

Abstract
BACKGROUNDS & AIMS
Chronic stress alters the hypothalamic–pituitary–adrenal axis, increases gut motility, and increases perception of visceral pain. We investigated whether epigenetic mechanisms regulate chronic stress-induced visceral pain in the peripheral nervous systems of rats.
METHODS
Male rats were subjected to 1 hr water-avoidance stress each day, or given daily subcutaneous injections of corticosterone, for 10 consecutive days. L4–L5 and L6–S2 dorsal root ganglia (DRG) were collected and compared between stressed and control rats (placed for 1 hour each day in a tank without water). Levels of cannabinoid receptor 1 (CNR1), DNA (cytosine-5-)-methyltransferase 1 (DNMT1), transient receptor potential vanilloid type 1 (TRPV1), and EP300 were knocked down in DRG neurons in situ with small interfering RNAs. We measured DNA methylation and histone acetylation at genes encoding the glucocorticoid receptor (NR3C1), CNR1, and TRPV1. Visceral pain was measured in response to colorectal distention.
RESULTS
Chronic stress was associated with increased methylation of the Nr3c1 promoter and reduced expression of this gene in L6–S2, but not L4–L5, DRGs. Stress was also associated with upregulation in DNMT1-associated methylation of the Cnr1 promoter and downregulation of glucocorticoid receptor-mediated expression of CNR1 in L6–S2, but not L4–L5, DRGs. Concurrently, chronic stress increased expression of the histone acetyltransferase EP300 and increased histone acetylation at the Trpv1 promoter and expression of the TRPV1 receptor in L6–S2 DRG neurons. Knockdown of DNMT1 and EP300 in L6–S2 DRG neurons of rats reduced DNA methylation and histone acetylation, respectively, and prevented chronic stress-induced increases in visceral pain.
CONCLUSIONS
Chronic stress increases DNA methylation and histone acetylation of genes that regulate visceral pain sensation in the peripheral nervous system of rats. Blocking epigenetic regulatory pathways in specific regions of the spinal cord might be developed to treat patients with chronic abdominal pain.
Keywords: Visceral hyperalgesia, HPA, epigenetic modification, pain sensitivity
Introduction
In human and animal models, chronic stress is commonly associated with enhanced abdominal pain (visceral hyperalgesia) and altered bowel function.1, 2 Despite considerable clinical and pre-clinical research, the pathophysiology of chronic stress-induced visceral hyperalgesia remains poorly understood. Alterations in CRF signaling, the substance P/neurokinin-1 receptor system, a disrupted intestinal epithelial barrier function and changes in the microbiota correlate with peripheral nociceptor sensitization.2, 3 Our previous studies revealed that chronic stress induces reciprocal changes in the anti-nociceptive endocannabinoid receptor 1 (CNR1; down-regulation) and pro-nociceptive endovanilloid TRPV1 (up-regulation) pain pathways in nociceptive L6-S2 DRG neurons that innervate the pelvic organs including the colon based on retrograde labeling and functional studies.4, 5 The CNR1 and TRPV1 pain pathways interact;6 activation of CNR1 inhibits TRPV1 function.7 How chronic stress modulates these pathways is unknown.
Epigenetics refers to a variety of processes which can have long-term effects on gene expression programs without changes in DNA sequence. Interest in epigenetic regulatory pathways has emerged rapidly recently because of their apparent significance in key physiological processes including cell cycle regulation. Important processes in epigenetic control include DNA methylation, catalyzed by DNA methyltransferases (DNMTs) including DNMT1 which is responsible for the maintenance of methylation patterns and DNMT3a and DNMT3b which are responsible for de novo methylation patterns, resulting in gene silencing,8, 9 and histone acetylation catalyzed by acetyltransferases that promote gene transcription.10 DNA methylation and histone acetylation are considered “stable yet reversible” because the relevant enzymes are inducible and may be altered by biochemical and environmental modulation.11, 12
Studies of epigenetic regulation in response to stress have been primarily focused on the central nervous system. For example, chronic stress alters the level of circulating glucocorticoids and DNA methylation at CpG islands on the glucocorticoid receptor (NR3C1) gene promoter in specific regions of the brain, resulting in altered NR3C1 expression and function in the HPA axis.13, 14 Little is presently known about the role of epigenetic mechanisms in modulating peripheral sensory nerve function. A subset of patients with irritable bowel syndrome (IBS) demonstrate altered expression of specific noncoding microRNAs in intestinal mucosa that inhibit glutamine synthetase, supporting a potential role for epigenetic pathways in this disorder.15 In the current study, we investigated a potential role for epigenetic regulatory pathways in chronic stress-enhanced abdominal pain (visceral hyperalgesia).
Materials and Methods
Animals
Male Sprague–Dawley rats (200–220 g) were obtained from Charles River Laboratories (Wilmington, MA). Animals were housed in an animal facility that was maintained at 22°C with an automatic 12 h light/dark cycle. The animals were given a standard laboratory diet and tap water was available ad libitum. All experimental procedures were performed in accordance with US National Institutes of Health guidelines and were approved by the University Committee on Use and Care of Animals at the University of Michigan. The experimenter was blinded to animal treatments during the assessment of the behavioral (pain) response.
Chronic water avoidance stress
Repeated exposure of adult rats to water avoidance (WA) stress was conducted as described previously.5, 16 The rats were placed on a glass platform in the middle of a Plexiglas tank filled with water (25°C) to 1 cm below the height of the platform. The animals were maintained on the tank for 1 hour in the morning daily for 10 consecutive days. The control (sham-stress) rats were placed similarly for 1 hour daily for 10 days in a tank without water.
Gene knockdown using small interfering RNA (siRNA)
For knockdown of the targeted genes in rat DRG neurons in situ, the pre-designed siRNA or the non-targeting negative control siRNA was dissolved in double-distilled water, diluted with the transfection reagent i-Fect (Neuromics, Edina, MN) to achieve a final concentration of 0.4 nM/10 μL, and injected intrathecally at the L6-S2 or L2–L3 spinal region every other day for a total of 5 injections in control and stressed rats during the 10-day stress phase. The following siRNAs were purchased from Ambion and used for gene knockdown: Nr3c1 (#s127819), Cnr1 (#s129266), Dnmt1 (#s136451), Trpv1 (#s136297), Ep300 (#s220365). Verification of region-specific gene knockdown at the spinal DRG levels was shown in the Supplemental Data using Trpv1 siRNA.
Visceromotor response (VMR) to colorectal distension (CRD)
Measurement of the VMR to CRD was conducted in separate groups of rats on day 11 following the completion of the 10-day WA stress procedure, as described previously.4, 5 Briefly, the VMR was quantified by measuring electromyographic activity in the external oblique musculature in the awake animals. CRD was conducted to constant pressures of 10, 20, 40, and 60 mm Hg by a custom-made distension control device. The responses were considered stable if there was less than 20% variability between 2 consecutive trials of CRD at 60 mm Hg. The increase in the area under curve (AUC), which is the sum of all recorded data points multiplied by the sample interval (in seconds) after baseline subtraction, was presented as the overall response during the course of the CRD test.
Methylated DNA immunoprecipitation (MeDIP) assay
MeDIP assay was performed using the EpiQuik™ Tissue MeDIP Kit (Epigentek, Farmingdale, NY), according to the manufacturer’s instruction. Briefly, the genomic DNA was sheared to random fragments between 200 bp to 1,000 bp. Immunoprecipitation was performed using monoclonal antibody against 5-methylcytosine (Epigentek) for the sample or normal mouse IgG as the negative control. The beads were treated with proteinase K for 1 h at 65°C and the methylated DNA was recovered by phenol-chloroform extraction followed by ethanol precipitation. Q-PCR amplification was performed. The relative changes in the methylation levels were normalized to the input DNA and Gapdh.
Chromatin immunoprecipitation (ChIP) assay
Chromatin immunoprecipitation was performed using a chromatin immunoprecipitation kit (EMD Millipore, Billerica, MA). In brief, DRG samples were cross-linked using 1% formaldehyde and terminated by incubation with 0.125 M glycine for 5 min. The cell lysate was incubated for 10 min at 4°C and the crude nuclear extract was collected by centrifugation at 600×g for 5 min at 4°C. The DNA was sonicated to random fragments between 200 bp to 500 bp. The chromatin was subjected to immunoprecipitation using the following antibodies: NR3C1 (#sc-1004; Santa Cruz Biotechnology, Dallas TX), DNMT1 (#13479; Cayman Chemical, Ann Arbor, MI) and acetyl-histone H3 (#P-2012; Epigentek). Normal mouse or rabbit IgG was used as a control. DNA was finally eluted in elution buffer and used for PCR or real-time PCR amplification using the same primer sets with MeDIP-qPCR.
Statistical analysis
To examine the VMR in response to CRD pressures, the electromyographic amplitudes, represented by calculating the AUC, were normalized as percentage of baseline response for the highest pressure (60 mm Hg) for each rat and then averaged for each group of rats. The effects of stress and/or pharmacologic treatment on the VMR to CRD was analyzed using a repeated-measures two-way ANOVA followed by Bonferroni post-test comparisons. Unpaired Student t test was used to examine the data for the protein/RNA expression obtained from Western blot, immunohistochemistry and qPCR. Results were expressed as means ± s.e.m. P < 0.05 was considered statistically significant.
Results
Chronic WA stress induced region-specific increase in DNA methylation of Nr3c1 promoter and decrease in NR3C1 expression in DRG neurons
Chronic WA stress induced a significant increase in CpG site (P1) methylation of the glucocorticoid receptor gene (Nr3c1) exon 17 promoter region (1738 to 1794) in nociceptive L6-S2 DRG neurons innervating the pelvic viscera (Figure 1A; P<0.05), similar to observations in the brain under stress conditions.17 No significant changes of CpG methylation at this promoter region was observed in L4–L5 DRG neurons that contribute to somatosensory innervation of the lower extremities (Supplementary Figure 1A). Methylated DNA immunoprecipitation (MeDIP) followed by PCR showed a significant increase in DNA methylation of the Nr3c1 promoter region near the transcription start site (TSS) in L6-S2 (P<0.01) but not L4–L5 DRG neurons (Figure 1B and C). Consistently, Nr3c1 mRNA and protein were decreased in L6-S2 but not L4–L5 DRG neurons of stressed rats (Figure 1D and E), predominantly in DRG neurons with a small soma, which correlate with peripherin-positive C-fibers that transmit pain (nociceptive) signals (P<0.05), but not in DRG neurons with large soma, which correlate with NF-200 positive A-fibers that transmit mechanical sensation (Figure 1F and Supplementary Figure 1B and C). These data indicate that chronic stress induces region-specific DNA methylation of the Nr3c1 promoter in L6-S2 but not L4–L5 DRGs, which is associated with region- and cell-specific down-regulation of NR3C1 expression in the subpopulation of nociceptive DRG neurons.
Figure 1.

Chronic water avoidance (WA) stress induced region-specific DNA methylation at the Nr3c1 promoter region and down-regulation in NR3C1 expression. (A) Pyrosequencing analysis of DNA methylation in the Nr3c1 exon 17 promoter region of L6-S2 DRG neurons from stressed and control rats. PCR neg: no input DNA; pos: MeDIP positive control using input DNA prepared from lumbar DRGs of control rats; neg: MeDIP negative control using normal mouse IgG; input: 4% input DNA from relevant DRGs for normalization; IP: MeDIP using anti-mouse 5-methylcytosine antibody. (B) Region-specific changes in DNA methylation of the Nr3c1 exon 17 promoter region measured by MeDIP and PCR. (C) Percentage change in Nr3c1 promoter methylation in control and stressed rats. (D) Relative mRNA levels of Nr3c1 in DRG neurons from control and stressed rats determined by qPCR. (E) Western blots for NR3C1 in different spinal (DRG) regions. (F) Quantification of the percentage of neurons that stained positive for NR3C1 in small vs. large L6-S2 DRG neurons. Data are expressed as mean ± stand error, n = 6 in each group. *P < 0.05, **P < 0.01 between control and WA stress rats.
NR3C1 regulates Cnr1 transcription in DRG neuron-derived F11 cells in vitro
Two potential glucocorticoid response elements (GREs) at cannabinoid receptor 1 gene (Cnr1) promoter region were identified through sequence blasting. Using the synthesized oligonucleotides identical to Cnr1 promoter GREs for DNA pull-down assay in F11 cells, we observed that NR3C1 bound to these oligonucleotides and CORT treatment increased the amount of NR3C1 pulled down by these two oligonucleotide sequences as shown in Figure 2A. Luciferase assay showed two Cnr1 promoter constructs (−376 to +87 and −1022 to +87 from TSS) had higher transcription activities (Figure 2B). NR3C1 knockdown by siRNA down-regulated NR3C1 protein expression and significantly reduced Cnr1 transcriptions in three of the four Cnr1 constructs (Figure 2B and C). These results support a role for NR3C1 as a positive transcription factor in modulation of Cnr1 transcription.
Figure 2.

Cnr1 transcription was regulated by NR3C1 in F11 cells. (A) DNA pull-down assay in F11 cells using synthesized glucocorticoid response elements (GREs). GRE1 (−95 to −124) and GRE2 (−247 to −276) represent two GREs near the Cnr1 TSS. (B) Cnr1 promoter transcription activity in F11 cells that were transfected with four different Cnr1 constructs: −1545 to +87, −1022 to +87, −376 to +87, and −203 to +87 from TSS. Transfected F11 cells were treated with Nr3c1 siRNA or vehicle. (C) Western immunoblots for NR3C1 in transfected F11 cells treated with/without Nr3c1-siRNA. * P<0.05; n = 4 for each treatment.
NR3C1 modulates CNR1 expression and function in DRG neurons in situ
Chromatin immunoprecipitation (ChIP) analysis revealed the binding of NR3C1 to Cnr1 promoter region in rat L6-S2 DRG neurons. The relative amount of NR3C1 binding to Cnr1 promoter decreased substantially in L6-S2 but not L4–L5 DRGs of stressed rats compared with controls (Figure 3A and Supplementary Figure 2A). Treatment with NR3C1 antagonist RU486 during the stress phase prevented the decrease of NR3C1 binding to the Cnr1 promoter (Figure 3B and C) and reversed the down-regulation of Cnr1 mRNA in L6-S2 DRG neurons of stressed rats (Figure 3D). Region-specific delivery of Nr3c1 siRNA decreased NR3C1 expression and down-regulated CNR1 protein expression in L6-S2 DRGs in situ in control rats (Figure 3E). Knockdown of CNR1 by siRNA in L6-S2 DRG neurons of control rats decreased CNR1 expression (Supplementary Figure 2B) and significantly increased the visceral pain response to colorectal distension, similar to the enhanced level observed in stressed rats (Figure 3F).
Figure 3.

NR3C1 regulated Cnr1 expression and function in DRG neurons in chronic stress in a region-specific manner. (A) ChIP using anti-NR3C1 antibody followed by PCR for Cnr1 promoter (−145 to −42) in L6-S2 and L4–L5 DRG neurons from control and stressed rats. PCR neg: no input DNA; ChIP pos: ChIP using input DNA prepared from lumbar DRGs of control rats; ChIP neg: ChIP with normal rabbit IgG. Input: 4% input DNA from relevant DRGs for normalization. (B) ChIP analysis of NR3C1 binding to Cnr1 promoter in L6-S2 DRG neurons in RU486-treated stressed rats. (C) Percentage of NR3C1 binding to Cnr1 promoter in L6-S2 DRG neurons. (D) Q-PCR analysis of relative Cnr1 mRNA levels in L6-S2 DRG neurons. (E) Western blots for NR3C1 and CNR1 in L6-S2 DRG neurons of rats treated with Nr3c1 siRNA or random sequence siRNA (control) delivered in situ. (F) Visceral motor response to colorectal distension in stressed and control rats treated with Cnr1 siRNA or vehicle (veh) delivered in situ. *P < 0.05 for stressed rats compared to controls or RU486-treated stressed rats; #P < 0.05 for stressed rats or Cnr1 siRNA-treated controls compared to vehicle-treated controls; two-way ANOVA followed by Bonferroni test; n = 6–8 per group.
Chronic WA stress increased DNMT1 expression in L6-S2 DRG neurons
The screening of three DNA methyltransferases revealed a robust increase in DNMT1 in L6-S2 DRG neurons from stressed rats compared with controls; alterations of DNMT3a (decreased) and DNMT3b (increased) were less significant (Figure 4A). Increased DNMT1 was detected in nuclei (Figure 4B) and DNMT1 activity was significantly (P < 0.05) increased in L6-S2 DRG neurons in stressed rats (Figure 4C). Immunofluorescence staining demonstrated that the increased level of DNMT1 was evident in small soma L6-S2 DRG neurons (Supplementary Figure 3A and B). Retrograde labeling using CTB-FITC also showed that DNMT1 was significantly increased in L6-S2 DRG neurons that innervate the colon (Figure 4D and Supplementary Figure 3B). Co-immunoprecipitation experiments revealed that DNMT1 binds to methyl binding protein 2 (MeCP2), and the level of binding increased significantly in L6-S2 DRG neurons from stressed rats (Supplementary Figure 3C), supporting the activation of DNMT1 for CpG methylation under chronic stress conditions. Consistently, CORT treatment in isolated DRGs from healthy control rats increased DNMT1 protein expression in L6-S2 but not L4–L5 DRGs which was prevented by RU486 (Figure 4E).
Figure 4.
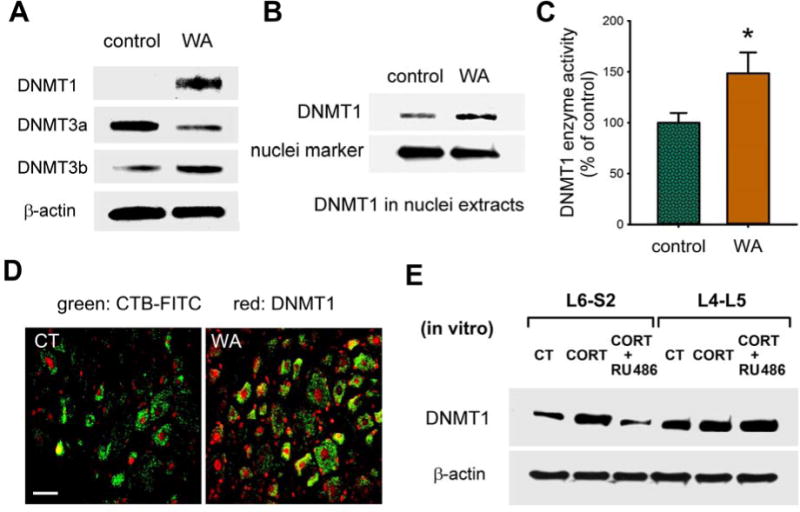
Increased DNMT1 expression and function in L6-S2 DRGs in chronic WA stress. (A) Western blots for DNMTs in L6-S2 DRG neurons from control and stressed rats. (B) Western immunoblots for DNMT1 in isolated nuclei fragmentation from L6-S2 DRGs of the stressed and control rats. (C) DNMT1 activity in nuclei of L6-S2 DRG neurons. (D) Immunofluorescence staining of DNMT1 (shown red) in L6-S2 DRG neurons innervating the colon. CTB-FITC (shown green) was used for retrograde labeling. Scale bar: 40 μm. (E) CORT treatment of isolated DRGs from control rats with/without RU486. n = 5 in each group.
DNMT1 mediates chronic WA stress-induced Cnr1 promoter methylation and function
Using MeDIP, we determined that methylation of the Cnr1 promoter region near the TSS was increased significantly (P < 0.05) in L6-S2 DRG neurons from stressed rats compared with controls, and this increase was prevented by RU486 treatment in situ (Figure 5A). In contrast, histone H3 acetylation was unchanged in this Cnr1 promoter region (Supplementary Figure 3D and E). Luciferase assay showed that pre-methylation of the Cnr1 promoter constructs inhibited Cnr1 transcription activity in both CORT-treated and untreated transfected cells (Figure 5B). Moreover, gene knockdown of DNMT1 at the L6-S2 level during the chronic stress phase prevented the increase in DNA methylation in the Cnr1 promoter region (Figure 5C), providing additional evidence that DNMT1 directly mediates methylation of the Cnr1 promoter in chronic stress. Finally, Dnmt1 siRNA delivered to L6-S2 level during the stress phase reduced DNMT1 expression, prevented CNR1 down-regulation caused by WA stress (Figure 5D), and significantly decreased (P < 0.05) the visceral motor response to colorectal distension at 40 and 60 mm Hg in stressed rats (Figure 5E), supporting an important role for DNMT1 in chronic stress–induced visceral hyperalgesia.
Figure 5.

DNMT1 mediates Cnr1 promoter methylation and visceral pain perception in chronic stress. (A) MeDIP of Cnr1 promoter region 1 (−376 to −146) and 2 (−145 to −42) in L6-S2 DRG neurons of rats with/without RU486. (B) Cnr1 promoter transcription activity in constructs treated with CORT or vehicle with/without pre-methylation. *P < 0.05 for CORT-treated compared to vehicle; #P < 0.01 for pre-methylated compared to control; ¤P < 0.01 for CORT-treated compared to pre-methylated CORT-treated. (C) MeDIP of Cnr1 promoter region 2 in L6-S2 DRG neurons from rats treated with/without Dnmt1 siRNA during WA stress. (D) Western blots for DNMT1 and CNR1 in L6-S2 DRG neurons from rats treated with/without Dnmt1 siRNA. (E) Visceral pain in stressed rats treated with Dnmt1 siRNA compared to vehicle. *P < 0.05 for stressed rats compared to controls; #P < 0.05 for stressed rats compared to Dnmt1 siRNA–treated stressed rats; two-way ANOVA followed by Bonferroni test. n = 6–8 in each group.
Up-regulation of histone acetyltransferase EP300 enhanced Trpv1 histone H3 acetylation and visceral pain in Chronic WA stress
The level of acetyl-histone H3 in the Trpv1 promoter region was significantly (P < 0.05) increased in L6-S2 but not L4–L5 DRG neurons of stressed rats, compared to controls, and blocked by RU486 treatment during the stress phase (Figure 6A and B). In contrast, the level of methylation in the same Trpv1 promoter region in stressed rats was unchanged and knockdown of DNMT1 by siRNA in L6-S2 DRGs did not significantly alter TRPV1 expression (Supplementary Figure 4A and B). Histone acetyltransferase EP300 was significantly increased in L6-S2 DRG neurons of stressed rats compared to controls (Figure 6C). Immunofluorescence staining confirmed this result, and that EP300 and TRPV1 co-localization was significantly (P < 0.05) increased in L6-S2 DRG neurons of stressed rats (Supplementary Figure 4C and D). Blocking glucocorticoid receptor with the antagonist RU486 prevented stress-induced up-regulation of EP300 expression in L6-S2 DRG neurons (Supplementary Figure 4E), supporting linkage to stress-axis. ChIP assay showed that EP300 binds directly to Trpv1 promoter, an action increased by chronic stress (Figure 6D). Targeted delivery of Ep300 siRNA to L6-S2 DRG levels eliminated EP300 binding to Trpv1 promoter in stressed rats (Figure 6D), reduced histone H3 acetylation of Trpv1 promoter (Figure 6E), and prevented the increase of TRPV1 expression caused by chronic WA stress (Supplementary Figure 4F). Assessment of visceral pain demonstrated that region-specific knockdown of EP300 significantly decreased (P < 0.05) visceral motor response to colorectal distension in stressed rats (Figure 6F). Furthermore, knockdown of TRPV1 with siRNA in L6-S2 but not L2–L3 DRG region had a similar preventive effect (Figure 6F and Supplementary Figure 5).
Figure 6.

Histone acetyltransferase EP300 regulates Trpv1 histone H3 acetylation, protein expression and visceral pain perception in chronic stress. (A) ChIP using anti–acetyl-histone H3 antibody followed by PCR for Trpv1 promoter in L6-S2 DRG neurons. (B) Percentage of H3 acetylation (normalized to control) at Trpv1 promoter in groups treated with/without RU486. *P < 0.05 for the stressed group compared to the control or RU486-treated group. (C) Western blots for EP300 in L6-S2 DRG neurons from control and stressed rats (n = 4 rats per group). (D) ChIP analysis using anti-EP300 antibody followed by PCR for Trpv1 promoter in L6-S2 DRG neurons from stressed rats treated with Ep300 siRNA compared to untreated. (E) ChIP analysis using anti–acetyl-histone H3 antibody followed by PCR for Trpv1 promoter in L6-S2 DRG neurons from stressed rats treated with Ep300 siRNA compared to untreated rats. (F) Visceral pain assessment in stressed rats treated with Ep300 siRNA or Trpv1 siRNA compared to untreated rats. *P < 0.05 for stressed rats compared to the control or siRNA-treated group; two-way ANOVA followed by Bonferroni test. n = 6–8 in each group.
Discussion
In this study, we report several novel findings which support the interpretation that epigenetic regulatory mechanisms play a pivotal role in chronic stress-induced visceral hyperalgesia, affecting nociceptive DRG neurons in a region-specific manner. Specifically, chronic WA stress induced DNA methylation of Nr3c1 promoter region resulting in down-regulation of its protein expression in a region- and cell (nociceptive)-specific manner in L6-S2 DRG neurons innervating the pelvic organs but not L4–L5 DRG neurons that contribute to the sciatic nerve somatosensory innervation of the lower extremities. NR3C1 acts as a positive transcription factor for the anti-nociceptive endocannabinoid CNR1 receptor in primary sensory DRG neurons. The “maintenance” DNA methyltransefase, DNMT1, is inducible under chronic stress and mediates de novo methylation of the Cnr1 promoter, and down-regulation of CNR1 receptor expression and function. Concurrently, chronic WA stress increased expression of histone acteyltransferase EP300 which enhanced histone H3 acetylation of pro-nociceptive endovanilloid Trpv1 promoter and receptor expression and function. Moreover, targeted delivery of siRNAs for either DNMT1 or EP300 to the L6-S2 spinal region during chronic stress blocked the changes observed in the specific epigenetic pathways and prevented the enhancement in abdominal pain (visceral hyperalgesia) (summarized in Figure 7).
Figure 7.
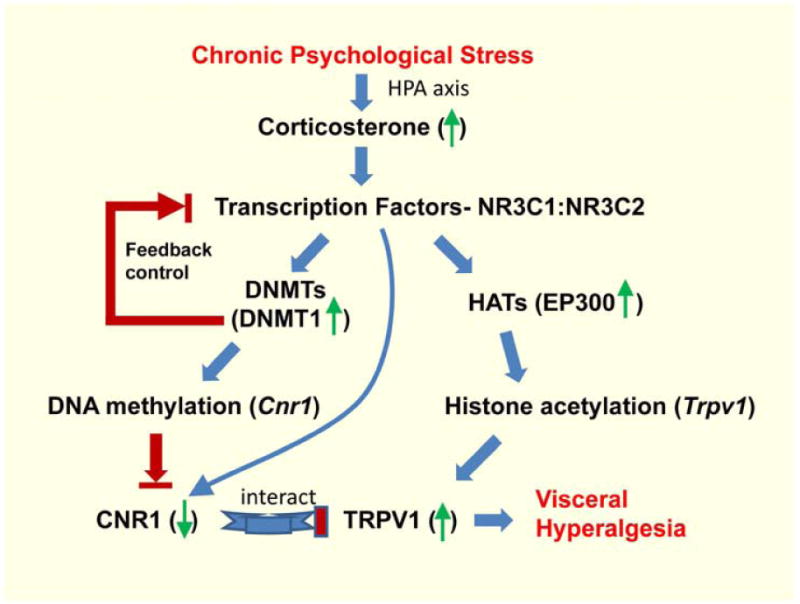
Proposed epigenetic regulation of CNR1 and TRPV1 pathways underlying chronic stress–induced visceral hyperalgesia in primary nociceptive DRG neurons. Chronic stress increases serum corticosterone, which activates glucocorticoid receptors (NR3C1) and mineralocorticoid receptors (NR3C2). NR3C1 and NR3C2 activation enhances transcription of DNA methyltransferases (DNMTs), i.e. DNMT1, and histone acetyltransferases (HATs), i.e. EP300. This leads to increased methylation of Cnr1 promoter region, culminating in decreased transcription factor (NR3C1) binding and thus reduced Cnr1 transcription and expression, and increased histone acetylation of Trpv1 promoter, resulting in increased Trpv1 transcription and function. These events occur preferentially in nociceptive DRG neurons innervating the GI tract.
Glucocorticoid receptor is an important target for epigenetic regulation in the HPA axis. Dysregulation in Nr3c1 transcription is associated with stress vulnerability. For example, increased DNA methylation in Nr3c1 promoter(s) is tightly linked to the down-regulation of NR3C1 expression in the brain that influences behavior, mood, learning and memory.18, 19 In primary nociceptive DRG neurons innervating the pelvic organs, we observed that chronic stress-induced visceral hyperalgesia is linked to significant increase in DNA methylation of Nr3c1 promoter and down-regulation of Nr3c1 transcription and expression in nociceptive DRG neurons. It has been shown that transcriptional control of NR3C1 appears to be tissue specific.20 Consistently, Nr3c1 promoter methylation increased preferentially in L6-S2 but not L4–L5 DRG neurons in stressed rats, supporting region-specificity of epigenetic regulation. Moreover, NR3C1 bound to GREs in the Cnr1 promoter and regulated Cnr1 transcription and expression in DRG neurons, supporting a modulator role of NR3C1 in the peripheral pain pathway. The evidence that chronic stress decreased NR3C1 binding to Cnr1 promoter in L6-S2 but not L4–L5 DRG neurons, together with the observation that knockdown of CNR1 in L6-S2 DRGs induced visceral hyperalgesia in control rats, further demonstrate region-specific modulation of peripheral pain pathways under conditions of chronic stress. The difference in the Nr3c1 promoter methylation pattern between L6-S2 and L4–L5 DRGs suggests that different levels of DRGs along the spinal cord may be developmentally “hard-wired”, which may cause the region-specific response under stress conditions. Alternatively, it is possible that the DRGs neurons innervating the pelvic organs (including the colon) are exposed to a macromolecular milieu that triggers a low-grade inflammatory response and activation of primary afferent pain pathways due to the impaired intestinal and/or bladder barrier function in chronic stress.21, 22 Nevertheless, our data support a novel and significant role for epigenetic regulation of peripheral pain pathways in chronic stress. Consistent with this, epigenetic gene regulation in the peripheral sensory system mediates pain perception in mouse models of neuropathic and inflammatory pain.23, 24 It should be noted that chronic stress is also associated with changes in receptor expression/function, trophic factors and signal transduction pathways “upstream” to the peripheral nervous system including the spinal cord and CNS. For example, a recent report revealed that DNA methylation of Nr3c1 and Crf genes are differentially altered in the amygdala under conditions of chronic stress that is associated with visceral hyperalgesia.25 This suggests that epigenetic regulatory pathways in the CNS also play a role in modulation of visceral pain. It is unresolved whether the changes observed in the CNS occur independently, or are a response to peripheral input.
Endocannabinoid receptors including CNR1 have been shown to be involved in nociception in models of acute, inflammatory and neuropathic pain.26 A recent report illustrates that the HPA-axis and the endocannabinoid system interact under stressful conditions,27 implicating a role for endocannabinoids in the modulation of visceral pain. Our previous studies demonstrate that chronic WA stress induces corticosterone-mediated enhanced endocannabinoid levels and down-regulation of CNR1 receptor in DRG neurons innervating the colon, and that treatment with a CNR1 (CB1) agonist prevented stress-induced visceral hyperalgesia.4, 5 It has been suggested that elevated endocannabinoid levels, possibly induced by enhanced glucocorticoids, reduces the CNR1 expression level.28 Data presented in the current study demonstrate that epigenetic regulation plays an important role in chronic stress-induced down-regulation of CNR1 expression and function in primary nociceptive DRG neurons. First, DNA methylation levels at Cnr1 promoter region were significantly increased in L6-S2 DRG neurons which were blocked by the corticoid receptor antagonist RU486. Second, in vitro methylation of Cnr1 promoter region significantly decreased Cnr1 transcription. Third, knockdown DNMT1 decreased Cnr1 promoter methylation and increased CNR1 receptor expression and function. In addition, NR3C1, as a transcription factor, positively mediated Cnr1 transcription and expression. Nr3c1 expression itself was regulated by its promoter DNA methylation status. It should be noted that DNMT1 is generally recognized to maintain methylation patterns.9,9 Our observations demonstrate that DNMT1 is inducible under conditions of chronic stress, mediates de novo methylation of the Cnr1 promoter and regulates stress-induced visceral hyperalgesia, indicating that DNMT1 is a potential therapeutic target in treatment of chronic stress-related visceral pain.
It is generally accepted that the endocannabinoid pathway and its receptors can modulate TRP receptor function.6, 7 Endocannabinoids, such as anandamide, are elevated in colonic DRGs in WA stress rats,4 and can directly activate TRPV1.29 On the other hand, activation of CNR1 activates inhibitory Gi/o proteins and leads to inhibition of adenylyl cyclase, reduces production of the second messenger cyclic adenosine monophosphate (cAMP), which subsequently inhibits TRPV1 function through down-regulation of protein kinase-dependent phosphorylation.30 Therefore, the down-regulation of CNR1 receptor expression observed in DRG neurons inverting the colon in chronic stress rats may reduce the inhibitory effect of CNR1 on TRPV1 function, culminating in enhanced visceral pain sensation.
Several members of the TRP receptor family including TRPV1, TRPV3, TRPV4, and TRPA1 have been implicated in neuropathic and visceral pain.31 Our previous study demonstrates that chronic stress increases TRPV1 expression and function in colonic DRG neurons.4, 5 Knockdown of TRPV1 in L6-S2 DRG neurons prevented chronic stress-induced visceral hyperalgesia supporting an important role of TRPV1 in chronic stress-induced visceral pain. Consistently, up-regulation of TRPV1 has been reported in a variety of gastrointestinal diseases associated with enhanced visceral pain,32 including the IBS in human patients.33 It has been suggested that intrinsic modulators can sensitize TRPV1 via phosphorylation and thereby enhance the probability of channel gating by heat or other stimuli.34 However, how the expression level of TRPV1 is up-regulated is largely unknown. We observed that histone acetyltransferase EP300 was significantly increased as well as its binding to Trpv1 promoter region that correlated to increased expression of TRPV1 observed in L6-S2 DRG neurons in WA stress rats. The increased expression of EP300 might be caused by elevated corticosterone level since glucocorticoids can up-regulate EP300 expression.35 Knockdown EP300 expression not only decreased the amount of EP300 binding to Trpv1 promoter region but also reduced histone H3 acetylation of the Trpv1 promoter and prevented visceral hyperalgesia in stress rats. These data indicate a novel role of EP300-mediated histone acetylation that contributes to chronic stress-induced TRPV1 receptor up-regulation and visceral hyperalgesia.
The study of epigenetic regulation of pain pathways underlying chronic stress-induced visceral hyperalgesia was primarily focused on the L6-S2 DRG neurons in this study. Other cell types such as satellite glial cells may also be affected by WA stress.36 L6-S2 DRG neurons innervate the pelvic organs including the colon and bladder. Indeed, it has been reported that chronic stress also alters the function urinary bladder.21 The enhanced bowel-bladder interaction under chronic stress conditions may elucidate a potential mechanism underlying stress-associated visceral pain syndrome.37
In summary, we have demonstrated that epigenetic regulatory mechanisms play a potentially pivotal role in chronic stress–induced visceral hyperalgesia, affecting nociceptive DRG neurons in a region- and cell-specific manner. DNMT1-mediated methylation of upstream promoter sites results in down-regulation of anti-nociceptive Cnr1 expression, which reduces CNR1-mediated inhibition of TRPV1 function; whereas, EP300-mediated histone H3 acetylation results in up-regulation of pro-nociceptive TRPV1 expression and function. These observations support the significance of concurrent epigenetic regulation of the CNR1 and TRPV1 pain pathways in the peripheral nervous system under conditions of chronic stress. These data advocate novel applications of gene silencing to treat chronic abdominal pain using interventions that target peripheral sensory pathways in a region-specific manner, thereby avoiding CNS side-effects.
Supplementary Material
Acknowledgments
We wish to thank Dana Dolinoy and Caren Weinhouse for providing pyrosequencing analysis. We also thank Fletcher A. White (Indiana University) for the gift of the F11 cell line. This research was supported by grant R01DK098205 to J.W.W. and the University of Michigan Gastrointestinal Peptide Center Grant 5P30DK034933 from the US National Institutes of Health. Pilot Feasibility grant F014289 to S.H. was supported by the Michigan Gastrointestinal Peptide Research Center.
Abbreviations: The abbreviations used in this paper
- CNR1
cannabinoid receptor 1
- ChIP
chromatin immunoprecipitation
- CORT
corticosterone
- CRD
colorectal distension
- CTB
chlorotoxin B
- CRF
corticotropin releasing factor
- DNMT1
DNA (cytosine-5-)-methyltransferase 1
- DRG
dorsal root ganglion
- EMG
electromyography
- NR3C1
glucocorticoid receptor
- GRE
glucocorticoid response element
- HPA
hypothalamic-pituitary-adrenal
- IBS
Irritable Bowel Syndrome
- MeDIP
methylated DNA immunoprecipitation
- NR3C2
mineralocorticoid receptor
- TRP
transient receptor potential
- TRPV1
transient receptor potential vanilloid type 1
- VMR
visceromotor response
- WA
water avoidance
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
S.H. and G.Z. contributed equally to this work. S.H. and G.Z. designed, performed and analyzed the experiments. S.H. and J.W.W. conceived the study, supervised the research, and wrote the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Reference List
- 1.Myer PA, Mannalithara A, Singh G, Singh G, Pasricha PJ, Ladabaum U. Clinical and economic burden of emergency department visits due to gastrointestinal diseases in the United States. Am J Gastroenterol. 2013;108:1496–1507. doi: 10.1038/ajg.2013.199. [DOI] [PubMed] [Google Scholar]
- 2.Sanger GJ, Chang L, Bountra C, Houghton LA. Challenges and prospects for pharmacotherapy in functional gastrointestinal disorders. Therap Adv Gastroenterol. 2010;3:291–305. doi: 10.1177/1756283X10369922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Larauche M, Mulak A, Tache Y. Stress and visceral pain: from animal models to clinical therapies. Exp Neurol. 2012;233:49–67. doi: 10.1016/j.expneurol.2011.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hong S, Fan J, Kemmerer ES, Evans S, Li Y, Wiley JW. Reciprocal changes in vanilloid (TRPV1) and endocannabinoid (CB1) receptors contribute to visceral hyperalgesia in the water avoidance stressed rat. Gut. 2009;58:202–210. doi: 10.1136/gut.2008.157594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hong S, Zheng G, Wu X, Snider NT, Owyang C, Wiley JW. Corticosterone Mediates Reciprocal Changes in CB 1 and TRPV1 Receptors in Primary Sensory Neurons in the Chronically Stressed Rat. Gastroenterology. 2011;140:627–637. doi: 10.1053/j.gastro.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Storr MA, Sharkey KA. The endocannabinoid system and gut-brain signalling. Curr Opin Pharmacol. 2007;7:575–582. doi: 10.1016/j.coph.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 7.Jeske NA, Patwardhan AM, Gamper N, Price TJ, Akopian AN, Hargreaves KM. Cannabinoid WIN 55,212–2 regulates TRPV1 phosphorylation in sensory neurons. J Biol Chem. 2006;281:32879–32890. doi: 10.1074/jbc.M603220200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet. 2000;9:2395–2402. doi: 10.1093/hmg/9.16.2395. [DOI] [PubMed] [Google Scholar]
- 9.Miranda TB, Jones PA. DNA methylation: the nuts and bolts of repression. J Cell Physiol. 2007;213:384–390. doi: 10.1002/jcp.21224. [DOI] [PubMed] [Google Scholar]
- 10.Karlic R, Chung HR, Lasserre J, Vlahovicek K, Vingron M. Histone modification levels are predictive for gene expression. Proc Natl Acad Sci U S A. 2010;107:2926–2931. doi: 10.1073/pnas.0909344107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zocchi L, Sassone-Corsi P. Joining the dots: from chromatin remodeling to neuronal plasticity. Curr Opin Neurobiol. 2010 doi: 10.1016/j.conb.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller CA, Sweatt JD. Covalent modification of DNA regulates memory formation. Neuron. 2007;53:857–869. doi: 10.1016/j.neuron.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 13.Weaver IC, Champagne FA, Brown SE, Dymov S, Sharma S, Meaney MJ, Szyf M. Reversal of maternal programming of stress responses in adult offspring through methyl supplementation: altering epigenetic marking later in life. J Neurosci. 2005;25:11045–11054. doi: 10.1523/JNEUROSCI.3652-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Witzmann SR, Turner JD, Meriaux SB, Meijer OC, Muller CP. Epigenetic regulation of the glucocorticoid receptor promoter 1(7) in adult rats. Epigenetics. 2012;7:1290–1301. doi: 10.4161/epi.22363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou Q, Verne GN. miRNA-based therapies for the irritable bowel syndrome. Expert Opin Biol Ther. 2011;11:991–995. doi: 10.1517/14712598.2011.577060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bradesi S, Schwetz I, Ennes HS, Lamy CM, Ohning G, Fanselow M, Pothoulakis C, McRoberts JA, Mayer EA. Repeated exposure to water avoidance stress in rats: a new model for sustained visceral hyperalgesia. Am J Physiol Gastrointest Liver Physiol. 2005;289:G42–G53. doi: 10.1152/ajpgi.00500.2004. [DOI] [PubMed] [Google Scholar]
- 17.Oberlander TF, Weinberg J, Papsdorf M, Grunau R, Misri S, Devlin AM. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics. 2008;3:97–106. doi: 10.4161/epi.3.2.6034. [DOI] [PubMed] [Google Scholar]
- 18.Denk F, McMahon SB. Chronic pain: emerging evidence for the involvement of epigenetics. Neuron. 2012;73:435–444. doi: 10.1016/j.neuron.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weaver IC, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7:847–854. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- 20.Kino T. Tissue glucocorticoid sensitivity: beyond stochastic regulation on the diverse actions of glucocorticoids. Horm Metab Res. 2007;39:420–424. doi: 10.1055/s-2007-980193. [DOI] [PubMed] [Google Scholar]
- 21.Smith AL, Leung J, Kun S, Zhang R, Karagiannides I, Raz S, Lee U, Glovatscka V, Pothoulakis C, Bradesi S, Mayer EA, Rodriguez LV. The effects of acute and chronic psychological stress on bladder function in a rodent model. Urology. 2011;78:967. doi: 10.1016/j.urology.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng G, Wu SP, Hu Y, Smith DE, Wiley JW, Hong S. Corticosterone mediates stress-related increased intestinal permeability in a region-specific manner. Neurogastroenterol Motil. 2013;25:e127–e139. doi: 10.1111/nmo.12066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chiechio S, Zammataro M, Morales ME, Busceti CL, Drago F, Gereau RW, Copani A, Nicoletti F. Epigenetic modulation of mGlu2 receptors by histone deacetylase inhibitors in the treatment of inflammatory pain. Mol Pharmacol. 2009;75:1014–1020. doi: 10.1124/mol.108.054346. [DOI] [PubMed] [Google Scholar]
- 24.Uchida H, Ma L, Ueda H. Epigenetic gene silencing underlies C-fiber dysfunctions in neuropathic pain. J Neurosci. 2010;30:4806–4814. doi: 10.1523/JNEUROSCI.5541-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tran L, Chaloner A, Sawalha AH, Greenwood Van-Meerveld B. Importance of epigenetic mechanisms in visceral pain induced by chronic water avoidance stress. Psychoneuroendocrinology. 2013;38:898–906. doi: 10.1016/j.psyneuen.2012.09.016. [DOI] [PubMed] [Google Scholar]
- 26.Walker JM, Huang SM. Cannabinoid analgesia. Pharmacol Ther. 2002;95:127–135. doi: 10.1016/s0163-7258(02)00252-8. [DOI] [PubMed] [Google Scholar]
- 27.Carrier EJ, Patel S, Hillard CJ. Endocannabinoids in neuroimmunology and stress. Curr Drug Targets CNS Neurol Disord. 2005;4:657–665. doi: 10.2174/156800705774933023. [DOI] [PubMed] [Google Scholar]
- 28.Hill MN, Carrier EJ, Ho WS, Shi L, Patel S, Gorzalka BB, Hillard CJ. Prolonged Glucocorticoid Treatment Decreases Cannabinoid CB1 Receptor Density in the Hippocampus. 18. 2008. pp. 221–226. [DOI] [PubMed] [Google Scholar]
- 29.Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sorgard M, Di M V, Julius D, Hogestatt ED. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature. 1999;400:452–457. doi: 10.1038/22761. [DOI] [PubMed] [Google Scholar]
- 30.Hermann H, De PL, Bisogno T, Schiano MA, Lutz B, Di M V. Dual effect of cannabinoid CB1 receptor stimulation on a vanilloid VR1 receptor-mediated response. Cell Mol Life Sci. 2003;60:607–616. doi: 10.1007/s000180300052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brederson JD, Kym PR, Szallasi A. Targeting TRP channels for pain relief. Eur J Pharmacol. 2013;716:61–76. doi: 10.1016/j.ejphar.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 32.Chan CL, Facer P, Davis JB, Smith GD, Egerton J, Bountra C, Williams NS, Anand P. Sensory fibres expressing capsaicin receptor TRPV1 in patients with rectal hypersensitivity and faecal urgency. Lancet. 2003;361:385–391. doi: 10.1016/s0140-6736(03)12392-6. [DOI] [PubMed] [Google Scholar]
- 33.Akbar A, Yiangou Y, Facer P, Walters JR, Anand P, Ghosh S. Increased capsaicin receptor TRPV1-expressing sensory fibres in irritable bowel syndrome and their correlation with abdominal pain. Gut. 2008;57:923–929. doi: 10.1136/gut.2007.138982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holzer P. TRPV1 and the gut: from a tasty receptor for a painful vanilloid to a key player in hyperalgesia. Eur J Pharmacol. 2004;500:231–241. doi: 10.1016/j.ejphar.2004.07.028. [DOI] [PubMed] [Google Scholar]
- 35.Yang H, Menconi MJ, Wei W, Petkova V, Hasselgren PO. Dexamethasone upregulates the expression of the nuclear cofactor p300 and its interaction with C/EBPbeta in cultured myotubes. J Cell Biochem. 2005;94:1058–1067. doi: 10.1002/jcb.20371. [DOI] [PubMed] [Google Scholar]
- 36.Golovatscka V, Ennes H, Mayer EA, Bradesi S. Chronic stress-induced changes in pro-inflammatory cytokines and spinal glia markers in the rat: a time course study. Neuroimmunomodulation. 2012;19:367–376. doi: 10.1159/000342092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peng HY, Hsieh MC, Lai CY, Chen GD, Huang YP, Lin TB. Glucocorticoid mediates water avoidance stress-sensitized colon-bladder cross-talk via RSK2/PSD-95/NR2B in rats. Am J Physiol Endocrinol Metab. 2012;303:E1094–E1106. doi: 10.1152/ajpendo.00235.2012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.