

Abstract
Hexavalent chromium is a human respiratory carcinogen that undergoes intracellular activation in vivo primarily via reduction with ascorbate. Replication of Cr-adducted DNA triggers mismatch repair that generates toxic DNA double-strand breaks (DSBs) as secondary lesions. Here, we examined the intranuclear distribution of chromate-induced breaks and a central DSB signaling branch targeting histone H2AX. Using ascorbate-restored cells (H460 human lung epithelial cells, normal human lung and normal mouse embryonic fibroblasts (MEFs)), we found that Cr(VI) produced a typical DSB-associated spectrum of H2AX modifications, including its Ser139-phosphorylated (known as γH2AX) and mono- and diubiquitinated forms. However, whereas canonical DSB signaling relies on ATM, the formation of γH2AX and its ubiquitinated products by Cr(VI) was dependent on ATR kinase. Based on the established mode of ATR activation, this suggests that Cr-induced DSB are not blunt-ended and likely contain single-stranded tails. Confocal imaging with markers of active and inactive chromatin revealed a selective formation of Cr-induced DSB in euchromatin of mouse and human cells. In contrast to DSB, Cr-DNA adducts were produced in both types of chromatin. The euchromatin targeting of Cr-induced DSB makes these lesions particularly dangerous by increasing the probability of deleting active tumor suppressors and producing oncogenic translocations. Accumulation of transcription-inhibiting ubiquitinated forms of γH2AX in euchromatin is expected to contribute to the ability of Cr(VI) to suppress upregulation of inducible genes.
Keywords: chromate, hexavalent chromium, γ-H2AX, ATR, euchromatin, cancer
DNA double-strand breaks (DSBs) are one of the most deleterious forms of DNA damage that induce a strong apoptotic response if not promptly repaired (Roos and Kaina, 2006). Misjoined DSB are the principal cause of chromosomal translocations, which represent one of the main mechanisms for activation of oncogenes in human cancers (Povirk, 2006). Inactivation of tumor suppressor genes via deletions is another procarcinogenic consequence of DSB production (Shrivastav et al., 2008; Wyman and Kanaar, 2006). In response to the formation of DSB, cells rapidly activate a stress signaling network targeting cell cycle progression and DNA repair. One of the earliest events occuring following DSB is the appearance of Ser139-phosphorylated histone H2AX, which is also known as γH2AX (Scully and Xie, 2013). H2AX constitutes approximately 25% of total histone H2A and differs from the canonical histone H2A by the presence of a COOH-terminal tail containing the SQ amino acid motif for phosphorylation by DNA damage-responsive kinases ATM, ATR, and DNAPK. ATM is generally responsible for the majority of γH2AX production at DSB sites, with DNAPK playing a secondary role (Burma et al., 2001). Phosphorylation of H2AX by ATR occurs in response to replication stress, which results in the generation of single-stranded DNA regions that provoke ATR activation (Scully and Xie, 2013). The formation of γH2AX eventually spreads over large chromatin regions, stimulating other histone modifications, chromatin remodeling, and recruitment of numerous DNA repair factors. Binding of MDC1 to γH2AX is responsible for the chromatin recruitment of the E3 ubiquitin ligase RNF168 that catalyzes mono- and diubiquitination of H2A/H2AX (Lukas et al., 2011). This K63-linked ubiquitination is necessary for chromatin retention of BRCA1 and 53BP1 that control the activation of the 2 major DSB repair pathways, homologous recombination and nonhomologous end-joining, respectively (Lukas et al., 2011; Panier and Boulton, 2014). Mice with knockouts of both H2ax alelles showed increased radiation sensitivity, growth retardation, immune defects, and chromosomal instability (Celeste et al., 2002).
Chromium(VI) is a recognized human respiratory carcinogen, which causes DNA damage after its intracellular activation through reduction to Cr(III) (Salnikow and Zhitkovich, 2008). Ascorbate (Asc) is the main reducer of Cr(VI) in the lung and other tissues in vivo (Standeven and Wetterhahn, 1991; Suzuki and Fukuda, 1990) whereas glutathione and cysteine are largely responsible for Cr(VI) metabolism in cultured cells due to their severe deficiency in this vitamin (Salnikow and Zhitkovich, 2008). Unlike 1-electron reduction by thiols, Cr(VI) metabolism by Asc involves the initial transfer of 2 electrons and consequently, lacks the formation of reactive Cr(V) intermediate (Stearns and Wetterhahn, 1994; Zhang and Lay, 1996). Restoration of physiological levels of Asc in cultured cells suppressed oxidative DNA damage by Cr(VI) (Reynolds et al., 2012) and abolished responses of the redox-sensitive dyes dichlorofluorescein (DCFH) and dihydrorhodamine (DeLoughery et al., 2014). Cr-DNA adducts are the most abundant form of DNA damage by Cr(VI) in mammalian cells (Quievryn et al., 2002; Zhitkovich et al., 1995). Short-term treatments with nonapoptotic doses of Cr(VI) have also been found to generate foci of the DSB marker γH2AX in human skin fibroblasts (Ha et al., 2004) and human colon and lung epithelial cells (Peterson-Roth et al., 2005; Reynolds et al., 2007). The production of γH2AX and 53BP1 foci by Cr(VI) in Asc-restored human cells reflected the presence of genuine DSB, as determined by direct measurements of these lesions by pulse-field gel electrophoresis (Reynolds et al., 2009; Zecevic et al., 2009). Replication of Cr-adducted DNA and a subsequent activation of mismatch repair were required for the generation of chromosomal DSB by Cr(VI) (Peterson-Roth et al., 2005; Reynolds et al., 2007, 2009). Despite their indirect mechanism of formation, DSB were responsible for the main clastogenic and cytotoxic effects of Cr(VI) in cells with physiological levels of Asc (Reynolds and Zhitkovich, 2007; Reynolds et al., 2007, 2009). Cr-induced DSB were more persistent in human cells lacking WRN helicase (Liu et al., 2009; Zecevic et al., 2009), which is involved in the initiation of homologous recombination repair (Zecevic et al., 2009). The unusual requirements for their formation and repair point to a possibility that Cr-elicited DSB could be produced in the nucleus nonrandom and/or trigger different stress signaling responses than those by the canonical DSB formed by ionizing radiation and other oxidants.
In this work, we investigated the intranuclear localization of Cr(VI)-induced DSB and identified a DNA damage-responsive kinase that was responsible for the critical upstream response to DSB, which is the formation of γH2AX. Importantly, our studies were performed in Asc-restored human and mouse cells.
MATERIALS AND METHODS
Chemicals
Dehydro-l-(+)-ascorbic acid dimer (DHA), potassium chromate (K2CrO4, 99% pure), and nitric acid (>99.999% pure) were purchased from Sigma-Aldrich. NU7026 and ETP46464 were from Calbiochem, 1,2-diamino-4,5-dimethoxybenzene dihydrochloride from Molecular Probes, bleomycin from LKT Labs, and KU55933 from BioVision. KU60019, VE821, and NU7441 were obtained from SelleckChem. All other reagents were purchased from Sigma-Aldrich.
Cells and treatments
All cells were obtained from the American Type Culture Collection. Human H460 lung epithelial line was cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum and penicillin/streptomycin in a humidified atmosphere of 95% air and 5% CO2. Human IMR90 normal lung fibroblasts were propagated in DMEM medium containing 10% serum and antibiotics in the atmosphere containing 5% O2 and 5% CO2. MEFs were also grown under 5% O2 and 5% CO2 in DMEM medium containing 10% serum. Growth of normal human and mouse fibroblasts in physiological 5% O2 improves their proliferative properties and inhibits the appearance of senescence, particularly for MEFs. H460 line has long been established for growth under atmospheric O2 levels, which does not impair its proliferation. Stock solutions of K2CrO4 [Cr(VI)] were prepared in deionized water and filter-sterilized before addition to cells. Treatments with Cr(VI) were done in the complete growth media following restoration of Asc levels in cells. All small-molecule inhibitors were added simultaneously with Cr(VI).
Restoration of Asc levels
Cells were incubated for 90 min with DHA in Krebs-HEPES buffer [30 mM HEPES (pH 7.5), 130 mM NaCl, 4 mM KH2PO4, 1 mM MgSO4, and 1 mM CaCl2] containing 0.5 mM d-glucose. We used 0.5 mM DHA for MEFs and 1 mM DHA for H460 and IMR90 cells. Cellular Asc was extracted by methanesulfonic acid and quantified by fluorescence after conjugation with 1,2-diamino-4,5-dimethoxybenzene (Reynolds and Zhitkovich, 2007). Volume of cells was determined from forward scattering measurements by flow cytometry (FACSCalibur, BD Biosciences).
Western blotting
Cells were collected by scraping and washed twice with ice-cold PBS at 1100 g for 5 min at 4°C. Total protein extracts were prepared by boiling cells for 10 min in a 2% SDS buffer (2% SDS, 50 mM Tris-HCl pH 6.8, 10% glycerol) supplemented with Halt Protease and Phosphatase Inhibitors (Thermo Scientific) and 10 mM N-ethylmaleimide. After cooling to the room temperature, proteins lysates were collected by centrifugation at 12 000 × g for 10 min. For detection of γH2AX and other proteins under 100 kDa size, samples were run on 10% SDS-PAGE gels and then electrotransferred onto PVDF membranes using PierceG2 Fast Blotter (Thermo Scientific). Larger proteins were separated on 6% or 10% gels and electroblotted onto PVDF membranes overnight by a wet transfer at 4°C. Membranes were incubated with primary antibody overnight at 4°C. The following primary antibodies were used: rabbit polyclonal anti-phospho DNAPK (S2056) (1:1000; Abcam), rabbit polyclonal anti-phospho KAP1 (S824) (1:1000, Bethyl), mouse monoclonal anti-MSH6 (1:1000, BD Biosciences), mouse monoclonal anti-γ-tubulin (1:2000, from Sigma-Aldrich), rabbit polyclonal anti-phospho-CHK1 (Ser317) (1:1000; Cell Signaling Technology), rabbit polyclonal anti-phospho-CHK2 (Thr68) (1:1000; Cell Signaling Technology), rabbit polyclonal anti-phospho-H2AX (Ser139) (1:1000; Cell Signaling Technology), rabbit polyclonal anti-fibrillarin (1:5000, Abcam), and rabbit polyclonal anti-histone H2A (1:1000, Millipore). Secondary antibodies were horseradish peroxidase-conjugated goat anti-mouse IgG (1:5000, Millipore) and goat anti-rabbit IgG (1:2000, Cell Signaling Technology).
Cytotoxicity
Cytotoxic effects of Cr(VI) were assessed by the determinations of the total metabolic activity of cell populations using the CellTiter-Glo luminescent cell viability assay (Promega), which quantifies cellular ATP. This assay integrates the number of viable cells and their metabolic activity, making it sensitive to all forms of cytotoxicity. Cells were seeded into black 96-well optical bottom cell culture plates (2000 cells/well for H460 and 1000 cells/well for IMR90) and allowed to attach overnight before treatments with Cr(VI). After 48 h post-Cr, the assay reagents were added to the plates and luminescence readings were taken after 10 min incubation at room temperature.
Clonogenic survival
A long-term reproductive viability of Cr(VI)-treated H460 cells was examined by the clonogenic assay, which assesses the ability of individual cells to retain proliferative activity that is manifested in the production of visible colonies. Cells were seeded onto 60-mm dishes (400 cells/dish) and grown overnight. Following restoration of Asc levels, cells were treated with Cr(VI) for 3 h and then cultured for 8 days. Cells were fixed with methanol and stained with a Giemsa solution (Sigma-Aldrich). Colonies containing 30 or more cells were counted.
Cellular accumulation of Cr(VI)
Uptake of Cr(VI) was measured by graphite furnace atomic absorption spectroscopy (GF-AAS) using nitric acid extracts of cells (Messer et al., 2006). H460 cells were seeded into 6-well plates (3 × 105 cells/well) and grown overnight. Cells were preloaded with Asc and then treated with Cr(VI) for 3 h. After removal of Cr(VI)-containing media, cell monolayers were washed twice with warm PBS and then cells were detached by incubation with trypsin-EDTA (Gibco 15400-054). After centrifugation at 800 × g for 5 min at 4°C, cells were washed 2 times with ice-cold PBS. The final cell pellets were resuspended in 50 µl of deionized water followed by the addition of 50 µl of 10% nitric acid. After freezing at −80°C, Cr was extracted by heating samples at 50°C for 60 min followed by recovery of metal-containing supernatants at 10 000 × g for 10 min at 4°C. The extracts were diluted with deionized water to 2% nitric acid prior to Cr determinations by GF-AAS (AAnalyst600 Atomic Absorption Spectrometer, Perkin-Elmer). Metal uptake was normalized per protein amounts in the cell pellets produced during nitric acid extraction.
Immunofluorescence
IMR90 or MEF cells were seeded at approximately 50% confluence on human fibronectin-coated coverslips. On the next day, cells were incubated with 1 mM DHA (IMR90) or 0.5 mM DHA (MEF) for 90 min and then treated with Cr(VI) in complete media. Following treatment of cells, Cr(VI)-containing media was aspirated, and cells were washed once with PBS. For experiments with γH2AX and 53BP1 staining, cells were directly fixed to the slides by one of the following methods: incubation with 3.7% paraformaldehyde for 15 min at room temperature or with methanol chilled to −20°C for 10 min at 4°C. Cells were permeabilized in 0.5% Triton X-100 for 15 min at room temperature and then blocked with 2% FBS in PBS at 37°C for 30 min. In experiments with XPA staining, cells were first extracted on the slides with a detergent solution (0.2% CHAPS, 150 mM NaCl, 20 mM Tris-HCl, pH 8.0, phosphatase/protease inhibitors) for 10 min at 4°C and then fixed with paraformaldehyde. Primary antibodies were diluted in 1% BSA, 0.5% Tween-20 in PBS. The following primary antibodies were used: mouse monoclonal for γH2AX (ab26350 from Abcam, 1:500 dilution), rabbit polyclonal for H3K9me3 (ab8898 from Abcam, 1:100 dilution), rabbit polyclonal for H3K4me3 (07-473 from Millipore, 1:500 dilution), rabbit polyclonal for 53BP1 (sc-22760 from Santa Cruz, 1:100 dilution), and mouse monoclonal for XPA (556453 from BD Biosciences, 1:250 dilution). Cells were incubated in primary antibody for 2 h at 37°C, and washed with PBS 3 times for 5 min at room temperature. The Alexa Fluor 488 goat anti-mouse (A11029, 1:500 dilution) and Alexa Fluor 568 goat anti-rabbit (A11036, 1:500 dilution) antibodies were from Life Technologies. Both secondary antibodies were prepared in the same buffer as primary antibodies. Cells were incubated with secondary antibody for 1 h at room temperature in the dark. Coverslips were then mounted on glass slides using Vectashield fluorescence mounting media with DAPI (H-1200). Cells were imaged either via the Nikon E-800 Eclipse fluorescent scope (×200 magnification, wide-field images) for cell counts, or on the Zeiss LSM710 confocal microscope (×630 magnification pictures, small fields of cells) for colocalization. Line traces were generated using the “Profile” function of Zeiss’ ZEN 2012 Blue Edition software.
Statistics
Statistical significance was evaluated by 2-tailed, unpaired t test.
RESULTS
Cytotoxicity and γH2AX Formation by Cr(VI) in Ascorbate (Asc)-Restored Cells
We selected H460 lung epithelial cells and IMR90 normal lung fibroblasts as our human cell models, which we have extensively characterized for toxic effects of Cr(VI) (Reynolds and Zhitkovich, 2007; Reynolds et al., 2009, 2012). Importantly, H460 cells showed normal signaling responses to DSB by ionizing radiation (Zhang et al., 2006). Nonimmortalized MEFs were used as a rodent cell model. Since Asc plays a key role in Cr(VI) metabolism in vivo but its levels are extremely low in cultured cells, we first determined conditions for restoration of its physiological concentrations. Under standard culture conditions, normal IMR90 and MEFs contained only 10–20 µM Asc (Fig. 1A), which corresponds to approximately 1%–2% of the physiological amounts of this vitamin in tissues (Kojo, 2004). Cellular levels of Asc in both types of cells were elevated to its physiological range of 1–2 mM by preincubation with dehydroascorbic acid (DHA) (Fig. 1A). In all experiments with Cr(VI) in this work, we employed cells with re-established Asc levels. For H460 cells, we elevated cellular Asc levels to approximately 1 mM (DeLoughery et al., 2014). Examination of cytotoxicity of Cr(VI) in H460 cells showed that a long-term survival assessed by clonogenic assay and the ATP-based cell viability measurements at 48 h post-exposure showed very similar levels of toxicity (Fig. 1B). Since IMR90 cells and MEFs are not immortalized and display poor growth under sparsely seeded conditions of the clonogenic procedure, we assessed toxicity of Cr(VI) in these cells by the ATP measurements at 48 h after 3 h- or 6 h-long exposures. Unlike H460 cells, 3 h treatments with Cr(VI) were only weakly toxic in IMR90 cells (16.5 ± 2.5% decrease in viability for the highest dose of 20 µM Cr) and essentially nontoxic in MEFs (Figs. 1C and 1D). Longer 6 h treatments with Cr(VI) produced stronger cytotoxic responses in both IMR90 and MEFs, although the decline in MEFs viability was still very modest (<20%). Despite a very close correlation between our short-term and long-term toxicity assays in H460 cells, we could not exclude a possibility that ATP-based viability measurements underestimated the extent of a long-term injury by Cr(VI) in IMR90 and MEFs. To mitigate this concern, our subsequent studies with Cr(VI) in these cells employed doses that caused less than 20% decreases in cellular ATP content at 48 h post-exposure.
FIG 1.

Ascorbate restoration and cytotoxicity of Cr(VI). A, Cellular Asc levels in control and DHA-treated cells (1 mM DHA for IMR90 and 0.5 mM DHA for MEFs). Data are means ± SD for 3 independent samples. B, Clonogenic survival and ATP-based cell viability measurements in Asc-restored H460 cells treated with Cr(VI) for 3 h. Cellular ATP levels were measured at 48 h after Cr(VI) exposure. Data are means ± SD for 2 independent experiments including 3 dishes per dose in each clonogenic assay and 4 wells per dose in the viability procedure. C, ATP-based viability measurements in Asc-restored IMR90 cells at 48 h after Cr(VI) exposures for 3 or 6 h. Data are means ± SD for 2 experiments that each included 4 wells per dose. D, Cell viability of Asc-restored MEFs at 48 h after Cr(VI) exposures for 3 or 6 h. Data are means ± SD for 2 experiments that each had 4 wells per dose.
To examine the formation of γH2AX by Cr(VI), we treated human cells with Cr(VI) for 3 h and MEFs for 6 h and then lysed them by boiling in 2% SDS in the presence of 10 mM N-ethylmaleimide, which inhibits protein deubiquitination. The longer Cr(VI) treatment of MEFs was chosen based on the absence of cytotoxicity after the shorter 3 h exposure (Fig. 1D), suggesting a slow uptake in these cells. Western blotting of total cell lysates found an increased formation of γH2AX and its mono- and diubiquitinated forms in Cr(VI)-treated H460 and IMR90 (Figs. 2A and 2B). The accumulation of ubiquitinated γH2AX was even more extensive in MEFs (Fig. 2C). Thus, Cr(VI) induces a DSB-associated pattern of histone H2AX modifications, which includes its Ser139 phosphorylation (γH2AX) and mono- and diubiquitination (Lukas et al., 2011; Scully and Xie, 2013), although the extent of γH2AX ubiquitination by Cr(VI) appears to be greater than that after ionizing radiation (Chen et al., 2013).
FIG 2.

Formation of Ser139-phosphorylated histone H2AX (γH2AX) in Cr(VI)-treated cells. Asc levels in cells were restored to the physiological range before the addition of Cr(VI). γH2AX-Ub1—monoubiquitinated and γH2AX-Ub2—diubiquitinated forms of γH2AX. Fibrillarin and tubulin were used as loading controls. A, Production of γH2AX after 3 h exposure to Cr(VI) in H460 and B, IMR90 cells. C, Western blot for γH2AX in MEFs treated with Cr(VI) for 6 h.
Search for a Kinase that Is Responsible for γH2AX Formation
In order to identify a DNA damage-responsive kinase that is required for H2AX phosphorylation by Cr(VI), we employed chemical inhibitors of ATM, ATR, and DNAPK. In the first series of experiments, we tested 2 inhibitors per kinase for their effective concentrations and target specificity. We used the radiomimetic drug bleomycin to cause oxidation-mediated DSB and activation of ATM and DNAPK. Treatments with camptothecin were employed to produce stalling of replication forks by trapped covalent complexes of topoisomerase I with DNA and trigger ATR and ATM activation. As expected, 2 selected inhibitors of ATM (KU60019 and KU55933) strongly suppressed γH2AX production by bleomycin (Fig. 3A). The residual γH2AX was probably formed by DNAPK (Burma et al., 2001), as both inhibitors completely abolished bleomycin-induced phosphorylation of the ATM target CHK2-Thr68 (Fig. 3B). The same inhibitors also eliminated phosphorylation of another ATM target KAP1-Ser824 in camptothecin-treated cells but did not change the levels of the ATR target, phosphorylated CHK1 at Ser317 (Fig. 3C). A robust activation of ATR kinase by camptothecin, as monitored by CHK1 phosphorylation, was fully suppressed by ETP46464 and VE821 but these inhibitors showed no effect on the ATM-dependent CHK2 phosphorylation (Fig. 3D). Next, we tested 3 concentrations of 2 DNAPK inhibitors (NU7026 and NU7441) for their off-target effects on ATM and ATR signaling in camptothecin-exposed cells. We found that only the highest dose of 60 µM NU7026 clearly diminished ATR activity as evidenced by a lower CHK1 phosphorylation (Fig. 3E). All doses of both inhibitors eliminated activating autophosphorylation of DNAPK at Ser2056 (Fig. 3F). In the subsequent experiments with Cr(VI), we used 30 µM NU7026 and 10 µM NU7441, which were fully effective in inhibiting DNAPK but lacked off-target inhibition of ATR activity in camptothecin-treated cells. Although our experiments with bleomycin and camptothecin employed regular H460 cultures, there are no apparent reasons to believe that Asc levels can alter selectivity of the tested inhibitors to the ATP-binding pockets of different kinases.
FIG 3.

Validation of activity and specificity of chemical inhibitors of; ATM, ATR, and DNAPK. H460 cells were treated with 1 µM camptothecin (CPT) or 20 µg/ml bleomycin for 1 h in the presence of the indicated inhibitors: ATM-i1—1 µM KU60019, ATM-i2—10 µM KU55933, ATR-i1—3 µM ETP46464, ATR-i2—10 µM VE821, DNAPK-i1—NU7026, DNAPK-i2—NU7441. MSH6, fibrillarin, and tubulin were used as loading controls. A, ATM-dependent formation of γH2AX and B, CHK2 phosphorylation in bleomycin-treated cells. C, Effects of ATM inhibitors on CPT-induced phosphorylation of KAP1 and CHK1. D, Effects of ATR inhibitors on CPT-induced phosphorylation of KAP1, CHK2, and CHK1. E, Effects of DNAPK inhibitors on CPT-induced phosphorylation of ATM (KAP1, CHK2) and ATR (CHK1) targets. F, Effects of DNAPK inhibitors on its autophosphorylation in bleomycin-treated cells.
Western blotting of Asc-restored H460 cells treated with 10 µM Cr(VI) (approximately LD75 dose, Fig. 1B) in the presence of the first set of kinase inhibitors showed unaffected levels of total γH2AX under conditions of suppressed ATM and DNAPK activities (Fig. 4A). In a striking contrast, inhibition of ATR by ETP46464 (ATR-i1) completely abolished Cr(VI)-induced formation of γH2AX and its ubiquitinated forms. The addition of this inhibitor had no effect on Cr(VI) uptake by H460 cells (Fig. 4B), excluding a possibility of different internal doses as the cause for the disappearance of γH2AX. To further test the involvement of specific kinases, we treated H460 cells with 2 lower concentrations of Cr(VI) (3 µM ∼ LD25, 8 µM ∼ LD65) in the presence of a second set of inhibitors. Again, inhibition of ATR fully eliminated the production of γH2AX (Fig. 4C). The independence of γH2AX formation by Cr(VI) on the canonical DSB-responsive kinase ATM in H460 cells could potentially reflect its transformed phenotype. Therefore, we next examined γH2AX production in Asc-restored IMR90 normal human lung fibroblasts. In experiments with IMR90, we scored percentage of cells containing γH2AX foci, which is a more sensitive marker of DSB than total γH2AX levels by Western blotting due to the ability to exclude cells displaying DSB-unrelated diffuse nuclear staining. We found that ATR-i1 completely eliminated γH2AX foci formation by 10 µM Cr(VI) (Fig. 5A). A less potent ATR inhibitor, ATR-i2, diminished the number of cells containing γH2AX foci by almost 4-fold. The employed dose of 10 µM Cr(VI) produced no changes in cell viability immediately after 3 h-long exposure (not shown) and caused less than 10% increase in cytotoxicity at 48 h post-exposure (Fig. 1C). Examination of IMR90 cells treated with essentially noncytotoxic 2 and 5 µM Cr(VI) showed a virtual absence of γH2AX formation in the presence of the ATR inhibitor-1 (Fig. 5B). Parallel scoring of samples incubated with the ATM inhibitor-1 found only a small decrease in the frequency of γH2AX-positive cells treated with 5 µM Cr(VI). Finally, we assessed γH2AX levels in Asc-restored MEFs incubated with Cr(VI) in the presence of ATR-i1 and ATR-i2 (Fig. 5C). Similar to the results in IMR90, we found that ATR-i1 totally abolished the formation of γH2AX by Cr(VI) whereas ATR-i2 eliminated a majority of H2AX phosphorylation. Thus, the use of 2 inhibitors for each of the 3 DNA damage-responsive kinases in 3 cell types showed that ATR played a central role in γH2AX formation triggered by Cr(VI)-induced DSB.
FIG 4.

Effect of ATM, ATR, and DNAPK inhibitors on H2AX phosphorylation in Cr(VI)-treated H460 cells. Asc-restored cells were treated for 3 h with Cr(VI). Histone H2A and tubulin served as loading controls. A, Formation of γH2AX and its ubiquitinated forms by 10 µM Cr(VI) in the presence of ATM-i1 (1 µM KU60019), DNAPK-i1 (30 µM NU7026), and ATR-i1 (3 µM ETP46464). “γH2AX-total” numbers indicate a total normalized intensity of all 3 bands after subtraction of the corresponding Cr-untreated controls. B, Uptake of Cr(VI) by H460 cells in the presence and absence of 3 µM ETP46464 (ATR-i1). Data are means ± SD for 3 independent samples. C, Western blot for γH2AX in H460 cells treated with Cr(VI) in the presence of a second set of inhibitors (ATM-i2—10 µM KU55933, DNAPK-i2—10 µM NU7441, ATR-i2—10 µM VE821). “γH2AX-total” numbers indicate a total normalized intensity of both γH2AX bands from 2 Western blots.
FIG 5.

Impact of ATR and ATM inhibitors on γH2AX formation in normal human and mouse fibroblasts. Asc-restored IMR90 and MEFs were treated with Cr(VI) for 3 h and immediately processed for immunostaining or lysed in 2% SDS for Westerns. Nuclei containing 5 or more γH2AX foci were scored as positive. A, Percentage of IMR90 cells containing γH2AX foci after exposure to 10 µM Cr(VI) in the presence or absence of ATR inhibitors. Data are means ± SD, n = 2 (**P < 0.005 relative to Cr-treated cells without inhibitors). B, Percentage of γH2AX-positive IMR90 cells treated with Cr(VI) in the presence of ATM and ATR inhibitors. Data are means ± SD, n = 3 (*P < 0.01 and ***P < 0.0001 relative to Cr-treated cells without inhibitors). C, Western blot for γH2AX in Cr(VI)-treated MEFs incubated with and without ATR inhibitors. Tubulin served as a loading control.
Intranuclear Localization of Cr-Induced DSB
Formation of DSB by Cr(VI) requires replication of Cr-damaged DNA, as evidenced by a complete elimination of DNA breakage in the presence of the DNA polymerase inhibitor aphidicolin (Reynolds et al., 2009). Costaining of Asc-restored human cells with γH2AX and cell cycle markers found that Cr-DSBs were predominantly found in G2 phase (Peterson-Roth et al., 2005; Reynolds et al., 2007, 2009). One explanation for replication dependence and G2 specificity of Cr-induced DSB could be DNA breakage in heterochromatin, which is known to replicate in late S phase. Therefore, we next proceeded to examine the presence of Cr-induced DSB in silenced (heterochromatin) and transcriptionally active chromatin (euchromatin). We first studied normal MEFs as these cells have clearly defined heterochromatic regions that are brightly stained by DNA dyes such as DAPI or Hoechst (Guenatri et al., 2004). As expected, we found that MEFs showed several DAPI chromocenters inside the nuclei (intranuclear heterochromatin) and increased DAPI staining along the nuclear membrane (peripheral heterochromatin) (Fig. 6A). DAPI-bright regions were costained with antibodies for histone H3 trimethylated at Lys9 (H3K9me3), which is a well-established marker of heterochromatin (Kimura, 2013). Cr-induced DSBs were completely excluded from MEFs heterochromatin, as evidenced by the absence of colocalization of γH2AX and H3K9me3 foci (Figs. 6A and 6B). Next, we examined the relationship between the distribution of γH2AX foci and the euchromatin marker histone H3 trimethylated at Lys4 (H3K4me3) (Kimura, 2013). We found that γH2AX localized to chromatin regions that were strongly enriched in H3K4me3 and excluded DAPI-bright areas (Figs. 6C and 6D). To confirm our findings with γH2AX, we investigated chromatin distribution of 53BPI foci, which are another established marker of DSB (Panier and Boulton, 2014), including Cr(VI)-induced DSB (Reynolds et al., 2007; Zecevic et al., 2009). Similar to γH2AX, 53BP1 foci were excluded from the DAPI-bright regions (Figs. 6E and 6F), further supporting the absence of DSB in MEFs heterochromatin. Finally, using immunostaining for H3K9me3 and H3K4me3, we determined that Cr-induced γH2AX foci were also present in euchromatic but not heterochromatic regions of IMR90 normal human lung fibroblasts (Figs. 7A–C).
FIG 6.

Costaining of Cr(VI)-treated MEFs for DSB and markers of euchromatin and heterochromatin. Asc-restored MEFs (1 mM cellular Asc) were treated with 10 µM Cr(VI) for 6 h and then fixed with paraformaldehyde and immunostained. More than 100 cells from 3 slides per treatment were analyzed for each type of staining. A, Representative confocal images of MEFs stained for γH2AX and the heterochromatic histone H3K9me3. B, Density scans along the arrow in the merged image of panel A. C, Representative confocal images of MEFs immunostained for γH2AX and the euchromatic histone H3K4me3. The arrow in the merged image shows a scanning path for line traces in panel D. D, Density scans through the left nucleus in the merged image of panel C. E, Representative confocal images of cells showing exclusion of 53BP1 foci from DAPI-bright heterochromatic foci. F, Density scans along a white arrow in the merged image of panel E.
FIG 7.

Costaining of γH2AX with markers of euchromatin and heterochromatin in IMR90 normal human fibroblasts. Asc-restored cells were treated with 10 µM Cr(VI) for 6 h and then fixed with paraformaldehyde and immunostained. More than 100 cells from 3 slides per treatment were analyzed for each type of staining. A, Representative confocal images of cells stained for γH2AX and histone H3K9me3. B, Representative confocal images of cells immunostained for γH2AX and histone H3K4me3. C, Density scans along the arrow in the merged image of panel B.
Formation of DSB in Cr(VI)-treated cells results from toxic processing of Cr-DNA adducts by mismatch repair (Peterson-Roth et al., 2005; Reynolds et al., 2009). One possibility for the observed nonrandom generation of DSB could be a preferential production of Cr-DNA adducts in the euchromatic regions. Cr-DNA adducts are efficiently recognized and removed in human cells by nucleotide excision repair (NER) (Reynolds et al., 2004). Cells lacking XPA and other essential NER proteins were severely deficient in repair of Cr-DNA adducts. Damage-containing chromatin recruits NER factors, which accumulate in a dose-dependent manner. Thus, the intranuclear distribution of Cr-DNA adducts can be monitored by immunostaining for the NER factors, such as XPA. To reveal a chromatin-bound fraction of XPA, we extracted control and Cr(VI)-treated IMR90 cells with a mild detergent prior to fixation with paraformaldehyde. The employed procedure produced almost no staining in controls but detected large amounts of chromatin-bound XPA in all Cr(VI)-treated cells (Fig. 8A). Confocal images of Cr(VI)-exposed cells showed a largely homogenous staining of XPA throughout the nuclei, indicating similar levels of Cr-DNA adduction in euchromatin and heterochromatin (Fig. 8B). One or two fainter areas of XPA staining visible in some nuclei corresponded to the DNA-poor nucleolar regions that are seen as “holes” in DAPI images.
FIG 8.
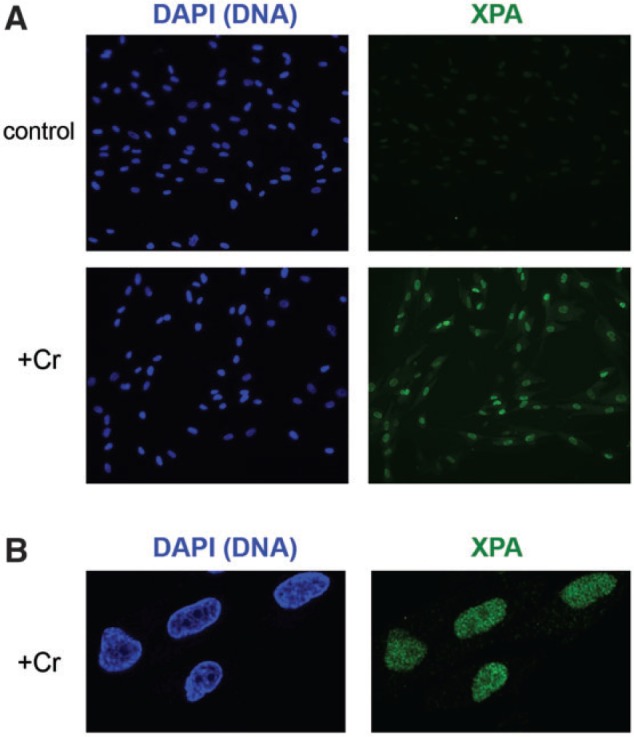
Chromatin binding of XPA in Cr(VI)-exposed IMR90 cells. Cells were treated with Cr(VI) as in Figure 7. Free nucleoplasmic XPA was removed by extraction with a 0.2% CHAPS solution prior to paraformaldehyde fixation and immunostaining for XPA. A, A wide-field view of control and Cr-treated IMR90 cells. B, Confocal images of Cr-treated cells.
DISCUSSION
Key Role of ATR in γH2AX Formation by Cr(VI)
Production of phosphorylated histone H2AX at Ser139 (known as γH2AX) is one of the earliest DSB-induced stress signaling events, which is commonly used for the detection and quantitation of DSB (Scully and Xie, 2013). The importance of γH2AX goes beyond its utility as a biochemical marker of DSB, as it initiates other histone modifications, chromatin recruitment of various regulators, and a large-scale chromatin remodeling, resulting in stress signaling amplification and activation of the major DSB repair pathways and checkpoint responses (Lukas et al., 2011; Scully and Xie, 2013). ATM is commonly viewed as the main kinase generating γH2AX in response to DSB, with DNAPK playing a distant secondary role (Burma et al., 2001; Scully and Xie, 2013). In this work, we found that Cr(VI)-induced DSB also caused extensive H2AX phosphorylation but it was principally mediated by ATR in both human and mouse cells. A previous study comparing 2 human fibroblast lines from genetically unrelated individuals found a significantly lower formation of γH2AX by Cr(VI) in ATM-null cells (Ha et al., 2004). Although the dosing regimen in this earlier work (3 h exposure, 6 µM Cr) was similar to our conditions (3 h exposure, 2–10 µM Cr), the application of selective inhibitors allowed us to test the involvement of the individual kinases in the same cells. Another important difference of our work was the use of cells with physiological levels of Asc, which plays a key role in Cr(VI) metabolism in vivo (Suzuki and Fukuda, 1990; Standeven and Wetterhahn, 1991). Restoration of cellular Asc suppresses oxidative DNA damage and production of reactive Cr(V) complexes by Cr(VI) (DeLoughery et al., 2014; Reynolds et al., 2012), which could have been responsible for the activation of ATM in Asc-deficient cultures (Ha et al., 2004). It is possible that ATM plays some role in either H2AX phosphorylation or other signaling responses at late post-exposure times, potentially being activated by apoptotic DNA damage or secondary lesions formed by processed DSB. The focus of our present work was on the initial responses to DSB, which we found to primarily involve ATR-dependent histone H2AX phosphorylation. One possible explanation for a selective ATR activation instead of ATM could be the structure of DSB ends. A classic model of DSB signaling is based on studies of blunt-ended DSB that are bound by the Mre11-Rad50-NBS1 complex, which triggers recruitment and activation of ATM (Lukas et al., 2011; Scully and Xie, 2013). Formation of DSB in Cr-treated cells requires activity of mismatch repair (Peterson-Roth et al., 2005; Reynolds et al., 2007, 2009), which operates by excision of one strand producing tracks of single-stranded DNA (Jiricny, 2013). Thus, it is likely that a majority if not all of Cr-induced DSB contain single-stranded tails, which are potent activators of ATR (Lukas et al., 2011) but they would block recruitment of the Mre11-Rad50-NBS1 complex and consequently, prevent ATM stimulation.
Euchromatin Localization of DSB
Our studies with confocal microscopy imaging of normal mouse and human cells costained with markers of DSB (γH2AX, 53BP1) and transcriptionally active or silenced chromatin showed that Cr-induced DSBs were present only in euchromatin. The formation of Cr-DNA adducts, which give rise to DSB via mismatch repair-dependent processing of replicated DNA (Peterson-Roth et al., 2005; Reynolds et al., 2007, 2009), appeared to be relatively evenly distributed throughout the nucleus, as monitored by the distribution of the DNA adduct-binding NER factor XPA. The nucleolus was the only nuclear region that had diminished amounts of chromatin-associated XPA, which simply reflected low concentrations of DNA inside this organelle (evident by very weak DAPI staining). Unlike Cr(VI), other agents generating replication-dependent, albeit mismatch repair-independent, DSB produced breakage in both euchromatic and heterochromatic areas (Cowell et al., 2007). Studies on ionizing radiation-treated cells have found that the initial DSB formation occurred in both euchromatin and heterochromatin. At later times, heterochromatin domains containing DSB underwent decondensation associated with a movement of DSB and their marker γH2AX from the inside to the periphery of the heterochromatic regions (Chiolo et al., 2011). In Cr(VI)-treated cells, heterochromatin domains remained condensed and γH2AX or 53BP1 foci were absent in the middle or periphery of heterochromatic areas, further supporting the oxidation-independent mechanism of DNA breakage. One potential reason for the euchromatin selectivity of Cr-induced DSB can be related to the enrichment of transcriptionally active genomic regions in histone H3 trimethylated at lysine 36 (H3K36me3) (Kimura, 2013). This histone modification has recently been found to be necessary for mismatch repair activity in S and G2 cells (Li et al., 2013) and the formation of DSB by Cr(VI) requires a functional mismatch repair (Peterson-Roth et al., 2005; Reynolds et al., 2007, 2009). Collision of transcription-associated processes at the sites of mismatch repair-mediated excision tracks can be another potential cause of euchromatin localization of Cr-induced DSB. Based on their mechanism of formation (Macfie et al., 2010), it is possible that DNA-Cr-protein crosslinks are also preferentially formed in sterically accessible euchromatic DNA, which could cause mismatch repair-independent DSB via collapse of arrested replication forks.
Implications for Understanding of Cr(VI) Toxicity
Inhalation exposure to Cr(VI) under the current occupational standard is estimated to confer up to 23% lifetime risk of developing lethal lung cancer (Park et al., 2004), raising questions what factors contribute to such a high carcinogenic potency of this metal. DSB are powerful procarcinogenic lesions that produce chromosomal translocations and deletions of various sizes, which results in activation of oncogenes and loss of tumor-suppressor genes, respectively (Povirk, 2006; Wyman and Kanaar, 2006). For DSB that are distributed throughout the entire genome, deletions in heterochromatin-located genes and repeat sequences are not expected to cause growth-promoting consequences. Translocations and fusions between 2 repressed genes or repeat regions are also not very likely to generate an actively expressed gene with a transformation potential. The observed targeting of Cr-induced DSBs to euchromatin suggests that their production could be associated with particular high risks of carcinogenic translocations and inactivated tumor suppressors due to the involvement of actively expressed genes. As discussed above, H2AX phosphorylation by ATR in Cr(VI)-treated cells suggests the presence of single-stranded tails at the DSB ends, making them more likely to undergo error-prone repair generating deletions (Shrivastav et al., 2008). The abundance of DSB in euchromatin can also, at least in part, be responsible for the inability of Cr(VI)-treated cells to upregulate expression of inducible genes (Hamilton and Wetterhahn, 1989; Wei et al., 2004). The formation of DSB causes bidirectional transcriptional repression, which has been linked to the accumulation of ubiquitinated forms of H2A/H2AX (Chen et al., 2013; Lukas et al., 2011). Euchromatin accumulation of DSB and the observed presence of large amounts of mono- and diubiquitinated γH2AX are expected to prevent transcriptional upregulation of many genes in Cr(VI)-treated cells. A summary of our main findings and their toxicological implications are graphically presented in Figure 9.
FIG 9.
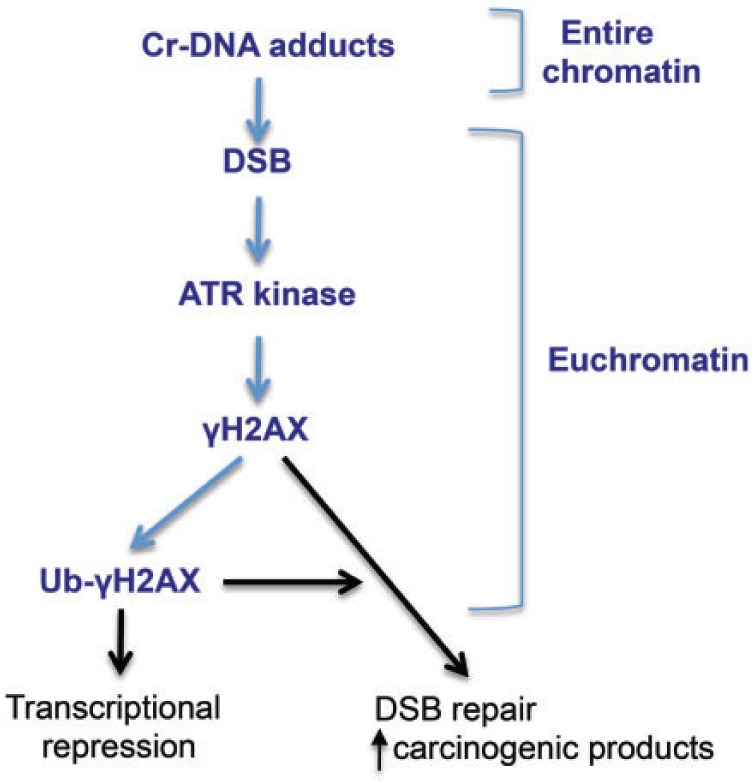
Summary of the main findings and their toxicological implications. Bold text identifies experimental findings whereas regular text and arrows indicate expected biological consequences.
FUNDING
The National Institute of Environmental Health Sciences (grant ES008786).
REFERENCES
- Burma S., Chen B. P., Murphy M., Kurimasa A., Chen D. J. (2001). ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 276, 42462–42467. [DOI] [PubMed] [Google Scholar]
- Celeste A., Petersen S., Romanienko P. J., Fernandez-Capetillo O., Chen H. T., Sedelnikova O. A., Reina-San-Martin B., Coppola V., Meffre E., Difilippantonio M. J., et al. (2002). Genomic instability in mice lacking histone H2AX. Science 296, 922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W. T., Alpert A., Leiter C., Gong F., Jackson S. P., Miller K. M. (2013). Systematic identification of functional residues in mammalian histone H2AX. Mol. Cell. Biol. 33, 11–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiolo I., Minoda A., Colmenares S. U., Polyzos A., Costes S. V., Karpen G. H. (2011). Double-strand breaks in heterochromatin move outside of a dynamic HP1α domain to complete recombinational repair. Cell 144, 732–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowell I. G., Sunter N. J., Singh P. B., Austin C. A., Durkacz B. W., Tilby M. J. (2007). γH2AX foci form preferentially in euchromatin after ionising-radiation. PLoS One 2, e1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLoughery Z., Luczak M. W., Zhitkovich A. (2014). Monitoring Cr intermediates and reactive oxygen species with fluorescent probes during chromate reduction. Chem. Res. Toxicol. 27, 843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenatri M., Bailly D., Maison C., Almouzni G. (2004). Mouse centric and pericentric satellite repeats form distinct functional heterochromatin. J. Cell. Biol. 166, 493–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha L., Ceryak S., Patierno S. R. (2004). Generation of S phase-dependent DNA double-strand breaks by Cr(VI) exposure: involvement of ATM in Cr(VI) induction of gamma-H2AX. Carcinogenesis 25, 2265–2274. [DOI] [PubMed] [Google Scholar]
- Hamilton J. W., Wetterhahn K. E. (1989). Differential effects of chromium(VI) on constitutive and inducible gene expression in chick embryo liver in vivo and correlation with chromium(VI)-induced DNA damage. Mol. Carcinog. 2, 274–286. [DOI] [PubMed] [Google Scholar]
- Jiricny J. (2013). Postreplicative mismatch repair. Cold Spring Harb. Perspect. Biol. 5, a012633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H. (2013). Histone modifications for human epigenome analysis. J. Hum. Genet. 58, 439–445. [DOI] [PubMed] [Google Scholar]
- Kojo S. (2004). Vitamin C: basic metabolism and its function as an index of oxidative stress. Curr. Med. Chem. 11, 1041–1064. [DOI] [PubMed] [Google Scholar]
- Li F., Mao G., Tong D., Huang J., Gu L., Yang W., Li G. M. (2013). The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSα. Cell 153, 590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F. J., Barchowsky A., Opresko P. L. (2009). The Werner syndrome protein functions in repair of Cr(VI)-induced replication-associated DNA damage. Toxicol. Sci. 110, 307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas J., Lukas C., Bartek J. (2011). More than a focus: the chromatin response to DNA damage and its genome integrity maintenance. Nat. Cell Biol. 13, 1161–1169. [DOI] [PubMed] [Google Scholar]
- Macfie A., Hagan E., Zhitkovich A. (2010). Mechanism of DNA-protein cross-linking by chromium. Chem. Res. Toxicol. 23, 341–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messer J., Reynolds M., Stoddard L., Zhitkovich A. (2006). Causes of DNA single-strand breaks during reduction of chromate by glutathione in vitro and in cells. Free Radic. Biol. Med. 40, 1981–1992. [DOI] [PubMed] [Google Scholar]
- Panier S., Boulton S. J. (2014). Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell. Biol. 15, 7–18. [DOI] [PubMed] [Google Scholar]
- Park R. M., Bena J. F., Stayner L. T., Smith R. J., Gibb H. J., Lees P. S. (2004). Hexavalent chromium and lung cancer in the chromate industry: a quantitative risk assessment. Risk Anal. 24, 1099–1108. [DOI] [PubMed] [Google Scholar]
- Peterson-Roth E., Reynolds M., Quievryn G., Zhitkovich A. (2005). Mismatch repair proteins are activators of toxic responses to chromium-DNA damage. Mol. Cell. Biol. 25, 3596–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Povirk L. F. (2006). Biochemical mechanisms of chromosomal translocations resulting from DNA double-stranded breaks. DNA Repair 5, 1199–1212. [DOI] [PubMed] [Google Scholar]
- Quievryn G., Messer J., Zhitkovich A. (2002). Carcinogenic chromium(VI) induces cross-linking of vitamin C to DNA in vitro and in human lung A549 cells. Biochemistry 41, 3156–3167. [DOI] [PubMed] [Google Scholar]
- Reynolds M., Peterson E., Quievryn G., Zhitkovich A. (2004). Human nucleotide excision repair efficiently removes chromium-DNA phosphate adducts and protects cells against chromate toxicity. J. Biol. Chem. 279, 30419–30424. [DOI] [PubMed] [Google Scholar]
- Reynolds M., Zhitkovich A. (2007). Cellular vitamin C increases chromate toxicity via a death program requiring mismatch repair but not p53. Carcinogenesis 28, 1613–1620. [DOI] [PubMed] [Google Scholar]
- Reynolds M., Stoddard L., Bespalov I., Zhitkovich A. (2007). Ascorbate acts as a highly potent inducer of chromate mutagenesis and clastogenesis: linkage to DNA breaks in G2 phase by mismatch repair. Nucleic Acids Res. 35, 465–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds M. F., Peterson-Roth E. C., Johnston T., Gurel V. M., Menard H. L., Zhitkovich A. (2009). Rapid DNA double-strand breaks resulting from processing of Cr-DNA crosslinks by both MutS dimers. Cancer Res. 69, 1071–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds M., Armknecht S., Johnston T., Zhitkovich A. (2012). Undetectable role of oxidative DNA damage in cell cycle, cytotoxic and clastogenic effects of Cr(VI) in human lung cells with restored ascorbate levels. Mutagenesis 27, 437–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos W. P., Kaina B. (2006). DNA damage-induced cell death by apoptosis. Trends Mol. Med. 12, 440–450. [DOI] [PubMed] [Google Scholar]
- Salnikow K., Zhitkovich A. (2008). Genetic and epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis: nickel, arsenic and chromium. Chem. Res. Toxicol. 21, 28–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully R., Xie A. (2013). Double strand break repair functions of histone H2AX. Mutat. Res. 750, 5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastav M., De Haro L. P., Nickoloff J. A. (2008). Regulation of DNA double-strand break repair pathway choice. Cell Res. 18, 134–147. [DOI] [PubMed] [Google Scholar]
- Standeven A. M., Wetterhahn K. E. (1991). Ascorbate is the principal reductant of chromium (VI) in rat liver and kidney ultrafiltrates. Carcinogenesis 12, 1733–1737. [DOI] [PubMed] [Google Scholar]
- Stearns D. M., Wetterhahn K. E. (1994). Reaction of Cr(VI) with ascorbate produces chromium(V), chromium(IV), and carbon-based radicals. Chem. Res. Toxicol. 7, 219–230. [DOI] [PubMed] [Google Scholar]
- Suzuki Y., Fukuda K. (1990). Reduction of hexavalent chromium by ascorbic acid and glutathione with special reference to the rat lung. Arch. Toxicol. 64, 169–176. [DOI] [PubMed] [Google Scholar]
- Wei Y. D., Tepperman K., Huang M. Y., Sartor M. A., Puga A. (2004). Chromium inhibits transcription from polycyclic aromatic hydrocarbon-inducible promoters by blocking the release of histone deacetylase and preventing the binding of p300 to chromatin. J. Biol. Chem. 279, 4110–4119. [DOI] [PubMed] [Google Scholar]
- Wyman C., Kanaar R. (2006). DNA double-strand break repair: all’s well that ends well. Annu. Rev. Genet. 40, 363–383. [DOI] [PubMed] [Google Scholar]
- Zecevic A., Menard H., Gurel V., Hagan E., DeCaro R., Zhitkovich A. (2009). WRN helicase promotes repair of DNA double-strand breaks caused by aberrant mismatch repair of chromium-DNA adducts. Cell Cycle 8, 2769–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Lay P. A. (1996). EPR studies of the reactions of Cr(VI) with L-ascorbic acid, L-dehydroascorbic acid, and 5,6-O-isopropylidene-L-ascorbic acid in water. Implications for chromium(VI) genotoxicity. J. Am. Chem. Soc. 118, 12624–12637. [Google Scholar]
- Zhang D., Zaugg K., Mak T. W., Elledge S. J. (2006). A role for the deubiquitinating enzyme USP28 in control of the DNA-damage response. Cell 126, 529–542. [DOI] [PubMed] [Google Scholar]
- Zhitkovich A., Voitkun V., Costa M. (1995). Glutathione and free amino acids form stable adducts with DNA following exposure of intact mammalian cells to chromate. Carcinogenesis 16, 907–913. [DOI] [PubMed] [Google Scholar]