

Abstract
In mammals, lactation is a rich source of nutrients and antibodies for newborn animals. However, millions of mothers each year experience an inability to breastfeed. Exposure to several environmental toxicants, including 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), has been strongly implicated in impaired mammary differentiation and lactation. TCDD and related polyhalogenated aromatic hydrocarbons are widespread industrial pollutants that activate the aryl hydrocarbon receptor (AHR). Despite many epidemiological and animal studies, the molecular mechanism through which AHR signaling blocks lactation remains unclear. We employed in vitro models of mammary differentiation to recapitulate lactogenesis in the presence of toxicants. We demonstrate AHR agonists directly block milk production in isolated mammary epithelial cells. Moreover, we define a novel role for the aryl hydrocarbon receptor repressor (AHRR) in mediating this response. Our mechanistic studies suggest AHRR is sufficient to block transcription of the milk gene β-casein. As TCDD is a prevalent environmental pollutant that affects women worldwide, our results have important public health implications for newborn nutrition.
Keywords: lactation, AHR, TCDD, AHRR, ARNT, mammary gland
Lactation is a critical biological process in mammals that provides both nutritional and immune support for offspring. However, an estimated 3–6 million human mothers worldwide suffer from impaired lactation each year (Lew et al., 2009). Several factors contribute to milk production and secretion, but a growing number of studies suggest certain environmental toxicants negatively impact the ability of women to initiate and sustain breastfeeding (Neville and Walsh, 1995). For example, maternal exposure to pesticides has been associated with shortened duration of lactation in both the United States (Rogan et al., 1987) and Mexico (Gladen and Rogan, 1995).
One specific xenobiotic known to affect lactation is 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). TCDD is a byproduct of natural processes, such as volcanic eruptions and forest fires, and industrial processes, including smelting, waste incineration, pesticide production, and combustion (Wong et al., 2012). Once produced, TCDD persists in the environment and contaminates air, soil, and food sources (Larsen, 2006; Travis and Hattemer-Frey, 1991). Although inhalation and skin exposure occur, ingestion of contaminated food sources is the primary route of exposure for humans (Carpenter, 2006). With an average half-life between 7 and 8 years in humans (Geyer et al., 2002), TCDD is a highly stable chemical that bioaccumulates in fat tissue. Thus, TCDD exposure amasses and persists overtime (Consonni et al., 2012).
Previous studies have demonstrated TCDD exerts its toxicity through activation of the aryl hydrocarbon receptor (AHR) (Fernandez-Salguero et al., 1996). As a ligand activated transcription factor, AHR is restricted to the cytoplasm in its unbound state. Once activated, AHR translocates to the nucleus and forms a transcriptionally active complex with the AHR nuclear translocator (ARNT) to alter expression of specific target genes (Tijet et al., 2006). Although the identity of endogenous AHR activators remains controversial, several exogenous chemicals have been shown to target AHR (Denison and Nagy, 2003). In particular, we previously identified a 1,2,4-bis-aryloxadiazole, referred to as 1023, as a novel AHR agonist (Basham et al., 2013, 2014).
An association between AHR activation and changes in milk production has been observed in animal studies. Specifically, pregnant mice exposed to TCDD in vivo produced lower levels of the milk proteins β-casein (Collins et al., 2009) and whey acidic protein (WAP) (Vorderstrasse et al., 2004), and were unable to nutritionally support their offspring (Vorderstrasse et al., 2004). Moreover, exposure of pregnant rats to TCDD led to severe defects in mammary gland differentiation and decreased pup size following lactation (Badesha et al., 1995; Fenton et al., 2002), suggesting TCDD exposure impaired functional development of the mammary gland. Together with epidemiological studies in humans, these observations support a strong link between AHR activation and diminished milk production.
Despite these studies, the molecular mechanism through which AHR signaling blocks milk production remains unclear. Moreover, reciprocal transplant studies with AHR-null mammary glands implicate both indirect, systemic effects and direct, cellular consequences of AHR signaling on alveolar differentiation (Lew et al., 2011). Indirectly, TCDD disrupts endocrine function by altering estrogen-mediated signaling (Matthews and Gustafsson, 2006; Safe et al., 1998). However, changes in circulating estradiol, progesterone, or prolactin levels were not observed in pregnant mice after in vivo TCDD exposure (Vorderstrasse et al., 2004), which suggests mammary tissue may respond directly to AHR agonists. Moreover, explant studies, where mammary glands were cultured ex vivo under hormonal stimulation with TCDD, showed decreased lobuloalveolar structures (Hushka et al., 1998). Together, these studies suggest AHR signaling contributes to impaired lactogenesis by directly targeting mammary tissue.
Based on these initial observations, we hypothesized that the epithelium had a significant role in mediating the response to TCDD. Thus, we aimed to identify direct epithelial mechanisms through which AHR activation blocks β-casein production. Using in vitro models of mammary morphogenesis and differentiation, we show that both environmental toxicants (TCDD) and novel AHR agonists (1023) block lactogenesis directly in mammary epithelial cells. Furthermore, we identify a new role for the aryl hydrocarbon receptor repressor (AHRR) in mediating this response. Our results support a model in which AHRR induction promotes formation of AHRR/ARNT heterodimers, which transcriptionally inhibit β-casein production.
MATERIALS AND METHODS
Mice
Mice were maintained following protocols reviewed and approved by the University of Utah Institutional Animal Care and Use Committee.
Isolation of Primary Mammary Epithelial Cells (MECs)
Organoids from the fourth inguinal mammary gland were isolated from virgin (8- to 12-week-old) female mice and processed to single epithelial cells as previously described (Basham et al., 2013).
Microarray Data
Data previously generated (Basham et al., 2013) were analyzed using GeneSifter software (Geospiza Inc, Seattle, WA). Microarray data analyzed for this publication can be obtained from the Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/, Accessed October 8, 2014.) using accession number GSE39249.
Chemical Compounds
TCDD (Cambridge Isotopes Laboratories, Inc., Andover, MA) was obtained as a dimethyl sulfoxide (DMSO) stock solution and used at a final concentration of 10 nM, which is within the range of exposures (0.1–100 nM) previously reported to be environmentally and pharmacologically relevant (Birnbaum and DeVito, 1995). The AHR agonist, 1023 was synthesized as previously described (Basham et al., 2013) and dissolved in DMSO (Sigma, St. Louis, MO) at a stock concentration of 10 mM. The AHR agonist, 1023 was used at a final concentration of 10 μM, which was selected based on a previous dose-response analysis in mammary epithelial cells (Basham et al., 2013), where 1023 exhibited an EC50 of 1.2 ± 0.050 μM. Based on these results, 10 μM was the minimum concentration required to achieve maximal effect.
HC11 Cell Culture and Induction with Lactogenic Hormones
HC11 cells were maintained at 37°C with 5% CO2 in HC11 culture media (RPMI [HyClone, Logan, UT], 10% fetal bovine serum [FBS] [HyClone, Logan, UT], ITS-X [Invitrogen, Carlsbad, CA], 1X penicillin/streptomycin/glutamine [Invitrogen, Carlsbad, CA], and 10 ng/ml murine epidermal growth factor [EGF] [BD Biosciences, San Jose, CA]). For induction with lactogenic hormones, 1 × 106 cells per well were seeded in 6-well tissue culture plates. Two-day confluent cultures were washed twice with EGF-free HC11 culture media and grown for 96 additional hours in EGF-free HC11 culture media containing 5 μg/ml ovine prolactin (Sigma, St. Louis, MO) and 1 μM dexamethasone (Sigma, St. Louis, MO). For compound treatments, FBS was reduced to 2% 12 h after seeding and cells were grown with 0.1% DMSO, 10 μM 1023 or 10 nM TCDD as indicated. Throughout each assay, media was replaced every 48 h.
AHR nuclear localization assay
In 12-well tissue culture plates, 60 000 HC11 cells stably expressing pEiZ-HA-AHR were seeded onto lysine-coated glass coverslips. Cells were seeded in HC11 culture media containing 10% charcoal-stripped FBS (CSFBS) (Sigma, St. Louis, MO). The media was replaced 12 h later with HC11 culture media containing 2% CSFBS. Cells were dosed 36 hours post-plating with 0.1% DMSO, 10 μM 1023, or 10 nM TCDD. Samples were stained 24 h after dosing. For each condition, a minimum of 100 cells per sample was scored.
Immunofluorescence
For hemagglutinin (HA) (4 μg/ml, Abcam, Cambridge, England, #ab9110) staining, HC11 cells were fixed with 4% paraformaldehyde for 15 min at room temperature (RT), washed once with 50 nM ammonium chloride (Sigma, St. Louis, MO), and permeablized with 0.2% Triton X-100 in phosphate buffered saline (PBS) for 8 min at RT. Cells were washed once in 1% bovine serum albumin (BSA) in PBS and blocked with 5% BSA and 1% normal goat serum in PBS for 10 min. Following block, samples were incubated with primary antibody diluted in 1% BSA in PBS for 1 h at RT. After primary antibody incubations, samples were washed 3 times with PBS and incubated with secondary antibody (1:1000 in 1% BSA in PBS, Invitrogen, Carlsbad, CA, Alexa series) for 1 h at RT. Nuclei were stained with DAPI (50 ng/ml, Molecular Probes, Eugene, OR) for 5 min at RT. Coverslips were mounted with ProLong Gold anti-fade reagent (Invitrogen, Carlsbad, CA).
Microscopy
Imaging was performed using an Olympus IX81-ZDC microscope with an ORCAER CCD camera and Slidebook 5.0.0.24 software (Intelligent Imaging Innovations, Inc, Denver, CO). A 60x Plan oil objective lens was used for representative images and a 40x U-Plan objective lens was used for images for quantification.
Western Blot
HC11 cells were washed twice and scraped in 1 ml cold PBS. Cells were pelleted at 1250 x g for 5 minutes, lysed in RIPA buffer (50 mM Tris-HCL pH 8.0, 150 mM NaCL, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100, 1 mM DTT, 1X protease/phosphatase inhibitor cocktail), and sonicated for 20 s. All processing steps occurred at 4°C or on ice. Whole-cell lysate (50 μg) was separated by 10% SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride (PVDF) membrane (Millipore, Billerica, MA) for detection of β-casein (1:100, Santa Cruz, Dallas, TX, #sc-17969), α-tubulin (1:1000, Sigma, St. Louis, MO, #T6199), HA (1:500, Covance, Princeton, NJ, #MMS-101R) or ARNT (1:500, BD Biosciences, San Jose, CA, #611078). Integrated intensity values were measured on a LI-COR Odyssey Scanner (Lincoln, NE) with median background subtraction and normalized to α-tubulin levels.
RNA Extraction, cDNA Synthesis, and Real-Time Polymerase Chain Reaction (qPCR)
RNA was isolated, converted to cDNA, and used for qPCR as previously described (Basham et al., 2013). Primer sequences were as follows: β-actin forward (5′-GGCTGTGCTGTCCCTGTATG-3′), β-actin reverse (5′-CAAGAAGGAAGGCTGGAAAA-3), β-casein forward (’-CAATCCCGTCCCAC AAAA-3), β-casein reverse (5′-TCCAGTTTCAGTCAGTTCAAAAA-3′), α-casein forward (5′-CAGAAGTGCTCTCTGTCTGTTCA-3′), α-casein reverse (5′-AGCATAGCAGCAGTGAGGAAA-3′), AHR forward (5′-CTTTGCTGAACTCGGCTTGC-3′), AHR reverse (5′-TTGCTGGGGGCACACCATCT-3′), Cyp1A1 forward (5′-GGTTAACCATGACCGGGAACT-3′), Cyp1A1 reverse (5′-TGCCCAAAC CAAAGAGAGTGA-3′), ARNT forward (5′-CTAAGAGACAGCTTTCAGCAGGT-3′), ARNT reverse (5′-AGGGTTTTGGAAGGTAAAGGAG-3′).
Plasmids and Molecular Cloning
Constructs expressing an shRNA against AHR or ARNT were previously generated (Basham et al., 2013) using the pLentiLox5.0-GFP (green fluorescent protein) vector (Cai et al., 2007) provided by Dr James Bear (University of North Carolina, Chapel Hill, NC). Lentiviral expression constructs were generated using the pEiZ plasmid (pHIV-Zsgreen, plasmid 18121, Addgene, Cambridge, MA) previously described (Welm et al., 2008). For generation of pEiZ-HA-AHR, a mouse AHR expression plasmid (pACTAG-HA-AHR) provided by Dr Oliver Hankinson (University of California, Los Angeles, CA) was used to PCR amplify HA-AHR with the addition of XbaI restriction sites. The following primers were used for amplification: forward (5′-TAAGCATCTAGAACCATGATCTTTTACCCATACGATGTTCCTG-3′), reverse (5′-TGCTTATCTAGAACCTCAACTCTGCACCTTGCTTAGGAAT-3′). The resulting PCR product was digested with XbaI and ligated into the same site in pEiZ. For generation of pEiZ-HA-AHRR, a mouse AHRR expression plasmid (pcDNA-mAhRR) (Karchner et al., 2002) provided by Dr Mark Hahn (Woods Hole Oceanographic Institution, Woods Hole, MA) was used to PCR amplify AHRR with addition of an N-terminal HA tag and EcoRI restriction sites. The following primers were used for amplification: forward (5′-CTAGAATTCCCACCATGAGCGTAGTCTGGGACGTCGTATGGGTAATGATGATTCCGTCTGGAGAGTGTACA-3′), reverse (5′-GACGAATTCACTCTAGGGTAGGAAAATTCCATCAGAGCC-3′). The resulting PCR product was subcloned into the pCR®2.1-TOPO® TA vector to create Topo-HA-AHRR, which was then digested with EcoRI, and ligated into the same site in pEiZ. For generation of pEiZ-HA-AHRRmutant, site-directed mutagenesis was performed using the Strategene QuikChange Site-Directed Mutagenesis Kit (Strategene, La Jolla, CA) and the Topo-HA-AHRR plasmid according to manufactures instructions. Lys-542, Lys-583, and Lys-660 were each mutated to an arginine residue in separate reactions using the following primer sequences: Lys-542-Arg sense (5′-CACTGGATGTGCCAATCAGGATGGAGAATGAATCTGG-3′), Lys-542-Arg antisense (5′-CCAGATTCATTCTCCATCCTGATTGGCACATCCAGTG-3′), Lys-583-Arg sense (5′-CCAGGATGCACCTGAGAACAGAGCCCGACTA-3′), Lys-583-Arg antisense (5′-TAGTCGGGCTCTGTTCTCAGGTGCATCCTGG-3′), Lys-660-Arg sense (5′-ACTGCAG AGCTCCTATTGTTAGGCGTGAGCCTC-3′), Lys-660-Arg antisense (5′-GAGGCTCACGCC TAACAATAGGAGCTCTGCAGT-3′). Following site-directed mutagenesis, Topo-HA-AHRRmutant was digested with EcoRI and the resulting fragment was ligated into pEiZ using the same site. For generation of pEiZ-HA-ARNT, an HA-tagged mouse ARNT expression plasmid (pACTAG-HA-ARNT) (Moffett et al., 1997) provided by Dr Oliver Hankinson (University of California, Los Angeles, CA) was used to PCR amplify the HA-ARNT fragment with additional EcoRI restriction sites. The resulting PCR product was subcloned into the pCR®2.1-TOPO® TA vector to create Topo-HA-ARNT, which was digested with EcoRI and ligated into the same site in pEiZ. The following plasmids for generating lentivirus have been previously described: pMDLg/pRRE (Dull et al., 1998), pRSV-Rev (Dull et al., 1998), and pVSV-G (Clontech, Mountain View, CA). The identity of each plasmid was confirmed by sequencing.
Production and Titration of Lentivirus
High titer lentivirus was produced, concentrated, and titrated as previously described (Basham et al., 2013).
Generation of Stable HC11 Cell Lines
For stable HC11 cell lines expressing an shRNA or expression construct, 1.25 × 106 cells were seeded in a 10-cm dish, cultured for 12 h, and transduced at a multiplicity of infection (MOI) of 20. For expression of HA-AHR, unsorted cells were used for immunofluorescence studies. For all other experiments, transduced cells were sorted 72 h later based on GFP expression to obtain stable lines.
RESULTS
AHR Agonists Inhibit Expression of Milk Genes in Mammary Epithelial Cells
To study the effect of AHR activation on lactation, we sought to recapitulate this process in vitro. Initially, we grew primary mammary epithelial cells (MECs) in our 3-dimensional branching assay (Basham et al., 2013) in the presence of 1023, an AHR agonist, and performed microarray analysis to determine differentially expressed genes. Compared with vehicle treatment (DMSO), cells grown with 10 μM 1023 showed downregulation of several genes important for milk production, including whey acidic protein and multiple casein genes (Table 1). Given the lack of stromal components in this system (Basham et al., 2013; Welm et al., 2008), these results suggested AHR activation blocked milk genes through a direct mechanism in mammary epithelial cells.
TABLE 1.
Genes Involved in Milk Production Downregulated in MECs Treated for 72 h With 10 µM 1023
| Gene Identifier | Gene Name | Gene Symbol | Ratioa |
|---|---|---|---|
| NM_007784 | Casein α s1 | Csn1 | −10.97 |
| NM_007786 | Casein κ | Csn3 | −8.24 |
| NM_011709 | Whey acidic protein | Wap | −7.88 |
| NM_009972 | Casein β | Csn2 | −6.84 |
aLog2 ratio for 1023-treated versus DMSO.
To validate these results, we next utilized the HC11 mammary epithelial cell line. HC11 cells were clonally derived from immortalized COMMA-1D epithelial cells (Ball et al., 1988), which were isolated from the mammary gland of mid-pregnant BALB/c mice (Danielson et al., 1984). Importantly, HC11 cells can be induced to differentiate with lactogenic hormones and produce milk proteins in culture (Ball et al., 1988; Doppler et al., 1990). First, we confirmed that 1023 and TCDD-activated AHR in this system. As AHR translocates to the nucleus following activation (Denison and Nagy, 2003; Heid et al., 2000; Pollenz et al., 1994), we assessed AHR localization following treatment with 1023 or TCDD. Specifically, we stably expressed an HA-tagged AHR construct in HC11 cells and performed immunofluorescence for HA after 24 h of drug treatment. We found that both 10 μM 1023 and 10 nM TCDD significantly increased the amount of nuclear AHR compared with DMSO (Figure 1A and B). Furthermore, we observed significant induction of the AHR response gene, cytochrome P4501A1 (Cyp1a1) (Whitlock, 1999), in parental HC11 cells following 48 h of compound treatment (Supplementary Figure 1A). Together, these experiments demonstrated 1023 and TCDD properly activated the AHR pathway in HC11 cells.
FIG. 1.

AHR activation blocks milk production in HC11 mammary cells. A, Mammary HC11 cells were transduced with lentivirus to generate a cell line stably expressing HA-tagged AHR. HC11 cells expressing HA-tagged AHR were treated with vehicle control (DMSO), 10 µM 1023, or 10 nM TCDD for 24 h. Immunofluorescence for HA was performed to measure nuclear translocation of AHR. Dashed line defines nuclei. Scale bar = 10 µm. B, Quantification of AHR nuclear translocation. Asterisk indicates a statistically significant difference compared with vehicle-treated cells (****P ≤ .0001). C, Mammary HC11 cells were treated with vehicle control (DMSO), 10 µM 1023, or 10 nM TCDD and induced with lactogenic hormones (LH) to produce milk proteins. HC11 cell lysates were probed for β-casein and α-tubulin (control). D, β-casein mRNA levels were measured in the same assay by qPCR and normalized to β-actin expression. Results are shown as normalized mean β-casein expression (±SEM). Asterisk indicates a statistically significant difference compared with vehicle-treated cells (****P ≤ .0001). E, Lentiviral shRNA constructs (shAHR#1 and shAHR#2) were used to stably knockdown AHR expression HC11 cells. Stable HC11 cell lines were treated, induced, and probed for β-casein and β-actin (control) as described above. Abbreviations: HA, hemagglutinin; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin; LH, lactogenic hormones; AHR, aryl hydrocarbon receptor; DMSO, dimethyl sulfoxide; qPCR, real-time polymerase chain reaction.
Next, we induced HC11 cells with lactogenic hormones and tested the effect of AHR activation on β-casein and α-casein expression. Using this assay, cells grown in the presence of 10 μM 1023 or 10 nM TCDD failed to produce the milk protein, β-casein, compared with vehicle-treated cells (DMSO) (Figure 1C). In agreement with our microarray data from primary MECs, mRNA expression of both β-casein and α-casein were also inhibited (Figure 1D and Supplementary Figure 1B), suggesting a transcriptional mechanism of gene regulation.
Previously, we identified and validated AHR as the biological target of 1023 (Basham et al., 2013). To confirm the observed inhibition of β-casein protein production was dependent on the AHR pathway, we used lentiviral constructs to generate stable HC11 cell lines expressing either a nonspecific shRNA (Control) or an shRNA against AHR (shAHR #1 and shAHR #2). In cells with ∼50% AHR knockdown (Supplementary Figure 1C), we observed a partial rescue in β-casein expression in the presence of AHR agonists (Figure 1D and Supplementary Figure 1D). These results suggested 1023 and TCDD inhibited β-casein production in mammary epithelial cells through AHR pathway activation.
The AHRR is Sufficient to Inhibit β-casein
Given our data in both primary MECs and mammary HC11 cells, we hypothesized AHR activation blocked β-casein production through a transcriptional mechanism. Moreover, we became interested in a potential role for AHRR in mediating this process. As a direct target of activated AHR (Mimura et al., 1999), AHRR is highly upregulated in primary MECs treated with 1023 (Basham et al., 2013) and has a known role as a transcriptional repressor. Specifically, AHRR shares high amino acid identity with the N-terminal portion of AHR, which contains both the basic helix-loop-helix (bHLH) and Per-ARNT-Sim “A” (PAS-A) domains (Mimura et al., 1999). These regions of the AHR protein facilitate DNA binding and ARNT dimerization, which allows AHRR to form a heterodimer with ARNT and bind xenobiotic-responsive elements (XREs) in the promoter region of AHR target genes. Subsequent recruitment of a co-repressor complex, which includes ANKRA2, HDAC4, and HDAC5, inhibits transcription of AHR target genes (Mimura et al., 1999; Oshima et al., 2007).
To test the effect of AHRR on β-casein expression, we first stably expressed a lentiviral HA-tagged AHRR construct in HC11 cells. We confirmed over-expression (Figure 2A) and validated the construct by measuring a well-characterized AHR response gene, Cyp1A1. Compared with control transduced cells, over-expression of AHRR decreased induction of Cyp1A1 after 48 h of treatment with 10 μM 1023 (Figure 2B). These results verified our tagged AHRR construct interacted with ARNT to functionally repress known target genes.
FIG. 2.

AHRR is sufficient to block milk production in HC11 mammary cells. HC11 cells were transduced with lentivirus to generate cell lines stably expressing HA-tagged AHRR (pEiZ-HA-AHRR) or a vector control (pEiZ). A, Western blot of lysates from stable cell lines probed for HA and α-tubulin (control). B, Stable HC11 cell lines were treated for 48 h with vehicle control (DMSO) or 10 µM 1023. Cyp1A1 mRNA levels were measured by qPCR and normalized to β-actin expression. Results are shown as normalized mean Cyp1A1 expression (± SEM). Asterisk indicates a statistically significant difference compared with treated vector control cells (****P ≤ .0001). C, Stable HC11 cell lines were induced with lactogenic hormones (LH) to produce milk proteins. β-Casein mRNA levels were measured by qPCR and normalized to β-actin expression. Results are shown as normalized mean β-casein expression (± SEM). Asterisk indicates a statistically significant difference compared with treated vector control cells (P ≤ .0001). D, Cell lysates were probed for β-casein and α-tubulin (control). E, An HC11 cell line was generated to express AHRR mutated to prevent SUMOylation in the C-terminal region (HA-AHRRmutant). All stable cell lines were induced with LH and probed for β-casein and α-tubulin (control) as before. Abbreviations: HA, hemagglutinin; AHRR, aryl hydrocarbon receptor repressor; LH, lactogenic hormones; DMSO, dimethyl sulfoxide; qPCR, real-time polymerase chain reaction.
Next, we tested whether AHRR expression was sufficient to block β-casein production in the absence of AHR agonists. In HC11 cells induced with lactogenic hormones, and without exposure to AHR agonists, β-casein production was repressed at both the mRNA (Figure 2C) and protein level (Figure 2D) in cells expressing HA-AHRR compared with control cells. These results demonstrated the presence of AHRR was sufficient to block β-casein expression.
AHRR contains a transcriptional repression domain within its C-terminal region, which consists of 3 conserved small ubiquitin-like modifier (SUMO)ylation sites at Lys-542, Lys-583, and Lys-660. Previous studies demonstrated these lysine residues are modified by SUMO-1 to facilitate interaction between AHRR and its co-repressor complex (Oshima et al., 2009). To genetically test the requirement of this interaction for AHRR to inhibit β-casein production, we mutated all 3 lysine residues to arginine (HA-AHRRmutant). Previously, these mutations in AHRR have been shown to allow interaction between AHRR and ARNT, but prevent interaction between AHRR and its co-repressor complex (Oshima et al., 2009). Compared with HC11 cells over-expressing wild-type AHRR, overexpression of the SUMOylation mutant signficantly rescued β-casein production (Figure 2E), suggesting AHRR requires interaction with its co-repressor complex to inhibit lactogenesis.
ARNT is Required for β-casein Production in Mammary Epithelial Cells
To elucidate the mechanism through which AHRR inhibits milk production, we examined the role of its major binding partner, ARNT, during normal lactation. Stable HC11 cell lines expressing a nonspecific lentiviral shRNA (Control) or a lentiviral shRNA against ARNT (shARNT #1 or shARNT #2) were generated (Figure 3A and B). Following induction with lactogenic hormones, knockdown of ARNT-inhibited β-casein expression at both the mRNA (Figure 3C) and protein level (Figure 3D), and expression of α-casein mRNA (data not shown). As loss of AHR in the absence of any chemical agonists had no effect on β-casein production in HC11 cells (Figure 1D), these results implicated an independent role for ARNT during milk production.
FIG. 3.

ARNT is required for milk production in HC11 mammary cells. A, Lentiviral shRNA constructs (shARNT#1 and shARNT#2) were used to stably knockdown ARNT expression in HC11 cells. ARNT expression was measured by qPCR, normalized to β-actin expression, and compared with HC11 cells stably expressing a control shRNA (Control). Results are shown as normalized mean ARNT expression (±SEM). Asterisk indicates a statistically significant difference compared with control shRNA cells (*P ≤ 0.05). B, ARNT protein levels were measured in cell lysates from stable lines. Western blot of lysates probed for ARNT and α-tubulin (control). C, Stable HC11 cell lines were induced with lactogenic hormones (LH) to produce milk proteins. β-casein mRNA levels were measured by qPCR and normalized to β-actin expression. Results are shown as normalized mean β-casein expression (±SEM). Asterisk indicates a statistically significant difference compared with treated control cells (P ≤ 0.0001). D, Western blot of lysates probed for β-casein and α-tubulin (control). Abbreviations: ARNT, aryl hydrocarbon receptor nuclear translocator; LH, lactogenic hormones; qPCR, real-time polymerase chain reaction.
Overexpression of ARNT Rescues β-casein Production in the Presence of AHR Agonists
Based on our results, we hypothesized ARNT participated in a transcriptionally active complex to promote expression of milk genes, such as β-casein, during lactogenesis. Furthermore, as ARNT was required for lactogenesis, we speculated AHRR might inhibit milk production through competitive interactions with ARNT. To test this hypothesis, we over-expressed ARNT prior to treatment with AHR agonists and measured β-casein levels to assess milk production. We reasoned over-expression of ARNT would promote transcriptionally active complexes and restore β-casein levels in the presence of AHR agonists. In HC11 cells stably over-expressing a lentiviral HA-ARNT construct (Figure 4A), we found β-casein expression partially restored in the presence of both 10 μM 1023 and 10 nM TCDD compared with control cells (Figure 4B). These results support a model in which AHRR blocks production of β-casein by acting as a transcriptional repressor to inhibit ARNT signaling.
FIG. 4.

ARNT overexpression rescues milk production in the presence of AHR agonists. A, A HA-tagged ARNT construct (pEiZ-HA-ARNT) was stably expressed in mammary HC11 cells using lentiviral transduction. An empty expression vector (pEiZ) was used to create a stable control HC11 cell line. Western blot of lysates probed for HA and α-tubulin (control). B, Stable HC11 cell lines were treated with vehicle control (DMSO), 10 μM 1023 or 10 nM TCDD and induced with lactogenic hormones (LH) to produce milk proteins. Western blot of lysates probed for β-casein and α-tubulin (control). Abbreviations: HA, hemagglutinin; ARNT, aryl hydrocarbon receptor nuclear translocator; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin; HA, hemagglutinin; AHR, aryl hydrocarbon receptor; DMSO, dimethyl sulfoxide.
DISCUSSION
Using an epithelial-based model of mammary lactation, we demonstrate AHR agonists directly block β-casein expression. Specifically, we showed AHRR, a robust downstream target of AHR signaling, was sufficient to inhibit the production of β-casein. Although well studied in the context of AHR activation, AHRR is also induced by other toxic insults. In particular, recent studies showed cigarette smoking, including secondhand exposure, caused significant demethylation and increased expression of AHRR (Philibert et al., 2012; Shenker et al., 2013). As women who smoke have consistently been shown to produce significantly less milk volume (Hopkinson et al., 1992; Vio et al., 1991) and breastfeed for a shorter duration (Andersen and Schioler, 1982; Hakansson and Carlsson, 1992; Schulte-Hobein et al., 1992; Vio et al., 1991; Whichelow, 1979), our results provide a potential molecular mechanism through which this toxicity occurs.
Following induction, AHRR forms a heterodimer complex with ARNT (Evans et al., 2008). As loss of ARNT significantly blocked β-casein production in mammary epithelial cells, our results implicate an important role for ARNT during lactation. These observations are consistent with the phenotype observed in conditional ARNT knockout mice, where ARNT deletion using the MMTV-Cre transgene resulted in impaired mammary function (Le Provost et al., 2002). Specifically, loss of ARNT led to incomplete alveolar development, smaller litter sizes, and 60% of dams that could not support their pups.
However, inactivation of ARNT using WAP-Cre resulted in normal mammary differentiation during pregnancy (Le Provost et al., 2002). Furthermore, transplantation of ARNT null mammary epithelium generated using MMTV-Cre into wild-type recipients normalized alveolar development (Le Provost et al., 2002). Although inconsistent with initial studies and our current findings, these incongruent results may be explained by differences in methodology. Specifically, WAP-Cre reduced ARNT levels in 80% of the mammary epithelium (Le Provost et al., 2002; Walton et al., 2001), resulting in residual ARNT expression that may facilitate normal development. In reciprocal transplant studies, the high selective pressure of transplantation may have induced compensation from other ARNT family members, including ARNT2. Expressed in the mammary gland (Martinez et al., 2008), ARNT2 is known to form functional complexes with AHR (Hirose et al., 1996) and may be capable of contributing to mammary development. Further in vivo studies will be required to explain these differences.
Our experiments with AHRR and ARNT suggest a model in which ARNT promotes milk production under lactogenic conditions (Figure 5). As a bHLH-PAS family member, ARNT requires dimerization to form a functional transcription complex. Although ARNT is known to interact with multiple proteins, we hypothesize that hypoxia inducible factor-1α (HIF1α) or single-minded 2 (SIM2) interact with ARNT to promote lactation. In previous studies, conditional knockout of ARNT prevented induction of known HIF1α target genes (Tomita et al., 2000), suggesting ARNT is critical for HIF1α signal transduction. Additionally, deletion of HIF1α in the mammary epithelium resulted in severe differentiation defects and failed lactation (Seagroves et al., 2003). Previous studies with SIM2 also support its potential role in cooperating with ARNT to regulate lactation. With SIM2 over-expression, precocious production of β-casein and WAP occurred in vitro and in vivo, and chromatin immunoprecipitation (ChIP) experiments showed SIM2 associated with the β-casein promoter. Conversely, loss of SIM2 inhibited milk production (Wellberg et al., 2010). Taken together, these experiments strongly implicate a role for HIF1α or SIM2 in promoting ARNT-mediated lactogenesis.
FIG. 5.
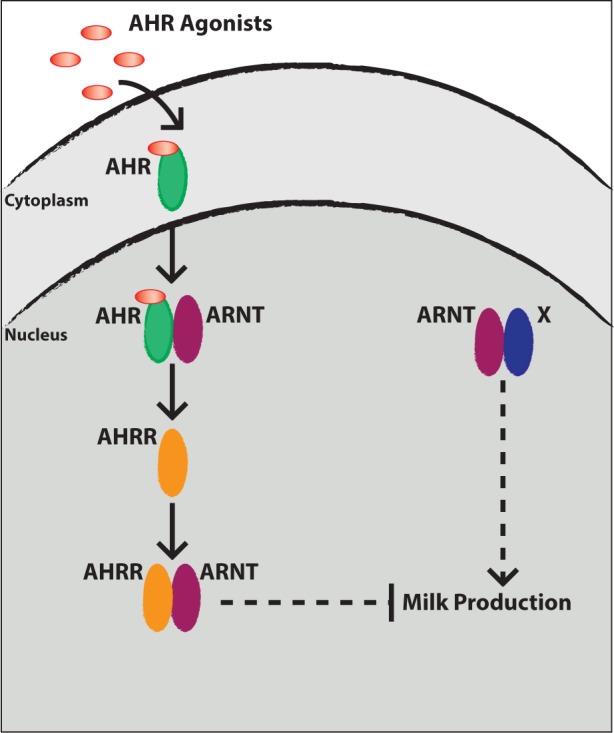
Working model for the mechanism through which AHR activation blocks milk production in mammary epithelial cells. (Right) In the absence of AHR agonists, we propose ARNT signaling promotes milk production during lactogenic stimulation. (Left) In the presence of AHR agonists, AHR translocates to the nucleus and forms a heterodimer with ARNT. AHR/ARNT heterodimers bind XRE sequences to increase transcription of AHR target genes, including AHRR. Once induced, AHRR competes with AHR for interaction with ARNT and forms a transcriptionally repressive complex, which we propose inhibits expression of milk proteins. Dashed lines indicate aspects of the model proposed by these data. Abbreviations: AHR, aryl hydrocarbon receptor; ARNT, aryl hydrocarbon receptor nuclear translocator; AHRR, aryl hydrocarbon receptor repressor; XRE, xenobiotic-responsive elements.
In response to toxic stimuli, our data suggest that AHRR/ARNT heterodimers form and are sufficient to block lactation. One potential mechanism of AHRR repression under these conditions is ARNT sequestration. Competition for a limited pool of ARNT has been observed previously, where ARNT availability regulated AHR signaling through HIF1α (Gradin et al., 1996) and SIM1 (Probst et al., 1997; Woods and Whitelaw, 2002). However, our experiments using an AHRR repression mutant that could bind ARNT, but not recruit a co-repressor complex, suggest this is not the dominant mechanism. Rather, our results favor a model in which AHRR SUMOylation and subsequent recruitment of a co-repressor complex that includes ANKRA2, HDAC4, and HDAC5 blocks transcription of genes important for milk production. Further experiments will be needed to determine whether activating ARNT complexes and repressive AHRR/ARNT heterodimers bind the same DNA response elements. Additionally, these studies will help determine whether ARNT-mediated complexes bind directly in the promoter region of milk target genes or whether they control activity of an intermediate factor(s). We have not identified AHR consensus elements within the mouse or human β-casein promoters, and since several milk genes are affected by AHR signaling, it is possible global rather than direct pathways attenuate lactation. Further studies will be necessary to determine the targets of AHR signaling that modulate lactogenesis.
Our data demonstrate AHR signaling directly disrupts milk protein production in isolated mammary epithelial cells. As industrial waste is one of the main sources of TCDD and other related polyhalogenated aromatic hydrocarbons, human exposure to these toxicants is highest in industrialized countries (Schecter et al., 2006a). However, acute exposure has occurred in several distinct populations worldwide, including exposure to Agent Orange herbicide in Vietnam (Schecter et al., 2006b), indigenous Canadian Inuit populations who consume contaminated marine species (Ayotte et al., 1996), and people living in Seveso, Italy during the 1976 industrial explosion (Warner et al., 2002), among others. Given the prevalence of these environmental pollutants and their ability to bio-accumulate over time, our study has substantial implications on public health, particularly with regard to the ability of women to breastfeed. Thus, future efforts to monitor TCDD exposure levels, analyze epidemiological data, and elucidate the molecular mechanism downstream of AHRR are needed to address this problem.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/
FUNDING
The National Institutes of Health [R01-CA143815, R01-CA140296, R01-GM090082]; the Department of Defense Breast Cancer Research Program [W81XWH-09-01-04310]; the National Institutes of Health Development Biology Training Grant [5T32 HD07491 to K.J.B.].
Supplementary Material
ACKNOWLEDGMENTS
We thank Drs James Bear (University of North Carolina, Chapel Hill, NC) and Thomas Marshall (University of Utah, Salt Lake City, UT) for providing the pLentiLox5.0-GFP vector; Dr Mark Hahn (Woods Hole Oceanographic Institution, Woods Hole, MA) for providing pcDNA-mAhRR, and Dr Oliver Hankinson (University of California, Los Angeles, CA) for providing pACTAG-HA-AHR and pACTAG-HA-ARNT. The authors declare that they have no conflicts of interest.
REFERENCES
- Andersen A. N., Schioler V. (1982). Influence of breast-feeding pattern on pituitary-ovarian axis of women in an industrialized community. Am. J. Obstet. Gynecol. 143, 673–677. [DOI] [PubMed] [Google Scholar]
- Ayotte P., Carrier G., Dewailly E. (1996). Health risk assessment for inuit newborns exposed to dioxin-like compounds through breast feeding. Chemosphere 32, 531–542. [DOI] [PubMed] [Google Scholar]
- Badesha J. S., Maliji G., Flaks B. (1995). Immunotoxic effects of exposure of rats to xenobiotics via maternal lactation. Part I 2,3,7,8-tetrachlorodibenzo-p-dioxin. Int. J. Exp. Pathol. 76, 425–439. [PMC free article] [PubMed] [Google Scholar]
- Ball R. K., Friis R. R., Schoenenberger C. A., Doppler W., Groner B. (1988). Prolactin regulation of beta-casein gene expression and of a cytosolic 120-kd protein in a cloned mouse mammary epithelial cell line. EMBO J. 7, 2089–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basham K. J., Bhonde V. R., Kieffer C., Mack J. B., Hess M., Welm B. E., Looper R. E. (2014). Bis-aryloxadiazoles as effective activators of the aryl hydrocarbon receptor. Bioorg. Med. Chem. Lett. 24, 2473–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basham K. J., Kieffer C., Shelton D. N., Leonard C. J., Bhonde V. R., Vankayalapati H., Milash B., Bearss D. J., Looper R. E., Welm B. E. (2013). Chemical genetic screen reveals a role for desmosomal adhesion in mammary branching morphogenesis. J. Biol. Chem. 288, 2261–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum L. S., DeVito M. J. (1995). Use of toxic equivalency factors for risk assessment for dioxins and related compounds. Toxicology 105, 391–401. [DOI] [PubMed] [Google Scholar]
- Cai L., Marshall T. W., Uetrecht A. C., Schafer D. A., Bear J. E. (2007). Coronin 1B coordinates Arp2/3 complex and cofilin activities at the leading edge. Cell 128, 915–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter D. O. (2006). Polychlorinated biphenyls (PCBs): Routes of exposure and effects on human health. Rev. Environ. Health 21, 1–23. [DOI] [PubMed] [Google Scholar]
- Collins L. L., Lew B. J., Lawrence B. P. (2009). TCDD exposure disrupts mammary epithelial cell differentiation and function. Reprod. Toxicol. 28, 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consonni D., Sindaco R., Bertazzi P. A. (2012). Blood levels of dioxins, furans, dioxin-like PCBs, and TEQs in general populations: A review, 1989-2010. Environ. Int. 44, 151–162. [DOI] [PubMed] [Google Scholar]
- Danielson K. G., Oborn C. J., Durban E. M., Butel J. S., Medina D. (1984). Epithelial mouse mammary cell line exhibiting normal morphogenesis in vivo and functional differentiation in vitro. Proc. Natl Acad. Sci. USA 81, 3756–3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denison M. S., Nagy S. R. (2003). Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu. Rev. Pharmacol. Toxicol. 43, 309–334. [DOI] [PubMed] [Google Scholar]
- Doppler W., Hock W., Hofer P., Groner B., Ball R. K. (1990). Prolactin and glucocorticoid hormones control transcription of the beta-casein gene by kinetically distinct mechanisms. Mol. Endocrinol. 4, 912–919. [DOI] [PubMed] [Google Scholar]
- Dull T., Zufferey R., Kelly M., Mandel R. J., Nguyen M., Trono D., Naldini L. (1998). A third-generation lentivirus vector with a conditional packaging system. J. Virol. 72, 8463–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans B. R., Karchner S. I., Allan L. L., Pollenz R. S., Tanguay R. L., Jenny M. J., Sherr D. H., Hahn M. E. (2008). Repression of aryl hydrocarbon receptor (AHR) signaling by AHR repressor: Role of DNA binding and competition for AHR nuclear translocator. Mol. Pharmacol. 73, 387–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton S. E., Hamm J. T., Birnbaum L. S., Youngblood G. L. (2002). Persistent abnormalities in the rat mammary gland following gestational and lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Toxicol. Sci. 67, 63–74. [DOI] [PubMed] [Google Scholar]
- Fernandez-Salguero P. M., Hilbert D. M., Rudikoff S., Ward J. M., Gonzalez F. J. (1996). Aryl-hydrocarbon receptor-deficient mice are resistant to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced toxicity. Toxicol. Appl. Pharmacol. 140, 173–179. [DOI] [PubMed] [Google Scholar]
- Geyer H. J., Schramm K. W., Feicht E. A., Behechti A., Steinberg C., Bruggemann R., Poiger H., Henkelmann B., Kettrup A. (2002). Half-lives of tetra-, penta-, hexa-, hepta-, and octachlorodibenzo-p-dioxin in rats, monkeys, and humans–a critical review. Chemosphere 48, 631–644. [DOI] [PubMed] [Google Scholar]
- Gladen B. C., Rogan W. J. (1995). DDE and shortened duration of lactation in a northern Mexican town. Am. J. Public Health 85, 504–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradin K., McGuire J., Wenger R. H., Kvietikova I., fhitelaw M. L., Toftgard R., Tora L., Gassmann M., Poellinger L. (1996). Functional interference between hypoxia and dioxin signal transduction pathways: Competition for recruitment of the Arnt transcription factor. Mol. Cell. Biol. 16, 5221–5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakansson A., Carlsson B. (1992). Maternal cigarette smoking, breast-feeding, and respiratory tract infections in infancy. A population-based cohort study. Scand. J. Prim. Health Care 10, 60–65. [DOI] [PubMed] [Google Scholar]
- Heid S. E., Pollenz R. S., Swanson H. I. (2000). Role of heat shock protein 90 dissociation in mediating agonist-induced activation of the aryl hydrocarbon receptor. Mol. Pharmacol. 57, 82–92. [PubMed] [Google Scholar]
- Hirose K., Morita M., Ema M., Mimura J., Hamada H., Fujii H., Saijo Y., Gotoh O., Sogawa K., Fujii-Kuriyama Y. (1996). cDNA cloning and tissue-specific expression of a novel basic helix-loop-helix/PAS factor (Arnt2) with close sequence similarity to the aryl hydrocarbon receptor nuclear translocator (Arnt). Mol. Cell. Biol. 16, 1706–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkinson J. M., Schanler R. J., Fraley J. K., Garza C. (1992). Milk production by mothers of premature infants: Influence of cigarette smoking. Pediatrics 90, 934–938. [PubMed] [Google Scholar]
- Hushka L. J., Williams J. S., Greenlee W. F. (1998). Characterization of 2,3,7,8-tetrachlorodibenzofuran-dependent suppression and AH receptor pathway gene expression in the developing mouse mammary gland. Toxicol. Appl. Pharmacol. 152, 200–210. [DOI] [PubMed] [Google Scholar]
- Karchner S. I., Franks D. G., Powell W. H., Hahn M. E. (2002). Regulatory interactions among three members of the vertebrate aryl hydrocarbon receptor family: AHR repressor, AHR1, and AHR2. J. Biol. Chem. 277, 6949–6959. [DOI] [PubMed] [Google Scholar]
- Larsen J. C. (2006). Risk assessments of polychlorinated dibenzo- p-dioxins, polychlorinated dibenzofurans, and dioxin-like polychlorinated biphenyls in food. Mol. Nutr. Food Res. 50, 885–896. [DOI] [PubMed] [Google Scholar]
- Le Provost F., Riedlinger G., Hee Yim S., Benedict J., Gonzalez F. J., Flaws J., Hennighausen L. (2002). The aryl hydrocarbon receptor (AhR) and its nuclear translocator (Arnt) are dispensable for normal mammary gland development but are required for fertility. Genesis 32, 231–239. [DOI] [PubMed] [Google Scholar]
- Lew B. J., Collins L. L., O'Reilly M. A., Lawrence B. P. (2009). Activation of the aryl hydrocarbon receptor during different critical windows in pregnancy alters mammary epithelial cell proliferation and differentiation. Toxicol. Sci. 111, 151–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew B. J., Manickam R., Lawrence B. P. (2011). Activation of the aryl hydrocarbon receptor during pregnancy in the mouse alters mammary development through direct effects on stromal and epithelial tissues. Biol. Reprod. 84, 1094–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez V., Kennedy S., Doolan P., Gammell P., Joyce H., Kenny E., Prakash Mehta J., Ryan E., O'Connor R., Crown J., et al. (2008). Drug metabolism-related genes as potential biomarkers: Analysis of expression in normal and tumour breast tissue. Breast Cancer Res. Treat. 110, 521–530. [DOI] [PubMed] [Google Scholar]
- Matthews J., Gustafsson J. A. (2006). Estrogen receptor and aryl hydrocarbon receptor signaling pathways. Nucl. Recept. Signal. 4, e016, 10.1621/nrs.04016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimura J., Ema M., Sogawa K., Fujii-Kuriyama Y. (1999). Identification of a novel mechanism of regulation of Ah (dioxin) receptor function. Genes Dev. 13, 20–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffett P., Reece M., Pelletier J. (1997). The murine Sim-2 gene product inhibits transcription by active repression and functional interference. Mol. Cell. Biol. 17, 4933–4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neville M. C., Walsh C. T. (1995). Effects of xenobiotics on milk secretion and composition. Am. J. Clin. Nutr. 61(Suppl. 3), 687S–694S. [DOI] [PubMed] [Google Scholar]
- Oshima M., Mimura J., Sekine H., Okawa H., Fujii-Kuriyama Y. (2009). SUMO modification regulates the transcriptional repressor function of aryl hydrocarbon receptor repressor. J. Biol. Chem. 284, 11017–11026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima M., Mimura J., Yamamoto M., Fujii-Kuriyama Y. (2007). Molecular mechanism of transcriptional repression of AhR repressor involving ANKRA2, HDAC4, and HDAC5. Biochem. Biophys. Res. Commun. 364, 276–282. [DOI] [PubMed] [Google Scholar]
- Philibert R. A., Beach S. R., Brody G. H. (2012). Demethylation of the aryl hydrocarbon receptor repressor as a biomarker for nascent smokers. Epigenetics 7, 1331–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollenz R. S., Sattler C. A., Poland A. (1994). The aryl hydrocarbon receptor and aryl hydrocarbon receptor nuclear translocator protein show distinct subcellular localizations in Hepa 1c1c7 cells by immunofluorescence microscopy. Mol. Pharmacol. 45, 428–438. [PubMed] [Google Scholar]
- Probst M. R., Fan C. M., Tessier-Lavigne M., Hankinson O. (1997). Two murine homologs of the Drosophila single-minded protein that interact with the mouse aryl hydrocarbon receptor nuclear translocator protein. J. Biol. Chem. 272, 4451–4457. [DOI] [PubMed] [Google Scholar]
- Rogan W. J., Gladen B. C., McKinney J. D., Carreras N., Hardy P., Thullen J., Tingelstad J., Tully M. (1987). Polychlorinated biphenyls (PCBs) and dichlorodiphenyl dichloroethene (DDE) in human milk: Effects on growth, morbidity, and duration of lactation. Am. J. Public Health 77, 1294–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safe S., Wang F., Porter W., Duan R., McDougal A. (1998). Ah receptor agonists as endocrine disruptors: Antiestrogenic activity and mechanisms. Toxicol. Lett. 102–103, 343–347. [DOI] [PubMed] [Google Scholar]
- Schecter A., Birnbaum L., Ryan J. J., Constable J. D. (2006a). Dioxins: An overview. Environ. Res. 101, 419–428. [DOI] [PubMed] [Google Scholar]
- Schecter A., Quynh H. T., Papke O., Tung K. C., Constable J. D. (2006b). Agent Orange, dioxins, and other chemicals of concern in Vietnam: Update 2006. J. Occup. Environ. Med. 48, 408–413. [DOI] [PubMed] [Google Scholar]
- Schulte-Hobein B., Schwartz-Bickenbach D., Abt S., Plum C., Nau H. (1992). Cigarette smoke exposure and development of infants throughout the first year of life: Influence of passive smoking and nursing on cotinine levels in breast milk and infant's urine. Acta. Paediatr. 81, 550–557. [DOI] [PubMed] [Google Scholar]
- Seagroves T. N., Hadsell D., McManaman J., Palmer C., Liao D., McNulty W., Welm B., Wagner K. U., Neville M., Johnson R. S. (2003). HIF1alpha is a critical regulator of secretory differentiation and activation, but not vascular expansion, in the mouse mammary gland. Development 130, 1713–1724. [DOI] [PubMed] [Google Scholar]
- Shenker N. S., Polidoro S., van Veldhoven K., Sacerdote C., Ricceri F., Birrell M. A., Belvisi M. G., Brown R., Vineis P., Flanagan J. M. (2013). Epigenome-wide association study in the European Prospective Investigation into Cancer and Nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking. Hum. Mol. Genet. 22, 843–851. [DOI] [PubMed] [Google Scholar]
- Tijet N., Boutros P. C., Moffat I. D., Okey A. B., Tuomisto J., Pohjanvirta R. (2006). Aryl hydrocarbon receptor regulates distinct dioxin-dependent and dioxin-independent gene batteries. Mol. Pharmacol. 69, 140–153. [DOI] [PubMed] [Google Scholar]
- Tomita S., Sinal C. J., Yim S. H., Gonzalez F. J. (2000). Conditional disruption of the aryl hydrocarbon receptor nuclear translocator (Arnt) gene leads to loss of target gene induction by the aryl hydrocarbon receptor and hypoxia-inducible factor 1alpha. Mol. Endocrinol. 14, 1674–1681. [DOI] [PubMed] [Google Scholar]
- Travis C. C., Hattemer-Frey H. A. (1991). Human exposure to dioxin. Sci. Total Environ. 104, 97–127. [DOI] [PubMed] [Google Scholar]
- Vio F., Salazar G., Infante C. (1991). Smoking during pregnancy and lactation and its effects on breast-milk volume. Am. J. Clin. Nutr. 54, 1011–1016. [DOI] [PubMed] [Google Scholar]
- Vorderstrasse B. A., Fenton S. E., Bohn A. A., Cundiff J. A., Lawrence B. P. (2004). A novel effect of dioxin: Exposure during pregnancy severely impairs mammary gland differentiation. Toxicol. Sci. 78, 248–257. [DOI] [PubMed] [Google Scholar]
- Walton K. D., Wagner K. U., Rucker E. B., 3rd, Shillingford J. M., Miyoshi K., Hennighausen L. (2001). Conditional deletion of the bcl-x gene from mouse mammary epithelium results in accelerated apoptosis during involution but does not compromise cell function during lactation. Mech. Dev. 109, 281–293. [DOI] [PubMed] [Google Scholar]
- Warner M., Eskenazi B., Mocarelli P., Gerthoux P. M., Samuels S., Needham L., Patterson D., Brambilla P. (2002). Serum dio concentrations and breast cancer risk in the Seveso Women's Health Study. Environ. Health Perspect. 110, 625–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellberg E., Metz R. P., Parker C., Porter W. W. (2010). The bHLH/PAS transcription factor singleminded 2s promotes mammary gland lactogenic differentiation. Development 137, 945–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welm B. E., Dijkgraaf G. J., Bledau A. S., Welm A. L., Werb Z. (2008). Lentiviral transduction of mammary stem cells for analysis of gene function during development and cancer. Cell Stem Cell 2, 90–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whichelow M. J. (1979). Breast feeding in Cambridge, England: Factors affecting the mother's milk supply. J. Adv. Nurs. 4, 253–261. [DOI] [PubMed] [Google Scholar]
- Whitlock J. P., Jr. (1999). Induction of cytochrome P4501A1. Annual review of pharmacology and toxicology 39, 103–25. [DOI] [PubMed] [Google Scholar]
- Wong M. H., Armour M. A., Naidu R., Man M. (2012). Persistent toxic substances: Sources, fates and effects. Rev. Environ. Health 27, 207–213. [DOI] [PubMed] [Google Scholar]
- Woods S. L., Whitelaw M. L. (2002). Differential activities of murine single minded 1 (SIM1) and SIM2 on a hypoxic response element. Cross-talk between basic helix-loop-helix/per-Arnt-Sim homology transcription factors. J. Biol. Chem. 277, 10236–10243. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.