

Abstract
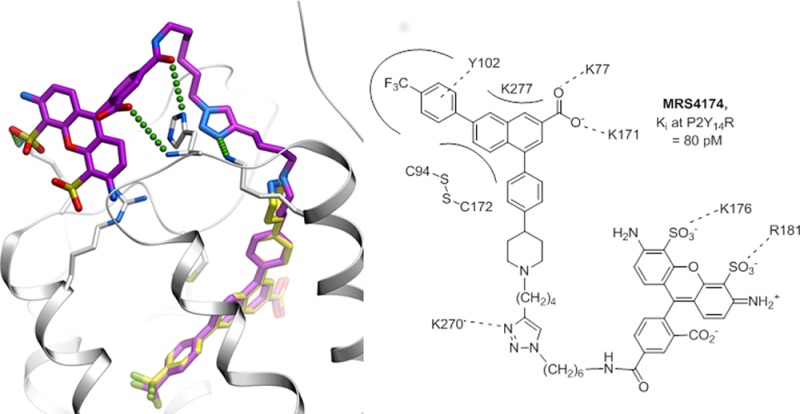
The P2Y14 receptor (P2Y14R), one of eight P2Y G protein-coupled receptors (GPCR), is involved in inflammatory, endocrine, and hypoxic processes and is an attractive pharmaceutical target. The goal of this research is to develop high-affinity P2Y14R fluorescent probes based on the potent and highly selective antagonist 4-(4-(piperidin-4-yl)-phenyl)-7-(4-(trifluoromethyl)-phenyl)-2-naphthoic acid (6, PPTN). A model of hP2Y14R based on recent hP2Y12R X-ray structures together with simulated antagonist docking suggested that the piperidine ring is suitable for fluorophore conjugation while preserving affinity. Chain-elongated alkynyl or amino derivatives of 6 for click or amide coupling were synthesized, and their antagonist activities were measured in hP2Y14R-expressing CHO cells. Moreover, a new Alexa Fluor 488 (AF488) containing derivative 30 (MRS4174, Ki = 80 pM) exhibited exceptionally high affinity, as compared to 13 nM for the alkyne precursor 22. A flow cytometry assay employing 30 as a fluorescent probe was used to quantify specific binding to P2Y14R. Known P2Y receptor ligands inhibited binding of 30 with properties consistent with their previously established receptor selectivities and affinities. These results illustrate that potency in this series of 2-naphthoic acid derivatives can be preserved by chain functionalization, leading to highly potent fluorescent molecular probes for P2Y14R. Such conjugates will be useful tools in expanding the SAR of this receptor, which still lacks chemical diversity in its collective ligands. This approach demonstrates the predictive power of GPCR homology modeling and the relevance of newly determined X-ray structures to GPCR medicinal chemistry.
G protein-coupled receptors (GPCRs) are important cell surface signaling proteins with characteristic seven membrane-spanning (7TM) helices. Eight GPCRs that respond to extracellular nucleotides comprise two subfamilies of P2Y receptors based on sequence and functional similarity: P2Y1 receptor (P2Y1R)-like P2Y1,2,4,6,11 and P2Y12R-like P2Y12–14. The cognate agonists of the human (h) P2Y receptors include ADP (P2Y1, P2Y12, P2Y13), ATP (P2Y2, P2Y11), uridine-5′-diphosphate 1 (UDP: P2Y6, P2Y14), UTP (P2Y2, P2Y4), and uridine-5′-diphosphoglucose 2 (UDPG: P2Y14) (Chart 1).1 P2Y receptors are widely distributed, with marked expression in a number of tissues including the central and peripheral nervous systems, lung, kidney, intestine, spleen, platelets, and immune cells.1
Chart 1. Representative P2Y14R Agonists (1–5) and Antagonists (6–8).

The P2Y14R is expressed on immune and epithelial cells and is involved in inflammation and hypoxic processes.2 Activation of P2Y14R contributes to mechanical pain hypersensitivity via microglial cells,3 enhances the mobility of neutrophils,4 and promotes the release of mediators from mast cells.5 Recent studies with P2Y14R knockout mice demonstrate the value of P2Y14R antagonism as a potential therapeutic goal for diabetes treatment.6
Development of selective and potent agonists and antagonists of P2Y14R is a highly important goal given the role of this signaling protein in health and disease. To date, the structure–activity relationships (SARs) of only three chemical classes have been extensively evaluated at the P2Y14R. The first class consists of synthetic nucleotide analogues of 2, including potent agonists 3–5 (Chart 1).7,8 The second class is made up of derivatives of 2-naphthoic acid, notably the highly potent and selective antagonist 4-(4-(piperidin-4-yl)-phenyl)-7-(4-(trifluoromethyl)-phenyl)-2-naphthoic acid 6 and its ester prodrug 7.9,10 The third class consists of analogues built on a pyrido[4,3-d]pyrimidine scaffold, with compound 8 as one of the most potent members.11 We have used the recently reported high-resolution crystal structures of the hP2Y12R in complex with an antagonist or full agonist12,13 to construct homology models of the hP2Y14R for ligand docking. This structural insight, which we show to be consistent with ligand pharmacology, has guided our molecular design process at this closely related receptor on the δ-branch of Family A GPCRs.
Most of the analogues of 2 are poor drug candidates due to their low bioavailability and stability; although they could be used in vitro, they are not optimal for in vivo or tissue-based assays. Various representative compounds of the 2-naphthoic acid class also display poor drug-like characteristics due to high molecular weight and high lipophilicity.14 Nevertheless, the chemical stability and high affinity and selectivity of 6 toward P2Y14R over other members of the P2Y family make derivatives of 6 potentially attractive candidates for further development of chemical probes for this receptor.15 We propose to use the 2-naphthoic acid series to design chain-extended analogues that contain fluorescent and other reporter groups for receptor detection and characterization. Such probes could be used in place of radioligands in pharmaceutical research and drug discovery.
Results and Discussion
A set of functionalized analogues of 6 was designed and prepared (Schemes 1 and 2) from the key ester intermediate 19, based on predictions from molecular modeling of modes of receptor binding. The strategy behind the selection of these target compounds is described in the following section. Coupling was accomplished by click coupling of an azide and alkyne by copper-catalyzed [2 + 3] cycloaddition16 or amide condensation.
Scheme 1. Synthesis of (A) N-Alkylethynyl Derivatives of 2-Naphthoic Acid Antagonist 6, (B) ω-Ethynylalkylamido Derivatives of 2-Naphthoic Acid Antagonist 6, (C) Terminal Amino and Protected Amino Derivatives of 2-Naphthoic Acid Antagonist 6, and (D) N-Methyl Analogue of 6.

Reagents and conditions for (A): (a) (1) 4-bromobutyne-1 (for 20), 5-bromopentyne-1 (for 21 and 23) or 6-bromohexyne-1 (for 22), K2CO3, DMF, 60–70 °C; (2) LiOH (0.5 M), CH3OH, H2O (20, 46%; 22, 57%; 23, 65%). Reagents and conditions for (B): (a) (1) 4-pentynoic acid (for 24) or 6-heptynoic acid (for 25), HATU, iPr2NEt, DMF, 25 °C; (2) LiOH (4 M), CH3OH, H2O (24, 40%; 25, 41%). Reagents and conditions for (C): (a) (1) tert-butyl (3-bromopropyl)carbamate, K2CO3, DMF, 60–70 °C; (2) LiOH (0.5 M), CH3OH, H2O (19%); (b) HCl (3 M), THF, H2O, 25 °C (26%). Reagents and conditions for (D): (a) CH2O (aqueous 37%), formic acid, 100 °C; (b) LiOH (0.5 M), CH3OH, H2O (55%).
Scheme 2. Synthesis of Fluorescent Conjugates of the 2-Naphthoic Acid P2Y14R Antagonists.

(A) AF488 as fluorophore. Reagents and conditions: CuSO4 (7.5% aqueous solution), sodium ascorbate (1 M aqueous solution), tBuOH, H2O, 25 °C (4%). (B) BODIPY as fluorophore. Reagents and conditions: CuSO4, sodium ascorbate, tBuOH, H2O, 25 °C (7.2%).
Molecular Modeling
To identify an energetically favorable binding pose of 6 and to find an appropriate site for fluorophore conjugation, we performed a series of docking studies using homology models of P2Y14R.12,13 Homology models of hP2Y14R were constructed on the basis of two recently solved high-resolution X-ray crystallographic structures of P2Y12R,12,13 which has 49% sequence identity in the 7TM domain to P2Y14R. The P2Y12R structure was solved both in complex with the non-nucleotide antagonist ethyl 6-(4-((benzylsulfonyl)carbamoyl)piperidin-1-yl)-5-cyano-2-methylnicotinate (34, AZD1283)17 and with the nucleotide agonist 2-methylthioadenosine 5′-diphosphate (35, 2-MeSADP), revealing two very distinct conformational states of the ligand binding pocket.12,13 Two homology models of P2Y14R based on either an agonist- or antagonist-bound structure of P2Y12R were generated as templates and compared for docking performance.
Docking into the P2Y14R model based on the P2Y12R–34 template did not result in high-score binding poses of the ligand, even when side chain flexibility of the receptor was taken into account. This is likely due to the fact that extracellular loops 1 and 2 (ECL1 and ECL2) are not resolved in the structure of the P2Y12R–34 template, leaving the expected binding pocket widely open and incomplete and rendering the homology model unsuitable for docking new ligands (see Discussion). On the other hand, flexible docking of compound 6 into a P2Y14R model based on the P2Y12R–35 template consistently predicted a high-scoring binding pose in which 6 fits snugly into the narrow cavity and partially overlaps with 35 (Figure 1A). In this pose, the trifluoromethylphenyl moiety of 6 is buried in the hydrophobic pocket closer to the central part of the receptor delineated by residues Val99, Asn156, His184, Ser187, Asn188, and Phe191. The carboxylate group forms an ionic bridge between positively charged amino groups of Lys77 (transmembrane helix 1, TM1) and Lys171 (ECL2) side chains. Most importantly, the 4-piperidylphenyl moiety of 6 in this model points toward the extracellular opening of the pocket so that its charged piperidine group is accessible to solvent. Hence, we concluded that the piperidine nitrogen in 6 is the most suitable location for attaching an elongated chain to link a fluorophore or other reporter group or pharmacophore. Figure 1B depicts one docked alkylated analogue of 6 (hexynyl derivative 22 shown in Figure 1 to bind with moderately high P2Y14R affinity) as an illustration of this concept.
Figure 1.
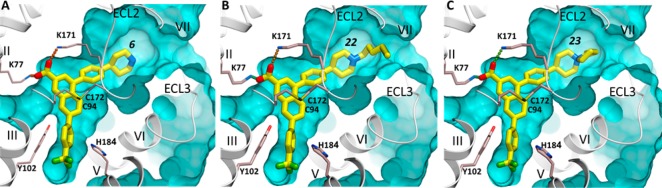
Docking of 6 and its derivatives at the hP2Y14R. (A) Predicted binding mode of the original scaffold 6. (B) Predicted binding mode of hexyne-containing derivative 22. (C) Predicted binding mode of cyclized azaspiro[4.5]decan-5-ium product 23. The docked compounds are shown with carbon atoms colored yellow. The hP2Y14R model, shown as gray ribbon and sticks with carbon atoms colored gray, was generated by homology using the P2Y12R crystal structure (PDB ID 4PXZ) and extensive conformational optimization. Ionic interactions of the carboxylic group with lysine side chains are traced as small spheres. The background part of the binding pocket is shown as a cyan surface.
The involvement of the carboxylate of 6 in strong polar interactions within the binding site is consistent with known SAR in this series: The inactivity of ester prodrug 7 and its congeners is indicative of the need for a free carboxylate.14 Thus, it was reported that replacement of the carboxylate functionality of 6 with other neutral (e.g., ester, amide) or negatively charged (e.g., acetic acid, phosphate) moieties led to a decrease in antagonist activity.9,14 The requirement for a free carboxylate excluded that site from consideration as a potential fluorophore conjugation site.
In the P2Y14R agonist series, we have already explored chain functionalization for conjugation to larger carrier moieties with retention or even enhancement of biological activity. In a series of nucleotide-based agonists, conversion of the carboxylate group of 36 (uridine-5′-diphosphoglucuronic acid) to N-alkyl amides preserves potency of the agonist.8 Additionally, conjugation of 36 to polyamidoamine (PAMAM) dendrimers yielded agonists with enhanced potency, depending on the density of pharmacophore substitution of the polymer.18
Thus, we sought to apply a functionalized congener approach to antagonists of the P2Y14R. Subsequent SAR exploration of the 2-naphthoic acid class of P2Y14R antagonists was performed to find an appropriate fluorophore–pharmacophore linker.
Chemical Synthesis
The key intermediate ethyl ester 19(10,14) containing a free piperidine nitrogen was prepared from protected amine 18 as previously described (Supporting Information Scheme 1).14,15
Intermediate 14 containing the naphthoic acid core was prepared in four steps from 9 and 10 (Supporting Information Scheme 1).19 In short, condensation between 9 and 10 in the presence of sodium hydride yielded (E)-benzylidenesuccinate 11. The tertiary butyl ester of 11 was saponified, and the resulting acid 12 was cyclized in the presence of acetic anhydride. Acetate 13 was further hydrolyzed to provide 14.
The aryl substituents at the naphthoate present in the structures of 6 and 19 were introduced via two consecutive Suzuki transformations in a way similar to that previously described (Supporting Information Scheme 1).10,15 The Suzuki coupling between 14 and p-trifluoromethylphenylboronic acid afforded 15. Phenol 15 was treated with triflic anhydride followed by bis(pinacolato)diboron to obtain boronate 17. The second Suzuki reaction between 17 and tert-butyl 4-(4-bromophenyl)-5,6-dihydropyridine-1(2H)-carboxylate resulted in compound 18, which contains the framework of 6. Hydrogenation of 18 followed by carbamate cleavage afforded desired compound 19.14
We began derivatization of 6 by alkylating the piperidine moiety of 19 (Scheme 1). Three homologous alkyl bromides bearing a terminal acetylene group were prepared from the corresponding alcohols20−23 and used in alkylation to prepare 20–22. It is interesting to note that the product of alkylation of 19 with 5-bromopent-1-yne, i.e., 21, was not isolated. Instead, a product identified as possessing the structure of 23 was obtained. The structural assignment in the case of cyclized 5-azaspiro[4.5]decan-5-ium product 23 was made on the basis of the 1H NMR spectra (Supporting Information). This cyclization was observed only during the attempted preparation of precursor 21, containing three methylene groups adjacent to the alkyne. The characteristic signal from the alkyne proton that would be expected in the case of 21 was absent in the proton NMR spectra. Additionally, two signals were found at 5.61 and 5.93 ppm, respectively, which are consistent with a 2-methylenepyrrolidinium fragment in the context of the structure.24,25
Acylated analogues 24 and 25 were prepared through standard amide coupling reactions of 19 with 4-pentynoic and 6-heptynoic acids, respectively, followed by the hydrolysis of the ethyl ester with lithium hydroxide (Scheme 2).
Two additional N-alkyl analogues 26 and 27 were prepared via alkylating 19 with tert-butyl (3-bromopropyl)carbamate26 followed by ester hydrolysis and carbamate cleavage. Analogue 27 contains an amino group that could be used to attach a fluorophore through acylation reactions. Compound 26 was included in the set of compounds as a test of how a bulky group such as tert-butyloxycarbonyl (Boc) or potentially a fluorophore would be tolerated in binding to the receptor.
The longest of the alkyne intermediates, 22, containing four methylene groups, was further treated with AF488 azide 29 containing a widely used fluorophore to furnish 30 as a fluorescent analogue of 6 (Scheme 2A). Similarly, analogue 32 was prepared by treating 22 with azide 31 carrying a boron-dipyrromethene (BODIPY) fluorescent moiety (Scheme 2B), which was prepared as reported.27 Compound 22 was chosen for further modification based on the observed hypothetical binding mode: the longer hexynyl chain reaches closer to the extracellular entrance to the binding pocket between ECL3 and helix VII (TM7, Figure 1B). We hypothesized that placing the alkyne at the entrance of the pocket, distal from the core pharmacophore binding site, would reduce the steric interference with ligand binding when conjugated to the fluorophore. The excitation and emission spectra of 30 and 32 show the expected maxima (30, 496 and 519 nm, respectively; 32, 626 and 642 nm, respectively) for these fluorophores (Supporting Information).
Biological Characterization
All synthetic analogues were evaluated functionally in Chinese hamster ovary (CHO) cells stably expressing hP2Y14R (P2Y14R-CHO cells, Table 1).15 The activity of each antagonist analogue at the P2Y14R was quantified by measuring its capacity to antagonize the agonist action of the native agonist uridine-5′-diphosphoglucose 2. Thus, 2-promoted inhibition of cyclic AMP accumulation was measured in the presence of an EC80 concentration (316 nM) of 2 alone or in the presence of 316 nM 2 and increasing concentrations (half-log concentrations between 10–10 and 10–5 M) of each newly synthesized molecule. IC50 values for 20 and 22–27 were determined from these inhibition curves, and Ki values were calculated using the Cheng–Prusoff equation.28
Table 1. Functional Evaluation of Antagonism by the 2-Naphthoic Acid Analogues in CHO Cells Stably Expressing hP2Y14Ra.
| compd | Ki (nM) |
|---|---|
| 6 | 0.3 ± 0.1 |
| 19 | 120 ± 8 |
| 20 | 2.7 ± 1.2 |
| 22 | 13.0 ± 1.1 |
| 23 | 2.0 ± 0.2 |
| 24 | 6.7 ± 2.2 |
| 25 | 8.3 ± 1.9 |
| 26 | 26.0 ± 6.0 |
| 27 | 14.0 ± 2.4 |
| 28 | 7.1 ± 1.6 |
| 30 | 0.08 ± 0.02 |
| 32 | >100 |
Antagonism of the inhibition of cyclic AMP accumulation induced by 2 (316 nM) acting at the hP2Y14R in the presence of 30 μM forskolin.
P2Y14R antagonistic activities for the N-substituted analogues of 6 remained in the low nanomolar range. The order of potency for these derivatives was as follows: short alkyne 20, cyclized 23 > N-acylpiperidines 24, 25 > long alkyne 22, aminopropyl derivative 27 > Boc-aminopropyl derivative 26. Ethyl ester 19 was 400-fold weaker as an antagonist than that of free carboxylate analogue 6. This confirms the tentative conclusion based on published work that the carboxylate is important for recognition of the 2-naphthoic acid series of P2Y14R antagonists.9,14N-Methyl derivative 28 displayed significant receptor affinity, emphasizing that the NH of the piperidine ring of 6 is not required for interaction with the P2Y14R and that this N is a suitable site for chain extension.
The high affinity of the cyclized azaspiro[4.5]decan-5-ium product 23 (Ki 2.0 nM) suggests some tolerance of steric bulk in that region. Because the amino group of the piperidine moiety of 23 is quaternized, it does not participate in direct H-bonding with the receptor. Docking of this compound to P2Y14R confirmed that the predicted orientation of the naphthoic acid scaffold can easily accommodate a rigid spiro ring (Figure 1C).
AF488 derivative 30 displayed enhanced potency as an antagonist with a Ki of 80 pM, compared to 0.3 nM for 6 and 13 nM for hexynyl intermediate 22 (Figure 2). In comparison, BODIPY derivative 32 was much weaker in its interaction with P2Y14R. Thus, 30 was identified as a very promising candidate fluorescent probe of P2Y14R.
Figure 2.
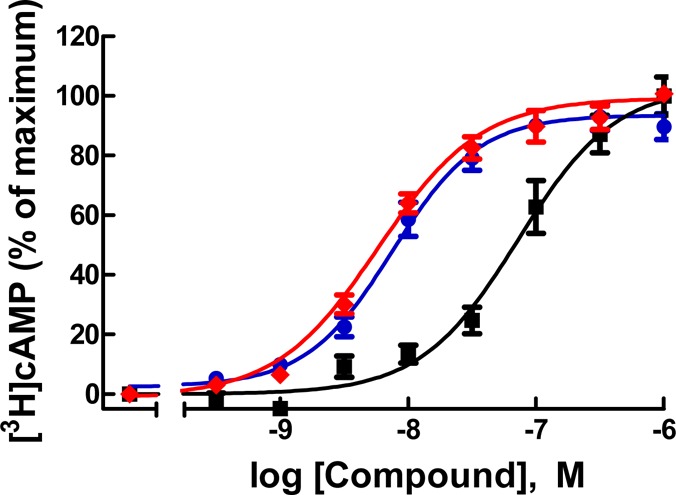
Concentration–response curves for 6 (blue), alkyne 22 (black), and conjugate 30 (red) in antagonism of the inhibition of cAMP formation via human P2Y14R in P2Y14R-CHO cells.
AF488 conjugate 30 bound specifically to the receptor in intact P2Y14R-CHO cells, as characterized using flow cytometry (FCM), a technique previously used in intact cell studies with fluorescent antagonists of other GPCRs.29 Cell labeling using the fluorescent probe at a fixed concentration of 10 nM was compared at two different incubation times; the level of specific binding of 30 was greater following a 30 min incubation compared to that at 20 min (Figure 3). A 30 min preincubation with agonists 1, 3, or 36 at 10 μM or antagonist 6 at 10 μM largely prevented the labeling in P2Y14R-CHO cells. The high fraction of specific binding of 30 is evident in Figure 4, which compares total cell fluorescence with exposure to various combinations of ligands. The residual nonspecific binding in the presence of 10 μM 6 is only 1.9% (20 min) or 1.1% (30 min) of the total binding of 10 nM 30 (Figure 4). Both a P2Y1R selective antagonist N6-methyl-2′-deoxyadenosine-3′,5′-bisphosphate (37, MRS2179)38 and a P2Y6R selective irreversibly binding antagonist N,N″-1,4-butanediylbis[N′-(3-isothiocyanatophenyl)]thiourea (38, MRS 2578) failed at 10 μM to prevent binding of 30. A weak, nonselective P2R antagonist suramin (at 10 μM) demonstrated only a minor reduction of the level of fluorescent cell labeling. The efficiency of inhibition of fluorescent labeling by competing ligands was in the order 6 > 3 > 1 (Figure 4), corresponding to their order of apparent affinities at the P2Y14R.8,15
Figure 3.

Binding of Alexa Fluor 488 (AF488) conjugate 30 (A–D) and boron-dipyrromethene (BODIPY) conjugate 32 (E, F) at the hP2Y14R as characterized using FCM. Fluorescent ligands were present at 10 nM, and preincubation with competing ligands was for 30 min at 37 °C. (A) Lack of nonspecific binding in control CHO cells of fluorescent antagonist 30. (B) Binding in P2Y14R-expressing CHO cells of fluorescent antagonist 30 alone or after preincubation with competing antagonist 6 (10 μM). (C) Binding in P2Y14R-expressing CHO cells of fluorescent agonist 30 alone or after preincubation with competing agonist 3 (5 μM). (D) Binding in P2Y14R-expressing CHO cells of fluorescent antagonist 30 alone or after preincubation with competing native agonist 1 (10 μM). (E) Binding in P2Y14R-expressing CHO cells of fluorescent antagonist 32 alone or after preincubation with competing agonists 3 and antagonist 6 (all, 10 μM). (F) Nonspecific binding in control CHO cells of fluorescent antagonist 32.
Figure 4.

Fluorescence ligand binding experiments using flowcytometry (FCM) in P2Y14R-CHO cells with 30 after 30 min preincubation at 37 °C with known P2Y14R agonists 1 and 3 or P2Y14R antagonist 6. Each column shows the brightness of each compound using 30 normalized to 100% for each time point and after correcting the mean fluorescence intensity values for autofluorescence. Results at 30 min expressed as mean ± SEM (n = 4) are 24.4 ± 11.5%, 1; 7.5 ± 0.8%, 3; 1.10 ± 0.06%, 6. *, P < 0.05, when compared to cells treated with only 30. No significant difference in mean fluorescence intensity was observed at 30 min in the presence of P2Y1R antagonist 37 (10 μM, 80.9 ± 8.6%, data not shown) or P2Y6R antagonist 38 (10 μM, 100.7 ± 10.5%, data not shown). Fluorescence in the presence of weak, nonselective P2R antagonist suramin (10 μM) was 82.1 ± 3.6% (data not shown).
Use of the BODIPY conjugate 32 was compared to 30 in experiments applying FCM. Although labeling of live P2Y14R-CHO cells by 32 was inhibited following preincubation with known P2Y14R ligands, it displayed significant nonspecific binding in control CHO cells, which increased upon prolonged incubation (Figure 3F). Thus, AF488 conjugate 30 had more desirable characteristics than 32 as a tracer for receptor detection and characterization and potentially in drug screening for activity at the P2Y14R.
The predicted binding mode of 30 in the P2Y14R, as characterized by the best conformational energy of the complex, is presented in Figure 5. Note that despite full flexibility of the ligand and the receptor pocket side chains the docking pose of the 2-naphthoic acid scaffold is almost identical to the one observed for 6, suggesting well-defined tight binding of this chemical scaffold. Beyond this scaffold, the triazole ring of the linker region of compound 30 interacts with the receptor through a salt bridge to the side chain of Lys270, which may contribute to the unusually high affinity of 30. Other interactions contributing to the binding affinity of 30 may be mediated by the AF488 moiety itself. Thus, the fluorophore moiety is predicted to reach back into the shallow groove in the ECL region of the receptor, where its carbonyl and carboxyl moieties make polar interactions with the side chain and main chain nitrogens of His264. One of its sulfonate groups also forms a salt bridge to the primary amine of Lys176. Such interactions of the AF488 substitution are unlikely to be strong or highly specific. Nonetheless, they favorably contribute to the binding of 30 and explain the 4-fold increase in affinity compared to that of 6 and the 160-fold increase compared to that of 22, which is the closest precursor of 30 studied here.
Figure 5.
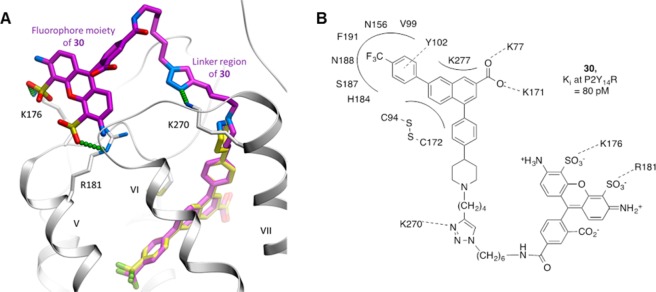
Predicted binding of compound 30 at the hP2Y14R. Compound 30 is shown as sticks with carbon atoms colored magenta. For comparison, the original scaffold 6 (the same conformation as in Figure 1A) is shown with carbon atoms colored light yellow. Predicted polar and ionic interactions of the triazole and AF488 fluorophore moieties of 30 with the receptor are traced by small green spheres. hP2Y14R is shown as a ribbon and sticks with carbon atoms colored gray.
In general, additional ligand tools and molecular probes are needed for characterizing and defining the action of P2YRs.38 Few radioligands for this family of receptors exist, and only a few subtypes can be targeted with selective agonists and/or antagonists.
This work is directed toward development of molecular probes for P2Y14R that are structurally derived from a newly reported potent and highly selective antagonist 6. In the absence of an atomic resolution structure of P2Y14R, we constructed homology models based on recently reported structures of the closely related P2Y12R12,13 and used them for virtual docking of P2Y14R-directed antagonists. Application of these models resulted in identification of the piperidine ring as a likely site for chain derivatization with preservation of affinity, and a series of analogues was synthesized to probe the effect of distal changes on antagonist potency. The structure of the P2Y12R template was recently solved both in complex with a non-nucleotide antagonist 34 and with the nucleotide agonist 35, revealing drastically different conformations of the receptor.12,13 The P2Y12R–34 antagonist complex structure (PDB ID 4NTJ) revealed a wide open binding pocket due to the unresolved ECL1 and ECL2 regions and the outward positions of helices VI (TM6) and VII (TM7). In contrast, TM6/TM7 are shifted toward the ligand in the agonist-bound P2Y12R structure (PDB ID 4PXZ), and the extracellular loops form a “lid” on top of ligand binding site participating in ligand coordination.
Our modeling and docking results suggested that the closed conformation of the extracellular region of P2Y14R is more suitable for binding compound 6, providing a reasonable steric fit for this high-affinity antagonist. The trifluoromethylphenyl group is bound within the largely hydrophobic subpocket, forming a π–π interaction with the Tyr102 side chain (a π–π interaction with ligands was found in all X-ray structures of P2Y12R). The carboxylate group forms strong charge–charge interactions with the positively charged side chains of Lys77 and Lys171, and the piperidinyl-phenyl moiety is sandwiched between TM7 and ECL2, with the piperidine nitrogen directed toward the extracellular side of the receptor. In contrast, the model based on the P2Y12R–34 template was not efficient in docking compound 6 or its derivatives, as it did not yield any high-scoring binding conformations. A possible explanation of this result is that the disordered conformational state of the extracellular region is specific to the P2Y12R complex with 34, an antagonist that apparently pushes TM6 outward and precludes a closed conformation of the loops.12 Moreover, our analysis suggests that some features of the extracellular region of the P2Y14R may favor a closed conformation of ECL2. Thus, despite high sequence similarity and gapless alignment between P2Y12R and P2Y14R (Supporting Information Figure 1), the ECL2 residues in P2Y14R carry only minimal electrostatic charge (+1) compared to the highly charged ECL2 in P2Y12R (+5 charge within a 15 residue stretch of this loop). While the excessive positive charge of ECL2 in the P2Y12R is thought to destabilize the loop in the absence of ATP/ADP analogues, the more neutral ECL2 in P2Y14R may favor the closed conformation even in an antagonist-bound or apo state. Predicted binding of compound 6 may further stabilize ECL2, since its carboxylate group forms ionic bridges with both Lys77 in TM2 and Lys171 in ECL2 in an extensive network of ionic interactions that also involves Lys277, Arg274, Asp81, and Glu278 (Supporting Information Figure 2). Our modeling therefore suggests novel insight into conformational states of the P2Y14R that are more reminiscent of the closed, rather than the open conformational state observed in the crystal structure of P2Y12R.
The placement of the piperidine moiety of docked 6 in a solvent-accessible pocket within the receptor initially led us to hypothesize that preservation of the protonated and charged state of this group might be important for retaining the affinity of target molecules for P2Y14R. However, biological characterization of the analogues synthesized revealed no advantage in potency for N-alkylated analogues of 6 in comparison tothat of N-acylated derivatives.
A goal of this research is to introduce reliable probes for P2Y14R to provide methods for fluorescent characterization of P2Y14R both in membranes and intact cells. The derivatives of 6 were furnished with alkynyl or amino groups, suitable for attachment of fluorescent moieties by click chemistry or amide coupling, respectively. Compounds 20, 22, 26, and 27 displayed potencies within an order of magnitude of that of the parent molecule 6, and these may be suitable for further conjugation with fluorescent dyes. Thus, our results demonstrate that P2Y14R potency can be preserved by chain functionalization, leading to fluorescent molecular probes for the receptor. Fluorescent ligands were reported previously for other P2YRs (P2Y2,4,6), but these are nucleotide agonist analogues that are subject to agonist-promoted internalization.27,30 Compound 30 is the first fluorescent antagonist probe of high affinity for a P2YR. It combines a hydrophilic fluorophore with a hydrophobic pharmacophore and consequently exhibits low nonspecific binding. The charged groups on the AF488 moiety are predicted to interact with charged amino acid residues on the ELs of the receptor to stabilize the high-affinity complex. It is likely that distal interactions on the ELs contribute to the exceptionally high affinity (80 pM) of 30. In contrast, BODIPY conjugate 32 is much more hydrophobic and displayed an unacceptable level of nonspecific binding in control CHO cells and a low affinity at the P2Y14R. More promising analogues now can be applied to the study of tissues and cells in disease models such as human neutrophils, where P2Y14R promotes chemotaxis,4 or in LAD2 human mast cells, where the receptor promotes release of inflammatory mediators.5 In addition to well-described agonist-mediated GPCR internalization, there are reports of antagonist-induced internalization.39 Fortunately, no such internalization has been observed in the FCM experiments (Figures 3A,B and 4). It could be seen that with incubation in the presence of parent antagonist 6 there is near complete reversal of binding of 30 (Figures 3B and 4), excluding the possibility of fluorescence uptake through receptor internalization in the time frame of the experiment. Additionally, the incubation of 30 with CHO cells lacking expression of P2Y14R for up to 2 h has shown no significant nonspecific binding (Figure 3A), thus excluding the possibility of passive transport of 30 through the membrane.
In conclusion, we used molecular modeling based on a closely related structural template of P2Y14R to identify the piperidine moiety of 6 as the most structurally permissive region of the antagonist for derivatization, with the objective of covalent chain attachment through a tertiary amino group. The pharmacological results for the newly synthesized compound series were consistent with the structural insights gained from P2Y12R-based homology modeling and docking at this closely related receptor. The resulting P2Y14R model with its ECLs defined conformationally allows prediction of specific interactions with antagonist analogues. This approach demonstrates the predictive power of GPCR homology modeling and the value of applying newly determined X-ray structures to the medicinal chemistry of GPCRs. Our results indicate that P2Y14R potency in this series of 2-naphthoic acid derivatives can be preserved by chain functionalization, leading to selective fluorescent molecular probes for the receptor. Indeed, AF488 conjugate 30 displayed highly favorable properties when used as a tracer in whole-cell binding assays of P2Y14R using FCM: pharmacological selectivities consistent with the apparent affinities of known P2Y ligands, unusually high receptor affinity of the tracer, and strikingly low nonspecific binding. The measurement of precise P2Y14R selectivity and in situ labeling, kinetics, and receptor saturation by 30 as well as development of routine screening methods will be the subject of future studies. Fluorescent ligand probes are of increasing interest as pharmacological tools for characterizing GPCRs.40 Such conjugates will be useful tools in expanding the SAR at this receptor, which still lacks chemical diversity in its collective ligands.
Methods
Homology Modeling
Homology modeling of hP2Y14R was performed using the recently solved high-resolution crystallographic structures of hP2Y12R,12,13 a closely related member of the P2YR subfamily. Within the 7TM domain portion of the receptor, the alignment between P2Y12R and P2Y14R sequences is fully continuous (gapless), with sequence identity as high as 49%, which facilitated reliable model building. The homology modeling was performed with the ICM-Pro molecular modeling package’s build model function using full energy-based conformational sampling and optimization of the side chains in internal coordinates.31−33 Both the closed conformation of the hP2Y12R extracellular region (PDB ID 4PXZ) and the open conformation (PDB ID 4NTJ) were used to generate the models for the docking procedure.
Docking
Compounds 6, 22, and 23 were docked into the models of hP2Y14R using ICM-Pro molecular modeling software.31 The ligand and residue side chains forming the binding pocket were treated as flexible, and their conformation was thoroughly sampled in 106 Monte Carlo (107 for compound 30) optimization steps to global convergence of the physics-based energy function.34 The docking procedure was repeated in 5 independent runs from different starting positions of the ligand outside of the pocket. Soft van der Waals potentials and a high temperature parameter were used to facilitate ligand sampling. The criteria for docking procedure convergence included a high score for the predicted binding of the ligand in the energy optimized complex (score < −30) and a consistently reproduced docking conformations (RMSD < 1.5 Å). Note that the hP2Y14R model based on the open receptor conformation did not satisfy either of these criteria for docking of small compounds 6, 22, and 23 and therefore was not used in docking of a larger compound 30. Starting positions for docking of compound 30 used optimal docking position of compound 6, while the position of fluorophore and linker was randomized.
Pharmacological Assays: Cell Culture and Reagents
CHO cells stably expressing hP2Y14R were generated as previously described,35 grown in F-12 medium with 10% FBS and 1% geneticin, and maintained at 37 °C in 5% CO2. 3-Isobutyl-1-methylxanthine (IBMX), forskolin, and 2 were purchased from Sigma-Aldrich. [3H]Adenine was purchased from American Radiolabeled Chemicals (St. Louis, MO). Geneticin, serum, and all cell culture medium were purchased from Gibco Life Technologies.
Quantification of Cyclic AMP Accumulation
P2Y14R-CHO cells were plated in 24-well plates approximately 24 h before the assay at 70 000 cells per well and were labeled 2 h before the assay with 1 mCi [3H]adenine/well in 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)-buffered serum-free DMEM. All assays were performed in the presence of 200 μM IBMX, a phosphodiesterase inhibitor, and were initiated by the addition of 30 μM forskolin. Tests of the antagonist activities of each of the newly synthesized molecules were carried out using an EC80 concentration (316 nM) of P2Y14R agonist 2 and a range of concentrations of the antagonist that usually spanned 5 orders of magnitude. Incubations were for 15 min at 37 °C and were terminated by aspiration of medium and addition of 500 μL of ice-cold 5% trichloroacetic acid. [3H]Cyclic-AMP was isolated by sequential Dowex and alumina chromatography as previously described.36,37
Data Analysis
Agonist potencies (EC50 values) were obtained from concentration–response curves by nonlinear regression analysis using the GraphPad software package Prism. All experiments were performed in triplicate assays and repeated at least three times. The results are presented as mean ± SEM from multiple experiments or, in the case of concentration–effect curves, from a single experiment carried out with triplicate assays that were representative of results from multiple experiments.
Cell Cultures for FCM
CHO-P2Y14R cells and wild-type CHO cells were grown in DMEM/F12 (1:1) with 10% FBS, 50 U/mL penicillin/streptomycin, and 2 mM l-glutamine. Cells were grown in 6-well plates (approximately 3 × 105 cells/well) and incubated at 37 °C in 5% CO2. When the cells reached 80% confluence, the medium was replaced with fresh preheated medium. Compound 30 was added in the presence or absence of the appropriate agonist or antagonist of P2Y14R, and the decrease in fluorescence intensity was measured by FCM.
Fluorescent Ligand Binding in CHO-P2Y14R Cells
CHO-P2Y14R cells were incubated with known agonists or antagonists of P2Y14R, such as agonists 1, 3, and 36 or antagonist 6. The cells were preincubated for 30 min with the known agonist or antagonist at 37 °C in 5% CO2. Two different incubation times (20 and 30 min) in the presence of fluorescent antagonist 30 were compared.
Following incubation with 30, the medium was removed, the cells were washed three times with ice-cold DPBS, and 1 mL of 0.2% EDTA was added to each well to detach the cells from the plate. After detaching, 1 mL of medium was added to neutralize the EDTA, and the cell suspension was incubated at 37 °C for 5–10 min. The cell suspensions were transferred to 5 mL polystyrene round-bottomed BD Falcon tubes and centrifuged for 5 min at 23 °C and 400g. After the supernatant was discarded, the cells were washed with 3 mL of DPBS and centrifuged again at 23 °C and 400g for 5 min. After centrifugation, the supernatant was discarded, and the cells were resuspended in 0.5 mL of DPBS for analysis by FCM.
FCM Analysis
The intensity of fluorescence emission of each sample was measured using FCM. Cell suspensions were vortexed briefly before analysis on a BD FACSCalibur flow cytometer. Samples were maintained in the dark during the analysis to avoid photobleaching. Mean fluorescent intensity was obtained in log mode using the FL-1 channel for 30 and FL-4 channel for 32, and 10 000 events were analyzed per sample. Data were collected and analyzed using BD Cell Quest Pro software. Data analysis was performed with GraphPad Prism 5 software. The mean autofluorescence of CHO cells was measured in the absence of the fluorescent ligand. The mean fluorescence intensity in the presence of fluorescent ligand was corrected by subtracting the autofluorescence.
Acknowledgments
Mass spectral measurements were carried out by J. Lloyd and N. Whittaker (NIDDK). We acknowledge support from the NIGMS Postdoctoral Research Associate (PRAT) Program and the Intramural Program of the National Institute of Diabetes and Digestive and Kidney Diseases. This work was supported by National Institutes of Health grant nos. GM38213 to T.K.H. and U54GM094618 to V.K. and R.C.S.
Glossary
Abbreviations
- AF488
Alexa Fluor 488
- BODIPY
boron-dipyrromethene
- CHO
Chinese hamster ovary
- DMEM
Dulbecco’s modified Eagle’s medium
- DMF
dimethylformamide
- DPPF
1,1′-bis(diphenylphosphino)ferrocene
- ECL
extracellular loop
- EDC
N-ethyl-N′-dimethylaminopropylcarbodiimide
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- PLC
phospholipase C
- PPTN
4-(4-(piperidin-4-yl)-phenyl)-7-(4-(trifluoromethyl)-phenyl)-2-naphthoic acid
- SAR
structure–activity relationship
- UDPG
uridine-5′-diphosphoglucose
- THF
tetrahydrofuran
- TM
transmembrane helix
Supporting Information Available
Coordinate file of the P2Y14R model complex with 30; scheme for synthesis of 19; synthesis procedures used for preparation of 19, 20, 22–28, 30, and 32; NMR and mass spectra of selected compounds; and fluorescence spectra of 30 and 32. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Boeynaems J.-M.; Communi D.; Robaye B. (2012) Overview of the pharmacology and physiological roles of P2Y receptors. Wiley Interdiscip. Rev.: Membr. Transp. Signaling 1, 581–588. [Google Scholar]
- Harden T. K., Sesma J. I., Fricks I. P., Lazarowski E. R.. Signalling and pharmacological properties of the P2Y14 receptor. Acta Physiol. 199, 149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K.; Yamanaka H.; Yanamoto F.; Okubo M.; Noguchi K. (2012) Multiple P2Y subtypes in spinal microglia are involved in neuropathic pain after peripheral nerve injury. Glia 60, 1529–1539. [DOI] [PubMed] [Google Scholar]
- Sesma J. I.; Kreda S. M.; Steinckwich-Besancon N.; Dang H.; García-Mata R.; Harden T. K.; Lazarowski E. R. (2012) The UDP-sugar-sensing P2Y14 receptor promotes Rho-mediated signaling and chemotaxis in human neutrophils. Am. J. Physiol.: Cell Physiol. 303, C490–C498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z.-G.; Ding Y.; Jacobson K. A. (2010) UDP-glucose acting at P2Y14 receptors is a mediator of mast cell degranulation. Biochem. Pharmacol. 79, 873–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J.; Morinaga H.; Oh D.; Li P.; Chen A.; Talukdar S.; Lazarowski E.; Olefsky J. M.; Kim J. J. (2012) GPR105 ablation prevents inflammation and improves insulin sensitivity in mice with diet-induced obesity. J. Immunol. 189, 1992–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko H.; Fricks I.; Ivanov A. A.; Harden T. K.; Jacobson K. A. (2007) Structure–activity relationship of uridine 5′-diphosphoglucose analogues as agonists of the human P2Y14 receptor. J. Med. Chem. 50, 2030–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko H.; Das A.; Carter R. L.; Fricks I. P.; Zhou Y.; Ivanov A. A.; Melman A.; Joshi B. V.; Kováč P.; Hajduch J.; Kirk K. L.; Harden T. K.; Jacobson K. A. (2009) Molecular recognition in the P2Y14 receptor: probing the structurally permissive terminal sugar moiety of uridine-5′-diphosphoglucose. Bioorg. Med. Chem. 17, 5298–5311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier J. Y.; Belley M.; Deschênes D.; Fournier J.-F.; Gagné S.; Gareau Y.; Hamel M.; Hénault M.; Hyjazie H.; Kargman S.; Lavallée G.; Levesque J.-F.; Li L.; Mamane Y.; Mancini J.; Morin N.; Mulrooney E.; Robichaud J.; Thérien M.; Tranmer G.; Wang Z.; Wu J.; Black W. C. (2011) The identification of 4,7-disubstituted naphthoic acid derivatives as UDP-competitive antagonists of P2Y14. Bioorg. Med. Chem. Lett. 21, 2836–2839. [DOI] [PubMed] [Google Scholar]
- Belley M., Deschenes D., Fortin R., Fournier J.-F., Gagne S., Gareau Y., Gauthier J. Y., Li L., Robichaud J., Therien M., Tranmer G. K., and Wang Z. (2009) Substituted 2-naphthoic acids as antagonists of GFPR105 activity. WO2009070873 A1, June 11, 2009.
- Guay D.; Beaulieu C.; Belley M.; Crane S. N.; DeLuca J.; Gareau Y.; Hamel M.; Henault M.; Hyjazie H.; Kargman S.; Chan C. C.; Xu L.; Gordon R.; Li L.; Mamane Y.; Morin N.; Mancini J.; Thérien M.; Tranmer G.; Truong V. L.; Wang Z.; Black W. C. (2011) Synthesis and SAR of pyrimidine-based, non-nucleotide P2Y14 receptor antagonists. Bioorg. Med. Chem. Lett. 21, 2832–2835. [DOI] [PubMed] [Google Scholar]
- Zhang K.; Zhang J.; Gao Z. G.; Zhang D.; Zhu L.; Han G. W.; Moss S. M.; Paoletta S.; Kiselev E.; Lu W.; Fenalti G.; Zhang W.; Müller C. E.; Yang H.; Cherezov V.; Katritch V.; Han G. W.; Jacobson K. A.; Stevens R. C.; Wu B.; Zhao Q. (2014) Structure of the human P2Y12 receptor in complex with an antithrombotic drug. Nature 509, 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K.; Zhang J.; Gao Z. G.; Paoletta S.; Zhang D.; Han G. W.; Li T.; Ma L.; Zhang W.; Müller C. E.; Yang H.; Jiang H.; Cherezov V.; Katritch V.; Jacobson K. A.; Stevens R. C.; Wu B.; Zhao Q. (2014) Agonist-bound structure of the human P2Y12R receptor. Nature 509, 119–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robichaud J.; Fournier J.-F.; Gagné S.; Gauthier J. Y.; Hamel M.; Han Y.; Hénault M.; Kargman S.; Levesque J.-F.; Mamane Y.; Mancini J.; Morin N.; Mulrooney E.; Wu J.; Black W. C. (2011) Applying the pro-drug approach to afford highly bioavailable antagonists of P2Y14. Bioorg. Med. Chem. Lett. 21, 4366–4368. [DOI] [PubMed] [Google Scholar]
- Barrett M. O.; Sesma J. I.; Ball C. B.; Jayasekara P. S.; Jacobson K. A.; Lazarowski E. R.; Harden T. K. (2013) A selective high-affinity antagonist of the P2Y14 receptor inhibits UDP-glucose–stimulated chemotaxis of human neutrophils. Mol. Pharmacol. 84, 41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thirumurugan P.; Matosiuk D.; Jozwiak K. (2013) Click chemistry for drug development and diverse chemical–biology applications. Chem. Rev. 113, 4905–4979. [DOI] [PubMed] [Google Scholar]
- Bach P.; Boström J.; Brickmann K.; van Giezen J. J. J.; Groneberg R. D.; Harvey D. M.; O’Sullivan M.; Zetterberg F. (2013) Synthesis, structure–property relationships and pharmacokinetic evaluation of ethyl 6-aminonicotinate sulfonylureas as antagonists of the P2Y12 receptor. Eur. J. Med. Chem. 65, 360–375. [DOI] [PubMed] [Google Scholar]
- Das A.; Zhou Y.; Ivanov A. A.; Carter R. L.; Harden T. K.; Jacobson K. A. (2009) Enhanced potency of nucleotide–dendrimer conjugates as agonists of the P2Y14 receptor: multivalent effect in G protein-coupled receptor recognition. Bioconjugate Chem. 20, 1650–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boger D. L.; McKie J. A.; Cai H.; Cacciari B.; Baraldi P. G. (1996) Synthesis and properties of substituted CBI analogs of cc-1065 and the duocarmycins incorporating the 7-methoxy-1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indol-4-one (MCBI) alkylation subunit: magnitude of electronic effects on the functional reactivity. J. Org. Chem. 61, 1710–1729. [DOI] [PubMed] [Google Scholar]
- Bélanger G.; Dupuis M.; Larouche-Gauthier R. (2012) Asymmetric total synthesis of (+)-virosine a via sequential nucleophilic cyclizations onto an activated formamide. J. Org. Chem. 77, 3215–3221. [DOI] [PubMed] [Google Scholar]
- Flahaut J.; Miginiac P. (1978) Synthèse d’alcools acétyléniques par alkylation d’hydroxy-ω-alkynes-1. Helv. Chim. Acta 61, 2275–2285. [Google Scholar]
- Negishi E.; Boardman L. D.; Sawada H.; Bagheri V.; Stoll A. T.; Tour J. M.; Rand C. L. (1988) Metal promoted cyclization. 18. Novel cyclialkylation reactions of (ω-halo-1-alkenyl)metal derivatives. Synthetic scope and mechanism. J. Am. Chem. Soc. 110, 5383–5396. [Google Scholar]
- Sharma S.; Oehlschlager A. C. (1989) Scope and mechanism of stannylalumination of 1-alkynes. J. Org. Chem. 54, 5064–5073. [Google Scholar]
- Baker M. V.; Brown D. H.; Skelton B. W.; White A. H. (2000) Intramolecular hydroamination of 1,4,7-tri(pent-4′-yn-1′-yl)-1,4,7-triazacyclononane: formation of an azoniaspiro-[4.8]-tridecane. Aust. J. Chem. 53, 791–797. [Google Scholar]
- Mayrargue J.; Vayssière M.; Miocque M. (1985) A new synthesis of quinuclidinium derivatives. Heterocycles 23, 2173–2175. [Google Scholar]
- Vernekar S. K. V.; Hallaq H. Y.; Clarkson G.; Thompson A. J.; Silvestri L.; Lummis S. C. R.; Lochner M. (2010) Toward biophysical probes for the 5-HT3 receptor: structure–activity relationship study of granisetron derivatives. J. Med. Chem. 53, 2324–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayasekara P. S.; Barrett M. O.; Ball C. B.; Brown K. A.; Hammes E.; Balasubramanian R.; Harden T. K.; Jacobson K. A. (2014) 4-Alkyloxyimino derivatives of uridine-5′-triphosphate: distal modification of potent agonists as a strategy for molecular probes of P2Y2, P2Y4 and P2Y6 receptors. J. Med. Chem. 57, 3874–3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y. C.; Prusoff W. H. (1973) Relationship between inhibition constant (K1) and concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic-reaction. Biochem. Pharmacol. 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- Kozma E.; Kumar T. S.; Federico S.; Phan K.; Balasubramanian R.; Gao Z. G.; Paoletta S.; Moro S.; Spalluto G.; Jacobson K. A. (2012) Novel fluorescent antagonist as a molecular probe in A3 adenosine receptor binding assays using flow cytometry. Biochem. Pharmacol. 83, 1552–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayasekara P. S.; Barrett M. O.; Ball C. B.; Brown K. A.; Kozma E.; Costanzi S.; Squarcialupi L.; Balasubramanian R.; Maruoka H.; Jacobson K. A. (2013) 4-Alkyloxyimino-cytosine nucleotides: tethering approaches to molecular probes for the P2Y6 receptor. MedChemComm. 4, 1156–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abagyan R. A., Orry A., Raush E., Budagyan L., and Totrov M. (2014) ICM, 3.8 ed., MolSoft LLC, La Jolla, CA.
- Abagyan R.; Batalov S.; Cardozo T.; Totrov M.; Webber J.; Zhou Y. (1997) Homology modeling with internal coordinate mechanics: deformation zone mapping and improvements of models via conformational search. Proteins 1, 29–37. [DOI] [PubMed] [Google Scholar]
- Cardozo T.; Totrov M.; Abagyan R. (1995) Homology modeling by the ICM method. Proteins 23, 403–414. [DOI] [PubMed] [Google Scholar]
- Totrov M.; Abagyan R. (1997) Flexible protein–ligand docking by global energy optimization in internal coordinates. Proteins 1, 215–220. [DOI] [PubMed] [Google Scholar]
- Fricks I. P.; Carter R. L.; Lazarowski E. R.; Harden T. K. (2009) Gi-dependent cell signaling responses of the human P2Y14 receptor in model cell systems. J. Pharmacol. Exp. Ther. 330, 162–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon Y.; Londos C.; Rodbell M. (1974) A highly sensitive adenylate cyclase assay. Anal. Biochem. 58, 541–548. [DOI] [PubMed] [Google Scholar]
- Harden T. K.; Scheer A. G.; Smith M. M. (1982) Differential modification of the interaction of cardiac muscarinic cholinergic and beta-adrenergic receptors with a guanine nucleotide binding components. Mol. Pharmacol. 21, 570–580. [PubMed] [Google Scholar]
- Jacobson K. A.; Jayasekara M. P. S.; Costanzi S. (2012) Molecular structure of P2Y receptors: mutagenesis, modeling, and chemical probes. Wiley Interdiscip. Rev.: Membr. Transp. Signaling 1, 815–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick N.; Narayan P.; Puett D. (2012) The endothelin subtype A receptor undergoes agonist- and antagonist-mediated internalization in the absence of signaling. Endocrinology 139, 3185–3192. [DOI] [PubMed] [Google Scholar]
- Ciruela F.; Jacobson K. A.; Fernández-Dueñas V. (2014) Portraying G protein-coupled receptors with fluorescent ligands. ACS Chem. Biol. 9, 1918–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.