ephrin-induced activation.41 Thus, the structure of the APY–EphA4 complex suggests that APY can effectively inhibit EphA4 signaling through multiple concerted mechanisms.
Figure 1.

APY binds to the ephrin-binding pocket of EphA4. Crystal structures of the EphA4 LBD (gray) in complex with (A,B) APY (orange); (C) part of the GH loop of ephrin-A2 (green, PDB 2WO3, ref (36)); (D) the ephrin-A2 receptor-binding domain; (E) part of the GH loop of ephrin-B2 (cyan, PDB 2WO2, ref (36)); or (F) the ephrin-B2 receptor-binding domain. In the upper panels, EphA4 LBD and ephrins are shown in surface representation. In the lower panels, the EphA4 LBD is shown in ribbon representation (gray) with APY in orange sticks (B) and the ephrins in green (D) or blue (F) ribbon representation. The DE, GH, and JK loops lining the ephrin-binding pocket in the EphA4 LBD are shown in darker shades of gray.
The structure also reveals a critical role for Gly8 in APY. This Gly resides at the apex of the circular portion of the peptide (Figure 2A,B), with torsion angles in the Ramachandran plot (φ = 115.9°; ψ = −17.6° for molecule A) that are only compatible with Gly among the natural amino acids. This enables formation of a “class 1, three-residue β-hairpin” structure that is essential for the correct alignment of the APY residues interacting with EphA4. Residues on one side of the peptide, including Tyr3, Tyr6, Trp10, and the disulfide bond, engage in hydrophobic interactions with the ephrin-binding pocket while the opposite side of the bound peptide remains mostly exposed to the solvent (Figure 2C; Supporting Information Figure 1). APY forms five hydrogen bonds with the EphA4 LBD (between the backbone NH of Tyr3APY and backbone carbonyl of Cys73EphA4; the side chain hydroxyl of Tyr3APY and backbone carbonyl of Pro112EphA4; the backbone NH of Tyr6APY and side chain carbonyl of Gln71EphA4; the backbone carbonyl of Tyr6APY and side chain NH of Gln71EphA4; and the side chain NH of Trp10APY and side chain hydroxyl of Thr104EphA4; Figure 2D; Supporting Information Figure 1). These interactions are consistent with previous mutagenesis data showing a critical role for Gln71EphA4, whose replacement with Ala abolished APY binding.32 In addition, the side chain of Gln71EphA4 is positioned by a hydrogen bond with the side chain of neighboring Thr69EphA4 (Supporting Information Figure 2A), in agreement with the loss of APY binding when Thr69EphA4 is replaced by Ala.32 The I59A and A193S mutations have also been shown to abolish APY binding,32 and the structure shows that both of these EphA4 residues also participate in hydrophobic interactions with the peptide (Supporting Information Figure 1). In contrast, replacement of Thr104EphA4 with Ala did not decrease APY binding,32 suggesting that the Ala may compensate for the loss of the hydrogen bond by contributing to the hydrophobic patch that accommodates Tyr6APY and Trp10APY.
Figure 2.

Detailed structure of APY bound to EphA4. (A) Detailed view of the structure of the APY peptide, which is shown in stick representation in orange with oxygens in red, nitrogens in blue, and disulfide bond in yellow. (B) The APY peptide has three intramolecular hydrogen bonds (dotted green lines, with distance in Å shown in black). The β-turn around Gly8 (indicated with thicker sticks) shows high strain, as indicated by the short unfavorable N–N distance between the Gly and Ser9 (dotted green line, with distance in Å shown in red). APY molecule A is shown, with all four molecules in the asymmetric unit shown in Supporting Information Figure 3. (C) The APY peptide is shown within the ephrin-binding pocket of EphA4. The hydrophobic peptide residues interacting with EphA4 are shown as spheres. EphA4 is shown in surface representation in gray with the DE, GH, and JK loops in darker shades of gray. N-ter, N-terminus. (D) There are five hydrogen bonds (dotted green lines, with distances in Å shown in black) between residues in APY (orange) and EphA4 (gray). Only EphA4 residues engaged in hydrogen bonds are shown.
Besides the interactions with EphA4, three intramolecular hydrogen bonds stabilize the β-hairpin conformation of the peptide (between the backbone NH of Val5 and backbone carbonyl of Ser11; the backbone carbonyl of Val5 and backbone NH of Ser11; and the backbone NH of Arg7 and backbone carbonyl of Ser9; Figure 2D; Supporting Information Figure 3). Additional internal peptide stabilization is provided by a network of hydrophobic interactions centered around Trp10APY, which includes an aromatic interaction between Tyr6APY and Trp10APY and hydrophobic interactions of Trp10APY with the side chains of the disulfide linked Cys4APY and Cys12APY (Figure 2C and Supporting Information Figure 2B,C). In summary, the crystal structure demonstrates that the APY peptide is an EphA4 competitive antagonist that also induces allosteric effects, with its cyclic scaffold representing a configuration well suited for occupying the dynamic ephrin-binding pocket of EphA4.
Structure-Guided Improvement of APY Peptide Antagonists
Given the key role of the β-hairpin structure of APY bound to EphA4, and the fact that the Gly8 at its apex is the only natural amino acid that can allow formation of this structure, we explored substitution of Gly8 with several unnatural amino acids that might allow for better positioning of the peptide for EphA4 binding while preserving the β-turn structure. We also recognized that amidation of the APY C terminus (Cys12) should result in formation of an additional hydrogen bond with the backbone carbonyl of Tyr3APY, further stabilizing the conformation of the bound peptide. Thus, we generated C-terminally amidated peptides and measured their ability to inhibit ephrin-A5 binding to EphA4 in ELISAs. As expected, the C-terminal amidation (leading to APY.am) increased the antagonistic potency of APY (∼10 fold), while replacements of Gly8 had variable effects (Figure 3A; Table 1). DAla8 did not substantially affect potency, consistent with the fact that D amino acids allow formation of the β-turn. In contrast, replacement of Gly8 with LAla8 carried out for comparison resulted in a ∼30 fold loss in potency, confirming the importance of the β-turn conformation. Further constraint of residue 8 through substitution with the achiral aminoisobutyric acid (Aib8) reduced potency by over 10-fold, possibly because of the additional methyl group, which could interfere with EphA4 binding.
Figure 3.
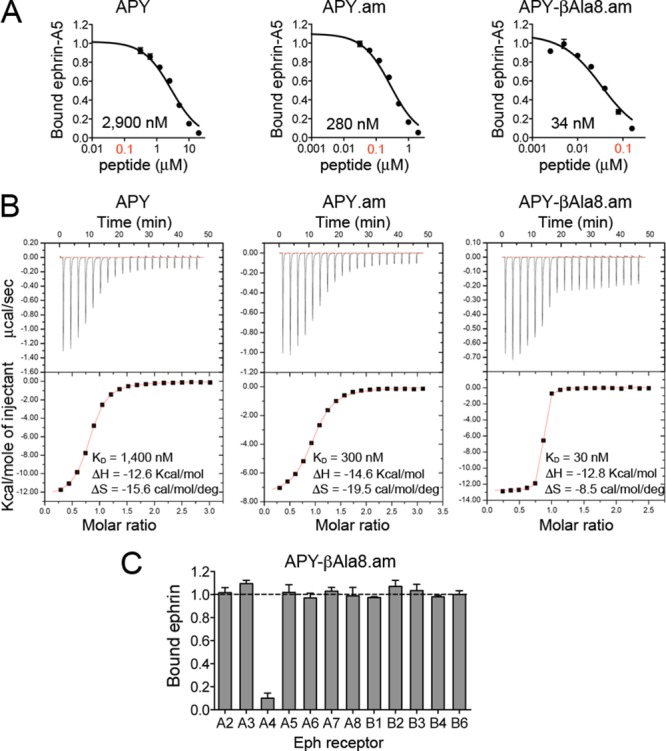
C-terminal amidation of APY and replacement of Gly8 with βAla yield the nanomolar EphA4 antagonist APY-βAla8.am. (A) Representative curves showing inhibition of ephrin-A5 AP binding to immobilized EphA4 Fc by APY derivatives in ELISAs. Bound ephrin-A5 values were normalized to those for bound ephrin-A5 in the absence of peptide and averages from triplicate measurements ± SE are shown. IC50 values are shown under each curve, and the 0.1 μM peptide concentration is in red font. (B) Isothermal titration calorimetry profiles for peptide binding to EphA4 (upper part of each panel) and plots of the integrated values for the reaction heats (after blank subtraction and normalization to the amount of injected peptide) versus EphA4/peptide molar ratio (lower part of each panel). (C) Eph receptor selectivity for APY-βAla8.am. ELISA measuring inhibition of ephrin-A5 AP binding to immobilized EphA Fc receptors and ephrin-B2 AP binding to EphB Fc receptors shows that 3.7 μM APY-βAla8.am selectively inhibits ephrin binding to EphA4. Bound ephrin is the signal in the presence of APY-βAla8.am normalized to the signal without peptide. Averages from triplicate measurements ± SE are shown.
Table 1. Potency of EphA4 Peptide Antagonists.
| peptide | IC50 ± SE (n)a by ELISA (nM) | KD by ITC (nM) |
|---|---|---|
| APY | 2,700 ± 300 (9) | 1400 |
| APY.am | 250 ± 20 (3) | 300 |
| APY-DAla8.am | 280 ± 30 (3) | 250 |
| APY-Ala8.am | 7,700 ± 400 (3) | 2500 |
| APY-Aib8.am | 2,600 ± 200 (3) | ndb |
| APY-βAla8.am | 31 ± 3 (13) | 30 |
n = number of experiments.
nd = not determined.
A critical observation was that the APY backbone structure appears to be slightly strained. For example, the hydrogen bonds are slightly longer than the ideal 2.9 Å length (Figure 2B and Supporting Information Figure 3). In addition, the conformation of the β-turn brings the amide groups of Gly8 and Ser9 into close proximity (2.6 Å), likely leading to electrostatic repulsion. To release the strain of the tight three-residue β-turn at the apex of the peptide, we inserted a methylene spacer into the backbone by replacing Gly8 with βAla. Remarkably, this increased the antagonistic potency of APY.am by ∼8 fold, corresponding to an IC50 of ∼30 nM for APY-βAla8.am (∼85 fold improvement over the original APY; Figure 3A; Table 1). Complementary determination of dissociation constant (KD) values for peptide binding to the EphA4 LBD using isothermal titration calorimetry (ITC) revealed binding affinities consistent with the IC50 values and confirmed that C-terminal amidation and replacement of Gly8 with βAla dramatically increase binding affinity, with a KD of ∼30 nM for APY-βAla8.am (Table 1, Figure 3, and Supporting Information Figure 4). This improvement in affinity makes APY-βAla8.am the most potent EphA4 antagonist developed to date. Importantly, the APY-βAla8.am peptide remains highly selective for EphA4 even at a concentration ∼100 fold higher than the IC50 value for inhibition of ephrin-A5-EphA4 binding (Figure 3C).
The Crystal Structure of APY-βAla8.am in Complex with EphA4 Shows Improved Binding Features
We next sought to better understand the molecular details underlying the dramatic increase in potency of APY-βAla8.am. The crystallization conditions used for the APY-EphA4 complex yielded initial crystals that were further refined to obtain diffracting crystals that enabled us to solve the structure of the new APY-βAla8.am-EphA4 complex at a resolution of 2.4 Å (Supporting Information Table 1). Comparison of bound APY-βAla8.am with APY revealed a very similar overall conformation (Figure 4A). As envisioned, an additional hydrogen bond links the amidated C terminus of APY-βAla8.am with the backbone carbonyl function of Tyr3, leading to an overall stabilization of the peptide and improving its interaction with EphA4 (Figure 4B). Furthermore, comparison of the four APY-βAla8.am-EphA4 complexes in the crystal asymmetric unit with their counterparts in the APY-EphA4 structure revealed additional features consistent with the optimized binding of APY-βAla8.am. These include a less strained β-turn due to a longer distance between the backbone NH of βAla8 and Ser9 in APY-βAla8.am compared to the corresponding distance between Gly8 and Ser9 in APY (Supporting Information Figure 3). Additionally, the two intrapeptide hydrogen bonds distal to the β-turn are less variable and more favorable in the four APY-βAla8.am molecules than in the APY molecules (average distance ± SD for the Val5 O-Ser11 N hydrogen bond is 3.03 ± 0.05 Å in the APY-βAla8.am molecules and 3.23 ± 0.36 Å in the APY molecules; average distance ± SD for the Val5 N-Ser11 O hydrogen bond is 3.03 ± 0.15 Å in the APY-βAla8.am molecules and 3.25 ± 0.31 Å in the APY molecules; Supporting Information Figure 3).
Figure 4.
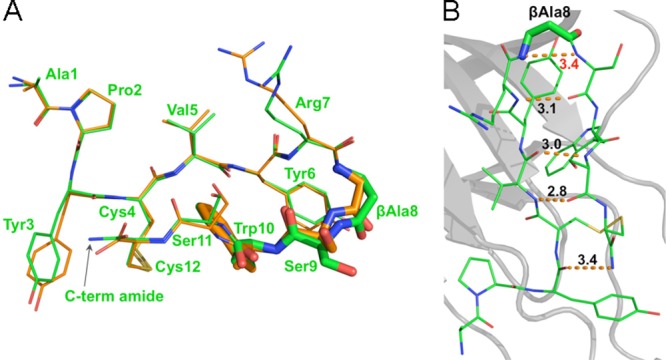
Crystal structure of APY-βAla8.am bound to EphA4 reveals critical differences from APY. (A) Overlay of APY-βAla8.am (green) and APY (orange) shows marked differences in the β-turn region, particular in the βAla8, Ser9, and Trp10 residues (highlighted by stick representation). Residues are labeled for the APY-βAla8.am peptide. (B) Intramolecular hydrogen bonds of APY-βAla8.am. Overall intramolecular hydrogen bond patterns and conformations are more favorable for APY-βAla8.am than for APY peptide, including the presence of an additional hydrogen bond between the C-terminal amide and Tyr3 of APY-βAla8.am. Molecule A is shown for both peptides, whereas all four molecules in the asymmetric unit are shown in Supporting Information Figure 3.
Secondary Phage Display Screens Reveal APY Features Important for EphA4 Binding
To further characterize the role of different APY residues, we constructed four secondary phage display libraries that retained Cys4 and Cys12 (essential for the cyclic conformation of APY) and Gly8 (essential for the β-turn) but had variable residues at several other positions (Supporting Information Table 2). While the absence of individual sequences should not be overinterpreted due to intrinsic biases in the phage display process, the sequences that are selected can provide useful insights into the molecular basis of peptide binding. To investigate whether the critical Tyr3, Tyr6, and Trp10 can be replaced by other amino acids, we isolated the EphA4-binding phage from libraries with variable residues at these positions. In line with the peptide-EphA4 structures, the sequences of 22 different binding peptides showed a preference for hydrophobic amino acids at position 3, including Leu (7 peptides), Tyr (5 peptides), Phe/Val (3 peptides each), and Ile/Trp/His (1 peptide each; Table 2, libraries 1 and 4). Sequencing of a randomly isolated phage documented the presence of at least 18 of the 20 amino acids at position 3 in libraries 1 and 4 (Supporting Information Table 3), supporting the importance of the selected amino acids for EphA4 binding rather than a bias in their library representation. To evaluate the approximate relative binding strength of the phage-displayed peptides, we used the KYL peptide antagonist28,32 to compete phage binding to EphA4 in ELISAs. The results suggest that peptides with Tyr, Phe, Trp, Val, Leu, His, and Trp (but not Ile) at position 3 can bind strongly to EphA4 because high KYL concentrations were needed to inhibit phage binding (Table 2, libraries 1 and 4).
Table 2. Peptides from Secondary Phage Display Screens.

Not from panning.
Clones with different DNA sequences. The sequence of the APY peptide (isolated from library 4) is boxed.
Same peptide sequence isolated from different libraries.
nd, not determined.
The Ala to Gly change at position 1 was not designed in the library and is thus due to an aberrant oligonucleotide.
The EphA4 binding peptides isolated from a library with variable position 6 (Table 2, library 1) showed that aromatic amino acids are strongly preferred at this position over the 14 other amino acids that were also documented at this position in library 1 (Supporting Information Table 3). Trp was the only amino acid identified at position 10 in the three peptides that were selected multiple times by panning library 2 on EphA4 (Table 2), consistent with the buried position of Trp10 in the binding interface (15 different amino acids were documented at position 10 in library 2; Supporting Information Table 3). The presence of an aromatic residue at position 6 and Trp at position 10 is consistent with the observed aromatic interaction between Tyr6 and Trp10 and the additional hydrophobic network of Trp10 with the disulfide bonded Cys4 and Cys12 (Figure 2C and Supporting Information Figure 2B,C), which are key features stabilizing the APY peptide.34
With regard to the APY N terminus (Ala-Pro), the EphA4-binding phage contained a variety of residues at position 1 (Table 2, library 4). At position 2, Pro was most frequent (8/12 peptides) with Ala present in the remaining peptides. However, only phage clones with Ala1/Glu1 and Pro2 exhibited strong binding to EphA4 (based on KYL competition ELISAs; Table 2, library 4). Thus, despite a lack of obviously important contacts in the crystal structure, the three residues outside the peptide macrocycle seem to be important for APY binding activity and tolerate limited sequence variability.
The crystal structures also show that Arg7, Ser9, and Ser11 of APY and APY-βAla8.am do not engage in direct interactions with EphA4 residues. Indeed, a wide variety of amino acids can occupy these positions in the EphA4-binding phage (Table 2, libraries 2 and 3). Furthermore, approximately half of the phage clones that were randomly chosen from library 3, in which residues 7, 9, and 11 are randomized, bound to EphA4 even though they were not isolated by panning on EphA4 (Supporting Information Table 3). Interestingly, all but one of the nonbinding peptides from this library have Pro at one of the randomized positions (Supporting Information Table 3). Thus, it appears that many amino acids but not Pro can occupy positions 7, 9, and 11 of the APY scaffold, likely due to the disrupting effects of Pro on the conformation of the circular portion of APY. These data indicate that residues forming the solvent-exposed side of APY derivatives bound to EphA4 could be modified without a substantial loss of binding affinity, for modifications such as peptide derivatization to improve pharmacokinetic properties.
APY-βAla8.am Inhibits EphA4 Activation in Cells at Submicromolar Concentrations
To assess the antagonistic potency of APY-βAla8.am in live cells stimulated with ephrin, we used human embryonal kidney (HEK293) cells stably expressing human EphA4. Stimulation of these cells with dimeric ephrin-A5 Fc promotes EphA4 phosphorylation on tyrosine residues, which is indicative of receptor activation. Quantifications of immunoblots revealed that APY-βAla8.am inhibits ephrin-A5-induced EphA4 tyrosine phosphorylation with an IC50 value of ∼0.5 μM, with almost complete inhibition at 1.6 μM. The higher IC50 for inhibition of ephrin-A5 Fc-induced EphA4 tyrosine phosphorylation in cells compared to inhibition of ephrin-A5 AP binding to EphA4 in ELISAs is likely due to the much higher ephrin concentration needed to activate EphA4 and possibly to other differences between the two types of experiments, in accordance with the Cheng–Prusoff equation.43 These results suggest that APY-βAla8.am is a ≥20 fold more potent EphA4 antagonist than APY and also KYL (Figure 5A and ref (28)), the peptide previously used in mouse models of nerve regeneration and ALS.3,10
Figure 5.

Inhibition of EphA4 activation in cells by APY-βAla8.am. (A) Inhibition of ephrin-A5-induced EphA4 tyrosine phosphorylation. EphA4 was immunoprecipitated from stably transfected HEK293 cells treated with ephrin-A5 Fc (+) or Fc control (−) in the presence of the indicated concentrations of APY-βAla8.am or APY. The immunoprecipitates were probed for phosphotyrosine (PTyr) and reprobed for EphA4. The graph on the right shows quantification of EphA4 tyrosine phosphorylation levels from the immunoblots of seven experiments, normalized to the phosphorylation level in the ephrin-A5/no peptide condition in each experiment. Averages from three to six measurements ± SE are shown in the graph. (B) Inhibition of EphA4-dependent growth cone collapse. Explants from embryonic day 6 chicken retina were pretreated with 0.3 μM APY-βAla8.am peptide, stimulated with ephrin-A5 Fc or Fc as a control, and stained to label actin filaments. The histogram shows the mean percentages of collapsed growth cones (∼70 to 500 per condition in each experiment). Error bars represent standard errors from three experiments. *, P < 0.05 compared to Fc without peptide by one-way ANOVA. (C) The APY-βAla8.am peptide does not have detectable cytotoxic effects. HT22 neuronal cells were grown in the presence of 30 μM peptide, or without peptide as a control, for 24 h, and cell viability was assessed using the MTT assay. Averages from six measurements ± SE are shown.
Impaired axon sprouting and lack of reinnervation are regarded as part of the pathology underlying neurodegenerative diseases such as ALS, ultimately causing neuronal cell death.10 We therefore also assessed the ability of APY-βAla8.am to inhibit the collapse of neuronal growth cones (enlarged structures at the leading tip of axons), since growth cone collapse is linked to the failure of injured axons to sprout and regenerate.3,4 We used nasal retinal explants, where ephrin-A5-induced collapse depends on EphA4 activation and can be blocked by 5 μM KYL peptide.4 The APY-βAla8.am peptide effectively blocked collapse at the much lower concentration of 0.3 μM (Figure 5B). The peptide did not detectably affect the morphology of growth cones in the absence of ephrin, consistent with a lack of nonspecific effects or toxicity. Indeed, a cell viability assay did not reveal significant cytotoxicity in the hippocampal neuron-derived HT22 cell line treated for 24 h with 30 μM APY-βAla8.am, a concentration 100 fold higher than that sufficient to inhibit growth cone collapse (Figure 5C). The KYL peptide has been the EphA4 antagonist of choice for promoting neural repair and counteracting neurodegeneration.3,4,10,11 The dramatically improved potency of APY-βAla8.am compared to KYL suggests that this derivative can serve as a new scaffold for the generation of greatly improved EphA4 antagonists exerting neuroprotective effects.
Conclusions
We developed a novel cyclic peptide antagonist, APY-βAla8.am, through structure-guided optimization of the APY peptide previously identified by phage display.28 APY-βAla8.am binds to EphA4 with nanomolar affinity while maintaining high receptor specificity. Importantly, APY-βAla8.am greatly surpasses in potency the KYL peptide that has been used in ALS, Alzheimer’s disease, and spinal cord injury models.3,10−12 The cyclic scaffold of APY-βAla8.am is a particularly valuable attribute, since cyclic peptides can show more favorable properties, including high binding affinity and metabolic stability.40 Future optimization efforts will aim at further improving the potency as well as several other properties of APY-βAla8.am, such as in vivo lifetime and efficacy.
Methods
EphA4 LBD Expression and Purification
The EphA4 LBD (residues 29–204 with Cys204 replaced by Ala) was cloned into the pETNKI-His-3C-LIC expression vector44 and expressed in E. coli origami 2(DE3) bacterial cells grown at 20 °C overnight. Cells were lysed by sonication and the EphA4 LDB was purified by Ni2+-affinity chromatography. The N-terminal His-tag was cleaved with 3C protease (leaving the three extra GPG residues at the EphA4 N terminus) and removed by gel filtration using a Superdex 200 column equilibrated in 100 mM NaCl, 10 mM Hepes, pH 7.9. Protein aliquots were flash frozen in liquid nitrogen and stored at −80 °C.
Crystallization and Structure Solution
For crystallization, the EphA4 LBD (∼20 mg mL–1 in 100 mM NaCl, 10 mM Hepes pH 7.9) was added to a 1.5 fold molar excess of peptide. Crystallization trials were conducted using the sitting-drop vapor diffusion method with commercial screens. Single diffraction quality crystals were obtained after a second round of screening using the Additive Screen HT (Hampton Research) in 0.2 M MgCl2, 0.1 M Tris pH 8.5, and 25% PEG3350 with the additives 4% 1,3-butanediol for APY or 3% 1,6-hexanediol for APY-βAla8.am. Crystals were cryoprotected in reservoir solution with the addition of 20% glycerol and cryocooled in a nitrogen stream at 100 K. Data sets were collected on a rotating anode X-ray generator (Rigaku) at 100 K and processed in iMosflm45 and with software from the CCP4 suite.46 Initial analysis suggested space group P2221, but multiple tests suggested crystal twinning, and finally P21 was determined as the real space group. Initial phases were obtained via molecular replacement using coordinates from PDB 2WO1 chain B36 as a search model. Noncrystallographic symmetry (NCS) restraints were used in early rounds of refinement, whereas later refinement stages included TLS (translation/libration/screw) and twin refinement (twin fractions: ∼0.35/0.65 for both structures). MolProbity47 was used for structure validation. Data collection and refinement statistics are summarized in Supporting Information Table 1.
Isothermal Titration Calorimetry
The EphA4 LBD and the peptides were diluted to obtain a final buffer containing 5% DMSO in 10 mM Hepes, pH 7.6, and 100 mM NaCl. ITC experiments were carried out at 296 K (23 °C) using an ITC200 calorimeter (Microcal). Two microliter aliquots of a 1 mM peptide solution were injected into the cell containing 205 μL EphA4 ligand-binding domain solution at a concentration of 65–95 μM. Experimental data were analyzed using the Origin software package (Microcal).
Inhibition of EphA4 Activation in Cells
HEK293AD stably expressing human EphA4 were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1 mM sodium pyruvate, and antibiotics. One hour prior to stimulation, the cells were starved in DMEM supplemented with 1 mM sodium pyruvate and antibiotics but without FBS. Cells were then incubated with different concentrations of APY or APY-βAla8.am for 20 min before the addition of 0.5 μg/mL ephrin-A5 Fc (corresponding to ∼3.5 nM of the dimer; R&D Systems) or Fc for another 20 min to activate EphA4. Cells were next washed in PBS and lysed in modified RIPA buffer (1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 20 mM Tris, 150 mM NaCl, 1 mM EDTA) containing 10 mM NaF, phosphatase inhibitor cocktails 1 and 2 (Sigma), and protease inhibitors. Protein concentrations were calculated using the BCA protein assay kit (Pierce). Cell lysates were immunoprecipitated with 4 μg affinity-purified polyclonal anti-EphA4 antibody generated using a peptide corresponding to the 11 C-terminal amino acids of EphA4.4 Immunoprecipitates were probed by immunoblotting with an antiphosphotyrosine-HRP antibody (BD Biosciences) and then reprobed with the anti-EphA4 antibody followed by a secondary HRP-conjugated antibody. Signal intensity of immunoblot bands was quantified by using the histogram function of Photoshop.
Growth Cone Collapse Assay
Explants from embryonic day 6 (E6) chicken nasal retinas were cultured on 35 mm glass-bottom MatTek plates precoated overnight with 200 μg/mL poly-d-lysine in PBS and then for 3 h with 20 μg/mL laminin in PBS at 37 °C. Explants were cultured overnight in DMEM-F12 containing 0.4% methylcellulose (Sigma-Aldrich), 0.45% glucose, N-2 supplement (Life Technologies), 2 mM l-glutamine (Life Technologies), 1 mM sodium pyruvate, 0.1% BSA, and antibiotics. The culture medium was then replaced with a medium without methylcellulose, and 3 h later the retinal explants were incubated with 0.3 μM APY-βAla8.am peptide for 30 min before stimulation with 1 μg/mL preclustered ephrin-A5 Fc or Fc as a control for 30 min in the continued presence of the peptide. Ephrin-A5 Fc was preclustered by incubating it for 30 min on ice with 1/10 polyclonal anti-Fc antibody (Jackson Laboratories). The explants were then fixed for 30 min in 3.7% formaldehyde, 4% sucrose in PBS at RT, permeabilized for 3 min with 0.1% Triton X-100 in PBS, and filamentous actin was stained with rhodamine-conjugated phalloidin (Life Technologies). Growth cones were photographed under a fluorescence microscope and scored in a blinded manner as collapsed when no filopodia were present at the tip of the neurite.
Acknowledgments
The authors thank A. Bobkov for performing the isothermal titration calorimetry experiments, W. Danho for helpful discussions, and the organizers of the 2013 APS/CCP4 School for help with model building and structure refinement. This work was supported by National Institutes of Health grants P01CA138390 (E.B.P. and S.J.R.) and R01GM098871 (P.E.D.), an EMBO Long-Term Postdoctoral Fellowship (B.C.L.), and DOD-BCRP Fellowship BC100466 (P.D.M.).
Glossary
Abbreviations
- ALS
amyotrophic lateral sclerosis
- AP
alkaline phosphatase
- GPI
glycosylphosphatidylinositol
- DMEM
Dulbecco’s Modified Eagle’s medium
- DMSO
dimethyl sulfoxide
- ELISA
enzyme-linked immunoabsorbent assay
- FBS
fetal bovine serum
- Fmoc
9-fluorenylmethoxycarbonyl
- HPLC
high-performance liquid chromatography
- HRP
horseradish peroxidase
- IC50
half maximal inhibitory concentration
- ITC
isothermal titration calorimetry
- KD
dissociation constant
- LBD
ligand-binding domain
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NCS
noncrystallographic symmetry
- TBS
Tris-buffered saline
- TLS
translation/libration/screw
Supporting Information Available
Supplementary methods, Tables 1–3, and Figures 1–4 are available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
Coordinates and structure factors have been deposited in the Protein Data Bank under accession numbers 4W50 (APY-EphA4 complex) and 4W4Z (APY-βAla8.am-EphA4 complex).
Author Present Address
∥ Biochemistry Department, University of Otago, 710 Cumberland Street, Dunedin 9054, New Zealand
Author Contributions
⊥ These authors contributed equally to this work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Pasquale E. B. (2008) Eph-ephrin bidirectional signaling in physiology and disease. Cell 133, 38–52. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Fu A. K.; Ip N. Y. (2012) Eph receptors at synapses: implications in neurodegenerative diseases. Cell Signal 24, 606–611. [DOI] [PubMed] [Google Scholar]
- Fabes J.; Anderson P.; Brennan C.; Bolsover S. (2007) Regeneration-enhancing effects of EphA4 blocking peptide following corticospinal tract injury in adult rat spinal cord. Eur. J. Neurosci. 26, 2496–2505. [DOI] [PubMed] [Google Scholar]
- Noberini R.; Koolpe M.; Peddibhotla S.; Dahl R.; Su Y.; Cosford N. D.; Roth G. P.; Pasquale E. B. (2008) Small molecules can selectively inhibit ephrin binding to the EphA4 and EphA2 receptors. J. Biol. Chem. 283, 29461–29472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai K. K.; Nguyen L. N.; Irie F.; Yamaguchi Y.; Pasquale E. B. (2003) Control of hippocampal dendritic spine morphology through ephrin-A3/EphA4 signaling. Nat. Neurosci. 6, 153–160. [DOI] [PubMed] [Google Scholar]
- Filosa A.; Paixao S.; Honsek S. D.; Carmona M. A.; Becker L.; Feddersen B.; Gaitanos L.; Rudhard Y.; et al. (2009) Neuron-glia communication via EphA4/ephrin-A3 modulates LTP through glial glutamate transport. Nat. Neurosci. 12, 1285–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona M. A.; Murai K. K.; Wang L.; Roberts A. J.; Pasquale E. B. (2009) Glial ephrin-A3 regulates hippocampal dendritic spine morphology and glutamate transport. Proc. Natl. Acad. Sci. U.S.A. 106, 12524–12529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldshmit Y.; Spanevello M. D.; Tajouri S.; Li L.; Rogers F.; Pearse M.; Galea M.; Bartlett P. F.; et al. (2011) EphA4 Blockers Promote Axonal Regeneration and Functional Recovery Following Spinal Cord Injury in Mice. PLoS One 6, e24636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanevello M. D.; Tajouri S. I.; Mirciov C.; Kurniawan N.; Pearse M. J.; Fabri L. J.; Owczarek C. M.; Hardy M. P.; et al. (2013) Acute delivery of EphA4-Fc improves functional recovery after contusive spinal cord injury in rats. J. Neurotrauma 30, 1023–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hoecke A.; Schoonaert L.; Lemmens R.; Timmers M.; Staats K. A.; Laird A. S.; Peeters E.; Philips T.; et al. (2012) EPHA4 is a disease modifier of amyotrophic lateral sclerosis in animal models and in humans. Nat. Med. 18, 1418–1422. [DOI] [PubMed] [Google Scholar]
- Vargas L. M.; Leal N.; Estrada L. D.; Gonzalez A.; Serrano F.; Araya K.; Gysling K.; Inestrosa N. C.; et al. (2014) EphA4 activation of c-Abl mediates synaptic loss and LTP blockade caused by amyloid-beta oligomers. PLoS One 9, e92309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu A. K.; Hung K. W.; Huang H.; Gu S.; Shen Y.; Cheng E. Y.; Ip F. C.; Huang X.; et al. (2014) Blockade of EphA4 signaling ameliorates hippocampal synaptic dysfunctions in mouse models of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 111, 9959–9964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmens R.; Jaspers T.; Robberecht W.; Thijs V. N. (2013) Modifying expression of EphA4 and its downstream targets improves functional recovery after stroke. Hum. Mol. Genet. 22, 2214–2220. [DOI] [PubMed] [Google Scholar]
- Ashida S.; Nakagawa H.; Katagiri T.; Furihata M.; Iiizumi M.; Anazawa Y.; Tsunoda T.; Takata R.; et al. (2004) Molecular features of the transition from prostatic intraepithelial neoplasia (PIN) to prostate cancer: genome-wide gene-expression profiles of prostate cancers and PINs. Cancer Res. 64, 5963–5972. [DOI] [PubMed] [Google Scholar]
- Iiizumi M.; Hosokawa M.; Takehara A.; Chung S.; Nakamura T.; Katagiri T.; Eguchi H.; Ohigashi H.; et al. (2006) EphA4 receptor, overexpressed in pancreatic ductal adenocarcinoma, promotes cancer cell growth. Cancer Sci. 97, 1211–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oki M.; Yamamoto H.; Taniguchi H.; Adachi Y.; Imai K.; Shinomura Y. (2008) Overexpression of the receptor tyrosine kinase EphA4 in human gastric cancers. World J. Gastroenterol. 14, 5650–5656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyner J. W.; Deininger M. W.; Loriaux M. M.; Chang B. H.; Gotlib J. R.; Willis S. G.; Erickson H.; Kovacsovics T.; et al. (2009) RNAi screen for rapid therapeutic target identification in leukemia patients. Proc. Natl. Acad. Sci. U.S.A. 106, 8695–8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y.; Luo Y. C.; Wan H. Y.; Wang J.; Zhang P. P.; Liu M.; Li X.; Li S.; Tang H. (2013) MicroRNA-10a is involved in the metastatic process by regulating Eph tyrosine kinase receptor A4-mediated epithelial-mesenchymal transition and adhesion in hepatoma cells. Hepatology 57, 667–677. [DOI] [PubMed] [Google Scholar]
- Brantley-Sieders D. M.; Jiang A.; Sarma K.; Badu-Nkansah A.; Walter D. L.; Shyr Y.; Chen J. (2011) Eph/ephrin profiling in human breast cancer reveals significant associations between expression level and clinical outcome. PLoS One 6, e24426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saintigny P.; Peng S.; Zhang L.; Sen B.; Wistuba I. I.; Lippman S. M.; Girard L.; Minna J. D.; et al. (2012) Global Evaluation of Eph receptors and ephrins in lung adenocarcinomas identifies EphA4 as an inhibitor of cell migration and invasion. Mol. Cancer Ther. 11, 2021–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukai J.; Yokote H.; Yamanaka R.; Arao T.; Nishio K.; Itakura T. (2008) EphA4 promotes cell proliferation and migration through a novel EphA4-FGFR1 signaling pathway in the human glioma U251 cell line. Mol. Cancer Ther. 7, 2768–2778. [DOI] [PubMed] [Google Scholar]
- Pasquale E. B. (2005) Eph receptor signalling casts a wide net on cell behaviour. Nat. Rev. Mol. Cell. Biol. 6, 462–475. [DOI] [PubMed] [Google Scholar]
- Pasquale E. B. (2010) Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat. Rev. Cancer 10, 165–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noberini R.; Lamberto I.; Pasquale E. B. (2012) Targeting Eph receptors with peptides and small molecules: progress and challenges. Semin. Cell Dev. Biol. 23, 51–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaman M. W.; Herrgard S.; Treiber D. K.; Gallant P.; Atteridge C. E.; Campbell B. T.; Chan K. W.; Ciceri P.; et al. (2008) A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 26, 127–132. [DOI] [PubMed] [Google Scholar]
- Parmentier-Batteur S.; Finger E. N.; Krishnan R.; Rajapakse H. A.; Sanders J. M.; Kandpal G.; Zhu H.; Moore K. P.; et al. (2011) Attenuation of scratch-induced reactive astrogliosis by novel EphA4 kinase inhibitors. J. Neurochem. 118, 1016–1031. [DOI] [PubMed] [Google Scholar]
- Tognolini M.; Hassan-Mohamed I.; Giorgio C.; Zanotti I.; Lodola A. (2014) Therapeutic perspectives of Eph-ephrin system modulation. Drug Discovery Today 19, 661–669. [DOI] [PubMed] [Google Scholar]
- Murai K. K.; Nguyen L. N.; Koolpe M.; McLennan R.; Krull C. E.; Pasquale E. B. (2003) Targeting the EphA4 receptor in the nervous system with biologically active peptides. Mol. Cell Neurosci. 24, 1000–1011. [DOI] [PubMed] [Google Scholar]
- Wu B.; Zhang Z.; Noberini R.; Barile E.; Giulianotti M.; Pinilla C.; Houghten R. A.; Pasquale E. B.; Pellecchia M. (2013) HTS by NMR of combinatorial libraries: a fragment-based approach to ligand discovery. Chem. Biol. 20, 19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharfe N.; Nikolic M.; Cimpeon L.; Van De Kratts A.; Freywald A.; Roifman C. M. (2008) EphA and ephrin-A proteins regulate integrin-mediated T lymphocyte interactions. Mol. Immunol. 45, 1208–1220. [DOI] [PubMed] [Google Scholar]
- Galimberti I.; Bednarek E.; Donato F.; Caroni P. (2010) EphA4 signaling in juveniles establishes topographic specificity of structural plasticity in the hippocampus. Neuron 65, 627–642. [DOI] [PubMed] [Google Scholar]
- Lamberto I.; Qin H.; Noberini R.; Premkumar L.; Bourgin C.; Riedl S. J.; Song J.; Pasquale E. B. (2012) Distinctive binding of three antagonistic peptides to the ephrin-binding pocket of the EphA4 receptor. Biochem. J. 445, 47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X.; Xu Y.; Yang Y.; Xi J.; Tian W.; Duggineni S.; Huang Z.; An J. (2013) Discovery and characterization of a novel cyclic peptide that effectively inhibits ephrin binding to the EphA4 receptor and displays anti-angiogenesis activity. PLoS One 8, e80183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogan A. A.; Thorn K. S. (1998) Anatomy of hot spots in protein interfaces. J. Mol. Biol. 280, 1–9. [DOI] [PubMed] [Google Scholar]
- Qin H.; Shi J.; Noberini R.; Pasquale E. B.; Song J. (2008) Crystal Structure and NMR Binding Reveal That Two Small Molecule Antagonists Target the High Affinity Ephrin-binding Channel of the EphA4 Receptor. J. Biol. Chem. 283, 29473–29484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowden T. A.; Aricescu A. R.; Nettleship J. E.; Siebold C.; Rahman-Huq N.; Owens R. J.; Stuart D. I.; Jones E. Y. (2009) Structural plasticity of Eph receptor A4 facilitates cross-class ephrin signaling. Structure 17, 1386–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin H.; Noberini R.; Huan X.; Shi J.; Pasquale E. B.; Song J. (2010) Structural characterization of the EphA4-Ephrin-B2 complex reveals new features enabling Eph-ephrin binding promiscuity. J. Biol. Chem. 285, 644–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singla N.; Goldgur Y.; Xu K.; Paavilainen S.; Nikolov D. B.; Himanen J. P. (2010) Crystal structure of the ligand-binding domain of the promiscuous EphA4 receptor reveals two distinct conformations. Biochem. Biophys. Res. Commun. 399, 555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiradake E.; Schaupp A.; del Toro Ruiz D.; Kaufmann R.; Mitakidis N.; Harlos K.; Aricescu A. R.; Klein R.; Jones E. Y. (2013) Structurally encoded intraclass differences in EphA clusters drive distinct cell responses. Nat. Struct. Mol. Biol. 20, 958–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craik D. J.; Fairlie D. P.; Liras S.; Price D. (2013) The future of peptide-based drugs. Chem. Biol. Drug Des. 81, 136–147. [DOI] [PubMed] [Google Scholar]
- Xu K.; Tzvetkova-Robev D.; Xu Y.; Goldgur Y.; Chan Y. P.; Himanen J. P.; Nikolov D. B. (2013) Insights into Eph receptor tyrosine kinase activation from crystal structures of the EphA4 ectodomain and its complex with ephrin-A5. Proc. Natl. Acad. Sci. U.S.A. 110, 14634–14639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin H.; Lim L.; Song J. (2012) Protein dynamics at Eph receptor-ligand interfaces as revealed by crystallography, NMR and MD simulations. BMC Biophys. 5, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y.; Prusoff W. H. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. (Amsterdam, Neth.) 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- Luna-Vargas M. P.; Christodoulou E.; Alfieri A.; van Dijk W. J.; Stadnik M.; Hibbert R. G.; Sahtoe D. D.; Clerici M.; et al. (2011) Enabling high-throughput ligation-independent cloning and protein expression for the family of ubiquitin specific proteases. J. Struct. Biol. 175, 113–119. [DOI] [PubMed] [Google Scholar]
- Battye T. G.; Kontogiannis L.; Johnson O.; Powell H. R.; Leslie A. G. (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr., D Biol. Crystallogr. 67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B. 3rd; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.