

Abstract
Two series of novel ether analogs of the sigma (σ) receptor ligand 1-[2-(3,4-dimethoxyphenyl)ethyl]-4-(3-phenylpropyl)piperazine (SA4503) have been prepared. In one series, the alkyl portion of the 4-methoxy group was replaced with allyl, propyl, bromoethyl, benzyl, phenethyl, and phenylpropyl moieties. In the second series, the 3,4-dimethoxy was replaced with cyclic methylenedioxy, ethylenedioxy and propylenedioxy groups. These ligands, along with 4-O-des-methyl SA4503, were evaluated for σ1 and σ2 receptor affinity, and compared to SA4503 and several known ether analogs. SA4503 and a subset of ether analogs were also evaluated for dopamine transporter (DAT) and serotonin transporter (SERT) affinity. The highest σ1 receptor affinities, Ki values of 1.75 nM – 4.63 nM, were observed for 4-O-des-methyl SA4503, SA4503 and the methylenedioxy analog. As steric bulk increased, σ1 receptor affinity decreased, but only to a point. Allyl, propyl and bromoethyl substitutions gave σ1 receptor Ki values in the 20 nM – 30 nM range, while bulkier analogs having phenylalkyl, and Z- and E-iodoallyl, ether substitutions showed higher σ1 affinities, with Ki values in the 13 nM – 21 nM range. Most ligands studied exhibited comparable σ1 and σ2 affinities, resulting in little to no subtype selectivity. SA4503, the fluoroethyl analog and the methylenedioxy congener showed modest six- to fourteen-fold selectivity for σ1 sites. DAT and SERT interactions proved much more sensitive than σ receptor interactions to these structural modifications. For example, the benzyl congener (σ1 Ki = 20.8 nM; σ2 Ki = 16.4 nM) showed over 100-fold higher DAT affinity (Ki = 121 nM) and 6-fold higher SERT affinity (Ki = 128 nM) than the parent SA4503 (DAT Ki = 12650 nM; SERT Ki = 760 nM). Thus, ether modifications to the SA4503 scaffold can provide polyfunctional ligands having a broader spectrum of possible pharmacological actions.
1. Introduction
Sigma (σ) receptors can be classified into distinct σ1 and σ2 subtypes that play prominent roles in certain cancers and in central nervous system (CNS) disorders. Malignancies often express high levels of σ1 and σ2 receptors with respect to the normal tissue, the σ2 receptors serve as markers of cell proliferation, and σ1 antagonists / σ2 agonists have potential for cancer therapy.1–3 In the CNS realm, σ1 receptor agonists improve cognition, show antidepressant and anxiolytic properties, and promote neuroregeneration.4–6 Moreover, selective σ1, and perhaps σ2, receptor antagonists show potential as medications for treatment of psychostimulant abuse.7–10 Compounds from many different chemical classes show varying degrees of affinity and selectivity for σ receptors, and structurally diverse ligands including progesterone, N,N′-dimethyltryptamine and sphingolipids have been suggested as endogenous ligands.11,12
The σ1 receptor has been cloned,13,14 while the primary structure of the σ2 receptor remains elusive.3 Protein biochemistry studies and homology modeling efforts are now revealing specific features of the σ1 receptor binding site at the amino acid level.15 Less is known about σ2 receptors, but recent studies show the orthosteric binding site to be located within the progesterone receptor membrane component 1 (PGRMC1) protein complex.3,16 Nonetheless, medicinal chemistry studies show a degree of commonality, with both σ1 and σ2 receptor pharmacophores simply described by an amine binding site flanked by two hydrophobic binding pockets.17,18 These basic models are fit well by N,N′-disubstituted piperazines having phenethyl and phenylpropyl groups to occupy the hydrophobic regions.
Two active series from this structural class of σ receptor ligands have attracted attention as potential pharmaceuticals. These are denoted as either “SA” compounds19,20 or “YZ” compounds (Fig. 1).21,22 They differ in the patterns and types of substituents on the phenethyl and phenylpropyl moieties, and typically display relatively high σ1 receptor binding affinity accompanied by modest selectivity against σ2 sites. Subtle structural differences influence their σ1 and σ2 receptor binding, as well as the behavioral pharmacology observed in animal models.22–27 SA4503 (Fig. 1), the most widely studied ligand from this class, is also known as cutamesine and is regarded as a σ1 receptor agonist.4–6,19 SA4503 causes dissociation of immunoglobulin binding protein from the σ1 receptor, a definitive biochemical test of stimulation.28 SA4503 has been evaluated in Phase II clinical trials for treatment of major depressive disorder29 and to aid in functional recovery after stroke.30,31 In the stroke trial,30 SA4503 proved well tolerated, and the data indicated improvements in individuals who received a 3 mg/d dosage for 28 days. Interestingly, SA4503 shows mixed properties in recent behavioral studies, and can either potentiate or attenuate certain effects of cocaine and methamphetamine in mice as a function of dose.23,24 By contrast, the YZ analogs profile as σ1 receptor antagonists based upon their ability to block cocaine-induced convulsions in mice.22
Figure 1.
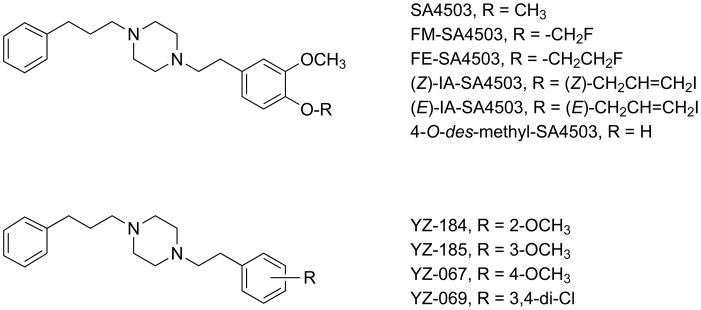
Representative “SA” and “YZ” sigma receptor ligands.
Although a number of different SA / YZ compounds have been studied, relatively little information is available regarding the effects of ether modifications on σ receptor binding. Findings come primarily from the development of radiolabeled SA4503 analogs for molecular imaging applications.32,33 The 4-methoxy position of SA4503 has been isotopically radiolabeled with carbon-11 for positron emission tomography (PET) studies of central and peripheral σ1 receptors in human beings and animal models of disease.34,35 The fluorine-18 labeled 4-fluoroethoxy36 (FE-SA4503, Fig. 1) and fluoromethoxy37 (FM-SA4503, Fig. 1) analogs have also been prepared, and used for PET studies of cerebral σ1 receptors in conscious monkeys.36,37 In our hands, SA4503 displays a 4.6 nM apparent affinity (Ki) for σ1 receptors with 14-fold selectivity for σ1 over σ2 sites.38 In side-by-side binding comparisons, the fluoroethyl modification caused a two-fold reduction in affinity for both σ1 and σ2 receptors while preserving the σ1 subtype selectivity.38 As previously discussed in detail,38 these σ receptor subtype selectivities are quite different from those initially reported for SA450319 and FE-SA4503,36 primarily as a consequence of the σ2 receptor binding results. In work aimed toward radioiodinated SA4503 analogs, the 4-methoxy group was replaced by (Z)- and (E)-iodoallyloxy substituents (IA-SA4503, Fig. 1).39 These modifications reduced σ1 affinity and increased σ2 affinity, leading to a pair of non-selective isomeric ligands having Ki values of 11 – 20 nM for both subtypes.
To gain a better understanding of the qualitative structure - affinity relationships associated with ether modifications of SA4503, we prepared nine novel analogs (Fig. 2) for a more systematic investigation. Two types of structural modifications were investigated. The first involved replacements of the 4-methoxy group with ethers of increasing steric bulk and hydrophobicity, as well as differing potential for π and other interactions (1 – 6). The second involved replacement of the 3,4-dimethoxy moiety with cyclic ethers of different ring sizes (7 – 9). Each compound was tested in vitro for affinity and selectivity of binding to σ1 and σ2 receptors using standard techniques. The σ receptor binding affinities of the known 4-O-des-methyl-SA450336b (Fig. 1) have not been reported, and this material was tested as well.
Figure 2.
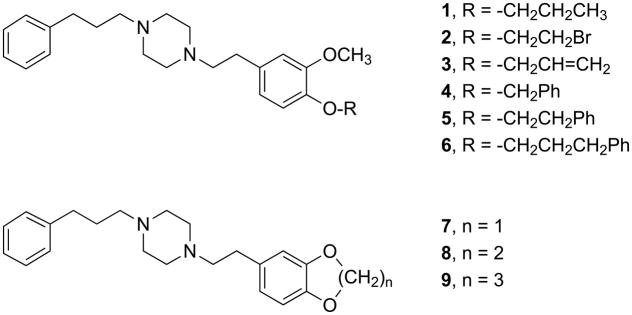
Novel SA4503 ether analogs 1–9.
In previous work, SA4503 showed low affinity for about 40 different receptors, ion channels and second messenger systems,19 although a significant secondary interaction with the vesicular acetylcholine transporter, Ki = 50 nM, has been observed.40 Interactions of SA4503 with dopamine transporters (DAT) and serotonin transporters (SERT) have not been reported to our knowledge. Several of the novel SA4503 ether analogs were designed to bear a structural resemblance to ligands such as GBR 12909 (vanoxerine) and certain rimcazole analogs that show high DAT and SERT affinities accompanied by weaker interactions with the σ receptor subtypes.41,42 Considering the importance of σ receptors and the DAT in mediating the effects of cocaine, compounds having actions at both sites are of interest from the viewpoint of psychostimulant medications development.41–43 As exemplified by fluvoxamine,4 ligands with actions at both σ receptors and the SERT are of interest as medications for treatment of depression and anxiety.4–6 Accordingly, we determined the affinities of SA4503 and a subset of the ether analogs for DAT and SERT with the aim of identifying structural modifications that would provide polyfunctional ligands having a broader spectrum of pharmacological actions.
2. Results and discussion
2.1. Chemistry
Target compounds 1 – 9 were prepared by the routes shown in Scheme 1. Direct reaction of the phenolate anion generated in situ from 4-O-des-methyl SA450336b with primary alkyl, allyl or benzyl bromides provided piperazines 1 – 6 (Scheme 1). Isolated yields from the Williamson ether synthesis ranged from 44 – 90% for 1, 2, 5 and 6 using phase transfer conditions, 40% KOH / tetra-n-butylammonium hydroxide (TBAH), at 50 °C. This set of reaction conditions emerged from studies on the synthesis of compound 1 by varying the type of base (NaOH, KOH) and the number of molar equivalents of base and alkylating agent. For the more reactive electrophile, allyl bromide, the reaction was performed from 0 °C to ambient temperature to give 3 in 57% yield. The phase transfer method was not amenable to use with benzyl bromide, and a number of products were formed that were not characterized. However, treatment of 4-O-des-methyl SA4503 with benzyl bromide and K2CO3 in absolute EtOH at ambient temperature overnight provided 4 in 35% yield. These reactions were not optimized further.
Scheme 1.

Synthesis of SA4503 ether analogs 1 – 6. Reagents and reaction conditions: (a) 1, 2, 5, 6: corresponding alkyl bromide, KOH / TBAH, 50 °C, 50 min; 3: allyl bromide, KOH / TBAH, 0 °C to ambient temperature, 50 min; 4: benzyl bromide, EtOH / K2CO3, 25 °C, N2 overnight.
Cyclic ethers 7 – 9 were synthesized by coupling substituted phenylacetic acids to 1-(3-phenylpropyl)piperazine to yield amide intermediates that were subsequently reduced with LiAlH4 (Scheme 2). The 3,4-(methylenedioxy)phenylacetic acid 10 required for preparation of 7 was commercially available. The known 3,4-(ethylenedioxy)phenylacetic acid 1144 and 3,4-(propylenedioxy)phenylacetic acid 1245 required for preparation of 8 and 9, respectively, were synthesized in 52% and 42% yields by treatment of 3,4-dihydroxyphenylacetic acid with K2CO3 and the appropriate alkyl dibromide in ethylene glycol at 120 °C for 4.5 h. Kashima and colleagues46 reported these conditions for the synthesis of cyclic ethers from catechols. This onestep route is a convenient alternative to the multi-step method of Sasamoto,44 and uses milder conditions than the method of Alker et al.45 that calls for KOH at reflux for 17 h. Coupling reactions were then performed using 1-hydroxybenzotriazole monohydrate (HOBt·H2O), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC·HCl) and 4-methylmorpholine in CH2Cl2 at 0 °C under N2 to give 13 – 15. These amides were isolated, characterized and then reduced with LiAlH4 in THF to give 7 – 9. Coupling reactions were accomplished in > 90% yields, while the reduction steps provided 33 – 85% isolated yields of the final targets.
Scheme 2.

Synthesis of SA4503 ether analogs 7 – 9. Reagents and reaction conditions: (a) BrCH2CH2Br, K2CO3 / HOCH2CH2OH, 120 °C, 4.5 h; (b) Br(CH2)3Br, K2CO3 / HOCH2CH2OH, 120 °C, 4.5 h; (c) 1-(3-phenylpropyl)piperazine, HOBt•H2O / EDC•HCl / 4-methylmorpholine, 0 °C, N2 overnight; (d) LiAlH4 / THF, 0 °C - ambient temperature, 7.5 h.
2.2. Sigma receptor binding
Table 1 shows binding affinities of 4-O-des-methyl SA4503 and novel ether analogs 1 – 9 for σ1 and σ2 receptors in guinea pig brain membranes. Published data, obtained using the same assay conditions, for SA4503 and FE-SA4503,38 as well as (Z)- and (E)-IA-SA4503,39 are included for comparison. Literature binding data for several YZ analogs and other reference ligands are also included. In addition, Table 1 shows a spot test of interactions with the DAT using a single high concentration (10000 nM) of SA4503 and analogs. This test was performed to prioritize the selection of compounds for more detailed studies (see Section 2.3).
Table 1.
σ Receptor affinity of SA4503 and novel ether analogsa,b, along with literature data for selected YZ and reference compounds.
| R | Sigma receptor binding | DATc | |||
|---|---|---|---|---|---|
| σ1 Ki (nM) | σ2 Ki (nM) | σ2 / σ1 | % Inh. | ||
| —H (des-methyl SA4503) | 1.75 ± 0.17 | 6.72 ± 0.21 | 3.8 | 20 | |
| —CH3 (SA4503)d | 4.63 ± 0.21 | 63.09 ± 4.33 | 13.6 | 23 | |
| —CH2CH2F (FE-SA4503)d | 8.03 ± 0.41 | 113.2 ± 11.7 | 14.1 | 19 | |
| —CH2F (FM-SA4503) e | 6.4 ± 1.3 | 250 ± 16 | 39.0 | -- | |
| (E)—CH2CH=CHI f | 16.91 ± 1.28 | 18.20 ± 1.99 | 1.1 | 69 | |
| (Z)—CH2CH= CHI f | 10.67 ± 1.32 | 16.71 ± 1.20 | 1.6 | 94 | |
| 1 | —CH2CH2CH3 | 25.60 ± 2.56 | 33.40 ± 1.11 | 1.3 | 43 |
| 2 | —CH2CH2Br | 25.37 ± 2.02 | 27.12 ± 0.96 | 1.1 | 38 |
| 3 | —CH2CH=CH2 | 30.13 ± 1.24 | 42.10 ± 0.66 | 1.4 | 44 |
| 4 | —CH2Ph | 20.79 ± 1.10 | 16.43 ± 1.13 | 0.8 | 93 |
| 5 | —CH2CH2Ph | 13.26 ± 0.82 | 16.57 ± 0.64 | 1.2 | 89 |
| 6 | —CH2CH2CH2Ph | 18.45 ± 0.48 | 23.31 ± 3.34 | 1.3 | 97 |
| 7 | —Methylenedioxy— | 2.55 ± 0.19 | 14.83 ± 0.43 | 5.8 | 42 |
| 8 | —Ethylenedioxy— | 10.20 ± 0.52 | 35.20 ± 1.69 | 3.4 | 33 |
| 9 | —Propylenedioxy— | 19.10 ± 3.11 | 52.10 ± 4.91 | 2.7 | 43 |
| YZ-184 (2-OCH3) g | 3.9 ± 0.6 | 7.5 ± 0.2 | 1.9 | -- | |
| YZ-185 (3-OCH3) g | 1.4 ± 0.2 | 10.2 ± 0.5 | 7.3 | -- | |
| YZ-067 (4-OCH3) g | 1.3 ± 0.3 | 28.6 ± 1.9 | 22 | -- | |
| YZ-069 (3,4-di-Cl) g | 2.2 ± 0.2 | 38.8 ± 1.6 | 24 | -- | |
| (+)-Pentazocine d | 1.62 ± 0.15 | 728.4 ± 48.8 | 450 | -- | |
| Ditolylguanidine (DTG) d | 35.45 ± 1.20 | 39.87 ± 2.61 | 1.1 | -- | |
Ki values are means ± SEM for 3 – 6 trials, each performed in duplicate.
1.0 nM [3H](+)-pentazocine (σ1), 3.0 nM [3H]ditolylguanidine / 200 nM (+)-pentazocine (σ2); guinea pig brain membranes.
% Inhibition for a 10000 nM concentration against [125I]RTI-121 specific binding in mouse striatal membranes. Binding data reported in
Ref. 38,
Ref. 37,
Ref. 39 and
Ref. 22.
Of the fourteen compounds studied, 4-O-des-methyl SA4503 showed the highest binding affinity for both σ1 and σ2 receptors (Table 1). The σ2 receptor binding is sensitive to this modification, with a ten-fold increase in affinity for 4-O-des-methyl SA4503 compared to SA4503. These findings are consistent with our recent report of a structurally similar phenol that showed much higher σ2 receptor affinity than the corresponding ligand bearing a methoxy group.47 4-O-des-methyl SA4503 is the only ligand tested that can be a hydrogen bond donor. By contrast, the hydrogen bond acceptors FE-SA4503 and FM-SA4503 showed the poorest σ2 receptor affinities of the series (Table 1). YZ-184 and YZ-185 have only hydrogen at the 4-position, but still display high σ2 receptor affinity, while YZ-067 and YZ-069 that have a chlorine or methoxy at the 4-position show a somewhat lower σ2 receptor affinity (Table 1). The data indicate that either having a hydroxyl group or no substituent at the 4-position promotes σ2 binding. Rhoades et al.48 have developed a three-dimensional model for the σ2 receptor pharmacophore, based upon ligands from seven different structural classes, that includes at least four hydrogen bond acceptor groups located at different positions of the binding pocket. Thus, the hydrogen bond donor properties of the phenol may contribute to high affinity σ2 binding.
For the SA4503 analogs described thus far, a smaller substituent volume leads to higher affinity binding to both σ1 and σ2 receptors that is accompanied by modest selectivities for the σ1 subtype. In this regard, oxygen substituents at both the 3- and 4-positions of the phenethyl ring are not essential for high affinity σ receptor binding. For example, monomethoxy analog YZ-185 binds to σ1 and σ2 receptors with Ki values of 1.4 nM and 10.2 nM, respectively (Table 1).21,22 The bulkier and more hydrophobic propyl, bromoethyl and allyl ether substitutions of analogs 1 – 3 proved detrimental to σ1 receptor binding, but slightly advantageous for σ2 receptor binding as compared to SA4503 (Table 1). These three compounds showed the poorest σ1 receptor affinities of the compounds tested, and displayed Ki values of 25 nM – 30 nM (Table 1). Their σ2 receptor affinities of 27 nM – 42 nM were also nearly equivalent, and engendered no appreciable subtype selectivity. The similarity of binding data across analogs 1 – 3 suggests that neither halogen bonding49 of bromine, nor π effects of the ethylene system, contribute significantly to the interactions. Further, these data indicate that the high electronegativity of fluorine, rather than a steric constraint, is responsible for the low σ2 receptor affinity of FE-SA4503 and FM-SA4503. Fluorine participation in hydrogen bonding acceptor or other dipole-dipole interactions50 that inhibit σ2 receptor binding fits with the notion that hydrogen bonding donor properties help promote the high affinity σ2 receptor binding of 4-O-des-methyl SA4503.
The homologous series of phenylalkyl ethers 4 – 6 had no appreciable subtype selectivity, and displayed a tight range of Ki values, 13 nM – 23 nM, for binding to both σ1 and σ2 sites (Table 1). As observed for analogs 1 – 3, incorporation of these bulkier and more hydrophobic substituents reduced σ1 but increased σ2 receptor affinity with respect to SA4503. The presence of an aromatic ring modestly increased the affinities of 4 – 6 for both σ1 and σ2 receptors as compared to alkyl and allyl analogs 1 – 3 (Table 1). Phenethyl ether analog 5 gave the highest overall σ1 and σ2 affinity of this series, but the chain length variations had small effects.
The previously reported (Z)- and (E)-iodoallyl ether analogs show σ1 and σ2 receptor affinities comparable to those of novel phenylalkyl ether analogs 4 – 6 (Table 1). Notably, the presence of iodine confers 2- to 3-fold higher σ1 and σ2 binding affinities for the iodoallyl analogs over the simple allyl analog 3. The similarity of σ receptor affinities for the (Z)- and (E)- configurations indicates little sensitivity to these particular geometric constraints. Thus, the extended ether analogs having bulky, hydrophobic and polarizable aromatic ring or iodine substituents are readily accommodated by the σ receptor binding pockets. While discrete π–π stacking interactions for the phenylalkyl ether moieties of 4 – 6 may contribute to binding, the overall data suggest that substituent bulk and hydrophobicity are main factors. These properties confer higher σ1 and σ2 receptor binding affinities for 4 – 6, as well as (Z)- and (E)- IA-SA4503, than observed for analogs 1 – 3 that have smaller molecular volumes.
Cyclic ether 7, with its compact methylenedioxy group, showed 2- to 4-fold increases in both σ1 and σ2 receptor affinity with respect to SA4503 (Table 1). Compared to each other, cyclic ethers 7 – 9 showed progressive 7.5- to 3.5-fold decreases in both σ1 and σ2 receptor affinities when moving from methylenedioxy (7) to the bulkier ethylenedioxy (8) and propylenedioxy (9) congeners. Thus, these three constrained ether analogs of SA4503 follow the pattern discussed earlier where a smaller substituent volume leads to relatively higher σ1 and σ2 receptor affinities accompanied by modest σ1 selectivity.
2.3. DAT and SERT binding
SA4503 and analogs varied widely in their potential to interact with the DAT (Table 1). Using a 10,000 nM concentration, their ability to inhibit [125I]-RTI-121 binding ranged from 19% to 97%. SA4503, FE-SA4503 and 4-O-des-methyl SA4503 exhibited the lowest inhibition (19% – 23%), while the alkyl / allyl ethers 1 – 3 and the cyclic ethers 7 – 9 showed a moderate level of inhibition (38% – 44%). Phenylalkyl ethers 4 – 6 and the (Z)- and (E)-iodoallyl ethers analogs gave the highest degree of inhibition (69% – 97%). Thus, the presence of large, hydrophobic ether substituents at the 4-O-position of the SA4503 phenethyl leads to stronger DAT binding.
Based upon these results, SA4503, as well as selected ether analogs displaying moderate (1, 3, 7) or high (4, 6, (Z)- and (E)-IA-SA4503) potential for DAT binding, were studied in more detail. Over a 100-fold range of DAT affinities was observed, while SERT affinities varied by only 7-fold (Table 2). SA4503 displayed the weakest interactions with both the DAT and SERT, with a decided 16-fold higher affinity for SERT over DAT. The DAT Ki of 12650 nM proved about 10-fold lower than the DAT Ki values previously reported for monomethoxy analogs YZ-185 and YZ-067 (Table 2).22 By contrast, SERT affinities for SA4503 (Ki = 760 nM) and YZ-185 (Ki = 985 nM)22 were quite similar. Somewhat surprisingly, the DAT affinity for SA4503 proved close to that previously reported by Matsumoto et al.22 for dichloro analog YZ-069 (Table 2). The DAT and SERT binding data for SA4503 were replicated, with no significant differences (n = 4; t-test, P > 0.05), using fresh serial dilutions prepared from powder.
Table 2.
DAT and SERT affinity of SA4503 and novel ether analogsa,b,c, along with literature data for selected YZ and reference compounds.
| R | DAT Ki (nM) b | DAT / σ1f | SERT Ki (nM) c | SERT / σ1f | |
|---|---|---|---|---|---|
| —CH3 (SA4503) | 12650 ± 832 | 2732 | 760 ± 31 | 164 | |
| (E) —CH2CH=CHI | 560 ± 57 | 33 | 174 ± 11 | 10.3 | |
| (Z) —CH2CH=CHI | 237 ± 13 | 22 | ND | ND | |
| 1 | —CH2CH2CH3 | 1821 ± 174 | 71 | 124 ± 13 | 4.8 |
| 3 | —CH2CH=CH2 | 2083 ± 156 | 69 | 295 ± 19 | 9.8 |
| 4 | —CH2Ph | 121 ± 16 | 6 | 128 ± 6 | 6.2 |
| 6 | —CH2CH2CH2Ph | 87 ± 5 | 5 | ND | ND |
| 7 | —Methylenedioxy— | 1840 ± 311 | 721 | 468 ± 41 | 184 |
| YZ-185 (3-OCH3) d | 1408 ± 164 | 1006 | 985 ± 126 | 703 | |
| YZ-067 (4-OCH3) d | 1466 ± 19 | 1128 | 1726 ± 57 | 1328 | |
| YZ-069 (3,4-di-Cl) d | 8196 ± 1005 | 3725 | 2865 ± 484 | 1302 | |
| GBR 12909 e | 12.0 ± 1.9 | 0.04 f | 24.1 ± 2.3 | 0.78 f |
Ki values are means ± SEM for n = 3 – 6 trials, each performed in duplicate, except for DAT assays of compounds 6 and (Z) —CH2CH=CHI, where n = 2. ND = not determined.
15 pM [125I]RTI, mouse striatal membranes.
0.3 nM [3H]paroxetine, mouse whole brain membranes. Binding data reported in
Ref. 22 and
Ref. 41.
All novel ether analogs tested proved more potent than SA4503 for binding to both DAT and SERT (Table 2). Ligands 1, 3 and 7 displayed nearly equal affinities, approximately 2000 nM, for DAT sites, while SERT binding showed more sensitivity to structure. Their SERT Ki values of 124 nM to 468 nM varied by 4-fold. Benzyl analog 4 and the (E)-iodoallyl congener displayed SERT Ki values of 128 nM and 174 nM, respectively (Table 2). Thus, further increases in ether substituent size and hydrophobicity preserved, but did not enhance, SERT affinity. By contrast, DAT affinities of the SA4503 ether analogs increased substantially in conjunction with increasing substituent size and hydrophobicity. Phenylalkyl ethers 4 and 6 gave the highest DAT affinities of the ligands tested, with Ki values of 121 nM and 87 nM (Table 2), and exhibited over 100-fold higher DAT affinity than SA4503. Considering the lower DAT affinity of the (Z)- and (E)-iodoallyl ether analogs (Table 2), the terminal aromatic rings on the ether substituents of 4 and 6 play an important role in this interaction.
We anticipated the possibility of finding moderately high DAT and SERT affinities for the phenylalkyl ether analogs of SA4503 based upon their structural resemblance to GBR 12909 (Fig. 3), a ligand that shows high DAT (Ki = 12 nM) and SERT (Ki = 24 nM) affinities accompanied by weaker interactions with both σ1 (Ki = 318 nM) and σ2 (Ki = 116 nM) sites.41 A more recent determination puts the apparent affinity of GBR 12909 for σ1 receptors at 50.8 nM.42 SA4503 and GBR 12909 are flexible molecules that can adopt hundreds of closely related low energy conformations.20,51 In both cases, the piperazine rings are thought to take a chair conformation, and the phenyl alkyl chains are fully extended. These features have been observed in crystal structures of SA4503 and analogs.20 One possible structural superimposition of GBR 12909 with 4, the benzyl ether analog of SA4503, is given in Fig. 3. Poses shown have the phenylpropyl piperazine moieties aligned, and were minimized using the MMFF94 force field routine of program Avogadro.52 Qualitatively, the overall molecular dimensions of the two compounds are comparable, and there is substantial spatial overlap between the terminal aromatic ring of the ether substituent of 4 and the diphenylmethoxy ring system of GBR 12909. GBR 12909,41 as well as the YZ-series of analogs,22 typically display low affinities, > 1000 nM, for the norepinephrine transporter. Consequently, the affinities of SA4503 and analogs were not determined for this site.
Figure 3.
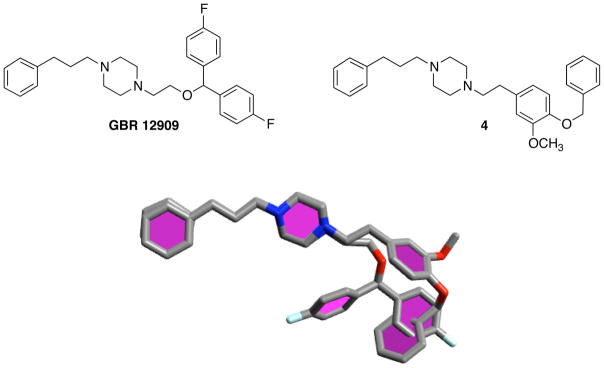
Comparison of GBR 12909 with 4, the benzyl ether analog of SA4503, and superimposition of one of many possible low energy conformers from each using program Avogadro.
3. Conclusions
SA4503, 4-O-des-methyl-SA4503 and two series of ether analogs have been prepared and evaluated for σ1 and σ2 receptor binding. The 14 compounds tested could be divided into two groups having different structure affinity relationships. The first group included 6 compounds, SA4503, 4-O-des-methyl SA4503, FE-SA4503 and cyclic ether analogs 7 – 9. These displayed relatively high σ1 receptor affinities, Ki values 1.75 nM – 19 nM, and variable σ2 receptor affinities, Ki values 6.7 nM – 113 nM, leading to modest σ1 receptor selectivities. The other group of eight SA4503 analogs had bulkier, hydrophobic ether substituents, and included alkyl / allyl ethers 1 – 3, phenylalkyl ethers 4 – 6 and the (Z)- and (E)-iodoallyl ether analogs. These compounds showed moderately high, nearly equal affinities for both σ1 and σ2 receptors, leading to ligands with little to no subtype selectivity. Considering there are two piperazine nitrogens in each ligand and two main hydrophobic binding pockets for each σ receptor subtype, at least eight different binding motifs are possible. Thus, one set of motifs may be favored for the binding of SA4503 and the compact analogs described above, while different motifs may be preferred as the ether substituents become larger and more hydrophobic. In this regard, some effects observed are likely to be unique to the exact position and type of substituent on the SA4503 scaffold. Diverse substituent pattern effects on σ receptor binding affinity and subtype selectivity are often found between studies of piperazines that have differing arylalkyl systems.17,18,20–22,53–58
SA4503 and a subset of ether analogs were also evaluated for DAT and SERT interactions. SA4503 displayed very weak binding to the DAT, Ki = 12650 nM. Affinity increased with progressively larger ether substitutions, and the phenylpropyl ether analog 6 showed a 145-fold higher DAT affinity of 87 nM. SERT binding varied 7-fold between SA4503 and analogs. Overall, monoamine transporter binding proved more sensitive than σ receptor binding to these structural modifications of SA4503.
Together, the data provide some insight into how manipulations of the substituents at the 4-position of the phenethyl side chain of SA4503 modulate σ receptor subtype affinity and selectivity. None of the novel ether analogs displayed higher σ receptor subtype affinity or selectivity than SA4503 or known congeners. On the other hand, ether substituents can be chosen that greatly increase DAT and SERT binding affinities, and the work identifies SA4503 as a scaffold for the preparation of ligands that can target multiple receptors. For instance, benzyl ether 4 shows moderately high affinities for σ1 and σ2 receptors, as well as for the DAT and SERT. Thus, certain ether analogs of SA4503 may well be polyfunctional in vivo, and would be of interest for behavioral testing in animal models of psychostimulant abuse, depression and anxiety.
4. Experimental section
4.1. Chemistry
4.1.1. General methods
Chemicals and solvents were reagent grade, and used as received from commercial sources. 4-O-des-methyl-SA4503 was prepared according to the literature procedure.36b 1H NMR spectra were obtained on Bruker ARX-250 (250 MHz), DRX-300 (300 MHz) or DRX-500 (500 MHz) spectrometers. Chemical shifts (1H) are reported for the free base in ppm (δ) relative to internal Me4Si in CDCl3. Splitting patterns are identified as singlet (s), broad singlet (br s), doublet (d), triplet (t), apparent pentet (app p), multiplet, (m), doublet of doublets of doublets, (ddd), doublet of doublets of triplets, (ddt). Elemental analyses were determined by Atlantic Microlab, Inc., Norcross GA. Analytical TLC was conducted on Polygram SIL G/UV254 plates, with additional visualization by staining with cerium molybdate. Preparative TLC was conducted on silica gel plates (20 × 20 cm, 1000 μm) from Analtech Inc., Newark DE. Column chromatography was performed using Silicycle® ultra pure silica gel (230–400 mesh) under N2 pressure. For storage and ease of handling, final targets were converted into the di-HCl salts. The free bases were dissolved in EtOH, concentrated HCl was added, the mixtures were evaporated to dryness, and the solid white products were recrystallized. Compounds 1 – 9 showed > 95% purity by reversed-phase HPLC analysis.
General procedure for alkylation of 4-O-des-methyl-SA4503 (1, 2, 3, 5 and 6)
Mixtures of 4-O-des-methyl-SA4503 (200 – 400 mg, 0.56 – 1.12 mmol), 40% KOH (1.1 – 2.2 mL), tetra-n-butylammonium hydroxide (1 M, CH3OH; 0.1 – 0.2 mL), and excess alkylating agent were heated at 50 °C for 50 min, and then diluted with H2O (30 mL) and extracted with CH2Cl2 (3 × 20 mL). The organic extracts were washed with saturated NaCl solution, dried over MgSO4, and concentrated under reduced pressure. The crude oily residues, pale yellow to dark brown in color, were purified by silica gel column chromatography (CHCl3 : CH3OH = 20 : 1).
1-(3-methoxy-4-propoxyphenethyl)-4-(3-phenylpropyl)piperazine (1)
Treatment of 4-O-des-methyl-SA4503 (200 mg, 0.56 mmol) with 40% KOH (1.1 mL), tetra-n-butylammonium hydroxide (0.1 mL) and 1-bromopropane (1200 mg, 9.74 mmol) according to the general procedure gave 1 (201 mg, 90%) 1H NMR: (250 Mz, CDCl3, δ) 1.03 (t, 3H, J = 7.5 Hz, CH3), 1.77–1.89 (m, 4H, CH2), 2.35–2.78 (m, 16H, CH2), 3.84 (s, 3H, OCH3), 3.94 (t, 2H, J = 7.5 Hz, OCH2), 6.70–6.80 (m, 3H, CH), 7.13–7.29 (m, 5H, CH). 13C NMR: (62.5 MHz, CDCl3, δ) 10.4, 22.5, 28.6, 33.2, 33.7, 53.2, 55.9, 57.97, 60.6, 70.6, 112.6, 113.2, 120.5, 125.7, 128.2, 128.3, 133.0, 142.1, 146.8, 149.3. Anal. Calcd for the di-HCl salt (C25H38Cl2N2O2): C, 63.96; H, 8.16; N, 5.97. Found: C, 63.91; H, 8.13; N, 5.91; m.p. 212 °C.
1-(4-(2-bromoethoxy)-3-methoxyphenethyl)-4-(3-phenylpropyl)piperazine (2)
Treatment of 4-O-des-methyl-SA4503 (400 mg, 1.13 mmol) with 40% KOH (2.2 mL), tetra-n-butylammonium hydroxide (0.2 mL) and 1,2-dibromoethane (690 mg, 3.66 mmol) according to the general procedure gave 2 (430 mg, 83%). 1H NMR: (250 Mz, CDCl3, δ) 1.77–1.89 (m, 2H, CH2), 2.35–2.78 (m, 16H, CH2), 3.62 (t, 2H, J = 7.5 Hz, CH2), 3.84 (s, 3H, CH3), 4.28 (t, 2H, J = 7.5 Hz, CH2), 6.70–6.84 (m, 3H, CH), 7.14–7.30 (m, 5H, CH). 13C NMR: (62.5 MHz, CDCl3, δ) 28.6, 29.0, 33.2, 33.7, 53.2, 56.0, 58.0, 60.5, 69.5, 113.0, 115.2, 120.7, 125.7, 128.2, 128.3, 134.7, 142.1, 145.7, 149.8; Anal. Calcd for the di-HCl salt (C24H35BrCl2N2O2): C, 53.94; H, 6.60; N, 5.24. Found: C, 53.94; H, 6.70; N, 5.24; mp 248–251 °C.
1-(4-allyloxy)-3-methoxyphenethyl)-4-(3-phenylpropyl)piperazine (3)
4-O-des-methyl-SA4503 (250 mg, 0.070 mmol) was treated with 40% KOH (1.4 mL) and tetra-n-butylammonium hydroxide (0.12 mL), followed by dropwise addition of allyl bromide (1500 mg, 12.40 mmol) at 0 °C. The mixture was then stirred at room temperature for 50 min, and worked up according to the general procedure to give 3 (160 mg, 57%). 1H NMR: (300 Mz, CDCl3, δ) 1.81–1.86 (app p, 2H, J = 7.5 Hz, CH2), 2.36–2.76 (m, 16H, CH2), 3.85 (s, 3H, OCH3), 4.55–4.58 (ddd, 2H, CH2, J = 5.4, 1.5, 1.5 Hz), 5.23–5.28 (ddt, 1H, J = 10.5, 1.5, 1.5 Hz), 5.35–5.42 (ddt, 1H, J = 17.1, 1.5, 1.5 Hz), 6.00–6.13 (ddt, 1H, J = 17.4, 10.5, 5.4 Hz), 6.70–6.81 (m, 3H, CH), 7.17–7.29 (m, 5H, CH). 13C NMR: (75 MHz, CDCl3, δ) 28.6, 33.2, 33.7, 53.2, 55.8, 58.0, 60.6, 69.9, 112.4, 113.6, 117.7, 120.4, 125.7, 128.3, 128.4, 133.4, 133.5, 142.1, 146.3, 149.3; Anal. Calcd for the di-HCl salt (C25H36Cl2N2O2): C, 64.23; H, 7.76; N, 5.99. Found: C, 64.33; H, 7.71; N, 5.96. m.p. 222 °C.
1-(4-(benzyloxy)-3-methoxyphenethyl)-4-(3-phenylpropyl)piperazine (4)
To a stirred solution of 4-O-des-methyl-SA4503 (250 mg, 0.70 mmol) in absolute EtOH (2.5 mL) was added K2CO3 (193 mg, 1.40 mmol) and benzyl bromide (192 mg, 1.12 mmol). The mixture was left under N2 overnight, and then filtered through Celite. Concentration under reduced pressure followed by preparative TLC (CHCl3 : CH3OH = 10 : 1) gave 4 as a pale yellow solid (110 mg, 35%). 1H NMR: (250 Mz, CDCl3, δ) 1.82–1.88 (m, 2H, CH2), 2.38–2.77 (m, 16H, CH2), 3.89 (s, 3H, CH3), 5.13 (s, 2H, CH2), 6.68–6.83 (m, 3H, CH), 7.19–7.46 (m, 10H, CH). 13C NMR: (62.5 MHz, CDCl3, δ) 28.6, 33.2, 33.7, 53.2, 55.9, 58.0, 60.6, 71.2, 112.6, 114.2,120.5, 125.70 127.2, 127.7, 128.2, 128.3, 128.4, 133.6, 137.4, 142.1, 146.5, 149.5. Anal. Calcd for the di-HCl salt (C29H38Cl2N2O2·0.5 H2O): C, 66.15; H, 7.47; N, 5.32. Found: C, 66.00; H, 7.34; N, 5.38. m.p. 213 °C.
1-(3-methoxy-4-phenylethoxyphenethyl)-4-(3-phenylpropyl)piperazine (5)
Treatment of 4-O-des-methyl-SA4503 (200 mg, 0.56 mmol) with 40% KOH (1.1 mL), tetra-n-butylammonium hydroxide (0.1 mL) and (2-bromoethyl)benzene (1800 mg, 9.733 mmol) according to the general procedure gave 5 (113 mg, 44%). 1H NMR: (250 Mz, CDCl3, δ) 1.82–1.89 (m, 2H, CH2), 2.38–2.78 (m, 16H, CH2), 3.17 (t, 2H, J = 7.5 Hz, CH2), 3.86 (s, 3H, CH3), 4.22 (t, 2H, J = 7.5 Hz, CH2), 6.74–6.83 (m, 3H, CH), 7.19–7.31 (m, 10H, CH). 13C NMR: (62.5 MHz, CDCl3, δ) 28.6, 33.2, 33.7, 35.8, 53.2, 56.0, 58.0, 60.6, 70.0, 112.8, 113.6, 120.7, 125.7, 125.8, 126.4, 127.3, 128.3, 128.4, 128.4, 129.0, 133.4, 138.1, 142.1, 146.6, 149.4. Anal. Calcd for the di-HCl salt (C30H40Cl2N2O2): C, 67.79; H, 7.58; N, 5.27. Found: C, 67.53; H, 7.64; N, 5.24. m.p. 252 °C.
1-(3-methoxy-4-(3-phenylpropoxy)phenethyl)-4-(3-phenylpropyl)piperazine (6)
Treatment of 4-O-des-methyl-SA4503 (200 mg, 0.56 mmol) with 40% KOH (1.1 mL), tetra-n-butylammonium hydroxide (0.1 mL) and 1-bromo-3-phenylpropane (1460 mg, 7.31 mmol) according to the general procedure gave 6 (214 mg, 81%). 1H NMR: (300 MHz, CDCl3, δ) 1.87 (app p, 2H, J = 6.0 Hz, CH2), 2.15 (app p, 2H, J =6.0 Hz, CH2), 2.43–2.85 (m, 18H, CH2), 3.86 (s, 3H, CH3), 3.99 (t, 2H, J = 6.0 Hz, CH2), 6.71–6.80 (m, 3H, CH), 7.16–7.31 (m, 10H, CH). 13CNMR: (75 MHz, CDCl3, δ) 28.2, 30.8, 31.3, 32.1, 32.2, 32.3, 33.0, 33.6, 34.4, 52.8, 56.0, 58.5, 61.5, 68.2, 112.7, 113.6, 120.6, 125.8, 125.9, 125.9, 128.4, 128.4, 128.4, 128.5, 132.8, 141.5, 141.8, 146.9, 149.5; Anal. Calcd for the di-HCl salt (C31H42Cl2N2O2): C, 68.24; H, 7.76; N, 5.13. Found: C, 67.94; H, 7.77; N, 5.12. m.p. 227 °C.
3,4-Ethylenedioxyphenylacetic acid (11) and 3,4-propylenedioxyphenylacetic acid (12)
To a solution of 3,4-dihydroxyphenylacetic acid (500 mg, 3.0 mmol) and either 1,2–dibromoethane (1.13 g, 6.0 mmol) or 1,3–dibromopropane (1210 mg, 6.0 mmol) in ethylene glycol (5 mL) was added anhydrous K2CO3 (1240 mg, 9.0 mmol). After heating at 120 °C for 4.5 h, the mixtures were cooled, diluted with H2O (50 mL), acidified to pH < 1 (2 N HCl), and extracted with EtOAc (3 x 20 mL). The organic layers were concentrated under reduced pressure, and residues were purified by column chromatography (EtOAc : Hexane = 50 : 50 for 11; 40 : 60 for 12) to give 1144 (301 mg, 52%) or 1245 (260 mg, 42%). 11 1H NMR: (300 MHz, CDCl3, δ) 3.56 (s, 2H, CH2), 4.23 (s, 4H, CH2), 6.75–6.87 (m, 3H, CH), 11.64 (br s, 1H, -COOH). 12 1H NMR: (250 MHz, CDCl3, δ) 2.19 (app p, 2H, J = 7.5 Hz, CH2), 3.55 (s, 2H, CH2), 4.21 (m, 4H, CH2), 6.81–6.95 (m, 3H, CH), 10.50 (br s, 1H, -COOH).
General procedure for phenylacetic acid coupling to 3-phenylpropylpiperazine (13 – 15)
Equimolar amounts of 3-phenylpropylpiperazine (0.83 – 1.55 mmol), HOBt·H2O, EDC·HCl and the substituted phenylacetic acid (10 – 12) were treated with 3 equivalents of 4-methylmorpholine in CH2Cl2 (7 – 15 mL) under N2 at 0 °C. Mixtures were stirred at ambient temperature overnight, and then evaporated to dryness under reduced pressure. Residues were diluted with saturated NaHCO3 and extracted with EtOAc. Organic layers were washed with saturated NaCl solution, dried over MgSO4, filtered and evaporated to dryness under reduced pressure. Residues were purified by column chromatography (CHCl3 : CH3OH = 20 : 1).
2-(3,4-methylenedioxy)phenyl-1-(4-(3-phenylpropyl)piperazin-1-yl)ethanone (13)
The general procedure was followed with 3-phenylpropylpiperazine (170 mg, 0.83 mmol) and 3,4-(methylenedioxy)phenylacetic acid (10, 150 mg, 0.83 mmol) to provide 13 (288 mg, 94%) 1H NMR: (300 MHz, CDCl3, δ) 1.76–1.81 (m, 2H, CH2), 2.25–2.39 (m, 6H, CH2), 2.62 (t, 2H, J = 6.0 Hz, CH2), 3.45 (t, 2H, J = 6.0 Hz, CH2), 3.63 (t, 4H, J = 6.0 Hz, CH2), 5.92 (s, 2H, OCH2O), 6.66–6.75 (m, 3H, CH), 7.15–7.27 (m, 5H, CH). 13C NMR: (75 MHz, CDCl3, δ) 28.3, 33.4, 40.4, 41.7, 46.0, 52.6, 53.0, 57.5, 100.9, 108.3, 109.0, 121.5, 125.7, 128.2, 128.3, 128.6, 141.8, 146.31, 147.8, 169.3. Anal. Calcd for C22H26N2O3·0.25 H2O: C, 71.23; H, 7.20; N, 7.55. Found: C, 71.41; H, 7.18; N, 7.54.
2-(3,4-ethylenedioxy)phenyl-1-(4-(3-phenylpropyl)piperazin-1-yl)ethanone (14)
The general procedure was followed using 3-phenylpropylpiperazine (316 mg, 1.55 mmol) and 3,4-(ethylenedioxy)phenylacetic acid (11, 301 mg, 1.55 mmol) to yield 14 (577 mg, 98%). 1H NMR: (250 MHz, CDCl3, δ) 1.78 (app p, 2H, J = 7.5 Hz, CH2), 2.24–2.39 (m, 6H, CH2), 2.62 (t, 2H, J = 7.5 Hz, CH2), 3.43 (t, 2H, J = 5.0 Hz, CH2), 3.63 (t, 2H, J = 5.0 Hz, CH2), 3.59 (s, 2H, CH2), 4.20 (s, 4H, OCH2CH2O), 6.66–6.81 (m, 3H, CH), 7.15–7.30 (m, 5H, CH). 13C NMR: (62.5 MHz, CDCl3, δ) 28.4, 33.4, 40.0, 41.7, 46.0, 52.7, 53.1, 57.5, 64.2, 64.3, 117.3, 121.5, 125.8, 128.2, 128.3, 128.3, 141.9, 142.3, 143.5, 169.4. Anal. Calcd for C23H28N2O3·0.5H2O: C, 70.93; H, 7.50; N, 7.19. Found: C, 70.73; H, 7.31; N, 7.20.
2-(3,4-propylenedioxy)phenyl-1-(4-(3-phenylpropyl)piperazin-1-yl)ethanone (15)
The general procedure was followed using 3-phenylpropylpiperazine (255 mg, 1.25 mmol) and 3,4-(ethylenedioxy)phenylacetic acid (12, 260 mg, 1.25 mmol) to yield 15 (443 mg, 90%) 1H NMR: (500MHz, CDCl3, δ) 1.63 (app p, 2H, J = 7.5 Hz, CH2), 1.98 (app p, 2H, J = 6.0 Hz, CH2), 2.09 (t, 2H, J = 5.0 Hz, CH2), 2.16 (t, 2H, J = 7.5 Hz, CH2), 2.21 (t, 2H, J = 5.0 Hz, CH2), 2.47 (t, 2H, J = 7.5 Hz, CH2), 3.27 (t, 2H, J = 5.0 Hz, CH2), 3.45 (s, 2H, CH2), 3.48 (t, 2H, J = 5.0 Hz, CH2), 3.98–4.01 (m, 4H, CH2), 6.62–6.77 (m, 3H, CH), 7.00–7.15 (m, 5H, CH). 13C NMR: (125 MHz, CDCl3, δ) 28.3, 31.8, 33.4, 39.8, 41.7, 46.0, 52.6, 53.0, 57.5, 70.4, 121.6, 121.7, 123.4, 125.7, 128.2, 128.3, 130.2, 141.9, 149.9, 151.1, 169.2. Anal. Calcd for C23H28N2O3·0.5H2O: C, 68.64; H, 7.19; N, 6.27. Found: C, 69.09; H, 7.31; N, 6.66.
General procedure for amide reduction (7 – 9)
Solutions of substituted amides (13 – 15, 0.79 – 1.27 mmol) in THF (7 – 12 mL) were added to a suspension of 3 equivalents of LiAlH4 in THF (7 – 12 mL) under N2 at 0 °C. Mixtures were then stirred at ambient temperature for 7.5 h, cooled in an ice bath, and quenched by sequential addition of EtOAc (3 mL) and HCl (2 N, 1 mL), followed by addition of solid NaHCO3 until CO2 evolution ceased. Mixtures were filtered, diluted with saturated NaHCO3 and extracted with EtOAc. Organic extracts were dried over MgSO4, filtered and evaporated to dryness under reduced pressure. Cyclic ether 7 was isolated by preparative TLC (CHCl3 : CH3OH = 10 : 1) while 8 and 9 were purified on silica gel columns (CHCl3 : CH3OH = 30 : 1).
1-(3,4-methylenedioxyphenethyl)-4-(3-phenylpropyl)piperazine (7)
The general procedure was followed using amide 13 (288 mg, 0.79 mmol) to give 7 as a yellow oil (90 mg, 33%). 1H NMR: (250 MHz, CDCl3, δ) 1.75–1.87 (m, 2H, CH2), 2.33–2.73 (m, 16H, CH2), 5.85 (s, 2H, OCH2O), 6.60–6.71 (m, 3H, CH), 7.15–7.28 (m, 5H, CH). 13C NMR: (75 MHz, CDCl3, δ) 28.6, 33.3, 33.6, 53.2, 57.9, 60.7, 100.7, 108.1, 109.1, 121.3, 125.7, 128.2, 128.3, 134.0, 142.1, 145.7, 147.5; Anal. Calcd for the di-HCl salt (C22H30Cl2N2O2): C, 62.12; H, 7.11; N, 6.59. Found: C, 61.85; H, 7.04; N, 6.55. m.p. 230 °C.
1-(3,4-ethylenedioxyphenethyl)-4-(3-phenylpropyl)piperazine (8)
The general procedure was followed using amide 14 (484 mg, 1.27 mmol) to give 8 as a yellow oil (340 mg, 73%). 1H NMR: (250 MHz, CDCl3, δ) 1.84 (app p, 2H, J = 7.5 Hz, CH2), 2.36–2.74 (m, 16H, CH2), 4.19 (s, 4H, OCH2CH2O), 6.65–6.80 (m, 3H, CH), 7.15–7.31 (m, 5H, CH). 13C NMR: (62.5 MHz, CDCl3, δ) 28.6, 33.3, 33.6, 53.2, 57.9, 60.7, 100.7, 108.1, 109.1, 121.3, 125.7, 128.2, 128.3, 134.0, 142.1, 145.7, 147.5; Anal. Calcd for the di-HCl salt (C23H32Cl2N2O2): C, 62.87; H, 7.34; N, 6.38. Found: C, 63.14; H, 7.49; N, 6.39. m.p. 230 °C.
1-(3,4-propylenedioxyphenethyl)-4-(3-phenylpropyl)piperazine (9)
The general procedure was followed using amide 15 (390 mg, 0.99 mmol) to give 9 as a yellow oil (321 mg, 85%). 1H NMR: (300 MHz, CDCl3, 1.84 (app p, 2H, J = 6.0 Hz, CH2), 2.15 (app p, 2H, J = 6.0 Hz, CH2), 2.37–2.75 (m, 16H, CH2), 4.16 (t, 4H, J = 6.0 Hz, -CH2O), 6.74–6.91 (m, 3H, CH), 7.15–7.30 (m, 5H, CH). 13C NMR: (75 MHz, CDCl3, δ) 28.6, 32.0, 32.7, 33.7, 53.2, 58.0, 60.5, 70.4, 121.3, 121.6, 123.4, 125.7, 128.3, 128.4, 135.6, 142.1, 149.4, 151.0; Anal. Calcd for the di-HCl salt (C24H34Cl2N2O2): C, 63.57; H, 7.56; N, 6.18. Found: C, 63.44; H, 7.71; N, 6.16. m.p. 212 °C.
4.2 Receptor Binding
Tritiated radioligands were obtained from PerkinElmer Life Sciences, Inc. (Boston, MA). [125I]RTI-121 was synthesized as described previously.59 Serial dilutions of test ligands were prepared in water containing 1% EtOH and 0.1% HOAc. Tritiated and [125I]-labeled radioligands were prepared in Tris-HCl buffers (50 mM). Tritium radioactivity was measured using a Wallac 1409 liquid scintillation counter and OptiPhase® HiSafe 2 cocktail at 44% efficiency. Fresh-frozen brains from male English Hartley guinea pigs were obtained from Rockland Immunochemicals, Inc. (Gilbertsville, PA) for σ receptor assays. Fresh male CD-1® mouse brains (Charles River Laboratories International, Inc., Wilmington, MA) were harvested, after animal euthanasia by cervical dislocation, as needed for the DAT and SERT assays. Studies involving living animals were performed humanely in compliance with the Guide for the Care and Use of Laboratory Animals promulgated by the U.S. National Institutes of Health, and with prior approvals from the Institutional Animal Care and Use Committees of the University of Missouri and the Harry S. Truman Memorial Veterans’ Hospital.
Assays for σ receptor binding were performed using 1.0 nM [3H](+)-pentazocine (σ1), 3.0 nM [3H]ditolylguanidine ([3H]DTG) / 200 nM (+)-pentazocine (σ2), and membranes from whole male guinea pig brains as previously described in detail.38 Non-specific binding was defined by haloperidol (1.0 μM; σ1) or by DTG (100 μM; σ2). Binding assays for DAT and SERT were performed using literature procedures with minor modifications. In brief, DAT assays used [125I]RTI at 15 pM and mouse striatal membranes, with non-specific binding defined by 0.5 μM GBR 12909.60 SERT assays used [3H]paroxetine at 0.3 nM and mouse whole brain membranes, with non-specific binding defined by 10 μM fluoxetine.61 Experiments were conducted in duplicate, and replicated three to six times. Data were analyzed by non-linear curve-fitting using a sigmoidal four-parameter logistic fit (Prism 6.0c; GraphPad Software, Inc., La Jolla, CA). Ki values were derived from inhibition IC50 data by the Cheng-Prusoff relationship,62 using reported Kd values of 2.3 nM for [3H](+)-pentazocine,38 23.9 nM for [3H]DTG,38 0.12 nM for [125I]RTI,60 and 0.13 nM for [3H]paroxetine.61
Acknowledgments
We thank the National Cancer Institute (P50 CA103130: Center for Single Photon-Emitting Cancer Imaging Agents) and the National Institute for Drug Abuse (1RC1DA028477: Development of Anti-Cocaine Medications) for partial support of this research. We thank the National Science Foundation (NSF CHE-0353891: Research Experience for Undergraduates (REU) - Site for an Introduction to Radiochemistry Research) for supporting Dr. Ryan M. Peterson, and the University of Missouri Life Sciences Undergraduate Research Opportunity Scholars program for supporting Dr. Sarah A. Lord (née Violand). We thank Mr. Michael W. Lever for assistance with Avogadro. We thank NSF CHE-95-31247 for NMR instrumentation support. We also acknowledge resources and facilities provided by the Harry S. Truman Memorial Veterans’ Hospital. This work does not represent the views of the U. S. Department of Veterans Affairs or the United States Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.van Waarde A, Rybczynska AA, Ramakrishnan N, Ishiwata K, Elsinga PH, Dierckx RA. Curr Pharm Des. 2010;16:3519–3537. doi: 10.2174/138161210793563365. [DOI] [PubMed] [Google Scholar]
- 2.Megalizzi V, Le Mercier M, Decaestecker C. Med Res Rev. 2012;32:410–427. doi: 10.1002/med.20218. [DOI] [PubMed] [Google Scholar]
- 3.Mach RH, Zeng C, Hawkins WG. J Med Chem. 2013;56:7137–7160. doi: 10.1021/jm301545c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Ishikawa M, Hashimoto K. J Receptor Ligand Channel Res. 2010;3:25–36. [Google Scholar]; (b) Hayashi T, Tsai SY, Mori T, Fujimoto M, Su TP. Expert Opin Ther Targets. 2011;15:557–577. doi: 10.1517/14728222.2011.560837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banister SD, Kassiou M. Curr Pharm Des. 2012;18:884–901. doi: 10.2174/138161212799436539. [DOI] [PubMed] [Google Scholar]
- 6.Niitsu T, Iyo M, Hashimoto K. Curr Pharm Des. 2012;18:875–883. doi: 10.2174/138161212799436476. [DOI] [PubMed] [Google Scholar]
- 7.Narayanan S, Mesangeau C, Poupaert JH, McCurdy CR. Curr Top Med Chem. 2011;11:1128–1150. doi: 10.2174/156802611795371323. [DOI] [PubMed] [Google Scholar]
- 8.Robson MJ, Noorbakhsh B, Seminerio MJ, Matsumoto RR. Curr Pharm Des. 2012;18:902–919. doi: 10.2174/138161212799436601. [DOI] [PubMed] [Google Scholar]
- 9.Lever JR, Miller DK, Fergason-Cantrell EA, Green CL, Watkinson LD, Carmack TL, Lever SZ. J Pharmacol Exp Ther. 2014;351:153–163. doi: 10.1124/jpet.114.216671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lever JR, Miller DK, Green CL, Fergason-Cantrell EA, Watkinson LD, Carmack TL, Fan KH, Lever SZ. Synapse. 2014;68:73–84. doi: 10.1002/syn.21717. [DOI] [PubMed] [Google Scholar]
- 11.(a) Su TP, London ED, Jaffe JH. Science. 1988;240:219–221. doi: 10.1126/science.2832949. [DOI] [PubMed] [Google Scholar]; (b) Johannessen M, Fontanilla D, Mavlyutov T, Ruoho AE, Jackson MB. Am J Physiol Cell Physiol. 2011;300:C328–C337. doi: 10.1152/ajpcell.00383.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruoho AE, Chu UB, Ramachandran S, Fontanilla D, Mavlyutov T, Hajipour AR. Curr Pharm Des. 2012;18:920–929. doi: 10.2174/138161212799436584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kekuda R, Prasad PD, Fei YJ, Leibach FH, Ganapathy V. Biochem Biophys Res Commun. 1996;229:553–558. doi: 10.1006/bbrc.1996.1842. [DOI] [PubMed] [Google Scholar]
- 14.Hanner M, Moebius FF, Flandorfer A, Knaus HG, Striessnig J, Kempner E, Glossmann H. Proc Natl Acad Sci USA. 1996;93:8072–8077. doi: 10.1073/pnas.93.15.8072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brune S, Schepmann D, Klempnauer KH, Marson D, Dal Col V, Laurini E, Fermeglia M, Wünsch B, Pricl S. Biochemistry. 2014;53:2993–3003. doi: 10.1021/bi401575g. [DOI] [PubMed] [Google Scholar]
- 16.Xu J, Zeng C, Chu W, Pan F, Rothfuss JM, Zhang F, Tu Z, Zhou D, Zeng D, Vangveravong S, Johnston F, Spitzer D, Chang KC, Hotchkiss RS, Hawkins WG, Wheeler KT, Mach RH. Nat Commun. 2011;2:380. doi: 10.1038/ncomms1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Glennon RA. Mini-Rev Med Chem. 2005;5:927–940. doi: 10.2174/138955705774329519. [DOI] [PubMed] [Google Scholar]; (b) Glennon RA. Brazil J Pharm Sci. 2005;41:1–12. [Google Scholar]
- 18.Abate C, Mosier PD, Berardi F, Glennon RA. Cent Nerv Syst Agents Med Chem. 2009;9:246–257. doi: 10.2174/1871524910909030246. [DOI] [PubMed] [Google Scholar]
- 19.Matsuno K, Nakazawa M, Okamoto K, Kawashima Y, Mita S. Eur J Pharmacol. 1996;306:271–279. doi: 10.1016/0014-2999(96)00201-4. [DOI] [PubMed] [Google Scholar]
- 20.Fujimura K, Matsumoto J, Niwa M, Kobayashi T, Kawashima Y, In Y, Ishida T. Bioorg Med Chem. 1997;5:1675–1683. doi: 10.1016/s0968-0896(97)00093-x. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y, Williams W, Torrence-Campbell C, Bowen WD, Rice KC. J Med Chem. 1998;41:4950–4957. doi: 10.1021/jm980143k. [DOI] [PubMed] [Google Scholar]
- 22.Matsumoto RR, Potelleret FH, Mack A, Pouw B, Zhang Y, Bowen WD. Pharmacol Biochem Behav. 2004;77:775–781. doi: 10.1016/j.pbb.2004.01.014. [DOI] [PubMed] [Google Scholar]
- 23.Rodvelt KR, Lever SZ, Lever JR, Blount LR, Fan KH, Miller DK. Pharmacol Biochem Behav. 2011;97:676–682. doi: 10.1016/j.pbb.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodvelt KR, Oelrichs CE, Blount LR, Fan KH, Lever SZ, Lever JR, Miller DK. Drug Alcohol Depend. 2011;116:203–210. doi: 10.1016/j.drugalcdep.2010.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mori T, Rahmadi M, Yoshizawa K, Itoh T, Shibasaki M, Suzuki T. Addict Biol. 2014;19:362–369. doi: 10.1111/j.1369-1600.2012.00488.x. [DOI] [PubMed] [Google Scholar]
- 26.Sage AS, Oelrichs CE, Davis DC, Fan K-H, Nahas RI, Lever SZ, Lever JR, Miller DK. Pharmacol Biochem Behav. 2013;110:201–207. doi: 10.1016/j.pbb.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 27.Sage AS, Vannest SC, Fan K-H, Will MJ, Lever SZ, Lever JR, Miller DK. ISRN Pharmacol. 2013:Article ID 546314, 7. doi: 10.1155/2013/546314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fujimoto M, Hayashi T, Urfer R, Mita S, Su TP. Synapse. 2012;66:630–639. doi: 10.1002/syn.21549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Appelberg B. Helsinki University; [accessed July 17, 2014]. Available from: http://clinicaltrials.gov/show/NCT00551109. [Google Scholar]
- 30.Urfer R, Moebius HJ, Skoloudnik D, Santamarina E, Sato W, Mita S, Muir KW. doi: 10.1161/STROKEAHA.114.005835. http://stroke.ahajournals.org/content/early/2014/09/30/STROKEAHA.114.005835. [DOI] [PubMed]
- 31.Ruscher K, Shamloo M, Rickhag M, Ladunga I, Soriano L, Gisselsson L, Toresson H, Ruslim-Litrus L, Oksenberg D, Urfer R, Johansson BB, Nikolich K, Wieloch T. Brain. 2011;134:732–746. doi: 10.1093/brain/awq367. [DOI] [PubMed] [Google Scholar]
- 32.Collier TL, Waterhouse RN, Kassiou M. Curr Pharm Des. 2007;13:51–72. doi: 10.2174/138161207779313740. [DOI] [PubMed] [Google Scholar]
- 33.Banister SD, Manoli M, Kassiou M. J Labelled Comp Radiopharm. 2013;56:215–224. doi: 10.1002/jlcr.3010. [DOI] [PubMed] [Google Scholar]
- 34.van Waarde A, Buursma AR, Hospers GA, Kawamura K, Kobayashi T, Ishii K, Oda K, Ishiwata K, Vaalburg W, Elsinga PH. J Nucl Med. 2004;45:1939–1945. [PubMed] [Google Scholar]
- 35.Toyohara J, Sakata M, Ishiwata K. Cent Nerv Syst Agents Med Chem. 2009;9:190–196. doi: 10.2174/1871524910909030190. [DOI] [PubMed] [Google Scholar]
- 36.(a) Elsinga PH, Kawamura K, Kobayashi T, Tsukada H, Senda M, Vaalburg W, Ishiwata K. Synapse. 2002;43:259–267. doi: 10.1002/syn.10045. [DOI] [PubMed] [Google Scholar]; (b) Kawamura K, Elsinga PH, Kobayashi T, Ishii S, Wang WF, Matsuno K, Vaalburg W, Ishiwata K. Nucl Med Biol. 2003;30:273–284. doi: 10.1016/s0969-8051(02)00439-0. [DOI] [PubMed] [Google Scholar]
- 37.Kawamura K, Tsukada H, Shiba K, Tsuji C, Harada N, Kimura Y, Ishiwata K. Nucl Med Biol. 2007;34:571–577. doi: 10.1016/j.nucmedbio.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 38.Lever JR, Gustafson JL, Xu R, Allmon RL, Lever SZ. Synapse. 2006;59:350–358. doi: 10.1002/syn.20253. [DOI] [PubMed] [Google Scholar]
- 39.Xu R, Watkinson LD, Carmack TL, Lever JR, Lever SZ. Med Chem. 2012;2:131–136. [Google Scholar]
- 40.Shiba K, Ogawa K, Ishiwata K, Yajima K, Mori H. Bioorg Med Chem. 2006;14:2620–2626. doi: 10.1016/j.bmc.2005.11.044. [DOI] [PubMed] [Google Scholar]
- 41.Husbands SM, Izenwasser S, Kopajtic T, Bowen WD, Vilner BJ, Katz JL, Newman AH. J Med Chem. 1999;42:4446–4455. doi: 10.1021/jm9902943. [DOI] [PubMed] [Google Scholar]
- 42.Cao J, Kopajtic T, Katz JL, Newman AH. Bioorg Med Chem Lett. 2008;18:5238–5241. doi: 10.1016/j.bmcl.2008.08.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hiranita T, Soto PL, Kohut SJ, Kopajtic T, Cao J, Newman AH, Tanda G, Katz JL. J Pharmacol Exp Ther. 2011;339:662–677. doi: 10.1124/jpet.111.185025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sasamoto M. Chem Pharm Bull. 1960;8:324–329. [Google Scholar]
- 45.Alker D, Cross PE, Wallis RM. 5,089,505. US Patent. 1992
- 46.Kashima C, Tomotake A, Omote Y. J Org Chem. 1987;52:5616–5621. [Google Scholar]
- 47.Lever SZ, Xu R, Fan KH, Fergason-Cantrell EA, Carmack TL, Watkinson LD, Lever JR. Nucl Med Biol. 2012;39:401–414. doi: 10.1016/j.nucmedbio.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 48.Rhoades DJ, Kinder DH, Mahfouz TM. Med Chem. 2014;10:98–121. doi: 10.2174/1573406409999131119103621. [DOI] [PubMed] [Google Scholar]
- 49.Lu Y, Shi T, Wang Y, Yang H, Yan X, Luo X, Jiang H, Zhu W. J Med Chem. 2009;52:2854–2862. doi: 10.1021/jm9000133. [DOI] [PubMed] [Google Scholar]
- 50.Bissantz C, Kuhn B, Stahl M. J Med Chem. 2010;53:5061–5084. doi: 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pandit D, Roosma W, Misra M, Gilbert KM, Skawinski WJ, Venanzi CA. J Mol Model. 2011;17:181–200. doi: 10.1007/s00894-010-0712-x. [DOI] [PubMed] [Google Scholar]
- 52.Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchison GR. J Cheminform. 2012;4:1–17. doi: 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Foster A, Wu H, Chen W, Williams W, Bowen WD, Matsumoto RR, Coop A. Bioorg Med Chem Lett. 2003;13:749–751. doi: 10.1016/s0960-894x(02)01034-x. [DOI] [PubMed] [Google Scholar]
- 54.Nahas RI, Lever JR, Lever SZ. Bioorg Med Chem. 2008;16:755–761. doi: 10.1016/j.bmc.2007.10.037. [DOI] [PubMed] [Google Scholar]
- 55.Moussa IA, Banister SD, Beinat C, Giboureau N, Reynolds AJ, Kassiou M. J Med Chem. 2010;53:6228–6239. doi: 10.1021/jm100639f. [DOI] [PubMed] [Google Scholar]
- 56.Younes SY, Labssita Y, Baziard-Mouysset G, Payard M, Rettori MM, Renard P, Pfeiffer B, Caignard D. Eur J Med Chem. 2000;35:107–121. doi: 10.1016/s0223-5234(00)00113-6. [DOI] [PubMed] [Google Scholar]
- 57.Abate C, Niso M, Marottoli R, Riganti C, Ghigo D, Ferorelli S, Ossato G, Perrone R, Lacivita E, Lamb DC, Berardi F. J Med Chem. 2014;57:3314–3323. doi: 10.1021/jm401874n. [DOI] [PubMed] [Google Scholar]
- 58.Berardi F, Abate C, Ferorelli S, Uricchio V, Colabufo NA, Niso M, Perrone R. J Med Chem. 2009;52:7817–7828. doi: 10.1021/jm9007505. [DOI] [PubMed] [Google Scholar]
- 59.Lever JR, Scheffel U, Stathis M, Seltzman HH, Wyrick CD, Abraham P, Parham K, Thomas BF, Boja JW, Kuhar MJ, Carroll FI. Nucl Med Biol. 1996;23:277–284. doi: 10.1016/0969-8051(95)02074-8. [DOI] [PubMed] [Google Scholar]
- 60.Boja JW, Cadet JL, Kopajtic TA, Lever J, Seltzman HH, Wyrick CD, Lewin AH, Abraham P, Carroll FI. Mol Pharmacol. 1995;47:779–786. [PubMed] [Google Scholar]
- 61.Hirano K, Kimura R, Sugimoto Y, Yamada J, Uchida S, Kato Y, Hashimoto H, Yamada S. Br J Pharmacol. 2005;144:695–702. doi: 10.1038/sj.bjp.0706108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cheng Y, Prusoff WH. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]