

Abstract
The crystal structures of three nuclear receptor (NR) complexes have emerged to reveal their multi-domain architectures on DNA. These pictures provide unprecedented views of interfacial couplings between the DNA binding domains (DBDs) and ligand binding domains (LBDs). The detailed pictures contrast with previous interpretations of low-resolution electron microscopy (EM) and small angle X-ray scattering (SAXS) data, which had suggested a common architecture consisting of disconnected DBDs and LBDs. Re-visiting both historical and recent interpretations of NR architecture, we invoke new principles underlying higher order quaternary organization and the allosteric transmission of signals between domains. We also discuss how NR architectures are being probed in living cells to understand dimerization and DNA binding events in real time.
A brief history of single domain structures
Nuclear receptors (NRs) are metazoan transcription factors that regulate metabolism, development, homeostasis and reproduction. In humans, the 48 NRs can be divided into four groups based on their receptor dimerization patterns and DNA-type preferences. The first group forms homodimers and binds to DNA inverted repeats, and includes steroid receptors such as GR, ER, PR, AR, and MR. A second group heterodimerizes with RXR and binds to DNA direct-repeats, and includes receptors such as PPAR, RAR, VDR, and TR. A third group consists of homodimers that bind to DNA direct-repeats, such as HNF-4α and Rev-Erb. The fourth group contains monomers that bind to extended single DNA half-sites, including receptors such as ROR and NURR family members [1–3]. Consensus half-sites are typically 5′-AGGTCA-3′ sequences for non-steroid receptors, and 5′-AGAACA-3′ sequences for steroid receptors.
When viewed from their N- to their C-terminus, NR polypeptides exhibit a modular organization consisting of five to six segments, designated A–F. Only two domains had been well characterized through high-resolution structural methodologies. These are the DNA binding domain (DBD) that specifically contacts response elements, and the ligand-binding domain (LBD) that recognizes endogenous small-molecule ligands and coregulator regions [4–6]. Crystallographic studies on DBD-DNA complexes have revealed the basis for half-site recognition, and the roles of inter-half-site spacing and half-site repeat nature as selectivity features [2]. Crystallography later revealed how ligands are bound in the LBD structures, beginning with the thyroid hormone receptor (TR) and retinoic acid receptor (RAR) [6–8]. The binding of different types of ligands to a single NR was subsequently shown for the estrogen receptor (ER) through a series of detailed structure-function studies [9, 10].
Most NR LBDs have the capacity to bind coactivator segments with LXXLL sequences, and corepressor segments with LXXXLXXX[I/L] sequences (where L = leucine, I=isoleucine, and X= any amino acid) [11, 12]. These short elements interact at the LBD surface in a manner that depends on the ligand occupied inside the LBD pocket. Components of coregulator complexes modify the histone tails in chromatin, favoring either the activation or repression of target genes [13]. Early crystallographic studies addressed how coactivator LXXLL segments recognize the surfaces of LBDs, focusing on PPARγ and ER LBDs [10, 14].
These and subsequent structural studies of isolated DBDs and LBDs provided us with a deep understanding of the molecular interactions within each of these domains [6]. However, our understanding was incomplete because these studies did not reveal how the many different domains and segments of a NR cooperate in the context of a quaternary architecture with functional relevance. These missing insights prevented the field from fully considering allosteric communications, such as how ligand binding may lead to changes in DNA binding and vice-versa. Now, three published reports reveal the detailed, higher order molecular architectures of NR complexes using X-ray crystallography [15–17]. These pictures are showing us surprisingly complex domain-domain interconnections, also providing new insights about how signals can be communicated between domains in an allosteric fashion. Previously, a different picture was proposed for full-length nuclear receptors, based only on solution based and low-resolution techniques. That picture was based on the two conserved domains (DBDs and LBDs) of NRs having no direct contacts with one another when the receptor was bound to its DNA element, being organized instead as isolated beads (domains) positioned on opposite ends of an extended string (hinge region). That disconnected domain architecture now seems inconsistent with both the recent set of crystallographic findings based on multiple NR complexes, and the larger body of structural, biophysical, and cell-based studies that support NR quaternary structures on DNA that have highly coupled DBD-LBD interfaces for allosteric communications between these domains. In this review, we discuss both the historical and newly reported findings that mechanistically examine how NR-DNA complexes use their complex molecular architectures to sense and transmit signals through their domains.
Carefully revisiting the mousetrap
One of the critical early goals of NR structural biology was to define the LBD conformations that could be reliably described as both the inactive and active states. To this end, the structure of the unliganded RXRα LBD structure was compared to a subsequent structure of the RARγ LBD with the activating ligand all-trans retinoic acid [8]. That comparison led the authors to propose a mousetrap mechanism for ligand-activation of NRs [8]. As shown in Figure 1a, ligand-binding was suggested to induce an altered position in Helix-12 (H12). H12 was described as a stable helix located away from the LBD body in the apo-state (deemed to be the inactive conformation). Upon ligand binding, H12 moves to a new position on the surface the LBD, entrapping the ligand (active conformation), hence it is dubbed the ‘mousetrap’ mechanism. However, further analysis of the mousetrap mechanism using those original crystallographic coordinates suggests that this interpretation may have been misguided (shown Figure 1b). The H12 position in the apo-state is positioned through artificial crystal packing interactions.
Figure 1.

Revisiting the “Mousetrap” mechanism. (a) The original mechanism was based on a structural comparison between unliganded retinoid X receptor alpha (RXRα) ligand binding domain (LBD) and liganded retinoic acid receptor gamma (RARγ) LBD, and later generalized [8, 57, 58]. Activating ligands would induce a large conformational shift in the helix-12 (H12) position of the LBD. H12 moves from an “inactive” position away from the LBD to an active conformation on the LBD. (PDB ID 1LBD and 2LBD). (b) Re-interpreting helix-12 position in the context of its original crystallographic coordinates. The position of helix-12 can be seen to be set by crystal packing forces (PDB ID 1LBD) [57]. (c) Crystal structure of estrogen-related receptor gamma (ERRγ) LBD showing a similar crystallographic artifact with respect to H12 (PDB ID 2GPV) [27]. The second subunit in the same ERR structure, not discussed in the report, shows no stable H12 position, suggesting instead that inactive state should have been alternatively characterized as one with significant H12 dynamics [27]. (d) An alternate model for ligand-induced activation. This mechanism is based on ligand-induced stabilization of H12 dynamics [18]. Instead of large movement in H12 between two stable positions, a highly dynamic and unstructured LBD segment gains structural order and a helical conformation when activating ligands are inside the LBD pocket. The stabilized conformation then facilitates the direct binding of coactivator derived LXXLL peptide segments to the LBD surface, as seen in the ER LBD structure [10].
An alternative, better-supported model for ligand activation, proposed by Schwabe and colleagues, was derived from their fluorescence spectroscopic studies [18]. This mechanism, known as helix-12 dynamic stabilization, instead characterizes the inactive LBD state as one with relatively high mobility and lack of structural order in “H12”. A disorder-to-order transition is induced with binding of activating ligands. Similarly, Nuclear Magnetic Resonance (NMR) studies of several NRs revealed ligand-induced stabilization of NR LBDs and the correspondent H12s[19–22]. Supporting data for this alternate model is strong, as the evidence comes from not only the RXRα-LBD, but also other NR LBDs and even a full-length NR and [23–26]. These reports used hydrogen-deuterium exchange mass-spectrometry (H/D ex MS) studies, and consistently found faster hydrogen exchange in “H12” in the apo-state, confirming its relative lack of structural order. This is inconsistent with the idea of H12 as a stable helix, as had been proposed in the mousetrap model. Instead, the binding of activating ligands produces a stable helical conformation in H12 residues, and also adds global stability to the LBD fold.
The so-called “inactive” H12 appears similarly misinterpreted in the ERR LBD structure [27]. Here again the authors describe the location of helix-12 as both ordered and positioned away from the receptor LBD, so as to be consistent with their original notion of the mousetrap mechanism [27]. However, as indicated in Figure 1c, the position of H12 is again strongly influenced by crystal packing interactions. Intriguingly, a second ERR subunit in the asymmetric unit, ignored in that report, shows a disordered state (not visible electron density) predicted by the helix-12 dynamic stabilization model [27].
A related question has been how ligands enter and exit the LBD. One hypothesis, that different parts of the NR LBD body can harbor a gate for ligand entry or exit from the ligand binding cavity, goes back to the first liganded NR LBD structure[7]. The H1–H3 loop and neighboring β-sheets were speculated to be a ligand entry site to the TR LBD in work from the Fletterick lab [7]. This hypothesis received further support from molecular dynamics simulations of the NR LBDs. The entry/exit channel for the ligand was initially believed to be only on the side of the receptor LBD where H12 is located[28], but several other competing ligand exit pathways were also identified[29–31], including one predicted by the analysis of the earlier TR LBD structure [7]. Only subtle protein conformational adaptations were shown to be required for ligand binding to the TR LBD irrespective of the entrance pathway, further indicating that H12 might not be the only, or even preferred route for ligand association/dissociation with the NR LBDs[32]. Consistent with the previous studies of radioactive estradiol dissociation from ER preparations [33], molecular dynamics (MD) simulations of ligand dissociation from the ER LBD revealed that preferred pathways of ligand dissociation from the LBD are mediated by the NR quaternary state [34]. Thus combined evidence gathered from crystallographic structures, NMR, MD simulations and biochemical studies all call for the questioning of the mouse trap model mechanism of NR activation.
Illusions of allostery
Most NR LBDs can bind alternatively to receptor-specific coactivators or corepressors, with the ligand acting as the switch for their coregulator exchange. For dimeric NRs, an important question has been whether two coregulators motifs bind equivalently to both subunits. Establishing the true binding stoichiometry between coregulator motifs and receptor dimers has proved to be particularly confusing in the case of RAR containing dimers Studies with isolated LBDs of RXR heterodimers (such as RXR-RAR) were interpreted to indicate that only one subunit in some RXR heterodimers can bind to the coativator derived LXXLL motif [35]. A combination of small angle X-ray scattering (SAXS) and X-ray crystallographic studies were applied in a study to understand how this asymmetric binding of coregulators is established with isolated RAR LBDs [35]. However, in this study the homodimer of RARβ LBD was used instead of the functional RXR-RAR heterodimer, and the authors proposed an allosteric mechanism to account for 1:2 stoichiometry of coactivator binding to RAR-RXR based on the LBD homodimer of RARβ. They postulated that the binding of the first LXXLL sends a signal across the dimer surface of to prevent the binding of the second motif, therefore allostery was invoked (Figure 2a) [35].
Figure 2.
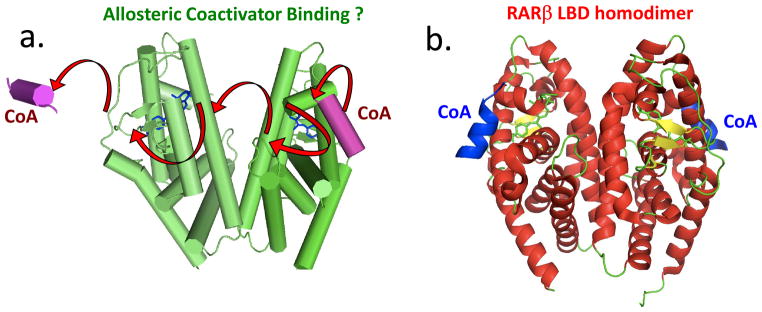
An allosteric control mechanism based on the crystal structure of the isolated RARβ LBD homodimer and small angle X-ray scattering (SAXS) solution data [35]. (a) The authors attempt to explain why only one coactivator peptide (shown in purple) might bind to RXR-RAR heterodimers, but the study used only the RARβ homodimer. Arrows indicated the authors’ suggested pathway for signal passage from one LBD to the adjacent LBD, to block coactivator binding. Solution-based and SAXS data suggest a 1:2 stoichiometry of coactivator: LBD binding [35] (b) A close examination of the deposited RARβ LBD X-ray coordinates show instead that each subunit of the homodimer has a bound coactivator peptide, inconsistent with the proposed allosteric model and SAXS interpretation (PDB ID 4DM6) [35].
The proposed allosteric mechanism, however, faces serious questions. To start with, no RARβ homodimers have been observed to be physiologically functional to date. Instead, RARs have only been described to function as heterodimers with RXR[36]. Therefore, conclusions drawn about allostery using RARβ homodimers may not be directly relevant to other nuclear receptors. Moreover, using the RARβ LBD homodimer as a proxy for the RAR-RXR LBD heterodimer is also not well-justified from the structural viewpoint given the buried solvent exposed area between the two RAR subunits is 17% larger than that formed between RAR and RXR [35]. The authors note that the RARβ recombinant protein used for the study was obtained from E. coli, where the monomeric form was predominantly seen, with only a minor fraction eluting as a homodimer [35]. This observation should have served as a strong warning that the RARβ homodimer is not a stable species in solution, and indeed that homodimer is not known to form in cells either. SAXS and X-ray crystallographic studies were, nevertheless applied in parallel to study the coactivator binding mode of the homodimeric RARβ, and both data sets were used to develop the notion that a single steroid receptor coactivator-1 (SRC-1) peptide binds to one side of the RARβ homodimer.
However, that interpretation is highly problematic, as the crystal structure of the RARβ homodimeric and the SAXS data are clearly inconsistent with each other. As shown in Figure 2b, the structural coordinates of this RARβ homodimer obtained from the PDB database indicated two SRC-1 coactivator peptides are bound to the RARβ homodimer, one on each subunit and both bound in the canonical fashion. Thus, the crystallographic coordinates do not support the SAXS data interpretations or the proposed mechanism for allostery that has been suggested (compare Figures 2a and 2b) [35]. Here, as with the mousetrap mechanism, we have carefully examined the crystal structures and propose that they do not strongly support the mechanisms suggested. Thus, we argue that the published interpretations should not be generalized to other NRs.
The low resolution disconnect
Innovations in single particle cryo-electron microscopy (EM) studies are increasingly allowing for near-atomic resolution of macromolecules [37]. Samples are imaged in a frozen-hydrated state at low temperatures, and neither staining nor chemical fixation is required. Macromolecules are embedded within the ice layer, and randomly oriented particles are photographed in different relative views. Large numbers of these 2D images are merged to generate 3D density maps or molecular envelopes. A clear advantage of this method is the lack of requirement for crystallizing macromolecules, which is by comparison tedious, time consuming and often unsuccessful. Another advantage is the samples for cryo-EM can be studied in a wider-range of buffers and salts, not necessarily those that produce crystals. The recent advances in resolution have come from the usage of direct electron detectors, powerful algorithms to correct for radiation-induced motion of particles, and improved methods in image processing.
At this time, however, EM studies applied to nuclear receptor complexes have only allowed interpretations in the 12–13 Å resolution range. These studies were applied to RXR-VDR and RXR-RAR complexes on DNA [38, 39]. Given the substantial resolution limits, these studies could not provide direct validation of the known critical features of full-length complexes, such as their ligand binding interactions, specific DNA-protein interactions, or LBD coregulator interactions. However, these EM low resolution pictures were used for large scale constructions of receptor-DNA complexes. This was done by placing the previously determined high resolution crystallographic structures of isolated LBDs and DBDs into the broad, low resolution envelopes that are obtained [38, 39].
Close examination of the RXR-VDR full-length complex EM data reveals several inconsistencies that question the accuracy of these reconstructions (Figure 3) [38]. The missing connections between LBDs and DBDs, became the foundation for a proposed “common architecture” [39]. But even more surprisingly, there is a clear disconnect in these images between the LBDs of each of RXR and VDR. This lack of LBD-LBD interactions contradicts decades of crystallographic studies that had been carried out on a number of LBD-LBD heterodimers involving RXR. The EM picture would suggest the RXR-VDR complex that was imaged may not be physiologically relevant, because it fails to show the properly dimerized functional state [40, 41]. Indeed, the standard distance and type of LBD-LBD dimerization interfaces that RXR uses have been established repeatedly by multiple investigators, and should involve closely interfaced helix 10/11 regions from each LBD[6, 40, 41]. Similar EM data for RXR-RAR has not yet been deposited in public repositories, and so we could not examine or display their picture [39].
Figure 3.
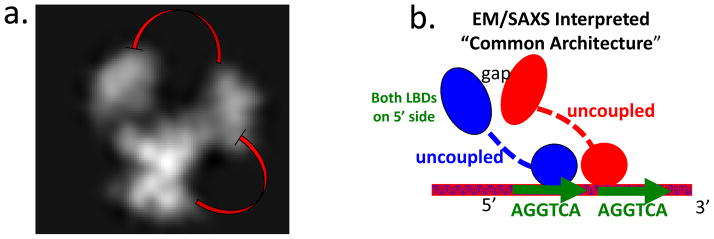

The features of the “common architecture” proposed for NRs on direct-repeats [39]. (a) The original electon microscopy (EM) data deposited for the full-length RXR-VDR complex (EM database EMD-1985) ([38]. The LBDs are distantly located from the DBDs. A significant and surprising gap is also apparent between the LBDs of VDR and RXR. This displacement of the two LBDS is inconsistent with previously described LBD-LBD interfaces observed for RXR-LXR, RXR-RAR, and RXR-PPAR complexes. (b) The EM/SAXS based proposes a concept of a common architecture for nuclear receptor full-length complexes that was supported by studies by the same authors based on fluorescence resonance energy transfer (FRET) [39]. That architecture consists of LBDs and DBDs being distally positioned from each other. The LBDs are to be positioned far to the 5′ side DNA half-sites, according to this proposed common architecture [38, 39, 44]. (c–e) The high-resolution crystal structures of multi-domain complexes of PPARγ-RXRγ on DR1 DNA, HNF-4α homodimer on direct-repeat with one base pair spacing (DR1) DNA, and RXRα-LXRβ bound to direct-repeat with four base pair spacing (DR4) DNA (PDB IDs 3E00, 4IQR, 4NQA)[15–17]. Each of these complexes displays multiple LBD-DBD interactions, contradicting the key features of the proposed “common architecture”. Red arcs indicate the actual domain-domain surfaces that are in direct physical contact in each of the high-resolution structures. None of these structures shows the LBDs positioned far to the 5′ side of the direct-repeats, as suggested by EM/SAXS studies.
The EM-derived RXR-VDR structure also appears difficult to reconcile with evidence regarding allostery from H/D ex MS studies on the same complex. The H/D ex MS studies have indicated a means of allosteric communication between the DBD and LBD in that full-length complex. Specifically, it was demonstrated that DNA binding is allosterically transmitted to the AF2 surface in the intact VDR RXR complex [42]. This type of allostery cannot be simply explained by the SAXS-generated description of the DBD and LBD of VDR, which shows absolutely no close contacts or interactions between these domains. Indeed, allostery has to be mechanistically reconciled with a signal being transmitted efficiently from one domain to a distal domain. Additional data presented in the study employing H/D ex MS further shows the lack of structured H12 moieties in the absence of ligand, also inconsistent with the notion of mouse-trap model [42].
The low-resolution EM images are also unable to address whether the DNA response element is bound specifically (with the AGGTCA half-sites engaged by the contact residues in the DBD). This is a critical question that should be verified, because NR quaternary structures are highly dependent on DNA binding. Even lower resolution information was then derived from a SAXS study [43]. As we have pointed out here, a previous correlation of a SAXS experiment with a crystallographic study failed to show agreement for the RARβ LBD, even in the hands of the same investigators [43]. Importantly, SAXS studies provide ab initio information about a protein envelope in solution and are not subjected to the influence of crystallographic packing artifacts, but the poor resolution (and thus limited informational content) and small number of structural restraints do not allow for singular or unambiguous assignment of polypeptide chains, providing only general information on the overall shape of a macromolecule. Moreover, using the SAXS technique alone does not allow one to visualize many the critical interfaces of full-length receptors (NR dimer, LBD-ligand, and DBD-DNA interfaces) and thus it should be complemented by additional experimental studies. Owing to these shortcomings, SAXS could be particularly vulnerable to interpretative errors. As an example, the SAXS data were interpreted by the study authors to show coactivator LXXLL binding to only one side of the RXR-PPAR heterodimer [43]. This interpretation has been inconsistent with multiple previous high-resolution crystal structures of RXR-PPAR LBD, which showed coactivator LXXLL motifs bind to both subunits equivalently [15, 41].
The common architecture myth
A structure of PPAR-RXR on DNA was never generated by EM [43]. Indeed, no PDB coordinate set has been ever deposited for a “solution structure” of this complex. Instead, the EM images on the RXR-VDR and RXR-RAR complexes were used in conjunction with a SAXS study of PPAR-RXR to produce a suggested picture of the PPAR-RXR/DNA “solution structure”[43]. These low-resolution data were further used to propose a broadly generalized “common architecture” for all NR dimers on direct-repeat DNAs [39].
The suggested common architecture has several key elements. The overall architecture was described as “open”, with the domains far apart (domains on a string) [39]. In particular, the DBDs were far out of contact range from the LBDs [44]. The LBDs were expected to lie along the far 5′ side (upstream) of the DBDs [44]. An obvious outcome of such a loosely organized architecture would be the preclusion of allosteric communications between DBDs and LBDs. Yet, multiple studies with a variety of different NRs had already established that DBDs allosterically communicate with LBDs [3, 42, 45, 46]. But, the single most ambitious aspect of the common architecture was its suggested generality [39]. The generalization to other NRs on direct-repeats, however, has since proven premature and problematic.
The common architecture concept was proposed based on the EM and SAXS studies of just one group, and other data emerging from crystallography, an independently reported SAXS study, multiple H/D ex MS studies, and mutagenesis studies on the full-length PPAR-RXR DNA complex were not integrated in constructing that architectural model. For example, an independently reported SAXS study on PPAR-RXR/DNA showed altogether different findings [43, 47]. Instead, the authors found that while the PPAR-RXR complex without DNA did have an open conformation, the binding of the heterodimer to the DNA generated a notably more compact state [47]. An H/D ex MS study carried out in the same report [47], and another H/D ex MS study independently conducted by another group was only consistent with PPAR-RXR DNA complex being in a notably more compact state [15]. The distinct patterns of protection on the PPAR-LBD between helix-2 and helix-3 were especially consistent with their being solvent protected, and indeed the observation from the PPAR-RXR/DNA crystal structure indicated that this region was buried by direct interfacial contacts with the RXR DBD [15, 47]. Mutagenesis studies were also suggestive of the compact state of PPAR-RXR in its DNA-bound form[15].
Crystallography delivers resolution and clarity
The first crystal structure showed how the PPAR-RXR full complex was highly organized and compacted on its consensus DR1 DNA [15]. A second structure followed five years later for the HNF-4α homodimer on DR1, and it also showed a compact state[16]. Most recently, the structure of the multi-domain RXR-LXR heterodimer on DR4 was reported [17]. The resolutions for all of these structures are in the range of 2.8–3.2 Å, much higher than the EM or SAXS studies. All crystal structures allowed the polypeptide chains to be unambiguously traced in each case, without the need to “glue” whole domains from previously reported crystal structures, as was done in the EM and SAXS studies.
In each crystal structure, it could be clearly established that the heterodimers are indeed specifically bound to their DNA sequences in the appropriate manner, and interact with bound ligands and coactivator segments on both subunits, in full agreement with previous studies. Every visualized interaction, such as the LBD-LBD interface, the DBD-DNA interface, the LBD-ligand interface, and the LBD-coregulator interface, has proved consistent with known modes of interaction established in the past twenty years of NR studies. At the same time, these three structures provide new insights about how quaternary arrangements are formed through domain-domain interfaces.
The quaternary features of the full-length PPAR-RXR structure have also been independently validated by H/D ex MS studies carried out on the same complex [15]. The HNF-4α domain-domain interfaces were thoroughly validated by multiple mutational, DNA-binding and transcriptional studies [16]. All of these studies support the DBD-LBD interactions in the crystal structures. Similarly, amino acid substitution at the interfacial face of the LBD of PPAR (F347) manifested in significant changes in DNA binding by the entire complex [15]. F347 is a particularly interesting, as the structure shows that it has two alternate positions, one of which is in close contact the RXR DBD on DNA. This toggle switching by F347 suggests that it may function as a key regulator for the subunit assembly process on DNA. The RXR-LXR and RXR-PPAR crystal structures are also consistent with previously established modes of DNA-binding asymmetry, with the RXR-DBD subunit occupying the downstream position in the RXR-PPAR complex, and occupying the upstream position in the RXR-LXR complex [15, 17].
All three crystal structures clearly contradict the basic rules of the “common architecture” proposed from the EM and SAXS interpretations (Figure 3a–e). For example, instead of DBDs being distant to LBDs, the crystal structures in every case show clearly connected DBD-LBD surfaces (Figure 3c-3). Whereas the early SAXS/EM data suggested LBDs were far upstream of DBDs, the crystal structures show that RXR-PPAR and RXR-LXR have their LBDs downstream of their DBDs, and both HNF-4α LBDs are firmly attached atop a DBD. Whereas the architecture was postulated to be shared among many NRs, each of the crystal structures showed a unique quaternary organization (Figure 3c–e). In the case of RXR-LXR, two distinct architectures were observed on DNA. This observation suggests a degree of structural flexibility in terms of how domains can reconfigure in the context of the full-length receptors. Both LXR-RXR structures still show clearly connected domain-domain contact surfaces within their architectures [17]. However, neither structure is an “open” configuration with disconnected DBDs and LBDs.
The architectural uniqueness among the crystal structures is also striking when one compares the RXRα-PPARγ structure to the HNF-4α structure, given that both use the DR1 response elements [15, 16]. Their uniqueness in quaternary structure comes from different sizes and sequences of the hinge regions of RXR, PPAR and HNF-4α. Furthermore, the non-conserved surface residues on the outside of each of these receptor LBDs are major determinants for how DBD-LBD interfaces form in each case. So far, the picture of the A/B domains contained in the N-terminal regions of receptors is one of disordered sequences without sequence conservation [16]. The RXR-LXR and HNF-4α structures did not include the A/B domains in the crystallized constructs [16, 17]. It remains difficult to ascertain whether other nuclear receptors have A/B domains that directly interact with other portions of their polypeptides, or if the disordered conformations of the A/B regions instead provide binding templates for interacting proteins.
A new guide to receptor architecture and allostery
Here, we describe a new set of architectural rules that are consistent with the multi-domain NR crystal structures, and with the larger body of solution-based and biophysical findings reported over the past two decades for both full-length receptors and their isolated domains. The NR quaternary architectures of NR dimers on DNA: a) use at least one type LBD-DBD physical interface that is DNA-dependent; b) use a DBD-DBD interface that is always DNA-dependent, and particularly sensitive to the size of the inter-half-site spacing within the response element; c) use LBD-LBD interfaces that form as had been previously shown with LBD structural studies, that is, with tight interfaces involving helix 10/11 elements of each LBD in a pseudo two-fold symmetric fashion; d) have the potential to form heterologous DBD-LBD interfaces employing different partners of the receptor heterodimer (this was seen in all three crystal structures), and e) have quaternary structures that are unique, despite the DBDs being highly conserved in sequence and the LBDs having similar overall folds.
It is clear that DNA binding is necessary for establishing most of the domain-domain interactions described in this section, except for the LBD-LBD interactions. This implies that the same receptor complex on two DNA elements (each with different inter-half-site spacing) would show altogether different quaternary organizations. When receptors are functionally organized on DNA, allosteric transmission pathways are established for transmitting signals through the domain-domain connections (Figure 4).
Figure 4.

Visualizing a pathway for allostery transmission within the HNF-4α homodimer-DNA complex. Shown is the crystal structure with the domain-domain junctions inside the red-circle [16]. Signals in the LBD, including post-translational modifications such as phosphorylation and methylation, as well as disease-linked mutations in the LBDs, can all be allosterically transmitted across the quaternary structure to weaken DNA binding [16].
Tightly knit domain-domain connections can allow for efficient transmission of a variety of different signals through the architecture. These signals can be post-translational modifications, ligand binding, DNA binding, and coregulator binding. Given the allosteric communication pathways appear in the context of full quaternary structures, new approaches should now be considered for therapeutic drug discovery. Past screening efforts have identified small-molecule receptor modulators using only the isolated LBDs, and not considering that allosterically acting ligands could be identified if the entire receptor complex is used in the screen.
How receptors organize in cells
In solution conditions where receptor architectures are studied, differing reagents can lead to uneven comparisons in the findings. Solution conditions such as pH, ionic conditions (affecting electrostatic interaction), glycerol concentration and detergent usage can have dramatic effects on the behaviours of complexes. Divalent cations can greatly impact DNA induced heterodimerization, as shown by EMSA studies and by Bioluminescence resonance energy transfer (BRET) [48, 49]. Some proteins lose significant DNA binding affinity as the ionic strength is increased. Indeed, every assay has a “biochemical bias”, where a particular conformation of NR is enriched prior to its characterization. Crystallographic studies should also be interpreted with reasonable caution, as the reagents and conditions used to achieve crystallization can impart unusual effects on the conformations and interactions that are subsequently observed. For instance, crystal packing can produce an artificial constraint on the observed conformations, and as shown in this article in the case of RXR H12, should be analysed particularly carefully in regions that are deemed to be novel or functionally important.
EMSA studies have been commonly used to establish NR binding to DNA. However this method can also bias the way we view NR-DNA interactions. In these studies, non-specific DNA sequences are often used to complex non-specific DNA binding proteins that could compete with the probe. However, non-specific DNAs can also be recognized in ways that interfere with NR dimerization, and could be reminiscent of infrequent (or random) DNA hits during a dynamic nuclear search pathway in living cells. It is possible that the EM and SAXS data were capturing the structures of complexes in a non-specific or random-DNA binding mode, explaining the lack of domain-domain interactions in the protein architectures. As discussed above, those receptor interactions could not be established within the EM/SAXS resolutions as being specific versus non-specific. In the crystal structures, direct visualizations confirmed highly specific DNA binding in each case, with the expected contacts visualized between amino acids and AGGTCA elements.
With those considerations in mind, Fluorescence recovery after photobleaching (FRAP) studies are now examining the NR cycle between the DNA-bound form and a diffuse pool of proteins in the nucleoplasm [50–52]. Additionally, BRET and Single Molecular Microscopy show that frequent low affinity DNA binding of MR and GR facilitates their search for specific sites, and that ligands can change the mobility of GR [53]. Large-scale chromatin immunoprecipitation (ChIP) combined with massively parallel sequencing (ChIP-Seq) studies to identify protein binding sites are beginning to show us that a significant proportion of endogenous sites might be due to the frequent non-specific DNA interactions that occur during the dynamic search pathway. Interestingly, nearly 1/3 of sites do not appear to contain a recognized canonical DR RE and may correspond to non-specific DNA binding events [54]. To distinguish receptor bound sites from those sites that are actually transcriptionally regulated by the same receptors, one study used a powerful approach of combining ChIP-seq data from PPARγ, RXR and RNA polymerase II (RNAPII) during adipocyte differentiation [55]. This kind of combined analyses allows a comprehensive and high-resolution genome-wide map of PPARγ:RXR target site binding to be differentiated in terms of transcriptionally productive versus non-productive binding events [55]. The binding mode of receptors could also involve tethering to other transcription factors that are occupying genomic DNA [56].
Important unanswered questions about nature of NR binding to DNA in vivo remain. For example, it is still unclear whether the DNA site fosters the two NR polypeptides to form into their architectural dimer, or if NR polypeptides are instead preformed dimers during their nuclear search for their specific DNA sites. Real time methods have now been applied to monitor the kinetics of NR polypeptide heterodimerization, as well as antagonist, agonist, and coregulator modulations of these kinetics. Using a BRET assay, one study suggests that the binding of a specific DNA element is an apparently significant driver of RXRα-PPARγ and RXR-TR heterodimerization, requiring the DBD integrity of both heteropartners [49]. This finding initially questioned the presence of a stable NR heterodimer in the absence of DNA in cells. However, a low and specific BRET signal suggested that heterodimers were still present in solution for RXRα-PPARγ and RXR-TR complexes even in absence of DNA, consistent with what had been suggested by others [47]. These observations indicate that low levels of pre-formed dimers exist to initiate interactions with DNA, but the DNA is still the overriding driver for receptor dimerization.
BRET studies have further examined the importance of the mutational site F347, which was observed crystallographically to locate on the PPAR LBD at its direct interface with the RXR-LBD. The F347 mutation in the PPARγ LBD, discussed earlier, was shown to stabilize heterodimerization between the PPARγ LBD and RXRα DBD [15]. The RXRα-PPARγ crystal structure shows the DNA-assembled state of the heterodimer, and it is clear that DBD-LBD interactions seen are entirely dependent on, and established by, the response elements, as pointed out in the crystallographic studies [15, 16]. Consistent with this requirement for DNA, an SAXS study did not find the RXR-PPAR interface in the heterodimer when it was unbound to DNA [47]. The authors suggest that a NR heterodimer may adopt different conformational states in solution. The importance of this F347 residue for heterodimerization has been tested in a BRET assay [49]. When mutated, rather than outright preventing DNA binding as suggested by EMSA experiments, the kinetics of heterodimer formation were markedly slowed during DNA binding[49]. The mutation reduced basal heterodimerization off-DNA, and also in the presence of non-specific DNA[49]. Interestingly, in live cells the PPARγ F347A mutation also altered subnuclear localization of the PPAR-RXR heterodimer. Taken together, these types of physiological studies are beginning to reveal the NR heterodimerization mechanisms within cells.
Concluding remarks
The structural studies with NRs over the past few decades focused on isolated DBDs and LBDs to understand their individual properties. We have discussed here how certain commonly invoked models, such as the mousetrap mechanism and an allosteric coregulator binding mechanism, have not been well-validated. Misguided notions about NR structures arise due to incorrect structural interpretations, which are made clear by the lack of correlation between SAXS and crystallographic studies. These misguided notions are exacerbated in some cases by over-generalization of the structural interpretations. We propose that allosteric control in NRs should be appropriately studied using detailed pictures from high-resolution structures of multi-domain NRs in their functional states.
Three intensive crystallographic efforts have now revealed the quaternary architectures for RXRα-PPARγ, HNF-4α and RXR-LXR complexes on DNA. These pictures are substantially detailed compared to the previous low-resolution EM and SAXS envelopes. The low-resolution interpretations had postulated a common architecture for NRs with entirely disconnected LBD and DBD domains. The crystal structures show instead directly connected LBDs and DBDs, and a variety of quaternary structures that are unique, rather than being commonly shared.
One of the most striking observations from the crystallographic studies is that paths are established for allosteric signal communication through tightly coupled domain-domain couplings. Multidisciplinary approaches, which include the combined use of high-resolution protein crystallography, EM, SAXS and NMR spectroscopy with a variety of biophysical and biochemical techniques such as H/D ex MS, FRET, BRET, FRAP and molecular dynamics simulations might provide ever more clear and detailed insight into the molecular mechanisms of signals transduction in the full-length NR settings. Embracing this concept of allosteric signal transmission should enhance future small- molecule screening efforts, as only isolated LBDs have typically been used to identify receptor modulators. New cellular studies are also revealing the molecular architecture of NRs inside living cells, addressing the key physiological questions of when, where and how NRs form their dimerization and DNA binding complexes.
Highlights.
The mousetrap mechanism, as the earliest concept of “active” versus “inactive” LBD conformations, appears to be misconceived due to crystal packing artifacts and their misinterpretations.
An alternate mechanism better explains how ligands can activate receptors, and is based on stabilization of helix-12 dynamics by activating ligands.
A recently proposed mechanism invoking allostery to explain how isolated LBDs of RXR-RXR bind coactivators is confounding, as it is based on contradictory SAXS and crystallographic results.
Low-Resolution Electron Microscopy and SAXS studies of RXR-VDR and RXR-RAR full-length receptors show disconnected DBD-LBD interactions and other domain displacements that suggest they may not be functional complexes.
A common architecture proposed for NR dimers based on EM/SAXS data interpretations suggests that DBDs and LBDs cannot directly communicate.
Three high-resolution crystal structures each showing extensive DBD-LBD domain-domain interactions and unique architectures, contradict the key elements of the EM/SAXS suggested common architecture.
The multi-domain crystal structures have LBD and DBD structures agree with all previous structures of isolated domains, as well as their known modes of binding with DNA, ligands and coregulators.
The crystal structures are further validated by mutagenesis and solution based studies (including a separately reported SAXS study) mass-spectrometry studies, BRET studiers. They also show DNA binding polarity that was clearly anticipated.
Domain-Domain interactions in all three crystal structures suggest that signals can be allosterically transmitted across receptor architectures.
Newly reported studies based on FRET, FRAP and BRET are revealing how NRs organize their dimerization and DNA interactions in real time and inside living cells.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mangelsdorf DJ, et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rastinejad F, et al. Understanding nuclear receptor form and function using structural biology. Journal of molecular endocrinology. 2013;51:T1–T21. doi: 10.1530/JME-13-0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Helsen C, et al. Evidence for DNA-binding domain--ligand-binding domain communications in the androgen receptor. Molecular and cellular biology. 2012;32:3033–3043. doi: 10.1128/MCB.00151-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khorasanizadeh S, Rastinejad F. Nuclear-receptor interactions on DNA-response elements. Trends in biochemical sciences. 2001;26:384–390. doi: 10.1016/s0968-0004(01)01800-x. [DOI] [PubMed] [Google Scholar]
- 5.Nagy L, Schwabe JW. Mechanism of the nuclear receptor molecular switch. Trends in biochemical sciences. 2004;29:317–324. doi: 10.1016/j.tibs.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 6.Huang P, et al. Structural overview of the nuclear receptor superfamily: insights into physiology and therapeutics. Annu Rev Physiol. 2010;72:247–272. doi: 10.1146/annurev-physiol-021909-135917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wagner RL, et al. A structural role for hormone in the thyroid hormone receptor. Nature. 1995;378:690–697. doi: 10.1038/378690a0. [DOI] [PubMed] [Google Scholar]
- 8.Renaud JP, et al. Crystal structure of the RAR-gamma ligand-binding domain bound to all-trans retinoic acid. Nature. 1995;378:681–689. doi: 10.1038/378681a0. [DOI] [PubMed] [Google Scholar]
- 9.Brzozowski AM, et al. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 10.Shiau AK, et al. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 11.Darimont BD, et al. Structure and specificity of nuclear receptor-coactivator interactions. Genes & development. 1998;12:3343–3356. doi: 10.1101/gad.12.21.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Webb P, et al. The nuclear receptor corepressor (N-CoR) contains three isoleucine motifs (I/LXXII) that serve as receptor interaction domains (IDs) Mol Endocrinol. 2000;14:1976–1985. doi: 10.1210/mend.14.12.0566. [DOI] [PubMed] [Google Scholar]
- 13.McKenna NJ, et al. Nuclear receptor coregulators: cellular and molecular biology. Endocrine reviews. 1999;20:321–344. doi: 10.1210/edrv.20.3.0366. [DOI] [PubMed] [Google Scholar]
- 14.Nolte RT, et al. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature. 1998;395:137–143. doi: 10.1038/25931. [DOI] [PubMed] [Google Scholar]
- 15.Chandra V, et al. Structure of the intact PPAR-gamma-RXR- nuclear receptor complex on DNA. Nature. 2008;456:350–356. doi: 10.1038/nature07413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chandra V, et al. Multidomain integration in the structure of the HNF-4alpha nuclear receptor complex. Nature. 2013;495:394–398. doi: 10.1038/nature11966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lou X, et al. Structure of the retinoid X receptor alpha-liver X receptor beta (RXRalpha-LXRbeta) heterodimer on DNA. Nature structural & molecular biology. 2014;21:277–281. doi: 10.1038/nsmb.2778. [DOI] [PubMed] [Google Scholar]
- 18.Kallenberger BC, et al. A dynamic mechanism of nuclear receptor activation and its perturbation in a human disease. Nature structural biology. 2003;10:136–140. doi: 10.1038/nsb892. [DOI] [PubMed] [Google Scholar]
- 19.Johnson BA, et al. Ligand-induced stabilization of PPARgamma monitored by NMR spectroscopy: implications for nuclear receptor activation. Journal of molecular biology. 2000;298:187–194. doi: 10.1006/jmbi.2000.3636. [DOI] [PubMed] [Google Scholar]
- 20.Berger JP, et al. Distinct properties and advantages of a novel peroxisome proliferator-activated protein [gamma] selective modulator. Mol Endocrinol. 2003;17:662–676. doi: 10.1210/me.2002-0217. [DOI] [PubMed] [Google Scholar]
- 21.Lu J, et al. Analysis of ligand binding and protein dynamics of human retinoid X receptor alpha ligand-binding domain by nuclear magnetic resonance. Biochemistry. 2006;45:1629–1639. doi: 10.1021/bi051474j. [DOI] [PubMed] [Google Scholar]
- 22.Hughes TS, et al. Ligand and receptor dynamics contribute to the mechanism of graded PPARgamma agonism. Structure. 2012;20:139–150. doi: 10.1016/j.str.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yan X, et al. Dynamics and ligand-induced solvent accessibility changes in human retinoid X receptor homodimer determined by hydrogen deuterium exchange and mass spectrometry. Biochemistry. 2004;43:909–917. doi: 10.1021/bi030183c. [DOI] [PubMed] [Google Scholar]
- 24.Figueira AC, et al. Analysis of agonist and antagonist effects on thyroid hormone receptor conformation by hydrogen/deuterium exchange. Mol Endocrinol. 2011;25:15–31. doi: 10.1210/me.2010-0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hamuro Y, et al. Hydrogen/deuterium-exchange (H/D-Ex) of PPARgamma LBD in the presence of various modulators. Protein science: a publication of the Protein Society. 2006;15:1883–1892. doi: 10.1110/ps.062103006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dai SY, et al. Unique ligand binding patterns between estrogen receptor alpha and beta revealed by hydrogen-deuterium exchange. Biochemistry. 2009;48:9668–9676. doi: 10.1021/bi901149t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greschik H, et al. Structural basis for the deactivation of the estrogen-related receptor gamma by diethylstilbestrol or 4-hydroxytamoxifen and determinants of selectivity. The Journal of biological chemistry. 2004;279:33639–33646. doi: 10.1074/jbc.M402195200. [DOI] [PubMed] [Google Scholar]
- 28.Blondel A, et al. Retinoic acid receptor: a simulation analysis of retinoic acid binding and the resulting conformational changes. Journal of molecular biology. 1999;291:101–115. doi: 10.1006/jmbi.1999.2879. [DOI] [PubMed] [Google Scholar]
- 29.Kosztin D, et al. Unbinding of retinoic acid from its receptor studied by steered molecular dynamics. Biophysical journal. 1999;76:188–197. doi: 10.1016/S0006-3495(99)77188-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martinez L, et al. Molecular dynamics simulations reveal multiple pathways of ligand dissociation from thyroid hormone receptors. Biophysical journal. 2005;89:2011–2023. doi: 10.1529/biophysj.105.063818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martinez L, et al. Molecular dynamics simulations of ligand dissociation from thyroid hormone receptors: evidence of the likeliest escape pathway and its implications for the design of novel ligands. Journal of medicinal chemistry. 2006;49:23–26. doi: 10.1021/jm050805n. [DOI] [PubMed] [Google Scholar]
- 32.Martinez L, et al. Only subtle protein conformational adaptations are required for ligand binding to thyroid hormone receptors: simulations using a novel multipoint steered molecular dynamics approach. The journal of physical chemistry B. 2008;112:10741–10751. doi: 10.1021/jp803403c. [DOI] [PubMed] [Google Scholar]
- 33.Zhong L, Skafar DF. Mutations of tyrosine 537 in the human estrogen receptor-alpha selectively alter the receptor’s affinity for estradiol and the kinetics of the interaction. Biochemistry. 2002;41:4209–4217. doi: 10.1021/bi0121095. [DOI] [PubMed] [Google Scholar]
- 34.Sonoda MT, et al. Ligand dissociation from estrogen receptor is mediated by receptor dimerization: evidence from molecular dynamics simulations. Mol Endocrinol. 2008;22:1565–1578. doi: 10.1210/me.2007-0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Osz J, et al. Structural basis for a molecular allosteric control mechanism of cofactor binding to nuclear receptors. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E588–594. doi: 10.1073/pnas.1118192109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang P, et al. Retinoic acid actions through mammalian nuclear receptors. Chemical reviews. 2014;114:233–254. doi: 10.1021/cr400161b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li X, et al. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nature methods. 2013;10:584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Orlov I, et al. Structure of the full human RXR/VDR nuclear receptor heterodimer complex with its DR3 target DNA. The EMBO journal. 2012;31:291–300. doi: 10.1038/emboj.2011.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rochel N, et al. Common architecture of nuclear receptor heterodimers on DNA direct repeat elements with different spacings. Nature structural & molecular biology. 2011;18:564–570. doi: 10.1038/nsmb.2054. [DOI] [PubMed] [Google Scholar]
- 40.Pogenberg V, et al. Characterization of the interaction between retinoic acid receptor/retinoid X receptor (RAR/RXR) heterodimers and transcriptional coactivators through structural and fluorescence anisotropy studies. The Journal of biological chemistry. 2005;280:1625–1633. doi: 10.1074/jbc.M409302200. [DOI] [PubMed] [Google Scholar]
- 41.Gampe RT, Jr, et al. Asymmetry in the PPARgamma/RXRalpha crystal structure reveals the molecular basis of heterodimerization among nuclear receptors. Molecular cell. 2000;5:545–555. doi: 10.1016/s1097-2765(00)80448-7. [DOI] [PubMed] [Google Scholar]
- 42.Zhang J, et al. DNA binding alters coactivator interaction surfaces of the intact VDR-RXR complex. Nature structural & molecular biology. 2011;18:556–563. doi: 10.1038/nsmb.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Osz J, et al. Solution Structures of PPARgamma2/RXRalpha Complexes. PPAR research. 2012;2012:701412. doi: 10.1155/2012/701412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Helsen C, Claessens F. Looking at nuclear receptors from a new angle. Molecular and cellular endocrinology. 2014;382:97–106. doi: 10.1016/j.mce.2013.09.009. [DOI] [PubMed] [Google Scholar]
- 45.Hall JM, et al. Allosteric regulation of estrogen receptor structure, function, and coactivator recruitment by different estrogen response elements. Mol Endocrinol. 2002;16:469–486. doi: 10.1210/mend.16.3.0814. [DOI] [PubMed] [Google Scholar]
- 46.Meijsing SH, et al. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. 2009;324:407–410. doi: 10.1126/science.1164265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bernardes A, et al. Low-resolution molecular models reveal the oligomeric state of the PPAR and the conformational organization of its domains in solution. PloS one. 2012;7:e31852. doi: 10.1371/journal.pone.0031852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moll JR, et al. Magnesium is required for specific DNA binding of the CREB B-ZIP domain. Nucleic acids research. 2002;30:1240–1246. doi: 10.1093/nar/30.5.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mulero M, et al. Analysis of RXR/THR and RXR/PPARG2 heterodimerization by bioluminescence resonance energy transfer (BRET) PloS one. 2013;8:e84569. doi: 10.1371/journal.pone.0084569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Royen ME, et al. FRAP and FRET methods to study nuclear receptors in living cells. Methods in molecular biology. 2009;505:69–96. doi: 10.1007/978-1-60327-575-0_5. [DOI] [PubMed] [Google Scholar]
- 51.van Royen ME, et al. Compartmentalization of androgen receptor protein-protein interactions in living cells. The Journal of cell biology. 2007;177:63–72. doi: 10.1083/jcb.200609178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maruvada P, et al. Dynamic shuttling and intranuclear mobility of nuclear hormone receptors. The Journal of biological chemistry. 2003;278:12425–12432. doi: 10.1074/jbc.M202752200. [DOI] [PubMed] [Google Scholar]
- 53.Groeneweg FL, et al. Quantitation of glucocorticoid receptor DNA-binding dynamics by single-molecule microscopy and FRAP. PloS one. 2014;9:e90532. doi: 10.1371/journal.pone.0090532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Evans RM, Mangelsdorf DJ. Nuclear Receptors, RXR, and the Big Bang. Cell. 2014;157:255–266. doi: 10.1016/j.cell.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nielsen R, et al. Genome-wide profiling of PPARgamma:RXR and RNA polymerase II occupancy reveals temporal activation of distinct metabolic pathways and changes in RXR dimer composition during adipogenesis. Genes & development. 2008;22:2953–2967. doi: 10.1101/gad.501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Glass CK, Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nature reviews Immunology. 2010;10:365–376. doi: 10.1038/nri2748. [DOI] [PubMed] [Google Scholar]
- 57.Bourguet W, et al. Crystal structure of the ligand-binding domain of the human nuclear receptor RXR-alpha. Nature. 1995;375:377–382. doi: 10.1038/375377a0. [DOI] [PubMed] [Google Scholar]
- 58.Wurtz JM, et al. A canonical structure for the ligand-binding domain of nuclear receptors. Nature structural biology. 1996;3:87–94. doi: 10.1038/nsb0196-87. [DOI] [PubMed] [Google Scholar]