

Abstract
Inhibition of cholesterol ester transfer protein (CETP), a protein mediating transfer of neutral lipids between lipoproteins, has been proposed as a means to elevate atheroprotective HDL subpopulations and thereby reduce atherosclerosis. However, off-target and adverse effects of the inhibition have raised doubts about the molecular mechanism of CETP-HDL interaction. Recent experimental findings have demonstrated the penetration of CETP into HDL. However, atomic level resolution of CETP penetration into HDL, a prerequisite for a better understanding of CETP functionality and HDL atheroprotection, is missing. We constructed an HDL particle that mimics the actual human HDL mass composition and investigated for the first time, by large-scale atomistic molecular dynamics, the interaction of an upright CETP with a human HDL-mimicking model. The results demonstrated how CETP can penetrate the HDL particle surface, with the formation of an opening in the N barrel domain end of CETP, put in evidence the major anchoring role of a tryptophan-rich region of this domain, and unveiled the presence of a phenylalanine barrier controlling further access of HDL-derived lipids to the tunnel of CETP. The findings reveal novel atomistic details of the CETP-HDL interaction mechanism and can provide new insight into therapeutic strategies.
Keywords: atherosclerosis, cholesterol/trafficking, estrogen, high density lipoprotein/metabolism, lipid transfer protein, lipoproteins, cholesterol ester transfer protein, molecular dynamics
The well-recognized associations between the plasma level of HDL-cholesterol (HDL-C), HDL metabolism, and the atheroprotective functions of HDL (1) have been driving forces for research on HDL as a potential therapeutic target (2). Several therapies aimed at increasing HDL-C levels have been tested. Among them is the inhibition of cholesterol ester transfer protein (CETP) (2, 3), a plasma neutral lipid transfer protein that mediates exchange/transfer of neutral lipids between HDL and apoB-100-containing lipoproteins and is involved in the reverse cholesterol transport process. Despite an increase in the level of HDL-C, there was no decrease in cardiovascular risk. This outcome shows that the direct relationship between increase in HDL-C and decrease in the risk for cardiovascular disease is not clear and some of the characteristics of HDL and/or CETP are not yet known, preventing full comprehension of the mechanism by which CETP interacts with HDL to facilitate lipid transfer in the circulation. Our objective was then to clarify the process by which CETP binds and interacts with HDL.
Atomistic molecular dynamics (MD) and coarse graining dynamics studies (4) have previously been applied to clarify the lipid transfer from HDL mediated by CETP. The HDL model was a lipid-mimicking droplet of HDL containing phospholipids (POPC) and cholesteryl ester, lacking the HDL main protein, apoA-I. While these previous computational studies considered a match to match curvature CETP-HDL, a recent experimental study relying on advanced electron microscopy (EM) technique and 3D reconstruction demonstrated that CETP and HDL form a binary complex by partial penetration of CETP into the lipid-apolipoprotein monolayer surface of HDL, with CETP in an upright position (5). However, molecular and atomistic level resolution was not sufficient to clarify the penetration process or the atheroprotective role of HDL. In a match-to-match curvature model, as proposed earlier (4), atomistic simulations have shown that CETP stays at the POPC hydrophilic head group level and does not penetrate deeper in the POPC fatty acyl layer of HDL, revealing the inadequacy of the earlier model, assuming congruency of the curvatures of CETP and HDL with a penetration model. Therefore, a new approach is needed to shed light on the lipid exchange mechanism mediated by CETP.
In the present work, we study the CETP-HDL interaction by large-scale atomistic MD. For the first time, CETP in an upright position and a HDL model mimicking a human HDL lipid mass composition are used in the modeling of the CETP-HDL system (Fig. 1) and in atomistic MD simulations. We had three objectives: 1) To demonstrate penetration of CETP into HDL and provide atomistic details and structural features of the penetration. 2) To explore a possible lipid transfer mechanism mediated by CETP upon penetration. 3) β-Estradiol (E2) molecules are transported in plasma lipoproteins mostly as fatty acyl derivatives, HDL, in particular, is reported to be a good carrier for E2 fatty acyl ester (6, 7) and the HDL/E2 fatty acyl ester complex has an enhanced atheroprotective role, at least in vitro, particularly among women (6, 8, 9). Furthermore, experimental findings indicate that esterified estradiol molecules are transferred from HDL to LDL particles via a CETP-mediated process, the details of which are unknown (10). By modeling a HDL particle of actual human HDL mass composition in association with E2 fatty acyl ester, we can further both studies, with the aim of clarifying lipid exchange, such as cholesteryl ester [cholesteryl oleate (ChOE)] transfer from HDL to CETP, but also possible E2 fatty acyl ester transfer from HDL to CETP. Our third objective, therefore, was to investigate the role of E2 fatty acyl ester associated with HDL in the CETP-HDL interaction and the mediation of its transfer by CETP. We present details of the modeling of HDL, proceeding from a starting nascent model of HDL (11–13) to the construction of spherical HDL models mimicking actual human HDL mass composition, and then explore the interaction of these with CETP. A detailed atomistic study of the CETP-HDL interaction is presented and MD-derived results discussed.
Fig. 1.

Starting configurations of CETP interacting with a spherical HDL particle. On the left is the old view (4) of the CETP-HDL interaction mechanism with the CETP curvature toward HDL. The black dashed line highlights the curvature of CETP. On the right is the present study with upright CETP interacting with HDL, the N barrel domain end of CETP facing the HDL lipid surface (5), and close proximity to the apoA-I C-terminal end. Trp105, Trp106, and Trp162 in the N barrel domain end of CETP are displayed (cyan).
METHODS
Model of nascent HDL
We used the double superhelix (DSH) model of nascent HDL (11–13), RCSB Protein Data Bank accession number 3K2S, as a starting structure for HDL. The stoichiometry phospholipid (POPC):free cholesterol (FChO):protein (apoA-I) used in our study was 200:20:2 similar to (12).
Models of spherical HDL
The end structure of nascent HDL obtained at 100 ns MD simulation (discoidal HDL model) was used as the starting structure to build spherical HDL particles. In order to build spherical HDL models with actual human mass composition, the lipid compositions derived from MS-lipidomic analysis of HDL in subjects with low and normal HDL-C levels (14) were used. Lipids were added by slots outside the discoidal HDL model. ChOE molecules were first added to our discoidal HDL particle, and then 91 POPCs were randomly removed. The number of FChO initially present in our starting discoidal HDL particle was adjusted, and SM, triglyceride (TO), lysophosphatidylcholine (LysoPC), and missing ChOE were randomly inserted to satisfy the in vivo composition of lipids found in low and normal HDL-C subjects, respectively [see (14) for details of the lipid composition].
Model of CETP
The boomerang model of CETP bound to four lipids with 2.2 Å resolution (15), RCSB Protein Data Bank accession number 2OBD, was used in our MD simulations. In this model, two 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) molecules cap the N- and C-terminal domains (called N and C openings) of CETP, and two ChOE molecules reside inside the hydrophobic tunnel of CETP.
Systems studied
Configurations for all interacting systems were prepared with the Discovery Accelrys Studio package. All interacting systems investigated are summarized in Table 1.
TABLE 1.
Summary of the investigated systems and corresponding MD time-scale
| Interacting Subsystems | MD Scale (ns) | |
| Nascent HDL | — | ∼100 |
| CETPa | ChOE + DOPC | 50 |
| CETPa | 3TO + DOPC | 60 |
| CETPb | TO + DOPC | 50 |
| CETPc | Nascent HDL | 100 |
| Nascent discoidal HDL | Lipidsd | 200 |
| Normal HDL-C | 10 Estradiol oleate | 50 |
| CETPc | Normal HDL-C | 860 |
Lipid-free CETP was used.
CETP-bound ChOEs were kept inside the CETP tunnel.
Refers to lipid-bound CETP and means that CETP was handled with its four bound lipids (two ChOE and two DOPC).
Refers to the lipid composition (14) needed to construct low and normal HDL-C-mimicking particles.
Atomistic MD simulation parameters and force field
All the simulations were done using GROMACS versions 4.5.3, 4.5.6, and 4.6.3 (16). The nascent DSH model of HDL (12) was inserted in a polyhedral box of size 12 × 20 × 12 nm3. The CETP-discoidal nascent HDL system was inserted in a cubic box of size 18 × 18 × 18 nm3 and the CETP-normal HDL-C system in a polyhedral box of size 20 × 20 × 30 nm3. All systems were filled with explicit molecules of water. Charges of CETP and apoA-I were set in accordance with physiological pH 7.4. In all systems, a salt (NaCl) concentration of 200 mM was created. Our CETP-normal HDL-C system model solvated in about 380,000 water molecules and electrolyzed with 2,200 ions included altogether about 1.2 million atoms. The more realistic isothermal-isobaric (NpT) ensemble was used. We employed a Nose-Hoover thermostat (17, 18) to set the temperature to 310 K. In the case of CETP-normal HDL-C, a temperature of 330 K was set for ChOE lipids in an attempt to satisfy the liquid-state condition assumed for the HDL core (4). Pressure was set isotropically to 1.0 bar with the help of a Parrinello-Rahman barostat (19). Electrostatic interactions were assessed by the particle mesh Ewald technique with a real space cut-off of 1.0 nm. For van der Waals interactions, a cut-off of 1.0 nm was set. Water molecules were described using the simple point charge water model (20). Water bonds were constrained with the SETTLE (21) algorithm and nonwater bonds with the LINCS algorithm (22).
All protein, apoA-I, and CETP parameters were derived from GROMACS MD software (16) using the united atom force field GROMOS53a6. The lipid parameters were a mixture of the united atom force field GROMOS and Berger, Edholm, and Jahnig (23) parameters. POPC and ChOE force fields were derived from published reports (24–26). A force field for TO was kindly provided by the research group of Ilpo Vattulainen from Tampere University of Technology, Finland. The GROMOS force fields for DOPC and SM were retrieved from Lipidbook. The force fields for FChO and LysoPC were hand built on the basis of the force fields of ChOE and POPC, respectively. A GROMOS-compatible force field for E2 was derived from the PRODRG server (27). Charges were then modified according to previous reports (28, 29). Subsequently, a force field for E2 oleate was built by integrating the oleate part of ChOE into the E2 force field.
Before any MD simulations were carried out, all the simulated systems were energy minimized. Table 1 summarizes the MD simulation time-scale for each investigated system. The nascent DSH model was simulated for 100 ns and the end structure was used as an initial structure for nascent HDL to construct spherical HDL. An overall MD simulation time of 200 ns was used for mimicking normal HDL-C and low HDL-C spherical particles. For CETP interacting with normal HDL-C, the first 200 ns MD simulation was treated as a pre-equilibration simulation from which the starting geometry was derived. Considering the well-known slow diffusion of lipid molecules, the use of large-scale atomistic MD simulation was required and microsecond scale was therefore employed. Thus, 1,100 ns (about 1 μs) atomistic MD simulations were performed, equivalent to 6,500 h of calculations. Of the total MD scale, only the 860 ns simulation slot, capturing the main steps in the CETP-normal HDL-C interaction, is presented.
RESULTS
Structure of nascent HDL
To be able to construct spherical HDL particles mimicking actual human HDL mass composition, we first started by simulating a model for nascent HDL. We initiated with the DSH ellipsoidal nascent HDL model (11, 13) of stoichiometry POPC:FChO:apoA-I (200:20:2). A MD simulation of about 100 ns was first performed on the DSH nascent HDL model. A minimum MD simulation length of 80 ns was shown to be necessary for equilibration of the nascent HDL particle [see the root-mean-square deviation (RMSD) plot in Fig. 2A]. Our MD-derived results were then compared with existing data to have a preliminary assessment on our force field.
Fig. 2.

Conformational changes in the nascent HDL and apoA-I chains as a function of MD simulation time. A: RMSD plot of nascent HDL. B: Overall structure variations at t = 0, 60, and 100 ns. C: Comparison of the starting ellipsoidal model of nascent HDL (13) at t = 0 ns with our discoidal nascent HDL particle obtained at t = 100 ns. Top and edge positions of the discoidal nascent HDL particle are pointed out (arrows). D: Radius of gyration of nascent HDL as a function of simulation time.
Our MD-derived results showed structural changes of the starting ellipsoidal HDL model. The structures of the two apoA-I protein chains were analyzed, along with the particle shape and size (Fig. 2B–D). Our results showed that the overall helical structure of apoA-I is conserved along the MD simulation. In addition, the overall height of the nascent HDL particle decreases while the diameter increases (Fig. 2B). An overall decrease of 9 Å in the particle radius of gyration (ΔRg) was determined on our 100 ns MD simulation (Fig. 2D). Despite seeming agreement of these general changes experienced by the DSH model with (13), highlighting of the phosphate of the POPC head groups (Fig. 2C) offers a clear view of the significant reorganization experienced by the DSH nascent HDL model over our MD simulation. We obtained an organized double phospholipid layer-like structure (Fig. 2C, at t = 100 ns) opposite to the spheroidal packing of the lipid core of the DSH after a 60 ns simulation (13), proving the instability of the DSH model and reconfirming the inherent problems of the DSH model and of the parameters used in previous simulations (30). Our resulting discoidal bilayer-like nascent HDL model matches the standard model for HDL and proved to be in-line with earlier assessments of the DSH model (30). The present results demonstrate that our force field can generate relevant structural data, which is a prerequisite for further MD studies. Our discoidal nascent HDL model conserved, however, one feature of the DSH model, i.e., the lack of self-association of apoA-I end domains (30, 31). However, as earlier observed, this feature does not affect the conformational flexibility of the apoA-I double belt for further modeling of spherical HDL (30).
From nascent to spherical HDL particles
Based on stepwise modifications of the lipid composition of our discoidal-like nascent HDL particle (end structure obtained at t = 100 ns), we constructed two representative HDL particle models with actual human HDL mass compositions, low and normal HDL-C-mimicking particles (for details, see “Models of spherical HDL” in the Methods). After each addition of lipids outside the discoidal nascent HDL, the system was energy minimized, MD simulations were performed, and the lipids were left to relax until they fully penetrated the HDL. This procedure was repeated until the low and normal HDL-C compositions were fulfilled. The modification of the composition of the core of the HDL particle upon penetration of the lipids and the subsequent maturation of the nascent HDL particle were observed on 200 ns MD simulation. Figure 3 presents the RMSD on the last 35 ns MD simulation, indicating the pre-equilibration of low HDL-C (last 15 ns) and normal HDL-C (last 7 ns) particles. In addition, the Fig. 3 inset demonstrates the change in the HDL particle shapes and of overall apoA-I chain structures. The resulting low and normal HDL-C-mimicking particles displayed spherical-like structures. Moreover, upon maturation of the discoidal nascent HDL, MD showed that the polypeptide ends of the two apoA-I chains were primarily modified (Fig. 3). The polypeptide ends of apoA-I have earlier been identified as important regions for HDL formation during lipidation (32). Again, we observed high flexibility of apoA-I in these domains. Only the normal HDL-C mimicking particle was further investigated and is discussed below.
Fig. 3.

RMSD plot of normal and low cholesterol (HDL-C)-mimicking HDL particles and comparison of the geometrical and structural changes in HDL and in apoA-I. The nascent HDL particle end structure (t = 100 ns MD simulation) is compared with the end structures of our normal and low HDL-C-mimicking particles, obtained upon maturation of nascent HDL. Circles are used to highlight a geometrical change in the shape of the HDL particles. apoA-I chains are shown in their secondary structure.
E2 fatty acyl ester associated with HDL
To construct E2 fatty acyl ester associated with HDL, we added ten molecules of E2 oleate outside our normal HDL-C-mimicking particle and performed a MD simulation for about 50 ns. Seven of the ten molecules penetrated HDL via their oleate fatty acyl tail, whereas the more hydrophilic head of E2 oleate faced the hydrophilic water environment. A snapshot of the pre-equilibrated E2 oleate associated with a normal HDL-C particle is presented in Fig. 4 (see the last 7 ns of the RMSD plot); the hydrophilic heads of E2 oleates protruding the HDL surface are highlighted. This result confirmed HDL to be a good carrier for E2 fatty acyl esters. In addition, the effect of E2 fatty acyl ester on apoA-I secondary structure was probed using the DSSP tool (33). However, no significant changes in the apoA-I secondary structure were observed upon association of E2 oleate with HDL.
Fig. 4.

RMSD plot of E2 fatty acyl ester associated with a normal HDL-C-mimicking particle. In the inset, the snapshots display our normal HDL-C-mimicking particle before and after interaction with 10 molecules of E2 oleate. After interaction (on right), we obtained E2 oleate associated with a normal HDL-C particle. E2 oleate molecules, in van der Waals representation and white color, are zoomed in to show that only the head of E2 oleate stays outside the HDL structure, while the fatty acyl tail penetrates inside HDL. Lipids are displayed in van der Waals representation as follows: POPC (orange), FChO (blue), TO (purple), ChOE (red), SM (green), LysoPC (pink), and apoA-I chains appear in their secondary structure (yellow).
Insight into CETP structure, identification of CETP openings
We investigated CETP structure prior to any interaction study of CETP with HDL to check whether MD could identify the CETP openings proposed earlier (15), to probe CETP dynamics in the presence and absence of its bound lipids, and provide further atomistic information on CETP and its ability to transfer neutral lipids.
The interaction of CETP with different lipid mixtures of ChOE, DOPC, and TOs was investigated by performing MD simulations for a minimum of 50 ns per case (Fig. 5). In all conditions, we observed penetration of DOPC, TO, and/or ChOE into CETP during the simulation period. Condition i (Fig. 5) displays two openings, one for ChOE and one for DOPC. ChOE penetrated CETP with its head at the center of the CETP structure, the “neck” of CETP (15), while the DOPC tail penetrated CETP, only partially, at the C opening. In condition ii (Fig. 5), DOPC penetrated CETP with its lipid tails, while its head remained exposed to the water solvent, in this way plugging the tunnel at the N opening. TO penetration into CETP is also demonstrated: two of the three TO molecules penetrated CETP. The hydrophobic fatty acyl tails appear inside the tunnel, while the glycerol backbone of TO faces the N and C openings. In the last condition iii (Fig. 5), where two ChOE were prelocated in the tunnel of CETP, we obtained penetration of TO and DOPC, both by the same C opening. The combined results on conditions i and ii (Fig. 5) confirm that DOPC can penetrate either the N or the C opening, whereas ChOE enters by the neck opening. Thus, two distinct openings for ChOE and DOPC were identified in good agreement with reported results (15). In addition, our MD results show that the CETP tunnel can host ChOE and TO simultaneously, validating the hypothesis that CETP mediates neutral lipid transport via its tunnel compartment (3). MD simulation was successfully used to identify CETP openings and gave further information on the capacity of the tunnel to hold several lipids. A few test simulations demonstrated collapse of CETP if lipids did not penetrate the tunnel. MD showed that even partial lipid penetration into CETP prevents collapse of the tunnel. This collapse reconfirms the importance of the bound lipids for CETP stability and transfer activity (4). In view of the results, only CETP with its four bound lipids was further considered.
Fig. 5.
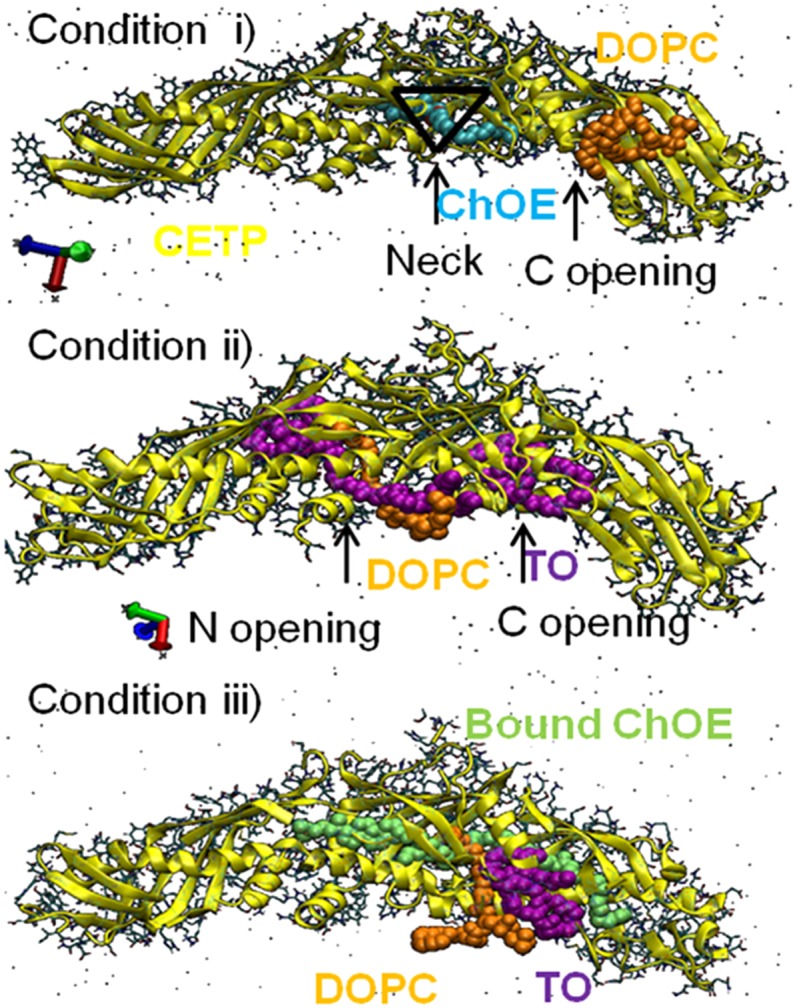
Identification of CETP openings. Condition i shows lipid-free CETP interacting with one ChOE and one DOPC. ChOE is shown accessing the CETP tunnel through the neck opening at center of the CETP structure (black triangle). Condition ii shows lipid-free CETP interacting with three TOs and one DOPC. The N and C openings accessed by TO and DOPC are indicated. Condition iii shows ChOE-bound CETP interacting with one TO and one DOPC.
Interaction of discoidal nascent HDL with CETP
Negative-stain EM experiments have demonstrated that CETP binds to the edge of nascent HDL and suggests proximity of apoA-I to CETP (34). We tested this hypothesis using our discoidal model of nascent HDL and we evaluated two configurations by MD, CETP interacting with the narrow top side of nascent HDL and CETP interacting with the longer edge of nascent HDL. Our MD simulation suggests that the longer edge position is preferred over the top position. CETP curvature proved to match the edge of nascent HDL perfectly, while barely attaching at the narrow top side (Fig. 6). This result fits well with the experimental data (34). In addition, independent of the considered starting position, CETP induced a change in the nascent HDL structure. In particular, we observed local geometrical changes from discoidal to spheroidal in the nascent HDL particle (Fig. 6). CETP engages in strong ionic interaction with POPC located at the surface of the nascent HDL, but it also interacts with apoA-I (Fig. 7). RMSD analysis showed that more than a third of the structural modifications in apoA-I are experienced by its C-terminal end (residues 220–243), a 20 amino acid polypeptide (Fig. 8). The mobility of apoA-I at the nascent HDL surface favors interaction with CETP and the participation of apoA-I in CETP-HDL interaction probably affects the structure of HDL. Consequently, we designed the interaction of our normal HDL-C-mimicking particle with upright CETP, taking into account the relative position of the apoA-I C-terminal end to CETP, as presented below.
Fig. 6.

Snapshots of CETP interacting with nascent HDL by its edge and top positions. The edge position is shown to be favored with perfect matching of CETP curvature with nascent HDL. A change in the shape of the nascent HDL is observed as pointed out by the double arrows. Color code: apoA-I C-terminal ends (purple); POPC phosphate groups (orange beads); proteins, CETP, and apoA-I are in their secondary structure representations (yellow); FChO in van der Waals representation (dark blue); and POPCs in bond representation (light blue).
Fig. 7.

Interaction of CETP with nascent HDL. A: Minimal distance CETP-POPC as a function of CETP residue for edge (left) and top (right) positions of CETP, respectively. Hydrophobic and electrostatic interactions are shown to be involved in the CETP-POPC interaction. B: Minimal distance apoA-I-CETP as a function of apoA-I residue for edge (left) and top (right) positions of CETP, respectively. apoA-I chains are proved to participate in the interaction of CETP with nascent HDL.
Fig. 8.

Structural changes in nascent HDL upon interaction with CETP. A: RMSD for the nascent HDL as a function of MD simulation time for edge (left) and top (right) positions of CETP. B: RMSD for apoA-I and its C-terminal end (residues 220–243) as a function of time for edge (left) and top (right) positions of CETP.
Interaction of a normal HDL-C particle with upright CETP
The interaction of spherical HDL with CETP was investigated with our newly designed normal HDL-C-mimicking particle. We considered an upright position for CETP, in which the N barrel domain end of CETP points toward HDL (5), and, as mentioned above, to take into account the possible role of apoA-I in the CETP-HDL interaction, we placed the C-terminal end region of apoA-I close to the N barrel domain end of CETP (Fig. 1). The results presented here refer to the main 860 ns MD run and show the major steps in the mechanism by which CETP and HDL interact.
Penetration of CETP into the HDL surface.
We plotted the radial distribution function [grdf (r)] for CETP and HDL lipids as a function of the simulation time (Fig. 9), with the center of mass of ChOE molecules taken as a reference. On the first 60 ns, the grdf (r) profiles suggest that CETP is on the HDL surface and did not penetrate deeper than the level of phosphate group (P) of POPC, therefore interacting with surface lipids. apoA-I chains, in particular their C-terminal ends, appear to have only a small contribution to CETP-HDL interaction in this time range (Fig. 10). The number of contacts and the persistence of the interaction between CETP and apoA-I increase after 60 ns, but the role of the apoA-I C-terminal end is not evidenced (Fig. 10). After 60 ns, a part of CETP slowly penetrates the HDL surface, and the grdf (r) indicates that CETP interacts with the POPC tails and adjacent ChOE molecules. Deeper penetration of CETP into HDL is observed after 230 ns, with the grdf (r) for CETP moving toward the grdf (r) for ChOE. After 290 ns, the grdf (r) for CETP remains fixed. CETP has reached its maximum penetration and interacts with the core ChOE molecules. As shown in Fig. 9, on the 640–860 ns slot, both helix X (residues 465–476) and helix A (residues 17–38) could be used as reference to delimit the parts of CETP inside and outside HDL. We measured the distance between helix A (residues 17–38), directly adjacent to the HDL surface, and the highest point at the free end of CETP and assessed a free end of 81 Å and a penetrating end of 38 Å, in agreement with (5). One comment is that the off-position of the grdf (r) for ChOE before 290 ns indicates a lack of compactness of the HDL core. This is corrected after 290 ns, HDL is fully relaxed and the core more compact.
Fig. 9.

The grdf (r) for CETP, phosphate group (P) of POPC, POPC, and ChOE. The grdf (r) for helix X and helix A of CETP are also presented. Helix A is closer to the HDL surface and is taken as reference to measure the penetrating end of CETP into HDL. The black arrows indicate the beginning of the grdf (r) profile for CETP.
Fig. 10.

The role of apoA-I in the interaction of upright CETP with a normal HDL-C-mimicking particle. Top plots: Minimum distance CETP-apoA-I as a function of apoA-I residue on several MD time length slots. apoA-I residues involved in the interaction with CETP are thus pointed out. Both apoA-I chains of HDL are represented separately. Bottom plots: Interaction of CETP with apoA-I as a function of time. Contacts are determined for distances lower than 0.6 nm, see dashed line in the top plots.
Tryptophan (Trp)105, Trp106, and Trp162 contained in flexible regions of CETP, flap 5 (Ω5 residues 90–110) and flap 6 (Ω6 residues 150–170) at the N barrel domain end of CETP, are shown to play an important anchoring role in the penetration and settling of CETP in HDL. Upon penetration, Ω5 and Ω6 are pulled apart giving rise to an opening (Fig. 11A). Examination of the distance between Trp106 and Trp162 (dTrp106-Trp162) allows an estimation of the size of the opening (Fig. 11B). The dTrp106-Trp162 is constant in the first ∼250 ns. This also corresponds to the time when the grdf (r) for CETP starts to be fixed and the penetration is maximum (Fig. 9). This suggests that Trps reach the ChOE core first, and it is only then that the formation of the opening can start. The dTrp106-Trp162 increases after 250 ns to reach a maximum at 550 ns. During that time the dTrp106-Trp162 doubles from 0.8 to 1.6 nm. Thereafter, the dTrp106-Trp162 decreases a little to reach a plateau at about 600 ns with a corresponding opening size of ∼1.1 nm. During the decreasing phase, CETP begins to stabilize on the ChOE core, and after 600 ns, a constant distance between the flaps is achieved and CETP obtains stability in HDL.
Fig. 11.

Atomistic view of CETP penetrating a normal HDL-C particle. A: CETP penetration as a function of simulation time. For clarity, only the ChOE core and apoA-I chains of HDL are shown. Trps (cyan) sit on the ChOE core of HDL. The dashed square is a magnification of the N barrel domain end of CETP. The white double arrows show the separation of Ω5 and Ω6 and evidence the formation of an opening. B: Size of the opening along 860 ns simulation. Snapshots of CETP at t = 150 ns and t = 540 ns are included. Color code: CETP and apoA-I (yellow ribbons); Ω4, residues 40–50 (light orange); Ω5, residues 90–110 (blue); Ω6, residues 150–170 (pink); ChOE core (red).
Lipid transfer mechanism mediated by CETP upon penetration into HDL.
CETP mediates the transfer of ChOE from HDL to apoB-100-containing lipoproteins, primarily to LDL (35). To understand how the transfer actually occurs in the CETP-HDL complex, we analyzed the dynamics of ChOE upon penetration of CETP inside the HDL particle. One of the 90 ChOE molecules constituting the HDL particle was selected and its dynamics followed as a function of MD simulation time (Fig. 12A). At t = 13 ns, the selected ChOE is located in the core of the HDL particle. At t = 350 ns, it has reached the opening formed between Trp106 and Trp162, and at t = 450 ns, it has penetrated the opening. At t = 540 ns, ChOE has moved further into the opening toward the tunnel of CETP. Figure 12A illustrates how the core ChOE molecules can be routed to the CETP opening. We have checked the repeatability of ChOE position change by monitoring ChOE dynamics from different starting configurations of CETP-HDL derived from the 200 ns pre-equilibration run. A closer view of the CETP-HDL complex (Fig. 12B) shows the dynamics of lipids in the opening. A group of lipids comprising TO, ChOE, and POPC have reached the opening, which seems to act as a storing space where lipids can be hosted.
Fig. 12.

Dynamics of a single molecule of ChOE as a function of MD simulation time. A: Lipid transfer from the core of HDL to the CETP opening. B: Trafficking of lipids in the CETP opening at t = 615 ns. C: Dynamics of ChOE and TO from 640 to 860 ns evidencing a barrier formed by Phe35, Phe93, and Phe147. Phosphate groups (P) of POPC (orange beads) indicate the shape of the HDL particle. Color code: CETP and apoA-I (yellow), apoA-I C-terminal (purple), Phe (ice-blue), Trp (cyan), ChOE (red), TO (purple), CETP-bound ChOE (lime), CETP-bound DOPC (gray), POPC (orange).
ChOE appears to compete with TO for access to the CETP tunnel. Throughout the MD simulation (Fig. 12C), ChOE is seen to be searching for a favorable path to the CETP tunnel, and the presence of TO appears to prevent its access. After 800 ns of MD simulation, however, a possible route for lipid transfer opens up. In fact, TO reveals a barrier between the opening and the tunnel of CETP, a barrier formed by an aromatic ring-rich region consisting mainly of phenylalanine (Phe) moieties, Phe35, Phe93, and Phe147. The role of TO in HDL may thus be strategic. At 860 ns (Fig. 12C), MD shows that, with its long hydrophobic chains, TO is able to open up the aromatic barrier to give ChOE access to the tunnel.
Role of E2 oleate in the CETP-HDL interaction.
To verify the role of E2 oleate associated with normal HDL-C in the interaction of upright CETP with HDL, we first monitored the minimal distance between E2 oleate and CETP to discern possible contacts. In particular, we looked at the ability of E2 oleate to interact with Ω5 at the N barrel domain end of CETP. Stability in the distance is reached after 100 ns MD simulation (Fig. 13), which is quite early on the MD scale relative to the opening formation at 250 ns (Fig. 11B). Hydrophobic interaction and H-bond formations are found to rule E2 oleate and CETP interaction. Close contacts are established between E2 oleate and amino acids of Ω5, in particular with the Trps at the beginning of the interaction (see inset Fig. 13). These contribute to the immobilization of CETP on HDL, allowing CETP to further penetrate HDL, and induce the formation of the opening at the N barrel domain end of CETP. By its quick ability to interact with CETP, E2 oleate and, therefore, E2 fatty acyl ester molecules help to stabilize CETP on/in HDL and have a role in anchoring CETP in HDL.
Fig. 13.

Interaction of E2 oleate with the N-terminal domain end of CETP. The minimal distance between E2 oleate and flap 5 (Ω5) is plotted on 600 ns MD simulation. The black arrow indicates when the distance is stable. In the inset, the distance of E2 oleate as a function of Ω5 residues is presented. The full line at 0.4 nm indicates the upper limit condition for favorable H-bond formation. Trp105 and Trp106 (gray squares) are found within the 0.4 nm upper limit.
DISCUSSION
Although structural consideration of both CETP and HDL would suggest simple key-lock interaction through a match-to-match curvature approach, this model does not agree with a penetration mechanism of CETP into HDL. An upright CETP-HDL approach was employed in the present study and MD simulation demonstrates the complexity of CETP-HDL interaction and confirms the partial penetration of CETP into HDL (5). Molecular and atomistic assessment of the interaction permitted by atomistic MD simulations proved that several regions of CETP and the structural composition of HDL are among the key metrics in the formation of the CETP-HDL complex, and in the functioning of lipid transfer. Though involving different mechanisms, the key-lock interaction model (4) and the penetration model with CETP in an upright position showed some common features. The first one is the central anchoring role of a Trp cluster from flap Ω5. In addition, the highest root-mean-square fluctuations within CETP have been found in the regions of flaps Ω4 and Ω5 (4), which are the regions in our model, together with Ω6, that prove to open up to create a path for lipid transfer at the N barrel domain end of CETP. Moreover, helix X was proposed earlier as a door for lipid transfer with opened and closed states (4). We show here instead, that the N barrel domain of CETP acts as a door for lipid transfer and we propose a penetration model where helix X is present and mainly in its closed state, which is most probably a necessary condition for proper lipid transfer functioning. This proposal agrees well with the earlier conclusion on the detrimental effect of the removal of helix X for cholesterol ester transfer activities (36).
We proposed a four step mechanism as key to the CETP-HDL interaction: 1) Immobilization of CETP on the HDL surface promoted by the interaction with POPC, apoA-I, and the presence of E2 fatty acyl esters. One remark here is that the recent EM studies of Zhang et al. (5) involved HDL containing possibly three apoA-I chains per particle. The number of apoA-I per HDL particle does not affect the ability of CETP to penetrate HDL, but we rather propose, according to our MD results (Fig. 10), that the number of apoA-I per HDL particle contributes to the interaction of CETP with HDL and could probably accelerate the process of penetration by immobilization of CETP on HDL. 2) Penetration of CETP into HDL with formation of an opening between the Trp-rich flap Ω5 and Ω6 at the N barrel domain end of CETP, Trps and E2 fatty acyl ester having the major anchoring role. 3) Transfer of ChOE from HDL to the N barrel domain opening of CETP. MD showed that ChOE can indeed migrate from the HDL core to the CETP opening. 4) Transfer of ChOE from the opening to the tunnel of CETP through an aromatic ring-rich region of Phe, acting as a barrier and penetrable with the assistance of TO. In addition, we suggest that CETP-bound DOPCs do not exchange, but instead play a central role in the circulation of CETP in the hydrophilic plasma. CETP-bound ChOEs may serve to stabilize the CETP tunnel. As shown in Fig. 5, the CETP tunnel can host several lipids simultaneously. As a consequence, CETP-bound ChOEs may easily be exchanged or remain in the tunnel when ChOE core molecules are passing through.
Several questions still remain as for the lipid transfer mechanism to LDL acceptor particles. If we hypothesize a ternary complex, HDL-CETP-LDL, involved in the lipid transfer/exchange between the lipoproteins and if the lipid exchange is a concerted action, then a lipid transfer from the N domain opening through the other end of the CETP tunnel and to LDL would be observed. Supplementary MD studies to verify that HDL core lipids can indeed circulate to the far end of CETP tunnel and therefore be ready to be transferred to LDL would help to clarify the local lipid transfer within CETP-HDL and exclude or lend support to the proposal of a concerted ternary complex lipid transfer process.
Although no CETP-mediated transfer of E2 fatty acyl ester was observed on the MD simulation scale, its potent anchoring role probably explains the enhanced atheroprotective role of HDL associated with E2 fatty acyl ester claimed earlier (9). It facilitates the CETP-HDL complex and the opening formations. If a ternary complex process, HDL-CETP-LDL, is involved in the transfer of E2 fatty acyl ester, then its role is multiple, as it should conserve its anchoring role and be transferred via CETP.
As mentioned in the Methods, the slow diffusion of lipids requires large-scale MD simulations; a ternary complex study would then be computationally demanding. However, modeling of HDL-CETP-LDL using a simplified model of the LDL particle would help to clarify the dynamics of the ternary complex and a full view of the lipid transfer/exchange process between lipoproteins might be obtained. Our work proposes a new direction for research that can further improve our understanding of the role of the CETP-HDL interaction in lipid transfer and reverse cholesterol transport processes.
Acknowledgments
The authors thank Ilpo Vattulainen and his group for the force field for TO. They thank the CSC IT Center for Science for allocating millions of CPU units and for the use of supercomputers.
Footnotes
Abbreviations:
- CETP
- cholesterol ester transfer protein
- ChOE
- cholesteryl oleate
- DOPC
- 1,2-dioleoyl-sn-glycero-3-phosphocholine
- DSH
- double superhelix
- dTrp106-Trp162
- distance between tryptophan 106 and tryptophan 162
- E2
- β-estradiol
- EM
- electron microscopy
- FChO
- free cholesterol
- grdf (r)
- radial distribution function
- HDL-C
- HDL-cholesterol
- LysoPC
- lysophosphatidylcholine
- MD
- molecular dynamics
- Phe
- phenylalanine
- RMSD
- root-mean-square deviation
- TO
- triglyceride
- Trp
- tryptophan
Financial support for this work was provided by the Research Council for Natural Sciences and Engineering and Academy of Finland Grants 1133184 (G.C-K. and M-L.R.) and 257545 (M.J.).
REFERENCES
- 1.Chapman M. J., Ginsberg H. N., Amarenco P., Andreotti F., Boren J., Catapano A. L., Descamps O. S., Fisher E., Kovanen P. T., Kuivenhoven J. A., et al. 2011. Triglyceride-rich lipoproteins and high-density lipoprotein cholesterol in patients at high risk of cardiovascular disease: evidence and guidance for management. Eur. Heart J. 32: 1345–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duffy D., Rader D. J. 2009. Update on strategies to increase HDL quantity and function. Nat. Rev. Cardiol. 6: 455–463. [DOI] [PubMed] [Google Scholar]
- 3.Hayek T., Azrolan N., Verdery R. B., Walsh A., Chajekshaul T., Agellon L. B., Tall A. R., Breslow J. L. 1993. Hypertriglyceridemia and cholesteryl ester transfer protein interact to dramatically alter high density lipoprotein levels, particle sizes, and metabolism - studies in transgenic mice. J. Clin. Invest. 92: 1143–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koivuniemi A., Vuorela T., Kovanen P. T., Vattulainen I., Hyvonen M. T. 2012. Lipid exchange mechanism of the cholesteryl ester transfer protein clarified by atomistic and coarse-grained simulations. PLOS Comput. Biol. 8: e1002299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang L., Yan F., Zhang S., Lei D., Charles M. A., Cavigiolio G., Oda M., Krauss R. M., Weisgraber K. H., Rye K. A., et al. 2012. Structural basis of transfer between lipoproteins by cholesteryl ester transfer protein. Nat. Chem. Biol. 8: 342–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tikkanen M. J., Vihma V., Jauhiainen M., Hockerstedt A., Helisten H., Kaamanen M. 2002. Lipoprotein-associated estrogens. Cardiovasc. Res. 56: 184–188. [DOI] [PubMed] [Google Scholar]
- 7.Vihma V., Tiitinen A., Ylikorkala O., Tikkanen M. J. 2003. Quantitative determination of estradiol fatty acid esters in lipoprotein fractions in human blood. J. Clin. Endocrinol. Metab. 88: 2552–2555. [DOI] [PubMed] [Google Scholar]
- 8.Höckerstedt A., Jauhiainen M., Tikkanen M. J. 2006. Estradiol fatty acid esterification is increased in high density lipoprotein subclass 3 isolated from hypertriglyceridemic subjects. Atherosclerosis. 185: 264–270. [DOI] [PubMed] [Google Scholar]
- 9.Badeau R. M., Metso J., Wahala K., Tikkanen M. J., Jauhiainen M. 2009. Human macrophage cholesterol efflux potential is enhanced by HDL-associated 17 beta-estradiol fatty acyl esters. J. Steroid Biochem. Mol. Biol. 116: 44–49. [DOI] [PubMed] [Google Scholar]
- 10.Helisten H., Hockerstedt A., Wahala K., Tiitinen A., Adlercreutz H., Jauhiainen M., Tikkanen M. J. 2001. Accumulation of high-density lipoprotein-derived estradiol-17 beta fatty acid esters in low-density lipoprotein particles. J. Clin. Endocrinol. Metab. 86: 1294–1300. [DOI] [PubMed] [Google Scholar]
- 11.Wu Z., Wagner M. A., Zheng L., Parks J. S., Shy J. M., 3rd, Smith J. D., Gogonea V., Hazen S. L. 2007. The refined structure of nascent HDL reveals a key functional domain for particle maturation and dysfunction. Nat. Struct. Mol. Biol. 14: 861–868. [DOI] [PubMed] [Google Scholar]
- 12.Wu Z., Gogonea V., Lee X., Wagner M. A., Li X. M., Huang Y., Undurti A., May R. P., Haertlein M., Moulin M., et al. 2009. Double superhelix model of high density lipoprotein. J. Biol. Chem. 284: 36605–36619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gogonea V., Wu Z. P., Lee X., Pipich V., Li X. M., Ioffe A. I., DiDonato J. A., Hazen S. L. 2010. Congruency between biophysical data from multiple platforms and molecular dynamics simulation of the double-super helix model of nascent high density lipoprotein. Biochemistry. 49: 7323–7343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yetukuri L., Soderlund S., Koivuniemi A., Seppanen-Laakso T., Niemela P. S., Hyvonen M., Taskinen M. R., Vattulainen I., Jauhiainen M., Oresic M. 2010. Composition and lipid spatial distribution of HDL particles in subjects with low and high HDL-cholesterol. J. Lipid Res. 51: 2341–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qiu X., Mistry A., Ammirati M. J., Chrunyk B. A., Clark R. W., Cong Y., Culp J. S., Danley D. E., Freeman T. B., Geoghegan K. F., et al. 2007. Crystal structure of cholesteryl ester transfer protein reveals a long tunnel and four bound lipid molecules. Nat. Struct. Mol. Biol. 14: 106–113. [DOI] [PubMed] [Google Scholar]
- 16.Pronk S., Pall S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M. R., Smith J. C., Kasson P. M., van der Spoel D., et al. 2013. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 29: 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nose S., Klein M. L. 1983. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 50: 1055–1076. [Google Scholar]
- 18.Hoover W. G. 1985. Canonical dynamics - equilibrium phase-space distributions. Phys. Rev. A. 31: 1695–1697. [DOI] [PubMed] [Google Scholar]
- 19.Shinoda W., Fukada T., Okazaki S., Okada I. 1995. Molecular dynamics simulation of the dipalmitoylphosphatidylcholine (DPPC) lipid bilayer in the fluid phase using the Nose-Parrinello-Rahman NPT ensemble. Chem. Phys. Lett. 232: 308–312. [Google Scholar]
- 20.Berendsen H. J. C., Postma J. P. M., van Gunsteren W. F., Hermans J. 1981. Interaction models for water in relation to protein hydration. In Intermolecular Forces. B. Pullman, editor. Reidel, Dordrecht, The Netherlands. 331–342. [Google Scholar]
- 21.Miyamoto S., Kollman P. A. 1992. Settle - an analytical version of the shake and rattle algorithm for rigid water models. J. Comput. Chem. 13: 952–962. [Google Scholar]
- 22.Hess B., Bekker H., Berendsen H. J. C., Fraaije J. G. E. M. 1997. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 18: 1463–1472. [Google Scholar]
- 23.Berger O., Edholm O., Jahnig F. 1997. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J. 72: 2002–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Höltje M., Förster T., Brandt B., Engels T., von Rybinski W., Höltje H. D. 2001. Molecular dynamics simulations of stratum corneum lipid models: fatty acids and cholesterol. Biochim. Biophys. Acta. 1511: 156–167. [DOI] [PubMed] [Google Scholar]
- 25.Tieleman D. P., Berendsen H. J. C. 1998. A molecular dynamics study of the pores formed by Escherichia coli OmpF porin in a fully hydrated palmitoyloleoylphosphatidylcholine bilayer. Biophys. J. 74: 2786–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koivuniemi A., Heikela M., Kovanen P. T., Vattulainen I., Hyvonen M. T. 2009. Atomistic simulations of phosphatidylcholines and cholesteryl esters in high density lipoprotein-sized lipid droplet and trilayer: clues to cholesteryl ester transport and storage. Biophys. J. 96: 4099–4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schüttelkopf A. W., van Aalten D. M. F. 2004. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 60: 1355–1363. [DOI] [PubMed] [Google Scholar]
- 28.Parrish D., Zhurova E. A., Kirschbaum K., Pinkerton A. A. 2006. Experimental charge density study of estrogens: 17beta-estradiol.urea. J. Phys. Chem. B. 110: 26442–26447. [DOI] [PubMed] [Google Scholar]
- 29.Oren I., Fleishman S. J., Kessel A., Ben-Tal N. 2004. Free diffusion of steroid hormones across biomembranes: A simplex search with implicit solvent model calculations. Biophys. J. 87: 768–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jones M. K., Zhang L., Catte A., Li L., Oda M. N., Ren G., Segrest J. P. 2010. Assessment of the validity of the double superhelix model for reconstituted high density lipoproteins: a combined computational-experimental approach. J. Biol. Chem. 285: 41161–41171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gu F., Jones M. K., Chen J., Patterson J. C., Catte A., Jerome W. G., Li L., Segrest J. P. 2010. Structures of discoidal high density lipoproteins: a combined computational-experimental approach. J. Biol. Chem. 285: 4652–4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zannis V. I., Chroni A., Krieger M. 2006. Role of apoA-I, ABCA1, LCAT, and SR-BI in the biogenesis of HDL. J. Mol. Med. 84: 276–294. [DOI] [PubMed] [Google Scholar]
- 33.Kabsch W., Sander C. 1983. Dictionary of protein secondary structure - pattern-recognition of hydrogen-bonded and geometrical features. Biopolymers. 22: 2577–2637. [DOI] [PubMed] [Google Scholar]
- 34.Bruce C., Davidson W. S., Kussie P., Lundkatz S., Phillips M. C., Ghosh R., Tall A. R. 1995. Molecular determinants of plasma cholesteryl ester transfer protein binding to high density lipoproteins. J. Biol. Chem. 270: 11532–11542. [DOI] [PubMed] [Google Scholar]
- 35.Tall A. R. 1993. Plasma cholesteryl ester transfer protein. J. Lipid Res. 34: 1255–1274. [PubMed] [Google Scholar]
- 36.Wang S., Deng L., Milne R. W., Tall A. R. 1992. Identification of a sequence within the C-terminal 26 amino acids of cholesteryl ester transfer protein responsible for binding a neutralizing monoclonal antibody and necessary for neutral lipid transfer activity. J. Biol. Chem. 267: 17487–17490. [PubMed] [Google Scholar]