

Abstract
We undertook a study of the mechanism by which Dr-positive bacteria invade epithelial cells. Our findings show that Dr-positive bacteria enter via a zipper-like mechanism that is independent of the Dr-induced mobilization of F-actin and of the signaling molecules that control Dr-induced F-actin rearrangements. We also observed that Dr-positive IH11128 bacteria entered cells that were positive for the caveola marker VIP21/caveolin (HeLa and Caco-2/Cav-1 cells) to the same extent as those that were not (parental Caco-2 cells). Using fluorescence labeling and confocal laser scanning microscopy, we provide evidence that during the adhesion step, the α5β1 integrin, which plays a pivotal role in Afa/Dr diffusely adhering Escherichia coli bacterial entry, is mobilized around adhering Dr-positive bacteria. We show that the receptor for Afa/Dr adhesins, glycosylphosphatidylinositol-anchored CD55; the raft marker, ganglioside GM1; and VIP21/caveolin are all recruited around adhering Dr-positive bacteria. We also observed that extracting membrane cholesterol with methyl-β-cyclodextrin (MBCD) did not affect the recruitment of CD55, GM1, or β1 integrin to adhering Dr-positive bacteria. In contrast, extracting or changing membrane-bound cholesterol by means of drugs that modify lipid rafts (MBCD, filipin III, or mevalonate plus lovastatin plus MBCD) inhibited the entry of Dr-positive IH11128 both into cells that expressed VIP21/caveolin (HeLa and Caco-2/Cav-1 cells) and into those that did not (parental Caco-2 cells). Finally, restoring cholesterol within the cell membrane of MBCD-treated cells restored Dr-positive IH11128 internalization.
Entering the cells that make up the epithelium is a significant way for a virulent bacterial pathogen to establish chronic or persistent infections (49). The presence of Dr-positive Escherichia coli correlates with chronic or recurrent urinary tract infections (12) and diarrhea in infants (4, 20, 65). Afa/Dr diffusely adhering E. coli (Afa/Dr DAEC) strains express a family of gene operons known as afa, dra, and daa that share a similar genetic pattern consisting of at least five genes, including genes A, B, C, D, and E (53). Gene E encodes the major structural proteins, which act as adhesins. The Afa/Dr adhesins include the afimbrial adhesin I (AfaE-I) (40) and adhesin III (AfaE-III) (42), the Dr hemagglutinin (51), the adhesin dr-II (60), and the fimbrial F1845 adhesin (4). Afa/Dr DAEC bacteria recognize membrane-associated glycosylphosphatidylinositol (GPI)-anchored proteins, decay-accelerating factor (CD55) (52), and carcinoembryonic antigen (CEA; CD66e) (24) on human epithelial cells by means of adhesins. We know that Afa/Dr DAEC strains penetrate into epithelial cells, although the bacterial factor(s) that triggers cell entry remains to be definitely identified. According to Le Bouguenec et al. (16, 17, 31), the AfaD protein encoded by the afaD gene acts as an invasin, whereas Nowicki et al. (21, 67), on the basis of evidence that a dra operon is needed for Dr-positive IH11128 bacterial cell entry, think that the DraE adhesin is sufficient to promote internalization, even though this strain expresses a DraD invasin (84). Afa/Dr DAEC uses both CD55 and the α5β1 integrin as receptor to enter epithelial cells, and it is one of the bacterial pathogens that exploit the microtubule network (21, 23). The results reported here confirm and extend previous findings on how uropathogenic Dr-positive IH11128 bacteria enter epithelial cells (23). By analyzing the initial steps of the infection process that depend on Dr adhesin, in particular, we tried to find out whether adhering Dr-positive bacteria recruited various lipid raft-associated molecules and whether this recruitment influenced the internalization of bacteria.
MATERIALS AND METHODS
Reagents and antibodies.
Methyl-β-cyclodextrin (MBCD), filipin III, H7, dl-mevalonic acid lactone (mevalonolactone), and lovastatin were supplied by Sigma-Aldrich Chimie SARL (L'Isle d'Abeau Chesnes, France). Erbstatin and U73122 were purchased from Biomol-Tebu (Paris, France).
A fluorescein isothiocyanate (FITC)-labeled mouse monoclonal antibody (MAb) directed against CD55 (decay-accelerating factor) (clone NaM16-4D3) was purchased from Bioatlantic (Nantes, France). A mouse MAb directed against VIP21/caveolin (caveolin-1) (clone 2297) was obtained from Transduction Laboratories (Lexington, Ky.). A mouse MAb directed against the integrin α5 subunit (CD49e) (clone SAM1) was purchased from Immunotech (Marseille, France). A rabbit polyclonal anti-β1 integrin (CD29) antibody (clone sc-8978) was obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, Calif.). A mouse MAb directed against β1 integrin (CD29) (clone MAR4) was purchased from Pharmingen (Paris, France). The FITC-labeled cholera toxin B subunit (CTB) was purchased from Sigma-Aldrich Chimie SARL. A rabbit immunoglobulin G anti-Dr adhesin antibody was generously provided by B. Nowicki (Texas University, Galveston). The Alexa Fluor 546-conjugated goat anti-rabbit and Alexa Fluor 488-conjugated goat anti-mouse antibodies used as secondary antibodies were from Molecular Probes, Inc. (Eugene, Oreg.).
Cell lines and culture.
Human cervical HeLa cells were cultured at 37°C in a 5% CO2-95% air atmosphere in RPMI 1640 with l-glutamine (Life Technologies) supplemented with 10% heat-inactivated (30 min at 56°C) fetal calf serum (FCS; Boehringer, Mannheim, Germany), as previously described (24). Cells were used for infection assays before they reached confluence, i.e., after 2 days in culture.
The parental Caco-2 cells were routinely grown in Dulbecco's modified Eagle's minimum essential medium (25 mM glucose) (Life Technologies, Cergy, France) supplemented with 15% heat-inactivated (30 min at 56°C) FCS and 1% nonessential amino acids (Life Technologies). Caveolin-1 cDNA-transfected Caco-2 cells (Caco-2/Cav-1) were cultured in the same medium as the parental Caco-2 cells, but in the presence of hygromycin (500 μg/ml). For maintenance purposes, the cell lines were passaged weekly, using 0.02% trypsin in Ca2+- and Mg2+-free phosphate-buffered saline (PBS) containing 3 mM EDTA. Experiments and cell maintenance were performed at 37°C in a 10% CO2-90% air atmosphere. The culture medium was changed daily. Cells were used before they reached confluence, i.e., after 5 days in culture.
Bacterial growth and cell infection.
The Dr-positive clinical isolate IH11128 was used (51). A recombinant E. coli strain expressing the Dr adhesin was obtained by transforming E. coli strain AAEC185 (6), which lacks type 1 pili, with the recombinant plasmid pCC90, which encodes the Dr adhesin (7). Mutant strains that carried the pCC90 plasmid, into which point mutations in draE were introduced by site-directed mutagenesis (7), were used. The E. coli HB101 strain, expressing type 1 pili, was used as a control.
Stock cultures were maintained in 10% glycerol at −80°C. Before the experiments, bacterial strains were transferred onto fresh Luria-Bertani agar (Difco Laboratories) and incubated at 37°C for 24 h. For each experiment, bacteria were subcultured in Luria-Bertani broth at 37°C for 18 h, with an appropriate antibiotic. On the day of the experiment, the bacteria were washed three times with sterile PBS and recovered with Hanks balanced salt solution (Invitrogen). Bacterial cells were counted in a Salumbini chamber and adjusted to an appropriate concentration.
The method used for bacterial infections of cultured epithelial cells has been described previously (23, 24). Briefly, the cells were washed twice with sterile PBS. E. coli bacteria were suspended in the cell culture medium, and the cells were infected with 108 CFU/ml. Infections with wild-type, Dr-positive IH11128 bacteria were conducted in the presence of 1% mannose to prevent type 1 fimbria-mediated binding. Infections with HB101 bacteria were conducted in the absence of 1% mannose in order to allow type 1 fimbria-mediated binding. The plates were incubated at 37°C in 10% CO2-90% air for 1 or 3 h. The monolayers were then washed three times with sterile PBS. All assays were conducted in triplicate with three successive cell passages.
Fluorescence labeling.
Cells were prepared on glass coverslips which were placed in 24-well tissue culture plates (TPP, ATGC, Paris, France). Immunolabeling was conducted without cell permeabilization, except for VIP21/caveolin labeling, in cells fixed with 3% paraformaldehyde for 15 min at room temperature, washed three times with PBS, and then treated with 50 mM NH4Cl for 10 min (to saturate the aldehyde function).
CD55 clustering was examined by direct immunofluorescence labeling using a FITC-labeled anti-CD55 MAb (diluted 1:100 in bovine serum albumin [BSA]-PBS). The recruitment of the ganglioside GM1 was investigated by direct fluorescence labeling using the FITC-labeled CTB (5 μg/ml) (34). The coverslips were incubated with the appropriate FITC-labeled reagent or antibody for 45 min at 22°C, and then the coverslips were washed three times with PBS. When VIP21/caveolin, β1 integrin, or the integrin α5 subunit was to be visualized, the infected cells were incubated with an anti-VIP21/caveolin MAb (diluted 1:100 in BSA-PBS), an anti-β1 integrin MAb (diluted 1:20 in BSA-PBS), or an anti-integrin α5 subunit MAb (diluted 1:5 in BSA-PBS), respectively, for 45 min at 22°C, washed three times with PBS, and then incubated with the appropriate secondary antibody, used at a dilution of 1:200 in 2% BSA-PBS, followed by three washes with PBS.
For the labeling of Dr-positive bacteria, a polyclonal anti-Dr antibody was used, and the bound antibody was revealed by using an Alexa Fluor 546-conjugated goat anti-rabbit antibody.
Preparations were mounted in Vectashield antifade mounting medium (Biosys SA, Compiègne, France). Specimens were examined by confocal analysis, which was conducted with a confocal laser scanning microscope (model LSM 510; Zeiss) equipped with an air-cooled argon ion laser at 488 nm and a helium neon laser at 543 nm and configured with an Axiovert 100 M instrument using a Plan Apochromat 63×/1.40 oil objective lens.
Scanning electron microscopy.
Monolayers were prepared on glass coverslips in 24-well tissue culture plates. After being infected, the cells were fixed in 2.5% glutaraldehyde in 0.1 M phosphate buffer for 30 min at room temperature. After being washed, the samples were postfixed in 2% OsO4 for 1 h and dehydrated in graded ethanol baths before critical-point drying with liquid CO2. Coverslips were then coated with gold-palladium and observed with a JEOL JSM-840-A scanning electronic microscope operated at 17 kV.
Quantification of cell-associated and intracellular bacteria.
E. coli cell association was determined by a quantitative determination of the numbers of bacteria associated with the infected cell monolayers. After being infected, the cells were washed three times with sterile PBS and lysed with sterile H2O. Appropriate dilutions were plated on tryptic soy agar (TSA) for determination of the numbers of viable cell-associated bacteria by counting of the bacterial colonies. Each cell association assay was conducted at least in triplicate, with three successive cell passages. The results were expressed as CFU of cell-associated bacteria per milliliter.
The internalization of E. coli was determined by the quantitative determination of the numbers of bacteria located within the infected cell monolayers by an aminoglycoside antibiotic assay. The concentration of gentamicin that reduced the bacterial count by 99.99% was determined in a preliminary experiment. After incubation, the monolayers were washed three time with sterile PBS and then incubated for 60 min in a medium containing 100 μg of gentamicin/ml. The bacteria that adhered to the cells were rapidly killed, whereas those located within cells were not. The monolayer was then washed with PBS and lysed with sterile H2O. Appropriate dilutions were plated on TSA for determination of the numbers of viable intracellular bacteria by counting of the bacterial colonies. The results were expressed as CFU of intracellular bacteria per milliliter or as percentages of the cell-associated bacteria.
Measurement of cell integrity and viability.
The cell integrity in all experiments using lipid-modifying drugs was determined by measuring the amount of lactate dehydrogenase (LDH) in the posttreatment culture medium (Enzyline LDH kit; Biomérieux, Dardilly, France). LDH is a cytoplasmic enzyme whose presence in the culture medium reflects the loss of plasma membrane integrity. The release of LDH into the culture medium of treated cells was determined by using a commercially available kit (Enzyline LDH kit; Biomérieux) according to the manufacturer's instructions. The control represented the spontaneous release of LDH activity into the supernatant of untreated cells, while the total release represented the LDH activity in the supernatant of cells lysed with H2O.
Cell viability was determined by measuring the numbers of living, metabolically active cells by the XTT assay (Cell Proliferation XTT-based kit II; Boehringer), which is based on the cleavage of the tetrazolium salt of sodium 3′-[1(phenyl-amino-carbonyl)-3,4-tetrazolium]-bis (4-methoxy-6-nitro) benzene sulfonic acid hydrate (XTT) by the dehydrogenase found in active mitochondria.
Treatment of cells with blockers of cell signaling molecules.
The drug concentrations used for cell treatments had previously been shown to block Afa/Dr DAEC-induced cell signaling (56, 57). A 25-μg/ml solution of the tyrosine protein kinase inhibitor erbstatin (methyl 2,5-dihydroxycinnamate) in dimethyl sulfoxide was added to the culture medium 60 min before infection. The aminosteroid U73122 (1-[6-[[17beta-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5- dione) (10 μM), a phospholipase Cγ (PLC-γ) inhibitor, was added to the culture medium 30 min before infection. A 50 μM solution of the protein kinase C (PKC) inhibitor H7 (1-[5-isoquinolinylsulfonyl]-2-methylpiperazine) was added to the culture medium 60 min prior to infection. All of these blockers remained present throughout the cell infection process. None of the treatments affected cell viability or cell integrity (not shown).
Treatment of cells with lipid-modifying drugs.
To investigate the role of lipid rafts in bacterial infection, we exposed the cells to no additive, MBCD (10 mM), or filipin III (5 μg/ml) for 60 min prior to bacterial infection. MBCD is an oligosaccharide molecule with a high affinity for sterols which effectively and rapidly extracts cholesterol from the plasma membrane (35), and filipin III is a molecule known to bind to cholesterol in the plasma membrane which impairs the invagination of caveolae (66). At the concentrations used, none of the treatments affected the integrity (LDH level in control cells, 16 ± 5 U/liter; in cells lysed with H2O, 2,995 ± 25 U/liter; in MBCD-treated cells, 19 ± 6 U/liter; in filipin III-treated cells, 18 ± 5 U/liter) or viability of the cells (not shown). The drug concentrations used had previously been shown to block lipid raft-dependent Dr-positive IH11128 cell entry (23).
Cholesterol biosynthesis in HeLa cells was inhibited, as was previously described (9). The cells were cultured in the presence of 250 μM dl-mevalonic acid lactone and 4 μM lovastatin in cell culture medium for 48 h before infection. Cholesterol was further extracted by treating the cells with MBCD (10 mM) for 60 min before infection.
The effect of MBCD was reversed in a subculture of MBCD-treated cells (10 mM, 60 min) in the presence of FCS (20%) for 3 h. As a control, MBCD-treated cells were also subcultured in the presence of RPMI medium.
Detergent extraction of cells.
For analysis of the detergent-resistant membrane (DRM), previously described detergent extraction and flotation protocols (9) were used. Cells were scraped into 1 ml of ice-cold 1% TNE-Triton X-100 (25 mM Tris-HCl [pH 7.5], 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, and 20 μg each of leupeptin, antipapain, pepstatin, and aprotinin/ml) and homogenized with five strokes in a tight-fitting Dounce homogenizer on ice. The detergent-insoluble fractions were purified through a sucrose gradient as follows. Samples were adjusted to 40% sucrose by adding 1 ml of 80% sucrose in 1% TNE-Triton X-100, layered under a sucrose gradient (3 ml of 35% sucrose, 2.5 ml of 25% sucrose, and 2.5 ml of 16% sucrose), and centrifuged at 39,000 rpm for 18 h in a swinging bucket rotor (model SW41; Beckman Instruments). The initial load and 10 1-ml fractions were collected for analysis. The enrichment profile of VIP21/caveolin in DRMs was analyzed by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis. Briefly, samples from the sucrose gradient were run in a 12.5% SDS gel and transferred to a polyvinylidene difluoride membrane (Amersham). The membranes were then subjected to Western blotting, using an anti-VIP21/caveolin antibody followed by a horseradish peroxidase-labeled secondary antibody which was then detected by enhanced chemiluminescence (Amersham).
Statistics.
The data are expressed as means ± standard deviations (SD) from at least three separate experiments of duplicates conducted with successive passages of cells. Statistical significance was assessed by Student's t test. Differences were considered significant at P values of <0.05.
RESULTS
The zipper-like internalization of Dr-positive IH11128 into HeLa cells is independent of the F-actin events that accompany Afa/Dr DAEC infection.
Cells infected for 3 h with Dr-positive E. coli and then processed for scanning electron microscopy analysis showed microvillus-like extensions emanating from the cell surface that were closely associated with adhering bacteria (Fig. 1A). In some adhering bacteria, the microvillus-like extensions emanating from the cell fused (Fig. 1B) or were enlarged (Fig. 1C) to form a zipper-like structure engulfing the adhering bacteria. This finding and previous data reporting a zipper-like internalization of AfaE-III-positive E. coli (31) suggest that this is probably the internalization mechanism used by all Afa/Dr DAEC strains to enter epithelial cells.
FIG. 1.

Scanning electron microscopic examination of the zipper-like internalization of Dr-positive IH11128 E. coli into HeLa cells. The cells were infected for 3 h at a concentration of 108 CFU/ml and were processed for scanning electron microscopy as described in Materials and Methods. The same zipper-like entry was observed for cells infected with the laboratory E. coli strain AAEC185, which lacks type 1 pili, transformed with the recombinant plasmid pCC90, which encodes Dr adhesin. (A) Microvillus-like extensions extend from the cell surface and are associated with bacteria. (B) Fused extensions forming a zipper-like structure. (C) Enlarged extensions forming a zipper-like structure.
The entry of pyelonephritogenic E. coli into HEp-2 cells has been reported to be linked to actin polymerization (85). Moreover, Goluszko et al. (22) have suggested that after the clustering of CD55, the mobilized cytoskeleton protein F-actin around adhering Dr-positive bacteria provides the motor force that is required for the bacteria to enter. To find out whether the zipper-like internalization of Dr-positive bacteria is an F-actin-dependent event, we used two mutant strains carrying the pCC90 plasmid, in which point mutations in draE were produced by site-directed mutagenesis (7). The pCC90-D54G mutant, in which Asp-54 was replaced with glycine, bound to CD55-positive CHO cells, but it promoted weak CD55 clustering activity around adhering bacteria (24). The pCC90-D54C mutant, in which Asp-54 was replaced with cysteine, exhibited less binding to CD55-positive CHO cells and had entirely lost its CD55 clustering activity (24). These two mutants were also found to have entirely lost the ability to promote F-actin disassembly in Caco-2 cells (56). F-actin mobilization around adhering Dr-positive bacteria was observed in HeLa cells infected with wild-type IH11128 bacteria, recombinant E. coli strain pCC90, and mutant strain pCC90-D54C (Table 1). In contrast, in cells infected with the pCC90-D54G mutant, no F-actin recruitment was observed around the adhering bacteria (Table 1). When we investigated the invasive capacity of wild-type Dr-positive IH11128 bacteria, recombinant E. coli pCC90, and mutant bacteria, we found that despite the differing mobilization activities of F-actin, there was no difference between their abilities to invade HeLa cells (Table 1). These findings suggest that the entry of Dr-positive bacteria into HeLa cells is not dependent on the mobilization of F-actin around adhering, Dr-positive bacteria.
TABLE 1.
Entry of Dr-positive and Dr mutant bacteria into HeLa cells
| Strain | F-actin mobilizationa | No. (%) of intracellular bacteriab |
|---|---|---|
| Wild-type IH11128 | Positive | 6.25 × 104 ± 2.23 × 104 (0.53) |
| Recombinant pCC90 | Positive | 7.45 × 104 ± 3.95 × 104 (0.63) |
| pCC90 D54C | Positive | 6.85 × 104 ± 3.42 × 104 (0.58) |
| pCC90 D54G | Negative | 1.04 × 104 ± 0.57 × 105 (0.88) |
For F-actin mobilization, the cells were infected for 3 h (108 CFU/ml). The cells were then processed to produce direct immunofluorescence labeling of F-actin with fluorescein-labeled phalloidin and for CLSM examination. F-actin recruitment around adhering bacteria forms a fine ring that outlines the entire circumference of the bacteria.
The internalized bacterial levels were determined as described previously (23). The results are expressed as the CFU per milliliter found intracellularly per well (n = 6) (data are means ± SD). A typical experiment was conducted over at least three successive passages of cells. Student's t test found no significant difference between the mutant strains and the recombinant and wild-type strains.
The disruption of apical F-actin in polarized HT-29 and Caco-2 enterocyte-like cells is followed by increased bacterial internalization (82). Afa/Dr DAEC promoted F-actin disassembly in intestinal INT407 and Caco-2 cells, a phenomenon regulated by a Ca2+-dependent signaling cascade including the Scr family of protein tyrosine kinases (PTKs), PLC-γ, and PKC (56, 57). Moreover, it was previously established that there is a strong link between the internalization of uropathogenic E. coli and host cell signaling involving tyrosine kinases in human kidney cells (55). We conducted experiments to find out whether PTKs, PLC-γ, and PKC are involved in the zipper-like internalization of Dr-positive bacteria into human cervical HeLa cells. To do this, we investigated whether inhibitors of these signaling molecules modified the internalization of Dr-positive E. coli. The results in Table 2 show that there was no change in Dr-positive IH11128 cell association or cell entry when the infection was conducted in the presence of blockers of signaling molecules.
TABLE 2.
Effect of signaling molecule inhibitors blocking Afa/Dr-induced F-actin disassembly on the association of Dr-positive IH11128 bacteria with HeLa cells and their penetration into HeLa cells
| Inhibitora | No. of cell-associated bacteriab | No. (%) of intracellular bacteriab |
|---|---|---|
| Control | 1.18 × 107 ± 0.56 × 107 | 5.02 × 104 ± 2.28 × 104 (0.42) |
| Erbstatin | 1.72 × 107 ± 0.66 × 107 | 9.10 × 104 ± 3.35 × 104 (0.52) |
| U73122 | 1.39 × 107 ± 0.31 × 107 | 1.55 × 105 ± 0.67 × 105 (1.09) |
| H7 | 1.39 × 107 ± 0.50 × 107 | 6.13 × 104 ± 2.35 × 104 (0.44) |
Erbstatin (25 μg/ml), U73122 (10 μM), and H7 (50 μM) were added to the culture medium 60 min prior to infection and remained present throughout the cell infection process. At the concentrations used, all of the blockers had previously been shown to be active, blocking the F1845- and Dr-induced disassembly of F-actin in INT407 cells (57) and Caco-2 cells (56). The cells were infected with Dr-positive IH11128 bacteria for 3 h at a concentration of 108 CFU/ml.
The levels of cell-associated bacteria and intracellular bacteria were determined as previously described (23). The results are expressed as CFU of cell-associated and intracellular bacteria per milliliter in each well (n = 6) (data are means ± SD). All assays were conducted in triplicate with three successive cell passages. Student's t test found no significant difference between the blockers and the control.
Recruitment of raft-associated molecules around adhering bacteria expressing Dr adhesin in epithelial cells.
As far as we were aware, it was not previously known whether raft-associated molecules play any role in the zipper-like mechanism of bacterial internalization. The nature, organization, composition, and role of lipid rafts, which are also known as DRMs, on the cell membrane have been recently revisited (27, 37, 72, 74). Some cells express rafts known as caveolae, which contain the caveola-specific marker VIP21/caveolin, whereas other cells express lipid rafts that do not express VIP21/caveolin. We tried to find out whether several known raft-associated molecules were mobilized around adhering bacteria during infection. For this purpose, we used a set of cultured epithelial cells, some of which expressed caveolae either constitutionally or newly and others of which did not. The expression of VIP21/caveolin was analyzed after the cells had been solubilized with Triton X-100. DRMs were purified in a sucrose flotation gradient (Fig. 2A), and VIP21/caveolin was detected by immunoblotting (Fig. 2B and C). The results show that VIP21/caveolin was considerably enriched in the low-density DRMs isolated from HeLa cells (Fig. 2B), whereas no VIP21/caveolin was found in the low-density DRMs isolated from parental Caco-2 cells (Fig. 2C), which is consistent with the results of a previous report (47). In contrast, VIP21/caveolin was highly enriched in low-density DRMs isolated from parental Caco-2 cells transfected with a cDNA encoding caveolin-1, which led to the appearance of caveolae (79) (Fig. 2C).
FIG. 2.

Expression of VIP21/caveolin in HeLa cells, undifferentiated parental Caco-2 cells, and caveolin-1 cDNA-transfected, undifferentiated Caco-2 cells. Cells were solubilized in Triton X-100, and DRMs were floated in a continuous sucrose gradient (39,000 rpm for 18 h in a swinging bucket rotor) (model SW41; Beckman Instruments). Fractions from the gradient were analyzed by SDS-polyacrylamide gel electrophoresis, followed by Western blot analysis to detect VIP21/caveolin (anti-VIP21/caveolin antibody followed by horseradish peroxidase-labeled secondary antibody, which was then detected by enhanced chemiluminescence) and Coomassie blue staining to detect proteins. (A) Fractions from the top to the bottom of the gradient correspond to a linear sucrose gradient of 16 to 32% sucrose. (B) Western blot showing VIP21/caveolin separated from solubilized membrane proteins of HeLa cells. (C) Western blot showing the presence of VIP21/caveolin in DRMs (18% sucrose) isolated from parental Caco-2 cells stably transfected with the VIP21/caveolin cDNA (Caco-2/Cav-1) (lane 1) and the absence of VIP21/caveolin in DRMs isolated from parental Caco-2 cells (lane 2).
The bacterial recruitment of lipid raft- and caveola-associated molecules (1, 72) was analyzed by confocal laser scanning microscopy (CLSM) of HeLa cells infected with the wild-type Dr-positive IH11128 bacteria (not shown) and the recombinant E. coli strain AAEC185, which lacks type 1 pili (6) and contains the recombinant plasmid pCC90 that encodes Dr adhesin (7) (Dr-positive bacteria) (Fig. 3). The lipid raft marker GM1 was revealed by direct labeling with FITC-labeled CTB (34). The specific caveola marker VIP21/caveolin was detected by indirect immunofluorescence labeling with an anti-VIP21/caveolin antibody. The labeling of CD55, VIP21/caveolin, and GM1 (Fig. 3A, E, and I, respectively) showed that these molecules are widely distributed at the cell surface of HeLa cells, giving rise to the characteristic diffuse punctate staining of raft-associated molecules (13). As previously reported (24), CD55 was recruited around adhering Dr-positive bacteria (Fig. 3B). Also, there was a considerable recruitment of both VIP21/caveolin and GM1 around adhering Dr-positive bacteria which formed fine rings around the circumference of adhering bacteria (Fig. 3F and J, respectively). The merged images of double-immunofluorescence staining of bacteria labeled with an anti-Dr adhesin and CD55, VIP21/caveolin antibody, or FITC-CTB (Fig. 3C, G, and K, respectively) and the CLSM analysis (0.5-μm-thick optical sections) of the fluorescence intensity along a transverse axis of double-labeled bacteria (Fig. 3D, H, and L, respectively) show that all of these raft markers colocalized with Dr adhesin expression by the bacteria. In contrast, for the rarely adhering E. coli AAEC185 bacteria randomly distributed at the cell surface, no GM1 clustering developed around the bacteria (Fig. 3M and N). In addition, a control consisting of HB101 bacteria infecting cells in the absence of 1% mannose in order to allow type 1 fimbria-mediated binding showed that no recruitment of GM1 developed around adhering bacteria (Fig. 3O and P).
FIG. 3.

Recruitment of CD55, VIP21/caveolin, and GM1 around adhering Dr-positive bacteria infecting epithelial HeLa cells. The cells were infected with the laboratory E. coli strain AAEC185, which lacks type 1 pili, transformed with the recombinant plasmid pCC90, which encodes Dr adhesin, for 1 h at a concentration of 107 CFU/ml. After infection, the samples were fixed and processed for double-immunofluorescence labeling with an anti-Dr adhesin antibody plus an anti-CD55 or anti-VIP21/caveolin antibody or FITC-CTB. Fluorescence was analyzed by CLSM. Optical sections (0.5-μm thick) that are representative of the apical parts of the cells are shown. (A, E, and I) Expression of CD55, VIP21/caveolin, and GM1, respectively, in uninfected HeLa cells. (B, F, and J) Recruitment of CD55, VIP21/caveolin, and GM1, respectively, around adhering Dr-positive bacteria infecting HeLa cells. Note that all of the adhering Dr-positive bacteria are positive for CD55, VIP21/caveolin, and GM1 labeling. The superimposition of double-immunofluorescence labeling for CD55, VIP21/caveolin, or GM1 (green) and Dr adhesin (red) appears in yellow in panels C, G, and K, respectively. The fluorescence intensities for CD55, VIP21/caveolin, or GM1 (green) and for Dr adhesin (red) were scanned across a transverse axis in a Dr-positive adhering bacterium (panels C, G, and K, white lines in white squares). (D, H, and L) Scanning intensities (green curves) of CD55, VIP21/caveolin, and GM1 colocalize with Dr adhesin (red curves) expressed by adhering bacteria. An identical mobilization of CD55, VIP21/caveolin, and GM1 was observed in cells infected with the Dr-positive IH11128 E. coli strain (not shown). No recruitment of GM1 was found around the rarely adhering E. coli strain AAEC185 (M and N). (O and P) No recruitment of GM1 occurred around HB101 bacteria adhering onto HeLa cells infected in the absence of mannose in order to allow type 1 fimbrial adhesion. It is important to note that no positive immunofluorescence was found around Dr-positive bacteria plated on a glass coverslip and subjected to immunofluorescence labeling with anti-CD55 and anti-VIP21/caveolin antibodies and FITC-CTB (not shown).
The above findings prompted us to investigate whether the raft marker GM1 was mobilized around adhering Dr-positive bacteria in parental Caco-2 cells that did not express VIP21/caveolin and whether GM1 and VIP21/caveolin were mobilized around adhering Dr-positive bacteria in cells that newly expressed caveolae (Caco-2/Cav-1 cells). Immunolabeling with an anti-CD55 antibody or FITC-CTB showed that CD55 and GM1 were recruited around Dr-positive bacteria adhering to parental Caco-2 cells and that CD55, VIP21/caveolin, and GM1 were recruited around adhering Dr-positive bacteria in parental Caco-2 cells transfected with a cDNA encoding caveolin-1 (not shown).
These results indicate, first, that Afa/Dr DAEC bacteria recruit GPI-anchored CD55 which is associated with lipid rafts that may or may not express VIP21/caveolin, and second, that the recruitment of the raft-associated molecules GM1 and VIP21/caveolin around adhering Afa/Dr DAEC bacteria is promoted by the Afa/Dr adhesin.
α5β1 integrin is recruited around adhering Dr-positive IH11128 bacteria.
Members of our laboratory recently reported that a polyclonal anti-α5β1 integrin antibody is able to block the entry of Dr-positive IH11128 bacteria into HeLa and undifferentiated parental Caco-2 cells, suggesting that this integrin may play a role in Afa/Dr DAEC internalization (23). We used immunofluorescence labeling and CLSM analysis to find out whether α5β1 integrin is recruited around adhering Dr-positive bacteria in infected HeLa cells. As shown in Fig. 4, in infected HeLa cells both the α5 integrin subunit (Fig. 4A) and β1 integrin (Fig. 4E) were recruited around adhering Dr-positive bacteria, forming fine rings around the circumference of cell-associated bacteria. CLSM analysis of the fluorescence intensity of double-labeled bacteria (Fig. 4C and G, white box) showed a strong colocalization between Dr adhesin (red curves) and the α5 integrin subunit or β1 integrin (green curves). We also tried to find out whether the β1 integrin was recruited around adhering Dr-positive bacteria in undifferentiated Caco-2/Cav-1 cells, which express VIP21/caveolin, and in undifferentiated parental Caco-2 cells, which do not. We found that the β1 integrin is also recruited around adhering Dr-positive bacteria (not shown).
FIG. 4.

The α5 integrin subunit and β1 integrin are recruited around adhering, Dr-positive bacteria infecting HeLa cells. The cells were infected as described in the legend to Fig. 3. After infection, the samples were fixed and processed for double-immunofluorescence labeling with an anti-α5 integrin subunit or anti-β1 integrin antibody (green fluorescence in panels A and E, respectively) and an anti-Dr adhesin antibody (red fluorescence in panels B and F). Note that all of the adhering Dr-positive bacteria were positive for α5 and β1 integrin labeling. The superimposition of both immunofluorescence labels appears in yellow (C and G). The fluorescence intensities for the α5 integrin subunit or β1 integrin (green) and for Dr adhesin (red) were scanned across a transverse axis in a Dr-positive adhering bacterium (panels C and G, white lines in white squares) and show that the α5 integrin subunit and β1 integrin colocalized with Dr adhesin expressed by the adhering bacteria (D and H, respectively). No labeling was found around Dr-positive bacteria plated on a glass coverslip and subjected to immunofluorescence labeling with an anti-α5 integrin subunit or anti-β1 integrin antibody (not shown). (I) HeLa cells treated with MBCD (10 mM) for 60 min before being infected show that β1 integrin remained recruited around adhering Dr-positive bacteria. The mobilization of the α5 integrin subunit and β1 integrin was the same in cells infected with the Dr-positive E. coli strain IH11128.
Do changes in the membrane-bound cholesterol level conversely affect the recruitment of molecules around adhering Dr-positive E. coli and bacterial internalization?
In order to find out whether a treatment with lipid-modifying drugs would have opposing effects on the recruitment of raft-associated molecules by adhering Dr-positive bacteria and on bacterial internalization, we treated the cells prior to infection with MBCD (10 mM) or filipin III (5 μg/ml). In addition, we conducted a combined cholesterol depletion-extraction treatment, in which we reduced cholesterol synthesis by culturing the cells prior to infection in the presence of lovastatin (4 μM) plus mevalonate (250 μM) and then subjected them to MBCD (10 mM) extraction (9).
We first investigated whether extracting the membrane-bound cholesterol with MBCD affected the expression of CD55, VIP21/caveolin, and GM1 and the recruitment of VIP21/caveolin and GM1 around adhering Dr-positive bacteria infecting HeLa cells. The distribution of CD55, GM1, and VIP21/caveolin at the cell surface was the same for MBCD-treated cells and untreated cells. After fluorescence labeling and CLSM analysis, we found that exposing the cells to MBCD prior to infection did not affect the recruitment of CD55, VIP21/caveolin, or GM1 around adhering Dr-positive bacteria. We further examined whether treatment with MBCD modified the recruitment of β1 integrin around adhering Dr-positive bacteria infecting HeLa cells. As we had already observed for CD55, GM1, and VIP21/caveolin, β1 integrin was still recruited around adhering, Dr-positive bacteria in MBCD-treated, infected cells (Fig. 4I).
We further investigated whether Dr-positive IH11128 bacteria were associated with or internalized within epithelial cells expressing lipid rafts that were positive or negative for VIP21/caveolin. The levels of cell-associated and internalized Dr-positive IH11128 bacteria were determined by quantitative bacterial counts (Table 3). Similar levels of cell-associated and internalized Dr-positive IH11128 bacteria were found in cells which did (HeLa cells) or did not (undifferentiated parental Caco-2 cells) constitutionally express VIP21/caveolin and in cells that newly expressed VIP21/caveolin (Caco-2/Cav-1 cells). We investigated whether drugs affecting lipid rafts and/or caveolae modified the entry of Dr-positive IH11128 into these cells. As shown in Table 3, a treatment with MBCD or filipin III inhibited the internalization of Dr-positive IH11128 bacteria to the same extent in all cell lines examined. Similarly, the combined cholesterol depletion-extraction treatment resulted in the inhibition of Dr-positive IH11128 internalization within HeLa cells. We noticed that filipin III, previously described as being specific to lipid rafts containing caveolae (2, 50, 54, 66, 83), blocked the internalization of Dr-positive IH11128 bacteria into undifferentiated parental Caco-2 cells, which did not express VIP21/caveolin, as well as into HeLa cells that constitutionally expressed VIP21/caveolin and into Caco-2 cells transfected with caveolin-1 cDNA, which newly expressed caveolae.
TABLE 3.
Effect of lipid-modifying drugs on cell entry of Dr-positive IH11128 bacteria
| Cell line | % (mean ± SD) of cell-associated bacteria found intracellularly with treatmentb
|
|||
|---|---|---|---|---|
| Untreated | MBCD | Filipin III | Lovastatin plus mevalonate plus MBCD extraction | |
| HeLa | 0.467 ± 0.152 | 0.004 ± 0.008* | 0.012 ± 0.007* | 0.009 ± 0.004* |
| Caco-2a | 0.424 ± 0.134 | 0.005 ± 0.008* | 0.021 ± 0.006* | ND |
| Caco-2/Cav-1a | 0.535 ± 0.145 | 0.014 ± 0.005* | 0.007 ± 0.008* | ND |
Undifferentiated cells (3 to 5 days in culture).
The cells were exposed for 60 min prior to infection to no additive, MBCD (10 mM), or filipin III (5 μg/ml). For the metabolic depletion of cholesterol, the cells were cultured in the presence of dl-mevalonic acid lactone (250 μM) and lovastatin (4 μM) in the cell culture medium for 48 h before infection, followed by exposure to MBCD (10 mM) for 1 h before infection. The cells were infected with Dr-positive IH11128 bacteria for 3 h at a concentration of 108 CFU/ml. The levels of cell-associated and intracellular bacteria were determined as described in Materials and Methods. The results are from six experiments.
, P < 0.01 versus untreated cells by Student's t test. ND, not determined. The levels of cell-associated bacteria were 1.78 × 107 ± 0.68 × 107, 1.67 × 107 ± 0.45 × 107, and 1.95 × 107 ± 0.58 × 107 CFU/ml in HeLa, parental Caco-2, and Caco-2/Cav-1 cells, respectively. The levels of cell-associated bacteria were not modified by the drug treatments (not shown).
To explain the results reported above, we hypothesized that the treatment of the cells with drugs that modify lipids could modify the association of signaling molecules that control bacterial internalization with the Afa/Dr DAEC-activated lipid rafts. We performed an experiment that was previously used to restore cholesterol within the cell membrane to lead to the reorganization of signaling molecules in the DRMs and the restoration of cellular responses (33, 38, 61). As reported in Fig. 5, the subculturing of MBCD-treated HeLa cells in the presence of FCS led to the restoration of Dr-positive IH11128 internalization to a level similar to that observed in untreated cells.
FIG. 5.
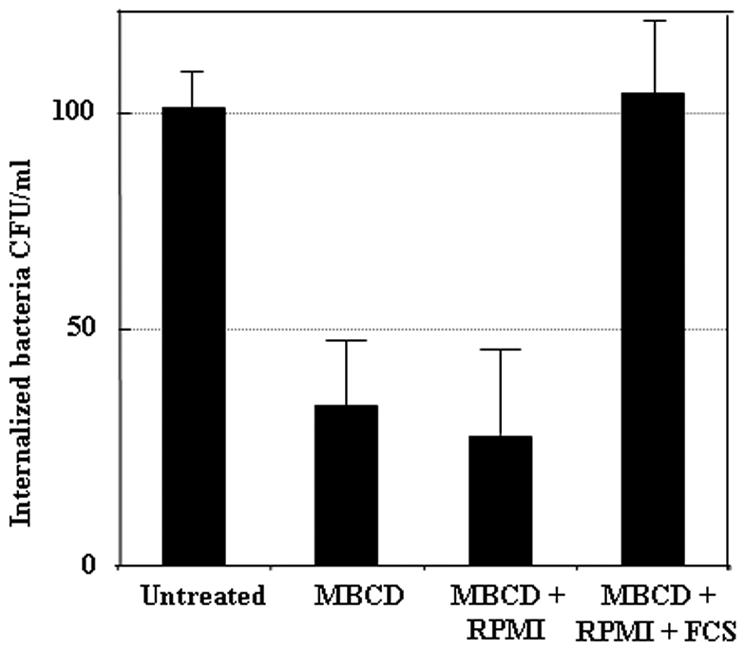
Reversal of inhibition of Dr-positive IH11128 internalization in HeLa cells treated with MBCD. The cells were exposed for 60 min prior to infection to no additive or to MBCD (10 mM). MBCD-treated cells were subcultured for 3 h in the presence of RPMI or FCS (20%). The cells were infected with Dr-positive IH11128 bacteria for 3 h at a concentration of 108 CFU/ml. The levels of intracellular bacteria were determined by a gentamicin assay. The results are expressed as CFU per milliliter of internalized bacteria ± SD (n = 4). P values were <0.01 for MBCD-treated cells and MBCD-plus-RPMI-treated cells versus untreated cells by Student's t test. P values were <0.01 for MBCD-plus-FCS-treated cells versus MBCD-treated cells.
DISCUSSION
The data reported here give new insights into the lipid raft-dependent internalization of Dr-positive bacteria within epithelial cells (21, 23). The results reported here indicate that (i) the internalization of Dr-positive bacteria occurs by a zipper-like mechanism which is independent of the Dr-induced mobilization of the microfilament cytoskeleton and of the signaling molecules that control the Dr-induced F-actin rearrangements; (ii) the lipid raft-dependent internalization of Dr-positive bacteria occurs in both cells that do and cells that do not express the marker of caveolae, VIP21/caveolin; (iii) during the adhesion step of infection, Dr-positive bacteria recruit CD55, GM1, VIP21/caveolin, and α5β1 integrin; and (iv) cholesterol, which has no apparent effect on the recruitment of CD55, VIP21/caveolin, GM1, or β1 integrin around adhering Dr-positive bacteria, is critical for bacterial internalization. Recent observations have highlighted the role of lipid rafts in microbial pathogenicity (11, 58, 64, 78). For example, cholesterol-rich domains are involved in the cell entry of Chlamydia trachomatis (32, 50), uropathogenic FimH-positive E. coli (2, 70, 71), and Mycobacterium kansasii (19, 59). Raft-associated molecular mechanisms are apparently engaged in mediating the cell signaling responses in the invasion process of Shigella flexneri, since bacterial entry is impaired after cholesterol depletion (41, 73). Moreover, cholesterol accumulates at bacterial entry sites and is retained by Salmonella-containing vacuoles after pathogen internalization (8, 18), and GPI-anchored proteins, GM1, and cholesterol are selectively incorporated into macropinosomes containing Brucella (81). Finally, during an enteropathogenic E. coli infection, the Ca2+-regulated membrane- and F-actin-binding protein annexin II is recruited into the cytoplasmic leaflet of CD44-containing lipid microdomains (86).
Pathogens entering host cells engage molecular mechanisms that vary widely from one pathogen to another (36). It was recently reported that uropathogenic Afa/Dr DAEC strains utilize α5β1 integrin as an internalization receptor (23). The data reported here reveal that the α5β1 integrin is recruited around adhering Dr-positive bacteria, together with raft-associated molecules, such as the ganglioside GM1 and the caveola marker VIP21/caveolin. Our results with Dr-positive E. coli and previous results with AfaE-III-positive E. coli (63) reveal that the α5β1 integrin is recruited around adhering Afa/Dr DAEC bacteria before internalization. The observations that α5β1 integrin is recruited around adhering Dr-positive bacteria and that it plays a pivotal role in internalization (23) are consistent with the zipper-like internalization observed in this study for Dr-positive bacteria and previously for AfaE-III-positive bacteria (31). The initial engulfment of Neisseria (5, 25), Listeria (28), Helicobacter (39), enteropathogenic E. coli (14), and Streptococcus (10) occurs via a zipper-like endocytosis mechanism. The prototype of zipper-like bacterial internalization is that of Yersinia, which, like that of Afa/Dr DAEC (23), involves the subversion of the α5β1 integrin (29) in a receptor-mediated mechanism that promotes the microfilament cytoskeleton-dependent advance of the pseudopod and involves receptor-ligand affinity, receptor clustering, signaling through focal adhesion kinase, and the stimulation of cytoskeletal rearrangements by small GTP-binding proteins (30). We observed that the zipper-like internalization of Dr-positive bacteria is independent of the events related to the microfilament cytoskeleton that accompany an Afa/Dr DAEC cell infection (3, 22, 56, 57). However, it is interesting that the mechanisms of cell entry used by uropathogenic FimH-positive and Dr-positive E. coli strains are different, although both of these pathogens use a raft-dependent internalization mechanism (2, 21, 23, 70, 71). Indeed, whereas the entry of FimH-positive E. coli into epithelial cells results from a massive cell membrane reorganization that is characteristic of a macropinocytic mechanism (2, 70, 71), Afa/Dr DAEC enters the cells by a zipper-like mechanism. Moreover, whereas cell entry by FimH-positive E. coli is accompanied by the activation of a cell signaling pathway involving protein tyrosine phosphorylation (46) and Rho-GTPase family members, namely Cdc42 and Rac1, controlling an F-actin-dependent process (45), we report here that the Dr-induced mobilization of F-actin and the signaling molecules controlling the Afa/Dr DAEC-induced F-actin events (56, 57) are not involved in the internalization of Dr-positive bacteria.
The data reported here reveal that raft-associated molecules, such as the ganglioside GM1 and the caveola marker VIP21/caveolin, are recruited around adhering Dr-positive bacteria together with α5β1 integrin. This suggests that the recognition of the GPI-anchored protein CD55 by Dr adhesin in turn activates the lipid rafts into which α5β1 integrin is recruited. Consistent with this, it has been observed that without cell stimulation, integrins are excluded from lipid rafts, but after activation, they are mobilized into the lipid raft compartment (15, 48, 77, 80). Moreover, lipid raft localization may increase integrin activity (26, 43). The prevailing current opinion is that different types of lipid rafts containing specific sets of molecules, which may or may not include GPI-anchored proteins that act by providing a platform for the concentration of signaling molecules, are present at the cell membrane (27, 37, 72, 74). It was previously reported that the recognition of CD55 by the Afa/Dr adhesins is accompanied by the activation of a signaling cascade including Src-family nonreceptor PTKs and PLC-γ (56, 57). Lipid rafts containing CD55 have been found associated with the Scr family of PTKs (44, 75). However, since GPI-anchored proteins localize only to the outer leaflet of the lipid bilayer, the signaling mechanism remains obscure. A growing body of evidence suggests that the kinases of the Scr family may be anchored to the plasma membrane as a result of the myristoylation or palmitoylation of their N-terminal residues. For instance, it has been reported that an association occurs between CD55 and the Src-like PTKs p56lck and p59fyn only when both palmitoylation of the amino-terminal cysteine residue(s) and myristylation of the amino-terminal glycine residue occur (68, 69). The disruption of membrane rafts by an MBCD treatment is generally followed by the exclusion of raft-associated molecules such as GPI-anchored proteins, GM1, and signaling molecules (13, 33, 38). In our experiments, when the membrane-bound cholesterol was extracted by an MBCD treatment at a concentration that was known to preserve the GPI-anchored proteins, we found that whereas the cell association of Dr-positive bacteria and the mobilization of CD55, GM1, VIP21/caveolin, and α5β1 integrin around adhering bacteria were not modified, the internalization of the bacteria was totally abolished. Moreover, an experiment conducted to replace the cellular cholesterol in MBCD-pretreated cells led to the restoration of the internalization of Dr-positive IH11128 bacteria. These results suggest that a cellular event occurs downstream of the mobilized raft-associated molecules to control bacterial internalization. It has been observed that cell signaling events are totally abolished by MBCD-induced lipid raft disruption (61, 62, 76). In light of this observation, it is tempting to speculate that the dissociation of rafts by MBCD treatment could break the connection between the GPI-anchored receptor, CD55, and the signaling molecules that control the internalization of Dr-positive bacteria. We are tempted to try to identify the signaling pathway that controls the bacterial internalization to find out how bacteria traffic intracellularly in order to survive. Since there is little information about how the zipper-like mechanism of internalization involving lipid rafts is regulated, the elucidation of the mechanism by which Dr-positive bacteria are internalized (21, 23) is clearly relevant for our understanding of such zipper-like mechanisms.
Acknowledgments
We thank André Le Bivic (NMDA-IBDM, Marseille, France) for his generous gift of the Caco-2/Cav-1 cell line.
Editor: V. J. DiRita
REFERENCES
- 1.Anderson, R. G. 1998. The caveolae membrane system. Annu. Rev. Biochem. 67:199-225. [DOI] [PubMed] [Google Scholar]
- 2.Baorto, D. M., Z. Gao, R. Malaviya, M. L. Dustin, A. van der Merwe, D. M. Lublin, and S. N. Abraham. 1997. Survival of FimH-expressing enterobacteria in macrophages relies on glycolipid traffic. Nature 389:636-639. [DOI] [PubMed] [Google Scholar]
- 3.Bernet-Camard, M. F., M. H. Coconnier, S. Hudault, and A. L. Servin. 1996. Pathogenicity of the diffusely adhering strain Escherichia coli C1845: F1845 adhesin-decay accelerating factor interaction, brush border microvillus injury, and actin disassembly in cultured human intestinal epithelial cells. Infect. Immun. 64:1918-1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bilge, S. S., C. R. Clausen, W. Lau, and S. L. Moseley. 1989. Molecular characterization of a fimbrial adhesin, F1845, mediating diffuse adherence of diarrhea-associated Escherichia coli to HEp-2 cells. J. Bacteriol. 171:4281-4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Billker, O., A. Popp, V. Brinkmann, G. Wenig, J. Schneider, E. Caron, and T. F. Meyer. 2002. Distinct mechanisms of internalization of Neisseria gonorrhoeae by members of the CEACAM receptor family involving Rac1- and Cdc42-dependent and -independent pathways. EMBO J. 21:560-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blomfield, I. C., M. S. McClain, J. A. Princ, P. J. Calie, and B. I. Eisenstein. 1991. Type 1 fimbriation and fimE mutants of Escherichia coli K-12. J. Bacteriol. 173:5298-5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carnoy, C., and S. L. Moseley. 1997. Mutational analysis of receptor binding mediated by the Dr family of Escherichia coli adhesins. Mol. Microbiol. 23:365-379. [DOI] [PubMed] [Google Scholar]
- 8.Catron, D. M., M. D. Sylvester, Y. Lange, M. Kadekoppala, B. D. Jones, D. M. Monack, S. Falkow, and K. Haldar. 2002. The Salmonella-containing vacuole is a major site of intracellular cholesterol accumulation and recruits the GPI-anchored protein CD55. Cell. Microbiol. 4:315-328. [DOI] [PubMed] [Google Scholar]
- 9.Coconnier, M. H., M. Lorrot, A. Barbat, C. Laboisse, and A. L. Servin. 2000. Listeriolysin O-induced stimulation of mucin exocytosis in polarized intestinal mucin-secreting cells: evidence for toxin recognition of membrane-associated lipids and subsequent toxin internalization through caveolae. Cell. Microbiol. 2:487-504. [DOI] [PubMed] [Google Scholar]
- 10.Dombek, P. E., D. Cue, J. Sedgewick, H. Lam, S. Ruschkowski, B. B. Finlay, and P. P. Cleary. 1999. High-frequency intracellular invasion of epithelial cells by serotype M1 group A streptococci: M1 protein-mediated invasion and cytoskeletal rearrangements. Mol. Microbiol. 31:859-870. [DOI] [PubMed] [Google Scholar]
- 11.Duncan, M. J., J. S. Shin, and S. N. Abraham. 2002. Microbial entry through caveolae: variations on a theme. Cell. Microbiol. 4:783-791. [DOI] [PubMed] [Google Scholar]
- 12.Foxman, B., B. Gillespie, J. Koopman, L. Zhang, K. Palin, P. Tallman, J. V. Marsh, S. Spear, J. D. Sobel, M. J. Marty, and C. F. Marrs. 2000. Risk factors for second urinary tract infection among college women. Am. J. Epidemiol. 151:1194-1205. [DOI] [PubMed] [Google Scholar]
- 13.Friedrichson, T., and T. V. Kurzchalia. 1998. Microdomains of GPI-anchored proteins in living cells revealed by crosslinking. Nature 394:802-805. [DOI] [PubMed] [Google Scholar]
- 14.Gabastou, J. M., S. Kerneis, M. F. Bernet-Camard, A. Barbat, M. H. Coconnier, J. B. Kaper, and A. L. Servin. 1995. Two stages of enteropathogenic Escherichia coli intestinal pathogenicity are up and down-regulated by the epithelial cell differentiation. Differentiation 59:127-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gagnoux-Palacios, L., M. Dans, W. Van't Hof, A. Mariotti, A. Pepe, G. Meneguzzi, M. D. Resh, and F. G. Giancotti. 2003. Compartmentalization of integrin alpha6beta4 signaling in lipid rafts. J. Cell Biol. 162:1189-1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garcia, M. I., P. Gounon, P. Courcoux, A. Labigne, and C. Le Bouguenec. 1996. The afimbrial adhesive sheath encoded by the afa-3 gene cluster of pathogenic Escherichia coli is composed of two adhesins. Mol. Microbiol. 19:683-693. [DOI] [PubMed] [Google Scholar]
- 17.Garcia, M. I., M. Jouve, J. P. Nataro, P. Gounon, and C. Le Bouguenec. 2000. Characterization of the AfaD-like family of invasins encoded by pathogenic Escherichia coli associated with intestinal and extra-intestinal infections. FEBS Lett. 479:111-117. [DOI] [PubMed] [Google Scholar]
- 18.Garner, M. J., R. D. Hayward, and V. Koronakis. 2002. The Salmonella pathogenicity island 1 secretion system directs cellular cholesterol redistribution during mammalian cell entry and intracellular trafficking. Cell. Microbiol. 4:153-165. [DOI] [PubMed] [Google Scholar]
- 19.Gatfield, J., and J. Pieters. 2000. Essential role for cholesterol in entry of mycobacteria into macrophages. Science 288:1647-1650. [DOI] [PubMed] [Google Scholar]
- 20.Giron, J. A., T. Jones, F. Millan-Velasco, E. Castro-Munoz, L. Zarate, J. Fry, G. Frankel, S. L. Moseley, B. Baudry, J. B. Kaper, et al. 1991. Diffuse-adhering Escherichia coli (DAEC) as a putative cause of diarrhea in Mayan children in Mexico. J. Infect. Dis. 163:507-513. [DOI] [PubMed] [Google Scholar]
- 21.Goluszko, P., V. Popov, R. Selvarangan, S. Nowicki, T. Pham, and B. J. Nowicki. 1997. Dr fimbriae operon of uropathogenic Escherichia coli mediate microtubule-dependent invasion to the HeLa epithelial cell line. J. Infect. Dis. 176:158-167. [DOI] [PubMed] [Google Scholar]
- 22.Goluszko, P., R. Selvarangan, V. Popov, T. Pham, J. W. Wen, and J. Singhal. 1999. Decay-accelerating factor and cytoskeleton redistribution pattern in HeLa cells infected with recombinant Escherichia coli strains expressing Dr family of adhesins. Infect. Immun. 67:3989-3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guignot, J., M. F. Bernet-Camard, C. Pous, L. Plancon, C. Le Bouguenec, and A. L. Servin. 2001. Polarized entry of uropathogenic Afa/Dr diffusely adhering Escherichia coli strain IH11128 into human epithelial cells: evidence for alpha5beta1 integrin recognition and subsequent internalization through a pathway involving caveolae and dynamic unstable microtubules. Infect. Immun. 69:1856-1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guignot, J., I. Peiffer, M. F. Bernet-Camard, D. M. Lublin, C. Carnoy, S. L. Moseley, and A. L. Servin. 2000. Recruitment of CD55 and CD66e brush border-associated glycosylphosphatidylinositol-anchored proteins by members of the Afa/Dr diffusely adhering family of Escherichia coli that infect the human polarized intestinal Caco-2/TC7 cells. Infect. Immun. 68:3554-3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hauck, C. R., and T. F. Meyer. 2003. “Small” talk: Opa proteins as mediators of Neisseria-host-cell communication. Curr. Opin. Microbiol. 6:43-49. [DOI] [PubMed] [Google Scholar]
- 26.Hogg, N., M. Laschinger, K. Giles, and A. McDowall. 2003. T-cell integrins: more than just sticking points. J. Cell Sci. 116:4695-4705. [DOI] [PubMed] [Google Scholar]
- 27.Ilangumaran, S., H. T. He, and D. C. Hoessli. 2000. Microdomains in lymphocyte signalling: beyond GPI-anchored proteins. Immunol. Today 21:2-7. [DOI] [PubMed] [Google Scholar]
- 28.Ireton, K., B. Payrastre, H. Chap, W. Ogawa, H. Sakaue, M. Kasuga, and P. Cossart. 1996. A role for phosphoinositide 3-kinase in bacterial invasion. Science 274:780-782. [DOI] [PubMed] [Google Scholar]
- 29.Isberg, R. R., and P. Barnes. 2001. Subversion of integrins by enteropathogenic Yersinia. J. Cell Sci. 114:21-28. [DOI] [PubMed] [Google Scholar]
- 30.Isberg, R. R., Z. Hamburger, and P. Dersch. 2000. Signaling and invasin-promoted uptake via integrin receptors. Microbes Infect. 2:793-801. [DOI] [PubMed] [Google Scholar]
- 31.Jouve, M., M. I. Garcia, P. Courcoux, A. Labigne, P. Gounon, and C. Le Bouguenec. 1997. Adhesion to and invasion of HeLa cells by pathogenic Escherichia coli carrying the afa-3 gene cluster are mediated by the AfaE and AfaD proteins, respectively. Infect. Immun. 65:4082-4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jutras, I., L. Abrami, and A. Dautry-Varsat. 2003. Entry of the lymphogranuloma venereum strain of Chlamydia trachomatis into host cells involves cholesterol-rich membrane domains. Infect. Immun. 71:260-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kabouridis, P. S., J. Janzen, A. L. Magee, and S. C. Ley. 2000. Cholesterol depletion disrupts lipid rafts and modulates the activity of multiple signaling pathways in T lymphocytes. Eur. J. Immunol. 30:954-963. [DOI] [PubMed] [Google Scholar]
- 34.Kenworthy, A. K., N. Petranova, and M. Edidin. 2000. High-resolution FRET microscopy of cholera toxin B-subunit and GPI-anchored proteins in cell plasma membranes. Mol. Biol. Cell 11:1645-1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kilsdonk, E. P., P. G. Yancey, G. W. Stoudt, F. W. Bangerter, W. J. Johnson, M. C. Phillips, and G. H. Rothblat. 1995. Cellular cholesterol efflux mediated by cyclodextrins. J. Biol. Chem. 270:17250-17256. [DOI] [PubMed] [Google Scholar]
- 36.Knodler, L. A., J. Celli, and B. B. Finlay. 2001. Pathogenic trickery: deception of host cell processes. Nat. Rev. Mol. Cell. Biol. 2:578-588. [DOI] [PubMed] [Google Scholar]
- 37.Kurzchalia, T. V., and R. G. Parton. 1999. Membrane microdomains and caveolae. Curr. Opin. Cell Biol. 11:424-431. [DOI] [PubMed] [Google Scholar]
- 38.Kwiatkowska, K., and A. Sobota. 2001. The clustered Fcgamma receptor II is recruited to Lyn-containing membrane domains and undergoes phosphorylation in a cholesterol-dependent manner. Eur. J. Immunol. 31:989-998. [DOI] [PubMed] [Google Scholar]
- 39.Kwok, T., S. Backert, H. Schwarz, J. Berger, and T. F. Meyer. 2002. Specific entry of Helicobacter pylori into cultured gastric epithelial cells via a zipper-like mechanism. Infect. Immun. 70:2108-2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Labigne-Roussel, A. F., D. Lark, G. Schoolnik, and S. Falkow. 1984. Cloning and expression of an afimbrial adhesin (AFA-I) responsible for P blood group-independent, mannose-resistant hemagglutination from a pyelonephritic Escherichia coli strain. Infect. Immun. 46:251-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lafont, F., G. Tran Van Nhieu, K. Hanada, P. Sansonetti, and F. G. van der Goot. 2002. Initial steps of Shigella infection depend on the cholesterol/sphingolipid raft-mediated CD44-IpaB interaction. EMBO J. 21:4449-4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Le Bouguenec, C., M. I. Garcia, V. Ouin, J. M. Desperrier, P. Gounon, and A. Labigne. 1993. Characterization of plasmid-borne afa-3 gene clusters encoding afimbrial adhesins expressed by Escherichia coli strains associated with intestinal or urinary tract infections. Infect. Immun. 61:5106-5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leitinger, B., and N. Hogg. 2002. The involvement of lipid rafts in the regulation of integrin function. J. Cell Sci. 115:963-972. [DOI] [PubMed] [Google Scholar]
- 44.Lisanti, M. P., M. Sargiacomo, and P. E. Scherer. 1999. Purification of caveolae-derived membrane microdomains containing lipid-anchored signaling molecules, such as GPI-anchored proteins, H-Ras, Src-family tyrosine kinases, eNOS, and G-protein-, beta-, and gamma-subunits. Methods Mol. Biol. 116:51-60. [DOI] [PubMed] [Google Scholar]
- 45.Martinez, J. J., and S. J. Hultgren. 2002. Requirement of Rho-family GTPases in the invasion of type 1-piliated uropathogenic Escherichia coli. Cell. Microbiol. 4:19-28. [DOI] [PubMed] [Google Scholar]
- 46.Martinez, J. J., M. A. Mulvey, J. D. Schilling, J. S. Pinkner, and S. J. Hultgren. 2000. Type 1 pilus-mediated bacterial invasion of bladder epithelial cells. EMBO J. 19:2803-2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mirre, C., L. Monlauzeur, M. Garcia, M. H. Delgrossi, and A. Le Bivic. 1996. Detergent-resistant membrane microdomains from Caco-2 cells do not contain caveolin. Am. J. Physiol. 271:C887-C894. [DOI] [PubMed] [Google Scholar]
- 48.Mitchell, J. S., O. Kanca, and B. W. McIntyre. 2002. Lipid microdomain clustering induces a redistribution of antigen recognition and adhesion molecules on human T lymphocytes. J. Immunol. 168:2737-2744. [DOI] [PubMed] [Google Scholar]
- 49.Mulvey, M. A. 2002. Adhesion and entry of uropathogenic Escherichia coli. Cell. Microbiol. 4:257-271. [DOI] [PubMed] [Google Scholar]
- 50.Norkin, L. C., S. A. Wolfrom, and E. S. Stuart. 2001. Association of caveolin with Chlamydia trachomatis inclusions at early and late stages of infection. Exp. Cell. Res. 266:229-238. [DOI] [PubMed] [Google Scholar]
- 51.Nowicki, B., J. P. Barrish, T. Korhonen, R. A. Hull, and S. I. Hull. 1987. Molecular cloning of the Escherichia coli O75X adhesin. Infect. Immun. 55:3168-3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nowicki, B., A. Hart, K. E. Coyne, D. M. Lublin, and S. Nowicki. 1993. Short consensus repeat-3 domain of recombinant decay-accelerating factor is recognized by Escherichia coli recombinant Dr adhesin in a model of a cell-cell interaction. J. Exp. Med. 178:2115-2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nowicki, B., R. Selvarangan, and S. Nowicki. 2001. Family of Escherichia coli Dr adhesins: decay-accelerating factor receptor recognition and invasiveness. J. Infect. Dis. 183(Suppl. 1):S24-S27. [DOI] [PubMed] [Google Scholar]
- 54.Orlandi, P. A., and P. H. Fishman. 1998. Filipin-dependent inhibition of cholera toxin: evidence for toxin internalization and activation through caveolae-like domains. J. Cell Biol. 141:905-915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Palmer, L. M., T. J. Reilly, S. J. Utsalo, and M. S. Donnenberg. 1997. Internalization of Escherichia coli by human renal epithelial cells is associated with tyrosine phosphorylation of specific host cell proteins. Infect. Immun. 65:2570-2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Peiffer, I., J. Guignot, A. Barbat, C. Carnoy, S. L. Moseley, B. J. Nowicki, A. L. Servin, and M. F. Bernet-Camard. 2000. Structural and functional lesions in brush border of human polarized intestinal Caco-2/TC7 cells infected by members of the Afa/Dr diffusely adhering family of Escherichia coli. Infect. Immun. 68:5979-5990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peiffer, I., A. L. Servin, and M. F. Bernet-Camard. 1998. Piracy of decay-accelerating factor (CD55) signal transduction by the diffusely adhering strain Escherichia coli C1845 promotes cytoskeletal F-actin rearrangements in cultured human intestinal INT407 cells. Infect. Immun. 66:4036-4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pelkmans, L., and A. Helenius. 2002. Endocytosis via caveolae. Traffic 3:311-320. [DOI] [PubMed] [Google Scholar]
- 59.Peyron, P., C. Bordier, E. N. N′Diaye, and I. Maridonneau-Parini. 2000. Nonopsonic phagocytosis of Mycobacterium kansasii by human neutrophils depends on cholesterol and is mediated by CR3 associated with glycosylphosphatidylinositol-anchored proteins. J. Immunol. 165:5186-5191. [DOI] [PubMed] [Google Scholar]
- 60.Pham, T. Q., P. Goluszko, V. Popov, S. Nowicki, and B. J. Nowicki. 1997. Molecular cloning and characterization of Dr-II, a nonfimbrial adhesin-I-like adhesin isolated from gestational pyelonephritis-associated Escherichia coli that binds to decay-accelerating factor. Infect. Immun. 65:4309-4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pike, L. J., and J. M. Miller. 1998. Cholesterol depletion delocalizes phosphatidylinositol bisphosphate and inhibits hormone-stimulated phosphatidylinositol turnover. J. Biol. Chem. 273:22298-22304. [DOI] [PubMed] [Google Scholar]
- 62.Pizzo, P., E. Giurisato, M. Tassi, A. Benedetti, T. Pozzan, and A. Viola. 2002. Lipid rafts and T cell receptor signaling: a critical re-evaluation. Eur. J. Immunol. 32:3082-3091. [DOI] [PubMed] [Google Scholar]
- 63.Plancon, L., L. Du Merle, S. Le Friec, P. Gounon, M. Jouve, J. Guignot, A. Servin, and C. Le Bouguenec. 2003. Recognition of the cellular beta1-chain integrin by the bacterial AfaD invasin is implicated in the internalization of afa-expressing pathogenic Escherichia coli strains. Cell. Microbiol. 5:681-693. [DOI] [PubMed] [Google Scholar]
- 64.Rosenberger, C. M., J. H. Brumell, and B. B. Finlay. 2000. Microbial pathogenesis: lipid rafts as pathogen portals. Curr. Biol. 10:R823-R825. [DOI] [PubMed] [Google Scholar]
- 65.Scaletsky, I. C., S. H. Fabbricotti, R. L. Carvalho, C. R. Nunes, H. S. Maranhao, M. B. Morais, and U. Fagundes-Neto. 2002. Diffusely adherent Escherichia coli as a cause of acute diarrhea in young children in Northeast Brazil: a case-control study. J. Clin. Microbiol. 40:645-648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schnitzer, J. E., P. Oh, E. Pinney, and J. Allard. 1994. Filipin-sensitive caveolae-mediated transport in endothelium: reduced transcytosis, scavenger endocytosis, and capillary permeability of select macromolecules. J. Cell Biol. 127:1217-1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Selvarangan, R., P. Goluszko, V. Popov, J. Singhal, T. Pham, D. M. Lublin, S. Nowicki, and B. Nowicki. 2000. Role of decay-accelerating factor domains and anchorage in internalization of Dr-fimbriated Escherichia coli. Infect. Immun. 68:1391-1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shenoy-Scaria, A. M., L. K. Gauen, J. Kwong, A. S. Shaw, and D. M. Lublin. 1993. Palmitylation of an amino-terminal cysteine motif of protein tyrosine kinases p56lck and p59fyn mediates interaction with glycosyl-phosphatidylinositol-anchored proteins. Mol. Cell. Biol. 13:6385-6392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shenoy-Scaria, A. M., J. Kwong, T. Fujita, M. W. Olszowy, A. S. Shaw, and D. M. Lublin. 1992. Signal transduction through decay-accelerating factor. Interaction of glycosyl-phosphatidylinositol anchor and protein tyrosine kinases p56lck and p59fyn 1. J. Immunol. 149:3535-3541. [PubMed] [Google Scholar]
- 70.Shin, J. S., and S. N. Abraham. 2001. Glycosylphosphatidylinositol-anchored receptor-mediated bacterial endocytosis. FEMS Microbiol. Lett. 197:131-138. [DOI] [PubMed] [Google Scholar]
- 71.Shin, J. S., Z. Gao, and S. N. Abraham. 2000. Involvement of cellular caveolae in bacterial entry into mast cells. Science 289:785-788. [DOI] [PubMed] [Google Scholar]
- 72.Simons, K., and D. Toomre. 2000. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell. Biol. 1:31-39. [DOI] [PubMed] [Google Scholar]
- 73.Skoudy, A., J. Mounier, A. Aruffo, H. Ohayon, P. Gounon, P. Sansonetti, and G. Tran Van Nhieu. 2000. CD44 binds to the Shigella IpaB protein and participates in bacterial invasion of epithelial cells. Cell. Microbiol. 2:19-33. [DOI] [PubMed] [Google Scholar]
- 74.Smart, E. J., G. A. Graf, M. A. McNiven, W. C. Sessa, J. A. Engelman, P. E. Scherer, T. Okamoto, and M. P. Lisanti. 1999. Caveolins, liquid-ordered domains, and signal transduction. Mol. Cell. Biol. 19:7289-7304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stefanova, I., V. Horejsi, I. J. Ansotegui, W. Knapp, and H. Stockinger. 1991. GPI-anchored cell-surface molecules complexed to protein tyrosine kinases. Science 254:1016-1019. [DOI] [PubMed] [Google Scholar]
- 76.Stulnig, T. M., M. Berger, T. Sigmund, H. Stockinger, V. Horejsi, and W. Waldhausl. 1997. Signal transduction via glycosyl phosphatidylinositol-anchored proteins in T cells is inhibited by lowering cellular cholesterol. J. Biol. Chem. 272:19242-19247. [DOI] [PubMed] [Google Scholar]
- 77.Upla, P., V. Marjomaki, P. Kankaanpaa, J. Ivaska, T. Hyypia, F. Gisou Van Der Goot, and J. Heino. 2004. Clustering induces a lateral redistribution of alpha2beta1 integrin from membrane rafts to caveolae and subsequent PKC-dependent internalization. Mol. Biol. Cell 15:625-636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.van der Goot, F. G., and T. Harder. 2001. Raft membrane domains: from a liquid-ordered membrane phase to a site of pathogen attack. Semin. Immunol. 13:89-97. [DOI] [PubMed] [Google Scholar]
- 79.Vogel, U., K. Sandvig, and B. van Deurs. 1998. Expression of caveolin-1 and polarized formation of invaginated caveolae in Caco-2 and MDCK II cells. J. Cell Sci. 111:825-832. [DOI] [PubMed] [Google Scholar]
- 80.Wary, K. K., A. Mariotti, C. Zurzolo, and F. G. Giancotti. 1998. A requirement for caveolin-1 and associated kinase Fyn in integrin signaling and anchorage-dependent cell growth. Cell 94:625-634. [DOI] [PubMed] [Google Scholar]
- 81.Watarai, M., S. Makino, Y. Fujii, K. Okamoto, and T. Shirahata. 2002. Modulation of Brucella-induced macropinocytosis by lipid rafts mediates intracellular replication. Cell. Microbiol. 4:341-355. [DOI] [PubMed] [Google Scholar]
- 82.Wells, C. L., E. M. van de Westerlo, R. P. Jechorek, H. M. Haines, and S. L. Erlandsen. 1998. Cytochalasin-induced actin disruption of polarized enterocytes can augment internalization of bacteria. Infect. Immun. 66:2410-2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wooldridge, K. G., P. H. Williams, and J. M. Ketley. 1996. Host signal transduction and endocytosis of Campylobacter jejuni. Microb. Pathog. 21:299-305. [DOI] [PubMed] [Google Scholar]
- 84.Zalewska, B., R. Piatek, H. Cieslinski, B. Nowicki, and J. Kur. 2001. Cloning, expression, and purification of the uropathogenic Escherichia coli invasin DraD. Protein Expr. Purif. 23:476-482. [DOI] [PubMed] [Google Scholar]
- 85.Zanetti, S., L. Sechi, A. Angioi, B. Perazzona, and G. Fadda. 1992. Entry of pyelonephritogenic Escherichia coli into HEp-2 cells due to actin polymerization. Microbiologica 15:117-123. [PubMed] [Google Scholar]
- 86.Zobiack, N., U. Rescher, S. Laarmann, S. Michgehl, M. A. Schmidt, and V. Gerke. 2002. Cell-surface attachment of pedestal-forming enteropathogenic E. coli induces a clustering of raft components and a recruitment of annexin 2. J. Cell Sci. 115:91-98. [DOI] [PubMed] [Google Scholar]