

Abstract
Many empirical studies estimating effective population size apply the temporal method that provides an estimate of the variance effective size through the amount of temporal allele frequency change under the assumption that the study population is completely isolated. This assumption is frequently violated, and the magnitude of the resulting bias is generally unknown. We studied how gene flow affects estimates of effective size obtained by the temporal method when sampling from a population system and provide analytical expressions for the expected estimate under an island model of migration. We show that the temporal method tends to systematically underestimate both local and global effective size when populations are connected by gene flow, and the bias is sometimes dramatic. The problem is particularly likely to occur when sampling from a subdivided population where high levels of gene flow obscure identification of subpopulation boundaries. In such situations, sampling in a manner that prevents biased estimates can be difficult. This phenomenon might partially explain the frequently reported unexpectedly low effective population sizes of marine populations that have raised concern regarding the genetic vulnerability of even exceptionally large populations.
Keywords: effective population size, subdivided populations, temporal method
Introduction
There is current concern over accelerating rates of loss of genetic variation in natural populations due to declining population size, potentially resulting in reduced capacity for adaptive change and future evolution (Frankel 1974; Traill et al. 2010). To obtain quantitative assessments of present rates of loss of genetic diversity, estimating the genetically effective size of natural populations has become increasingly common in the fields of conservation and evolutionary genetics (Palstra & Ruzzante 2008).
The expected rate of loss of heterozygosity of a population is 1/(2NeI) per generation, where NeI is the inbreeding effective size of the population. There is also a variance effective size (NeV) that reflects the amount of random gene frequency change from one generation to the next (genetic drift), but the two quantities NeI and NeV are identical when population size is constant (Crow & Kimura 1970). Many simplified models of population structure only use the concept ‘effective size’ (Ne) without making the distinction between NeI and NeV.
When assessing the effective size of natural populations, many studies apply the so-called temporal method that provides an estimate of the variance effective size (NeV) through the amount of allele frequency change over one or more generations (Waples 1989). The temporal method assumes that the study population is completely isolated and that any observed genetic change is entirely due to genetic drift caused by restricted effective size. In the real world, however, many or most populations are only partially isolated, that is, they belong to a population system and are connected to neighbouring ones through migration. Violation of the assumption of complete isolation constitutes a common source of bias, the direction and magnitude of which is generally unknown, and investigators applying the temporal method tend to ignore or minimize the effect of migration in the discussion of their results (Wang & Whitlock 2003; Leberg 2005; Wang 2005; Luikart et al. 2010; Waples & England 2011).
There are several challenges involved in the estimation of effective size of a population that belongs to a population system. First, allele frequency shifts caused by immigration into the focal population may erroneously be interpreted as genetic drift and thus bias the estimate of NeV (Wang & Whitlock 2003). Further, when genetic differentiation is weak between the subpopulations that constitute the population system (the global population), it may be difficult to identify population boundaries and target the focal population for sampling. Many marine organisms, for example, are characterized by high migration rates and low levels of divergence between populations (Ward et al. 1994). A sample from the wild may easily include multiple populations, and samples collected at different occasions may consist of individuals from more or less disjunct population segments. The present study was actually prompted by empirical results on brown trout (Salmo trutta) where the temporal method provided strikingly small estimates of NeV that appeared incompatible with a seemingly very large and genetically homogeneous population (Palm et al. 2003).
Finally, there are two effective sizes to be considered: the local population and the population system as a whole (NeV,tot). In the absence of extensive studies designed to delineate genetic population structure, it is difficult to tell the difference between a subdivided population and a randomly mating one (Ryman et al. 2006; Waples & Gaggiotti 2006). An investigator sampling from a seemingly large and genetically homogeneous population may actually be dealing with a population system without being aware of it. In such situations, an estimate of NeV can be strongly misleading because, depending on the subpopulations included in the sample, it may refer to a local subpopulation affected by immigration, the global population or something in between.
The problem of using the temporal method for estimating effective size of a population under migration has been addressed by Wang & Whitlock (2003). They devised an approach for simultaneous estimation of NeV and the immigration rate (m) that can be applied to situations where allele frequency estimates are available for the immigrant gene pool as well as for the focal population. They also discuss some of the general effects of ignoring migration when estimating NeV for the special case of a local population receiving immigrants from an infinitely large donor population.
There is, however, no theory that quantifies the expected bias in the estimate of NeV in terms of the characteristics of the global population and the number of subpopulations included in the sample. In this study, we provide expressions for the expected value of the estimate of NeV when applying the temporal method to samples from a population system where the component local populations (subpopulations) are connected by migration, focusing on the traditional island model of migration (Wright 1965). Rather than estimating migration rates, our main interest is to quantify the amount of bias of local and global NeV estimates when disregarding migration. We pay special attention to situations of high gene flow where population structure may be difficult to detect or delineate, and where the same or different subpopulations may be included in the samples used for measuring the temporal change of allele frequencies.
The temporal method
We consider an isolated population of a diploid organism with discrete generations, and NeV is estimated from allele frequency shifts over T generations (T ≥ 1) assuming that all genetic change is due to random genetic drift, ignoring the potential effects of migration, mutation and selection (Nei & Tajima 1981; Waples 1989; Wang & Whitlock 2003; Jorde & Ryman 2007). The temporal method can also be used for estimation of effective size in organisms with overlapping generations when appropriate demographic data are available in addition to those on allele frequency change (Jorde & Ryman 1995, 1996; Waples & Yokota 2007). For the purpose of the present discussion, however, we only consider populations with discrete generations.
Letting p denote the allele frequency in the first of two consecutive generations, the parametric drift variance (the sampling variance of p) is p(1 − p)/(2NeV). We can rewrite this as NeV = 1/(2F), where F is the drift variance standardized by p(1 − p). Similarly, when considering drift accumulated over multiple generations (T > 1), we have F≈T/(2NeV) for small values of T, and NeV≈T/(2F) (Waples 1989).
In a typical situation, the parametric value of the drift variance (F) is unknown, however, and must be replaced by an estimate (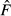 ) based on the allele frequency difference observed over the T generations between measurements. The expected value of
) based on the allele frequency difference observed over the T generations between measurements. The expected value of 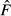 depends on how the allele frequency difference was measured and how the samples were drawn from the population (before or after reproduction), and those conditions determine the procedure for transforming
depends on how the allele frequency difference was measured and how the samples were drawn from the population (before or after reproduction), and those conditions determine the procedure for transforming 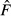 into a bias corrected estimator of genetic drift (F*) that can be used for assessing NeV. When T is not too large (say, T ≤ 10; Luikart et al. 1999), an estimate of NeV is obtained as
into a bias corrected estimator of genetic drift (F*) that can be used for assessing NeV. When T is not too large (say, T ≤ 10; Luikart et al. 1999), an estimate of NeV is obtained as 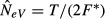 .
.
The most appropriate way for calculating 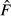 has been debated, and the early measures resulted in biased estimates of NeV when applied to small samples or skewed allele frequencies. A recently derived estimator eliminates those bias problems to a good approximation (Jorde & Ryman 2007).
has been debated, and the early measures resulted in biased estimates of NeV when applied to small samples or skewed allele frequencies. A recently derived estimator eliminates those bias problems to a good approximation (Jorde & Ryman 2007).
Methods
The derivations underlying the conclusions of this study are presented in Appendix S1. The main text only includes some of the key expressions and is meant to be readable without accessing Appendix S1.
We consider a diploid organism with discrete generations and a population system of s subpopulations, each of census size N that is identical to the variance effective size NeV (N = NeV), where mutation and selection are ignored and migration occurs as in an island model of migration (Appendix S1). In each subpopulation, a proportion m of the genes are derived from the population system as a whole (including the focal subpopulation), and the rest (1 − m) are from the subpopulation itself. Conceptually, each subpopulation in generation t contributes an infinite number of progeny to a migrant gene pool. In the next generation (t + 1), any particular subpopulation consists of a mixture of genes drawn from the migrant gene pool (proportion m) and from the focal subpopulation (proportion 1 − m). The population model is demographically deterministic and genetically stochastic where s, N and m are fixed quantities, whereas alleles are sampled binomially from both the focal subpopulation and the migrant gene pool, corresponding to the case of ‘stochastic migration and fixed migration rate’ discussed by Sved & Latter (1977). In the limiting case of m = 1, the global population corresponds to a Wright–Fisher model with sN individuals and 2sN genes. We refer to this situation (m = 1) as panmixia (Appendix S1, section 2).
Temporally spaced samples for estimation of NeV are taken after migration in two consecutive generations (t and t + 1), and those samples may include individuals from one or more subpopulations. The sampled subpopulations may, or may not, be the same at both occasions. Sampling from multiple or different subpopulations is meant to mimic mixed sample compositions obtained unintentionally for reasons such as a poorly known population structure or subpopulation boundaries that are difficult to identify. In other situations, the investigator might deliberately collect mixed samples in an attempt to assess global rather than local effective size.
Within subpopulations, the allele frequency change from one generation to the next is determined by the joint effects of drift and migration, and estimates of NeV will be biased if one assumes (erroneously) that the change is due to drift alone. We derive expressions (Appendix S1) describing how the expected variance of allele frequency shift over one generation is affected by characteristics of the population system, that is, the number of subpopulations (s), their effective size (N), the migration rate (m) and the amount of divergence among subpopulations (FST). We focus primarily on estimates of NeV based on allele frequency changes measured over a single generation (t to t + 1), because the logic for understanding short-term changes constitutes the basis for interpreting those observed over longer periods of time. Also, most empirical studies employing the temporal method measure change over only one or a few generations.
We conducted computer simulations to verify the analytical results with a slightly modified version of easypop (Balloux 2001), as described in Appendix S2. The original version of this software only reports genotypic data for the individuals in the final generation of the simulation. We modified the source code to allow output of multilocus genotypes in multiple and arbitrarily chosen generations, making it possible to mimic the drawing of temporally spaced ‘samples’ from optional generations.
The equations describing the expected value of 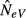 (Appendix S1) are derived without assuming migration–drift equilibrium (steady state). In the numerical examples, however, we have assumed approximate equilibrium conditions.
(Appendix S1) are derived without assuming migration–drift equilibrium (steady state). In the numerical examples, however, we have assumed approximate equilibrium conditions.
Results
For the infinite island model (s=∞) with ‘stochastic migration and fixed migration rate’, the equilibrium value for FST when migration and drift are in balance is
| 1 |
(Appendix S1, eqn A4; derived from Sved & Latter 1977), where m is the proportion of immigrants into each subpopulation and N (=NeV) is their effective size. As discussed in Appendix S1, FST is defined as FST = V/[P(1 − P)], where V is the variance of allele frequencies among subpopulations and P is the overall allele frequency of the global population.
When the number of subpopulations is finite, there is strictly speaking no equilibrium value for FST in the absence of mutation, as all subpopulations will eventually become fixed for the same allele whenever m > 0. However, long before such fixation occurs FST will approach a steady state (quasi-equilibrium) where (Appendix S1, eqn A5)
| 2 |
While local effective populations sizes remain N (=NeV) regardless of migration rates, the effective size of the total population (NeV,tot) is
| 3 |
(Appendix S1, eqn A18), which depends on the migration rate as well as on the degree of genetic differentiation among subpopulations (FST). The equilibrium value for this total NeV,tot is infinite when there is no migration (m = 0), because FST then approaches unity (cf. eqn 1) and the denominator for NeV,tot becomes zero. On the other hand, when migration occurs (m > 0), the total effective size is generally larger than, or approximately equal to, the summed effective sizes of the local subpopulations, sN (Fig.1a).
Figure 1.
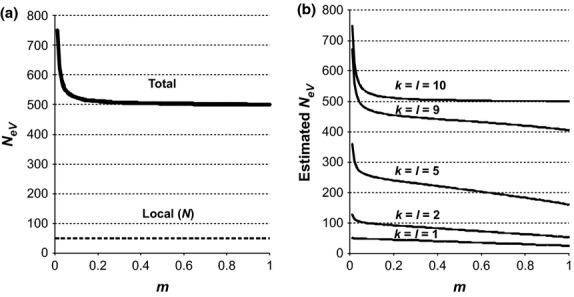
True (a) and expected estimated (b) variance effective size (NeV) at different migration rates (m) for an island model population system in approximate migration–drift equilibrium with s = 10 subpopulations of effective size N = 50. The expected estimates in (b) refer to a situation where the same 1, 2, 5, 9 or 10 subpopulations were sampled in two consecutive generations (k = l; see text for details).
Applying the above model to an empirical situation of NeV estimation, we must consider the expected result of a series of possible scenarios, particularly when sampling from a system of interconnected populations where population structure is unclear or unknown. For the temporal method, we consider two random samples of nt and nt+1 diploid individuals, respectively, that are drawn one generation apart from the pooled set of k subpopulations being available for sampling. Scoring the samples at a set of independently segregating gene loci, we denote the sample allele frequencies at the ith locus on the two occasions to be pt and pt+1, respectively. From these sample frequencies, we calculate a measure (estimate) of the amount of allele frequency change (assumed to represent genetic drift only) over the generation interval as
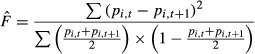 |
4 |
(Jorde & Ryman 2007), where the summations are over all loci (and over all alleles if there are more than two alleles per locus).
Under sampling plan II (see Nei & Tajima 1981; Waples 1989), the quantity (4) can be corrected for the expected contribution from sampling to yield an unbiased estimator of genetic drift (Jorde & Ryman 2007, eqn 13)
| 5 |
where 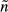 is the harmonic mean of the sample sizes nt and nt+1 in generations t and t + 1, respectively. With a single generation passing between the two sample events, an estimate of the variance effective size in generation t is then given by
is the harmonic mean of the sample sizes nt and nt+1 in generations t and t + 1, respectively. With a single generation passing between the two sample events, an estimate of the variance effective size in generation t is then given by
| 6 |
This study explores how this estimator of NeV behaves when migration occurs among subpopulations, and when individuals from different subpopulations enter the samples. For the purpose of this presentation, we assume that k out of the s subpopulations are sampled in each of two consecutive generations. Letting l denote the amount of overlap, that is, the number of subpopulations sampled at both occasions, we focus on the extreme cases of k = l (the same k subpopulations were sampled both times) and l = 0 (different subpopulations were sampled on each occasion). For large sample sizes (nt, nt+1→∞), we find that for the case when the same k subpopulations are sampled each generation (k = l), the expected value of eqn (6) is
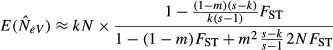 |
7 |
(Appendix S1, eqn A14), which holds for any value of FST (equilibrium or not). When equilibrium is attained, eqn (1) or (2) can be substituted for FST, and we evaluate eqn (7) for various numbers of actual (s) and sampled (k) subpopulations, and for different levels of migration (m).
Equation (7) indicates that when sample sizes are large and the same k subpopulations are sampled at both occasions (k = l), the expected estimate of NeV is the combined size of the subpopulations included in the sample (kN) multiplied by a ‘reduction factor’ that lowers the estimate below kN. The reducing effect of this correction grows stronger as m increases and k decreases (Fig.1b). Another implication of (7), which will be discussed in greater detail below, is that global effective size (NeV,tot) is systematically underestimated unless all the s subpopulations are included in both samples (Fig.1b).
When completely different subpopulations are included in the two samples (l = 0) the expected value of eqn (6) is
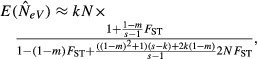 |
8 |
(Appendix S1, eqn A21), which also holds whether equilibrium has been attained or not. The general structure of this expression is similar to that of the previous one (7), except that the reducing effect of the ‘reduction factor’ is far more powerful (below). In the next few sections, we consider first the behaviour of the expected estimate of NeV for particular combinations of k and l at large sample sizes (eqn (7) and (8)), and next the effects of small sample sizes and of multiple generations between measurements.
Both samples include one single subpopulation
Here we consider first the case of an infinite island model (s=∞). When both samples only include individuals from one and the same subpopulation at both occasions (k =l = 1), we find by inserting k = 1 into eqn (7) and substituting eqn (2) for FST that under migration–drift equilibrium the estimated NeV is expected to go from 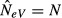 (when m = 0) to
(when m = 0) to 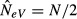 (when m = 1). That is, in the absence of migration, eqn (6) estimates the local effective population size (N), as it should.
(when m = 1). That is, in the absence of migration, eqn (6) estimates the local effective population size (N), as it should.
In contrast, with migration, the estimate 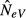 becomes lower than N and reaches N/2, or a slightly higher value under a finite island model (s < ∞), when all the inhabitants of an island are immigrants from the global population (m = 1). In other words, with high levels of migration, sampling a single subpopulation for estimating NeV is expected to downwardly bias the estimated local effective size, and even more so if the estimate is thought to represent the effective size of the total population (NeV,tot = sN). The reason for this latter, perhaps unexpected, finding is considered in detail in the Discussion. Here, we note that this behaviour of
becomes lower than N and reaches N/2, or a slightly higher value under a finite island model (s < ∞), when all the inhabitants of an island are immigrants from the global population (m = 1). In other words, with high levels of migration, sampling a single subpopulation for estimating NeV is expected to downwardly bias the estimated local effective size, and even more so if the estimate is thought to represent the effective size of the total population (NeV,tot = sN). The reason for this latter, perhaps unexpected, finding is considered in detail in the Discussion. Here, we note that this behaviour of 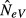 is verified by results of computer simulations, as depicted in Fig.2(a). For the finite model used as an example in Fig.2, with parameters s = 10 and N = 50, the expected estimate for m = 1 is
is verified by results of computer simulations, as depicted in Fig.2(a). For the finite model used as an example in Fig.2, with parameters s = 10 and N = 50, the expected estimate for m = 1 is 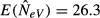 (eqn 2 and 7), that is, slightly higher than N/2 = 25 expected under an infinite island model.
(eqn 2 and 7), that is, slightly higher than N/2 = 25 expected under an infinite island model.
Figure 2.
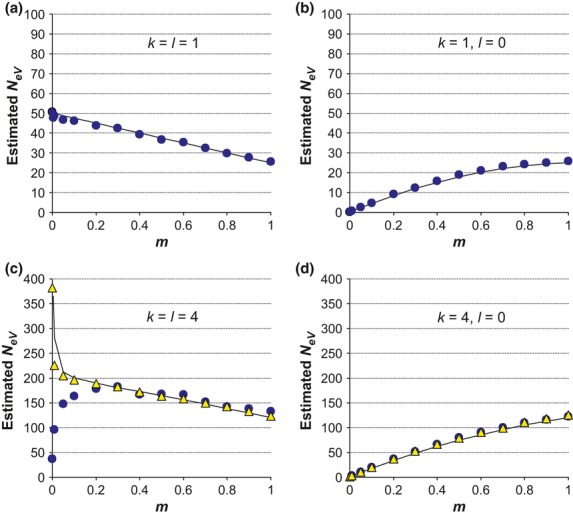
Expected and simulated estimates of NeV at different migration rates (m) for an island model population system in approximate migration–drift equilibrium with s = 10 subpopulations of effective size N = 50. The populations sampled in t and t + 1 are either the same (k = l) or different ones (l = 0), l symbolizing the amount of overlap. Solid lines represent derived expected values (eqn 7 and 8) and symbols simulated results. Note the different scales of the y-axes. (a, b) NeV was estimated by sampling from a single subpopulation in each generation (k = 1). The populations sampled in t and t + 1 are either the same (a; k = l = 1) or different ones (b; l = 0). Simulated sample size is n = 100 diploid individuals in each generation. (c, d) As above, except that k = 4, subpopulations are sampled in each generation and random sampling of a total of n = 80 (circles) or n = 800 (triangles) individuals from the four subpopulations available for sampling (corresponding to an average of n/k = 20 or n/k = 200 individuals sampled from each subpopulation). The four subpopulations sampled in t + 1 are either the same (c; k=l = 4) or different from those in t (d; l = 0).
Local NeV is always seriously underestimated when the samples include one subpopulation in generation t and another one in t + 1. For a large number of subpopulations in equilibrium, expression (8) reduces to 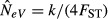 . For k = 1 and N = 50, this corresponds to an expected estimated NeV of 0.25 at m = 0 and to one of 25 at m = 1, in good agreement with computer simulations of a similar scenario (Fig.2(b), where s = 10 rather than s = ∞ and eqn 8 yields an expected
. For k = 1 and N = 50, this corresponds to an expected estimated NeV of 0.25 at m = 0 and to one of 25 at m = 1, in good agreement with computer simulations of a similar scenario (Fig.2(b), where s = 10 rather than s = ∞ and eqn 8 yields an expected 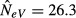 ). Hence, and as is to be expected, under panmixia (m = 1), it does not matter whether we sample the same or different subpopulations in t and t + 1, and the estimate will be biased downwards in either case. The reason for this downward bias is the high variance in allele frequencies between the two generations, which is expected when sampling two different subpopulations. As shown in Appendix S1, this symmetry holds for any value of k, that is, when m = 1, the expressions for
). Hence, and as is to be expected, under panmixia (m = 1), it does not matter whether we sample the same or different subpopulations in t and t + 1, and the estimate will be biased downwards in either case. The reason for this downward bias is the high variance in allele frequencies between the two generations, which is expected when sampling two different subpopulations. As shown in Appendix S1, this symmetry holds for any value of k, that is, when m = 1, the expressions for 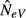 are identical for l = 0 and l = k (Appendix S1, eqn A16 and A23).
are identical for l = 0 and l = k (Appendix S1, eqn A16 and A23).
Both samples include multiple subpopulations
When each sample includes members from multiple subpopulations and the expected contribution from each subpopulation is the same at both occasions (k = l > 1), we predict from eqn (7) a larger NeV estimate than in the case of one and the same subpopulation being sampled (k = l = 1). Evaluation of eqn (7) reveals that at high migration rates, there is a downward bias such that the expected estimate is conspicuously smaller than kN, the combined size of the subpopulations sampled. Further, the expected value of 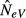 is smallest in the limiting case of m = 1 (Figs1b and 2c, solid line), similar to when sampling the same population in both generations. The analytical results are supported by the results from computer simulations when using large sample sizes (Fig.2c, triangles; see below for smaller sample sizes).
is smallest in the limiting case of m = 1 (Figs1b and 2c, solid line), similar to when sampling the same population in both generations. The analytical results are supported by the results from computer simulations when using large sample sizes (Fig.2c, triangles; see below for smaller sample sizes).
As a numerical example, we may consider the expected estimate for m = 1 in the case of a global population with s = 10 subpopulations of effective size N = NeV = 50 where four of them are sampled in each generation (k = l = 4; Fig.2c, solid line). At migration–drift equilibrium, we have FST = 0.009 (eqn 2), and inserting these values into eqn 7 (or the simplified A16) yields an expected estimate of 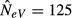 . Considered as an estimate for a local population, for example in a situation where including multiple subpopulations in the samples was accidental, the estimate is biased upwards as compared to the true value of N = 50, whereas it is biased downwards if considered as an estimate of the total population, if sampling multiple subpopulations was a deliberate attempt at estimating NeV,tot (sN = 500). As implied by eqn 7 (and A16), the magnitude of this latter systematic downward bias relates to the proportion of subpopulations sampled; it is most pronounced when sampling a single one (k = l = 1), and it disappears when the global population as a whole is available for sampling (k = l = s). This means that the estimate will approach the effective size of the total population in the limiting case of all subpopulations being sampled (k=s). In other words, and as noted above, unbiased estimation of the global effective size requires sampling from all the constituent subpopulations, and not just from a subset (cf. Fig.1b).
. Considered as an estimate for a local population, for example in a situation where including multiple subpopulations in the samples was accidental, the estimate is biased upwards as compared to the true value of N = 50, whereas it is biased downwards if considered as an estimate of the total population, if sampling multiple subpopulations was a deliberate attempt at estimating NeV,tot (sN = 500). As implied by eqn 7 (and A16), the magnitude of this latter systematic downward bias relates to the proportion of subpopulations sampled; it is most pronounced when sampling a single one (k = l = 1), and it disappears when the global population as a whole is available for sampling (k = l = s). This means that the estimate will approach the effective size of the total population in the limiting case of all subpopulations being sampled (k=s). In other words, and as noted above, unbiased estimation of the global effective size requires sampling from all the constituent subpopulations, and not just from a subset (cf. Fig.1b).
The extreme, and sometimes potentially unrealistic, scenario when multiple but completely different subpopulations are included in the two samples (k > 1, l = 0) yields predicted estimates that are similar to those considered above when a single subpopulation is sampled in generation t and another one in t + 1 (k = 1, l = 0). That is, the expected estimate at migration–drift equilibrium is approximated by k/4 at m = 0 and by kN/2, or a somewhat higher value when s is finite, at m = 1 (eqn 8 and A22–23), as verified by computer simulations (Fig.2d). Hence, when different subpopulations are sampled at the two occasions, whether a single one each time or several different ones, the quantity that is estimated by the temporal method is largely a reflection of the degree of genetic differentiation among populations and is therefore strongly dependent on migration rate. As an estimate of effective size, local or total, this quantity will be biased downwardly for all m.
There are of course intermediate situations of samples including contributions from multiple subpopulations, some of which are the same and others different, as could occur, for example, during a reanalysis of older samples taken on different occasions and not covering exactly the same geographical area. In general, such situations should lead to results intermediate between those for the extreme situations (l = 0 vs. l = k) considered here.
Effect of sample size
The expected value of estimated NeV is derived assuming that the number of genes sampled approaches infinity (Appendix S1), but as indicated by the simulation results, our equations are quite robust to violations of that assumption when both samples include a single subpopulation (k = 1; Fig.2a,b). When drawing finite samples from multiple subpopulations (k > 1), however, a variance component is introduced that is due to differences in the relative contribution from the different subpopulations, and this additional source of variation can cause a downward bias in the estimate of NeV.
As an example of the effect of sample size on the estimate of NeV, we may consider the limiting case of m = 0. The subpopulation allele frequencies will eventually become fixed at 0 or 1, and there is no allele frequency change due to genetic drift that can contribute information on effective size. Irrespective of the fixed allele frequencies, however, temporally spaced samples from a mixture of the same k subpopulations (k > 1) may exhibit different allele frequencies if the proportionate contribution from the different subpopulations varies between sampling occasions. Such an allele frequency difference between samples may be erroneously interpreted as a signal of genetic drift. It can be shown, for example, that when m = 0 the expected estimate of NeV at equilibrium is 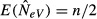 regardless of the true value of NeV, where n is the total number of individuals sampled from the same k subpopulations in each generation.
regardless of the true value of NeV, where n is the total number of individuals sampled from the same k subpopulations in each generation.
This bias is apparent in the results of the computer simulations depicted in Fig.2(c) for the case of a total population with s = 10 subpopulations where four of them are sampled (k=l = 4) in each generation. We simulated total sample sizes of n = 80 and n = 800, implying that an average of n/k = 20 and n/k = 200 individuals, respectively, are sampled from each subpopulation. As seen from the figure, the effect of sample size is negligible at high migration rates, as expected, because when m is large, the subpopulation allele frequencies are similar and the variance component due to sampling from multiple subpopulations is minor. The sample size is critical at low migration rates, however. Drawing a total of n = 800 individuals provides a fairly accurate picture of the expected estimate of effective size calculated from eqn (7), which approaches infinity as m goes towards zero (Fig.2c, triangles and solid line). In contrast, when m is small an additional downward bias occurs for n = 80, and in the limiting case of m = 0, when the total NeV of the four sampled subpopulations is infinity, the simulated estimate is only 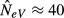 (Fig.2c, circles).
(Fig.2c, circles).
When different populations are sampled in t and t + 1, the effect of sample size (n) on the expected estimate of NeV is negligible (Fig.2d; k = 4, l = 0), the apparent reason being that differences in relative contribution from the various subpopulations is not an issue when different ones are sampled at the two occasions. Again, at m = 1, the expected estimate of NeV for l = 0 is identical to the corresponding quantity obtained when the same four subpopulations are included in both samples (k =l = 4; Fig.2c; Appendix S1, eqn A16 and A23).
Time between measurements
In general, estimates of local NeV are expected to grow progressively larger as the time between measurements increases, eventually approaching the NeV of the global population (Wang & Whitlock 2003). The reason is that within each subpopulation, migration continuously pulls deviating allele frequencies back towards the global mean. Therefore, when measured over extended periods of time, the allele frequency change within a subpopulation will tend to reflect the change of the global population rather than the shifts around the global mean that have occurred within the subpopulation.
The dependence of 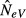 on global effective size introduces a bias when estimating local effective size and measuring genetic change over multiple generations. Our analytical results on the expected estimate of NeV refer to estimates obtained when measuring allele frequency changes in consecutive generations, and we used computer simulations to assess the effect of estimating NeV from changes accumulated over multiple generations. Similar to Fig.2, we set local effective size to NeV = N = 50 and simulated the sampling of the same (k = l = 1) and the same four (k = l = 4) subpopulations T = 1–10 generations apart, mimicking different sizes of the global population by setting the total number of subpopulations to s = 2, 10, and 500 with migration rates m = 0.1 and m = 1 (Fig.3).
on global effective size introduces a bias when estimating local effective size and measuring genetic change over multiple generations. Our analytical results on the expected estimate of NeV refer to estimates obtained when measuring allele frequency changes in consecutive generations, and we used computer simulations to assess the effect of estimating NeV from changes accumulated over multiple generations. Similar to Fig.2, we set local effective size to NeV = N = 50 and simulated the sampling of the same (k = l = 1) and the same four (k = l = 4) subpopulations T = 1–10 generations apart, mimicking different sizes of the global population by setting the total number of subpopulations to s = 2, 10, and 500 with migration rates m = 0.1 and m = 1 (Fig.3).
Figure 3.
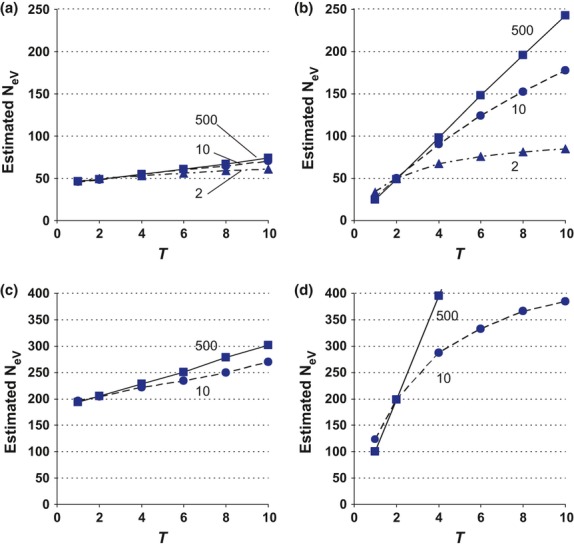
Simulated estimates of variance effective size when measuring allele frequency shifts over T = 1–10 generations and sampling the one and same (k = l = 1, top, a and b) or the same four (k = l = 4, bottom, c and d) subpopulations in population systems comprising s = 2, 10 or 500 subpopulations with migration rates m = 0.1 (left, a and c) and m = 1 (right, b and d). Within each panel, the curves are labelled with respect to s. The number of individuals sampled in each generation is n = 100 in (a) and (b), and n = 800 in c and d (cf. Fig.2). Note the different scales of the y-axes, and that four subpopulations cannot be sampled when s = 2 (bottom, c and d).
As expected, our estimates of NeV are consistently larger when measuring change over multiple generations than between consecutive ones (Fig.3). Over the intervals of T = 1–10 generations considered, however, the effect is relatively modest when migration rates are low to moderate (m ≤ 0.1; Fig.3a,c), whereas high migration rates may result in conspicuously larger estimates (Fig.3b,d). Also, the number of subpopulations constituting the global one strongly affects the estimate, resulting in more inflated estimates when the sampled population(s) belong to a large global population than to a smaller one.
We conjecture that the expected NeV converges to the global effective size regardless of the sampling scheme as the time interval T grows, although the convergence is slower the smaller the migration rate is. Indeed, this has been shown by Waples & Yokota (2007) with simulations and analytically by F. Olsson, O. Hössjer, L. Laikre, N. Ryman (unpublished data) for models with overlapping generations where subpopulations can represent age classes. This phenomenon is related to a coalescence interpretation of Wakeley (1999). If the ancestral lines of the genes sampled at the second time point are followed sufficiently many generations back in time, they will enter a collecting phase, quite independently of the starting configuration.
Discussion
There are several problems involved when using the temporal method for estimating local or global effective size in a population system, and particularly so when differentiation is weak (Fig.2). Sampling from one or more specific local populations is expected to underestimate their combined effective size when measuring change over a single generation. Measuring over longer periods can introduce an upward bias, the magnitude of which depends on the length of the sampling interval and the effective size of the global population (Fig.3).
When the parameter of interest is global effective size, all the subpopulations must be sampled to prevent underestimation, which may be difficult in many situations. This is true also in the limiting case of m = 1, if reproduction is spatially or temporally structured in a way that results in a distinct grouping of the breeders that constitute the unit for sampling efforts. Further, at high levels of differentiation (little migration), inadequate sample sizes may introduce a downward bias also when all the subpopulations are sampled. Thus, applying the temporal method for assessing the rate of loss of heterozygosity due to restricted effective size may seriously overestimate the ‘genetic risks’ when sampling from a population where the genetic structure is unclear or poorly known.
When assessing NeV from real data by means of the temporal method, the estimation error involves not only systematic bias, but also a random part. We found expressions for the systematic bias when either spatial substructure is ignored or when sampling is not uniform over the population. For single locus data sets, the random error is of the same order as the bias. However, whereas the bias formula remains the same regardless of the number of loci (see Section 5 of Appendix S1), the random error will decrease with the number of loci examined. In the following paragraphs, we discuss various aspects and possible implications of this systematic bias in more detail.
Bias with migration
We think our most surprising finding is that sampling from the same subpopulation in t and t + 1 when migration rates are so high that the global population behaves as a panmictic unit results in a downwardly biased estimate of local effective size. At first glance, this result may seem counterintuitive, because one might intuitively expect to estimate the effective size of the population system as a whole when sampling from an unstructured population. As we have shown, however, this is not correct (Fig.2a), and a strict proof is given in Appendix S1. A more intuitive explanation follows below, where we consider the limiting situations of m = 0 and m = 1 for the simplified conditions of an infinite island model (s = ∞) where effective and census size is the same for local populations (Fig.4).
Figure 4.
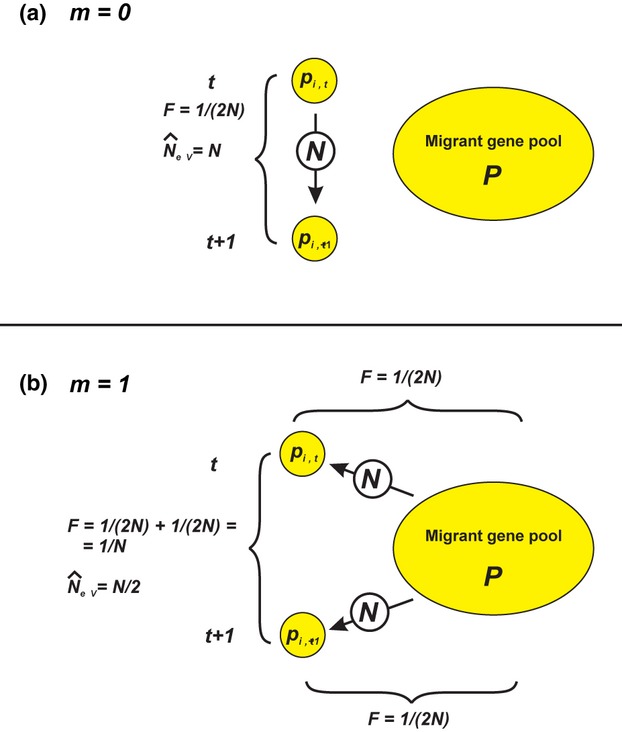
Estimation of NeV with the temporal method in a single subpopulation (k = l = 1) that follows the infinite island model of migration (s = ∞) where the effective and actual size of a subpopulation is the same. The difference (pi,t+1 − pi,t) is the allele frequency change between two consecutive generations (T = 1) in the ith subpopulation, and NeV is estimated as 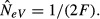 (a) With no gene flow (m = 0), the expected variance of the allele frequency change is pi,t (1 − pi,t)/(2N), the standardized variance (F) is F = 1/(2N), the expected estimate of NeV is
(a) With no gene flow (m = 0), the expected variance of the allele frequency change is pi,t (1 − pi,t)/(2N), the standardized variance (F) is F = 1/(2N), the expected estimate of NeV is 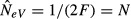 , and the temporal method provides an unbiased estimate of the local NeV. (b) When m = 1, the allele frequency change in two successive generations within a subpopulation results from two independent sampling events from the migrant gene pool, where allele frequency P is constant under the infinite island model. The total standardized variance of the allele frequency difference between the two successive generations within a subpopulation is the sum of the variances resulting from the two independent events. Thus, the temporal method estimates that the local NeV is only one-half of the actual local NeV.
, and the temporal method provides an unbiased estimate of the local NeV. (b) When m = 1, the allele frequency change in two successive generations within a subpopulation results from two independent sampling events from the migrant gene pool, where allele frequency P is constant under the infinite island model. The total standardized variance of the allele frequency difference between the two successive generations within a subpopulation is the sum of the variances resulting from the two independent events. Thus, the temporal method estimates that the local NeV is only one-half of the actual local NeV.
In the case of complete isolation (m = 0), each subpopulation behaves as an independent unit, and the assumptions of the basic model for estimating NeV of an isolated population are met (Fig.4a). All the individuals in the offspring generation (t + 1) represent progeny derived directly through sampling from the parental gene pool in the preceding generation (t), and there are no immigrants. The variance of the allele frequency change over one generation in the ith subpopulation is pi,t(1 − pi,t)/(2N), the standardized variance F is F = 1/(2N) and applying eqn (6) for estimation of NeV as 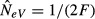 results in
results in 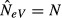 , as it should.
, as it should.
In contrast, when m = 1, every subpopulation is formed through sampling immigrants from an infinitely large migrant gene pool with a constant allele frequency P that equals the average allele frequency over all the subpopulations. Therefore, there is no direct parent–offspring relationship between the individuals of a particular subpopulation in generations t and t + 1 (Fig.4b). Because of the lack of such a relationship, we expect a larger variance of the allele frequency change pi,t+1 − pi,t, and hence a smaller estimate of NeV, when m = 1 than when m = 0. In each generation, the subpopulation allele frequencies (pi,t and pi,t+1) are expected to differ from P because of sampling. The sampling variance is P(1 − P)/(2N) in each generation, and the standardized variance is F = 1/(2N). The standardized variance of the allele frequency change pi,t+1 − pi,t from t to t + 1 is then the sum of the variances of each of the two sampling events (F = 1/N), and the expected estimate of NeV is then 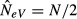 . Thus, when m = 1, the expected variance of the allele frequency change is twice as large as when m = 0, and we expect estimates of NeV to get progressively smaller as migration increases from 0 to 1, approaching half the effective size of the local subpopulation as m approaches unity. Expressed differently, the reason why we are not estimating global effective size when m = 1 is that the samples are drawn from samples from the global population, that is, from the subpopulations, rather than directly from the global population as whole.
. Thus, when m = 1, the expected variance of the allele frequency change is twice as large as when m = 0, and we expect estimates of NeV to get progressively smaller as migration increases from 0 to 1, approaching half the effective size of the local subpopulation as m approaches unity. Expressed differently, the reason why we are not estimating global effective size when m = 1 is that the samples are drawn from samples from the global population, that is, from the subpopulations, rather than directly from the global population as whole.
Bias of other methods
Waples & England (2011) have considered the estimation of effective size using the linkage disequilibrium (LD) method when there is gene flow (Waples & Do 2008, 2010). They assumed an island model of migration, the situation where subpopulation boundaries can be identified, and focused on estimation of local effective size when sampling from a single subpopulation (equivalent to the case of k = l = 1 in our present terminology for the temporal method; Figs1b and 2a).
They concluded that the LD estimator is robust to migration and accurately reflects local effective size as long as migration rate is of the order of ∼0.10 or less; they also concluded that the estimate converges on global effective size for increasing migration rates. Thus, for low-to-moderate migration rates, the temporal and LD methods seem to be similar with respect to bias when estimating local effective size, although the LD estimates are biased upwards and the temporal ones are biased downwards (Fig.2a). In contrast, the LD approach is associated with a much larger bias than the temporal method at high migration rates. The tendency for LD estimates to converge on global effective size implies that the (upward) bias may be indefinite, whereas the (downward) bias of the temporal method is never expected to exceed about 50%.
Application of the LD method to sampling from multiple subpopulations (k > 1) was not considered by Waples & England (2011). It is not clear what would be expected at high migration rates, and that situation needs to be evaluated. It appears intuitively, although, that sampling at random from the population system as a whole would result in downwardly biased estimates of global effective size at moderate-to-low migration rates. The reason is that allele frequency differences between subpopulations will contribute an additional component of LD that would result in low estimates.
This expectation of a downward bias is supported by our preliminary computer simulations with the same software as Waples & England (2011). For example, for the parameters used in Fig.1 (s = 10, N = NeV = 50, and k = l = s = 10), the true global effective size at m = 0.10 is approximately 520, whereas the LD estimates from ten replicate simulations averaged 200 with a range of 171–215 and a harmonic mean of 199. Thus, in the situation where the temporal method is expected to yield approximately unbiased estimates of global effective size when using reasonably large sample sizes, the LD approach seems to result in estimates that are biased downwards.
Loss of heterozygosity with migration
The difficulties associated with obtaining a reasonably unbiased estimate of global effective size (Fig.1b) have implications for assessments of genetic vulnerability and loss of genetic variation. Under an island model, the rate of loss of heterozygosity in a local subpopulation is determined by the effective size of the global population rather than that of the local one. The expected change of heterozygosity in a local subpopulation (HS) and in the global one as a whole (HT) can be obtained directly from recursion equations for gene identity (Nei 1975; Li 1976; Ryman & Leimar 2008). As an example, Fig.5 depicts the expected change of GST (equivalent to FST), HS and HT over the first t = 500 generations for a population system similar to that in Fig.2 with s = 10 partially isolated subpopulations of effective size N = 50 and a migration rate of m = 0.01 in the absence of mutation (the figure was produced using eqn 2–3 of Ryman & Leimar 2008).
Figure 5.
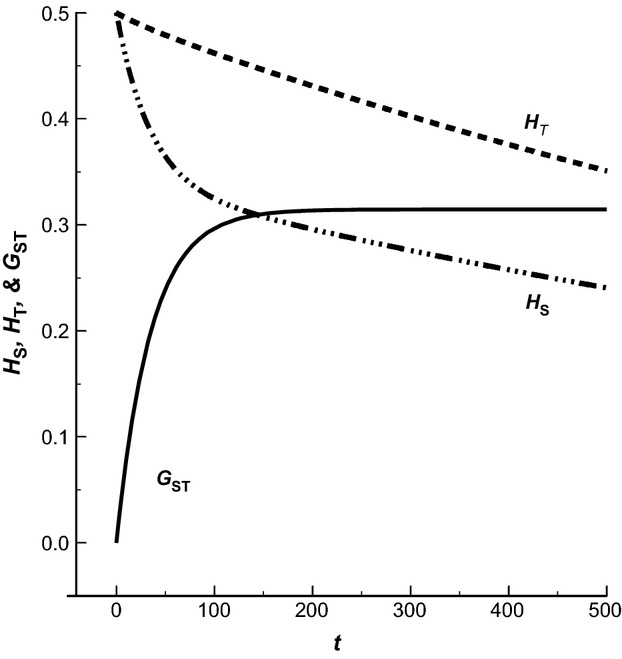
Change of GST, HS and HT over time (t; 500 generations) for s = 10 partially isolated populations (m = 0.01) of effective size N = 50. At t = 0, the ten populations are assumed to represent copies of a single population with HS = HT = 0.5.
In the early phases of divergence, HS declines at a faster rate than HT. As migration–drift equilibrium is approached, however, and GST approaches its steady state value (GST≈0.31 in this particular case), the decline of HS slows down and proceeds at the same rate as that of HT. The generality of the above observation of identical rates of decay for HS and HT when GST has reached steady state follows directly from the definition of GST = 1–HS/HT. For GST to stay constant, which is the definition of steady state, it is necessary for the two quantities HS and HT to change at the same rate, and this holds true also for mutation rates larger than zero.
The implication of the above relationship between HS and HT under steady state is that when migration occurs (m > 0) loss of heterozygosity of a local population is determined by the effective size of the population system as a whole (NeI,tot), which is typically much larger than that of a local population. For the parameters used for Fig.5, for example, and using NeV as a proxy for NeI, global NeI takes a value of NeI,tot = 750. From a conservation genetics perspective, this is the number relevant for assessing the loss of heterozygosity in a specific subpopulation rather than the local effective size under complete isolation (N = 50). As we have shown, necessary conditions for obtaining an estimate of global effective size that is not seriously biased downwards include proper information on the population structure and a rather extensive sampling effort. When those conditions are not met, there is an obvious risk for exaggerating the genetic ‘risk’ facing a local population.
Detecting substructure
As we have seen, the temporal method can drastically underestimate global effective size also at migration rates so high that they are expected to practically swamp subpopulation differences. In the example in Fig.2, for instance, where the number of subpopulations is s = 10 and local effective size is N = 50, a migration rate of, say, m = 0.5 would result in weak differentiation that could be difficult to detect also with extensive sampling. Under those conditions, the expected estimate of NeV based on a single subpopulation is 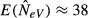 , that is, <10% of the global effective size, and using such an estimate drastically exaggerates the true contemporary rate of heterozygosity decline.
, that is, <10% of the global effective size, and using such an estimate drastically exaggerates the true contemporary rate of heterozygosity decline.
From a practical point of view, we consider the estimation problems easier to deal with when subpopulations are highly divergent. In such situations, a reasonably ambitious study of the genetic population structure in the targeted area should have a good chance of revealing the existence of multiple populations through, for example, spatial genetic differentiation or a significant heterozygote deficiency. Having established the occurrence of such heterogeneities, the investigator is alerted and can take appropriate action when designing the study and evaluating data.
Whether or not subpopulations can be readily detected, the best sampling strategy for estimating local effective size seems to be to sample individuals in connection with local breeding, as population mixture should be minimized at such times. There might still be bias due to migration among populations, but such bias is relatively modest reaching a maximum of 50% for the island model considered herein. If instead the global population is the target for analysis, sampling needs to include as many subpopulations as possible and in representative proportions in temporal replicates. This could be a challenging task for many species, and precision of the estimate may be low unless the global population is effectively small.
As mentioned above, the present study was actually prompted by the results from an empirical genetic monitoring study on brown trout (Salmo trutta) in central Sweden (Palm et al. 2003). We collected samples of approximately 100 fish annually for 20 years from a well defined and about 200 m long part of a stream, the collection site representing a subset of an approximately 3 km long and physically homogeneous section of the stream, without apparent barriers to migration, and where brown trout is abundant. Variance effective size was assessed from allozyme data using the temporal method, and we estimated an NeV of about 50 that stayed reasonably stable over the study period, as did average heterozygosity which was high in comparison with that observed for a series of independent, neighbouring populations. Subsequent sampling indicated strikingly uniform allele frequencies over the entire 3 km section of the stream (unpublished), largely compatible with a single randomly mating population, and we found it difficult to understand how a seemingly very large population with a high heterozygosity could yield NeV estimates of no more than ∼50. In the light of the present findings, however, this observation can be explained by sampling from one of a large number of small subpopulations or spawning aggregations with substantial migration between them. Suitable locations for spawning are probably to occur at several places over the stream section examined, and the nearly complete lack of differentiation suggests high migration rates between these subpopulations. In the most extreme case of m = 1, and assuming a migration pattern that can be approximated by that of an island model, we would expect that our estimate of NeV corresponds to half the effective size of the particular subpopulation targeted for sampling, that is, with a true local effective size of 100, we would expect an estimate ∼50. We do not know the number of subpopulations in the stream section studied, but even a relatively modest number of 5–10 subpopulations would correspond to a global effective size in the order of 500–1000 where loss of heterozygosity due to genetic drift would be negligible over extended periods of time.
Marine populations
Many marine species are characterized by large population sizes, high levels of genetic variation and weak differentiation (Ward et al. 1994; DeWoody & Avise 2000; Hare et al. 2011). A series of recent studies on exploited marine species report surprisingly small estimates of effective size, however. For example, Hauser & Carvalho (2008) reviewed 28 Ne assessments for marine fishes and found that estimates of effective size are between two and five orders of magnitude smaller than census size (NC), with an average Ne/NC ratio of 10−4, which is only a small fraction of those reported for most other species. They also noted that most point estimates were in the range from hundreds to low thousands, and within the range where loss of genetic variability could be a justified management concern.
The genetic structure of many marine species is typified by multiple subpopulations (spawning areas) with a high migration between them and sometimes a seemingly chaotic distribution of genetic variation (e.g. Broquet et al. 2013), potentially similar to the island migration model examined here where no apparent correlation between geographical and genetic distance is expected. It is generally assumed that the most important explanation to the strikingly small Ne/NC ratios observed in marine species is a high variance in reproductive success, which is possible in organisms with a potential for high fecundity and a long lifespan (e.g. Hedgecock 1994; Hedrick 2005; Gomez-Uchida & Banks 2006; Hedgecock et al. 2007; Hauser & Carvalho 2008; Palstra & Ruzzante 2008; Hedgecock & Pudovkin 2011). Hedgecock (1994) introduced the concept of ‘sweepstake reproductive success’ to illustrate the reproductive outcome in such organisms where a small number of breeders have the potential for spawning a large part of the following generation, making the comparison with a sweepstake lottery with few winners and many losers (Hedgecock & Pudovkin 2011).
There is ample evidence that sweepstake reproductive success is an important factor behind the extremely small Ne/NC ratios observed in many marine species (Hedgecock & Pudovkin 2011). The findings of the present analysis, however, suggest an additional explanation. Perhaps these marine species consist of multiple, weakly differentiated populations that, unknown to the investigators, were included in different temporal samples. The estimates of Ne would then have been downwardly biased estimates of NeV of local populations, rather than of global effective size (NeV,tot), resulting in a severe mismatch between the quantities compared when assessing Ne/NC. In the review by Hauser & Carvalho (2008), for example, 24 of the 28 estimates were obtained by the temporal method for estimating NeV.
Management implications
The tendency of NeV to overstate the impression of genetic vulnerability has crucial implications for management. Assuming, for example, that populations of NeI < 50 are considered at risk for excessive loss of genetic variation, and that a manager has difficulties to determine whether a population is isolated or not. How should an estimate of 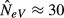 be dealt with? Automatically presuming that NeV exaggerates the genetic threat, and taking no action, may be harmful to the focal population, whereas erroneously assuming that heterozygosity is lost at a rate of
be dealt with? Automatically presuming that NeV exaggerates the genetic threat, and taking no action, may be harmful to the focal population, whereas erroneously assuming that heterozygosity is lost at a rate of 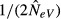 and launching a ‘rescue’ programme might divert resources from populations in stronger need of support. In such situations, immediate remedial action may not be warranted unless there are independent indications that this population has lost genetic variation, for example by exhibiting a lower heterozygosity or fewer alleles than neighbouring ones. An alternative step could be to initiate a genetic monitoring programme that keeps track of potential changes of the amount of genetic variation.
and launching a ‘rescue’ programme might divert resources from populations in stronger need of support. In such situations, immediate remedial action may not be warranted unless there are independent indications that this population has lost genetic variation, for example by exhibiting a lower heterozygosity or fewer alleles than neighbouring ones. An alternative step could be to initiate a genetic monitoring programme that keeps track of potential changes of the amount of genetic variation.
Acknowledgments
We thank Olof Leimar and Robin Waples for valuable comments and suggestions, and Thomas Broquet and four anonymous reviewers for comments on a previous version of the manuscript. NR thanks Francois Balloux for providing the C source code for easypop. The work was funded by grants from the Swedish Research Council (LL, NR), the Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning (Formas; LL, NR) and the Swedish Environmental Protection Agency (LL). OH's research was financed by the Swedish Research Council, contract nr. 621-2008-4946, and the Gustafsson Foundation for Research in Natural Sciences and Medicine. PEJ was supported by the Research Council of Norway. Parts of the analyses were carried out within the framework of the BaltGene project funded by BONUS Baltic Organisations’ Network for Funding Science EEIG. This article is based partially on work supported by the US National Science Foundation Grant DEB 074218 to FWA and was partially conducted as part of the Genetic Monitoring Working Group jointly supported by the National Evolutionary Synthesis Center (NSF #EF-0423641) and the National Center for Ecological Analysis and Synthesis, a Center funded by NSF (NSF #EF-0553768), the University of California, Santa Barbara and the State of California. FWA and NR acknowledge the hospitality of the Hawaii Institute of Marine Biology (HIMB) when working on the manuscript.
Appendix
N.R. and L.L. led the genetic monitoring project that initiated and provided the original idea to this paper, and N.R., F.W.A., and L.L. conceived the study and defined the basic questions. N.R. led the analytical work with input from all the coauthors. O.H. derived the mathematical expressions and wrote Appendix S1 with contributions from N.R. and P.E.J. N.R. conducted the computer simulations in close contact with P.E.J. All the authors participated in discussions that developed the work, and they also wrote various parts of the manuscript with N.R. leading the writing process.
Data accessibility
This study is theoretical and not based on empirical data. Simulations have been conducted using a software that is freely available (easypop; Balloux 2001).
Supporting Information
Appendix S1 Derivations and mathematical expressions for expected estimates of variance effective population size (NeV) when sampling from a population system assuming an island model of migration.
Appendix S2 Computer simulations to verify analytical results.
References
- Balloux F. EASYPOP (version 1.7): a computer program for population genetics simulations. Journal of Heredity. 2001;92:301–302. doi: 10.1093/jhered/92.3.301. [DOI] [PubMed] [Google Scholar]
- Broquet T, Viard F, Yearsley JM. Genetic drift and collective dispersal can result in chaotic genetic patchiness. Evolution. 2013;67:1660–1675. doi: 10.1111/j.1558-5646.2012.01826.x. [DOI] [PubMed] [Google Scholar]
- Crow JF, Kimura M. An Introduction to Population Genetics Theory. Caldwell, New Jersey: The Blackburn Press; 1970. [Google Scholar]
- DeWoody JA, Avise JC. Microsatellite variation in marine, freshwater and anadromous fishes compared with other animals. Journal of Fish Biology. 2000;56:461–473. [Google Scholar]
- Frankel OH. Genetic conservation—our evolutionary responsibility. Genetics. 1974;78:53–65. doi: 10.1093/genetics/78.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Uchida D, Banks M. Estimation of effective population size for the long-lived darkblotched rockfish Sebastes crameri. Journal of Heredity. 2006;97:603–606. doi: 10.1093/jhered/esl042. [DOI] [PubMed] [Google Scholar]
- Hare MP, Nunney L, Schwartz MK, et al. Understanding and estimating effective population size for practical application in marine species management. Conservation Biology. 2011;25:438–449. doi: 10.1111/j.1523-1739.2010.01637.x. [DOI] [PubMed] [Google Scholar]
- Hauser L, Carvalho GR. Paradigm shifts in marine fisheries genetics: ugly hypotheses slain by beautiful facts. Fish and Fisheries. 2008;9:333–362. [Google Scholar]
- Hedgecock D. Does variance in reproductive success limit effective population sizes of marine organisms? In: Beaumont AR, editor. Genetics and Evolution of Marine Organisms. London: Chapman and Hall; 1994. pp. 122–134. [Google Scholar]
- Hedgecock D, Pudovkin AI. Sweepstakes reproductive success in highly fecund marine fish and shellfish: a review and commentary. Bulletin of Marine Science. 2011;87:971–1002. [Google Scholar]
- Hedgecock D, Launey S, Pudovkin AI, et al. Small effective number of parents (Nb) inferred for a naturally spawned cohort of juvenile European flat oysters Ostrea edulis. Marine Biology. 2007;150:1182. [Google Scholar]
- Hedrick P. Large variance in reproductive success and the N-e/N ratio. Evolution. 2005;59:1596–1599. [PubMed] [Google Scholar]
- Jorde PE, Ryman N. Temporal allele frequency change and estimation of effective size in populations with overlapping generations. Genetics. 1995;139:1077–1090. doi: 10.1093/genetics/139.2.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorde PE, Ryman N. Demographic genetics of brown trout (Salmo trutta) and estimation of effective population size from temporal change of allele frequencies. Genetics. 1996;143:1369–1381. doi: 10.1093/genetics/143.3.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorde PE, Ryman N. Unbiased estimator for genetic drift and effective population size. Genetics. 2007;177:927–935. doi: 10.1534/genetics.107.075481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leberg P. Genetic approaches for estimating the effective size of populations. Journal of Wildlife Management. 2005;69:1385–1399. [Google Scholar]
- Li WH. Effect of migration on genetic distance. The American Naturalist. 1976;110:841–847. [Google Scholar]
- Luikart G, Cornuet JM, Allendorf FW. Temporal changes in allele frequencies provide estimates of population bottleneck size. Conservation Biology. 1999;13:523–530. [Google Scholar]
- Luikart G, Ryman N, Tallmon DA, Schwartz MK, Allendorf FW. Estimation of census and effective population sizes: the increasing usefulness of DNA-based approaches. Conservation Genetics. 2010;11:355–373. [Google Scholar]
- Nei M. Molecular Population Genetics and Evolution. Amsterdam, Netherlands: North-Holland; 1975. [PubMed] [Google Scholar]
- Nei M, Tajima F. Genetic drift and estimation of effective population size. Genetics. 1981;98:625–640. doi: 10.1093/genetics/98.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm S, Laikre L, Jorde PE, Ryman N. Effective population size and temporal genetic change in stream resident brown trout (Salmo trutta L.) Conservation Genetics. 2003;4:249–264. [Google Scholar]
- Palstra FP, Ruzzante DE. Genetic estimates of contemporary effective population size: what can they tell us about the importance of genetic stochasticity for wild population persistence? Molecular Ecology. 2008;17:3428–3447. doi: 10.1111/j.1365-294x.2008.03842.x. [DOI] [PubMed] [Google Scholar]
- Ryman N, Leimar O. Effect of mutation on genetic differentiation among nonequilibrium populations. Evolution. 2008;62:2250–2259. doi: 10.1111/j.1558-5646.2008.00453.x. [DOI] [PubMed] [Google Scholar]
- Ryman N, Palm S, André C, et al. Power for detecting genetic divergence: differences between statistical methods and marker loci. Molecular Ecology. 2006;15:231–245. doi: 10.1111/j.1365-294X.2006.02839.x. [DOI] [PubMed] [Google Scholar]
- Sved JA, Latter BDH. Migration and mutation in stochastic-models of gene frequency change.I. Island model. Journal of Mathematical Biology. 1977;5:61–73. doi: 10.1007/BF00275807. [DOI] [PubMed] [Google Scholar]
- Traill LW, Brook BW, Frankham RR, Bradshaw CJA. Pragmatic population viability targets in a rapidly changing world. Biological Conservation. 2010;143:28–34. [Google Scholar]
- Wakeley J. Nonequilibrium migration in human history. Genetics. 1999;153:1863–1871. doi: 10.1093/genetics/153.4.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JL. Estimation of effective population sizes from data on genetic markers. Philosophical Transactions of the Royal Society B-Biological Sciences. 2005;360:1395–1409. doi: 10.1098/rstb.2005.1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JL, Whitlock MC. Estimating effective population size and migration rates from genetic samples over space and time. Genetics. 2003;163:429–446. doi: 10.1093/genetics/163.1.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waples RS. A generalized approach for estimating effective population size from temporal changes in allele frequency. Genetics. 1989;121:379–391. doi: 10.1093/genetics/121.2.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waples RS, Do C. LdNe: a program for estimating effective population size from data on linkage disequilibrium. Molecular Ecology Resources. 2008;8:753–756. doi: 10.1111/j.1755-0998.2007.02061.x. [DOI] [PubMed] [Google Scholar]
- Waples RS, Do C. Linkage disequilibrium estimates of contemporary Ne using highly variable genetic markers: a largely untapped resource for applied conservation and evolution. Evolutionary Applications. 2010;3:244–262. doi: 10.1111/j.1752-4571.2009.00104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waples RS, England PR. Estimating contemporary effective population size on the basis of linkage disequilibrium in the face of migration. Genetics. 2011;189:633–644. doi: 10.1534/genetics.111.132233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waples RS, Gaggiotti O. What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity. Molecular Ecology. 2006;15:1419–1439. doi: 10.1111/j.1365-294X.2006.02890.x. [DOI] [PubMed] [Google Scholar]
- Waples RS, Yokota M. Temporal estimates of effective population size in species with overlapping generations. Genetics. 2007;175:219–233. doi: 10.1534/genetics.106.065300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward RD, Woodwark M, Skibinski DOF. A comparison of genetic diversity levels in marine, freshwater, and anadromous fishes. Journal of Fish Biology. 1994;44:213–232. [Google Scholar]
- Wright S. The interpretation of population structure by F-statistics with special regard to systems of mating. Evolution. 1965;19:395–420. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Derivations and mathematical expressions for expected estimates of variance effective population size (NeV) when sampling from a population system assuming an island model of migration.
Appendix S2 Computer simulations to verify analytical results.
Data Availability Statement
This study is theoretical and not based on empirical data. Simulations have been conducted using a software that is freely available (easypop; Balloux 2001).