

Abstract
The expression of fructosyltransferase (FTF), the enzyme that synthesizes fructan from sucrose, is regulated in the cariogenic bacterium Streptococcus mutans. However, the exact mechanism of FTF regulation is unknown. In this study, the role of a two-component regulatory system (covRS) in FTF expression was investigated. A CovR-defective mutant of S. mutans NG8 was constructed by homologous recombination. By use of immunoblotting, the mutant was shown to overexpress FTF in the absence of sucrose, while the wild type and a covRS-complemented mutant showed sucrose-inducible FTF expression. Reverse transcription-PCR showed that the ftf transcript levels were increased in the covR mutant, suggesting regulation at the transcriptional level. The covR mutant was also found to overproduce extracellular carbohydrate, and this phenotype was reversed by covRS complementation. Paper chromatographic studies and chemical tests showed that the extracellular carbohydrate contained glucose and glucuronic acid but not fructose. These results suggest that the extracellular carbohydrate was not fructan. The production of a glucose- and glucuronic acid-containing extracellular carbohydrate has not been reported for S. mutans and may be considered novel. In conclusion, the results indicate that the expression of FTF and a glucose- and glucuronic acid-containing carbohydrate was negatively regulated by the covRS two-component regulatory system in S. mutans.
Streptococcus mutans is the major etiological agent involved in dental caries (11, 20). The bacterium produces a number of sucrose-metabolizing enzymes that are critical in cariogenesis. These enzymes include fructosyltransferase (FTF) and three glucosyltransferases (GTFs). FTF synthesizes fructan polymers from the fructose moiety of sucrose. GTFs synthesize glucan polymers from the glucose moiety of sucrose. Water-insoluble glucan has been shown to mediate the attachment of S. mutans to the tooth surface (11, 20). The fructan polymer can serve as a carbohydrate reserve when nutrient availability is low. The role of FTF and GTFs in the virulence of S. mutans was demonstrated in studies showing that FTF-negative or GTF-negative mutants caused a reduced level of smooth-surface dental caries in the gnotobiotic rat model (23, 24). The expression of FTF and GTFs was previously shown to be influenced by environmental pH, growth rate, and carbon sources (14, 15, 18, 29). However, the exact mechanism of regulation of FTF expression is not known.
In bacteria, two-component regulatory systems are known to function in virulence, adaptation, and survival (12). A typical two-component system consists of a membrane-bound sensor histidine kinase and a cytoplasmic responder protein. The sensor protein senses changes in the environment, and the responder protein regulates gene expression. Relevant to this study, a two-component regulatory system named covRS (also termed csrRS) has been described for Streptococcus pyogenes (7, 17). CovRS is encoded by the covRSX operon, in which covR codes for the responder and covS codes for the sensor. The identity and function of CovX remain unclear. CovRS has been shown to negatively regulate capsular hyaluronic acid production, streptokinase, streptolysin S, mitogenic factor SpeMF, cysteine protease SpeB, and a Mac-1-like protein in S. pyogenes (9). In vitro studies have shown that CovR binds to the promoters of several of these virulence genes (8, 22). Recently, Biswas and Scott (2) showed that a positive regulator, RocA, activates covR expression in S. pyogenes. In contrast, the regulation of virulence genes in S. mutans is less well understood.
In this study, a two-component regulatory system encoded by the covRSX operon from S. mutans was isolated and insertionally inactivated. S. mutans CovRS was shown to negatively regulate FTF expression and the production of a novel glucose- and glucuronic acid-containing extracellular carbohydrate.
MATERIALS AND METHODS
Bacteria and growth conditions.
S. mutans NG8 (serotype c) and mutants were grown in Todd-Hewitt broth or chemically defined FMC-glucose medium (25) at 37°C without agitation. When needed, tetracycline and kanamycin were added to the medium at 10 and 500 μg/ml, respectively. Escherichia coli was cultured in Luria-Bertani medium at 37°C. Ampicillin, tetracycline, and kanamycin were used at 100, 15, and 50 μg/ml, respectively. For induction experiments, filter-sterilized sucrose was added to FMC-glucose medium to a final concentration of 0.5% (wt/vol). For growth experiments involving pH, FMC-glucose medium was adjusted to pHs 5.5, 6.0, and 7 with HCl before inoculation.
Cloning and sequencing of the cov operon from S. mutans NG8.
A 3,102-bp DNA fragment carrying the entire cov operon was amplified from the chromosome of S. mutans NG8 by PCR with Taq DNA polymerase and primers SL178 (AGCCCGGGTTCTAACATAAAGTTTA; SmaI site is underlined) and SL179 (ACGGTACCTAAAGTCTTCTGCCAGTTT; KpnI site is underlined). The primer sequences were derived from BLAST searching of the S. mutans UA159 genome at the University of Oklahoma (www.genome.ou.edu) by using the S. pyogenes covR gene as the query sequence (GenBank accession number AF082668). The forward primer, SL178, was designed to include a 133-bp upstream sequence from covR (Fig. 1A). The reverse primer, SL179, included 143 bp from the stop codon of covX. The PCR conditions were as previously described (13). The PCR product was digested with KpnI and SmaI and cloned into KpnI-HincII-digested pUC18. The resulting plasmid, pCovRSX/18, was maintained in E. coli XL-1 Blue. The covRSX operon was further subcloned as overlapping DNA fragments (1-kb SphI-MscI, 0.9-kb HpaI-HindII, and 1.1-kb HindIII-BglII fragments) in pUC18 (Fig. 1A). The cloned DNA fragments were sequenced with M13 forward and reverse primers (Applied Biosystems-Dalhousie University-National Research Council of Canada joint laboratory facilities).
FIG. 1.

covRSX operon of S. mutans NG8. (A) Organization of the genes within the operon. The restriction sites are HindIII (Hd), HpaI (H), MscI (M), SalI (S), and BglII (B). The lollipop indicates the putative Rho-independent terminator. The thick lines under the operon are the fragments subcloned for sequencing. (B) cov operon with the insertion of pVA981 (Tetr) in covR. The 10.5-, 7.5-, 2.6-, and 0.9-kb DNA fragments observed in Southern hybridization are indicated. The 10.5- and 7.5-kb fragments were generated because of the SalI and HindIII sites from pVA981. (C) Promoter region of the operon. Arrows indicate inverted repeats. The predicted transcriptional start site (T), Shine-Dalgarno (S.D.) sequence, and −10 and −35 sequences are shown in bold type.
Inactivation of covR and complementation.
A 400-bp DNA fragment coding for the internal region of CovR was amplified from the S. mutans NG8 genome by PCR with primers SL160 (TATGACACGATTACAGCCTT) and SL161 (CGGTGAGTTAACTCTACCTC). The PCR product was initially cloned into dephosphorylated SmaI-digested pUC18 (pCovR/18) and was subsequently subcloned as an EcoRI-ScaI fragment into the EcoRI-NruI sites of suicide vector pVA981 (Tetr) (26). The resulting plasmid, pCovR/981, was transformed into S. mutans NG8 via natural transformation as described previously (13). Transformants were selected on tetracycline-containing Todd-Hewitt agar.
To create a construct for complementation, DNA representing the cov operon from pCovRSX/18 was subcloned into the E. coli-streptococcus shuttle vector pDL276 (Kanr) (6). This procedure was achieved by ligating the 3.1-kb KpnI-SphI fragment from pCovRSX/18 into the same sites of pDL276. The resulting plasmid, pCovRSX/276, was introduced into the covR mutant by natural transformation. Transformants were selected on Todd-Hewitt agar containing tetracycline and kanamycin.
Southern hybridization.
Isolation of chromosomal DNA from S. mutans and blotting of HindIII- or SalI-digested DNA onto nylon membranes were performed as described previously (28). DNA probes were prepared from pVA981, the 533-bp MscI-HindIII covS internal fragment, and pCovR/18 by using an enhanced chemiluminescence direct labeling and detection system (Amersham Pharmacia Biotechnology Inc., Baie d'Urfé, Quebec, Canada). Southern hybridization and signal detection were performed according to the manufacturer's instructions.
RNA isolation and reverse transcription.
RNA was isolated from S. mutans by a previously described method with modifications (28). Briefly, 50 ml of prewarmed Todd-Hewitt broth was inoculated (1:1) with an overnight culture, and the mixture was incubated for 1.5 h at 37°C. Glycine (5% [wt/vol]) was added, and the mixture was further incubated for 1 h at 37°C. Cells were harvested by centrifugation, washed with cold phosphate-buffered saline, and resuspended in 0.8 ml of a buffer containing 50 mM glucose, 25 mM Tris [pH 8], 1 mM EDTA, and 1 kU of mutanolysin (Sigma-Aldrich Canada Ltd., Oakville, Ontario, Canada). After incubation at 37°C for 15 min, the cells were centrifuged, vortexed in 700 μl of Trizol reagent (Invitrogen Life Technologies, Burlington, Ontario, Canada) for 2 min, cooled on ice for 5 min, and extracted with chloroform. RNA was precipitated from the aqueous layer with isopropanol and washed with 70% ethanol. The pellet was resuspended in 200 μl of diethyl pyrocarbonate (DEPC)-treated water and treated with 20 U of RNase-free DNase (Invitrogen), followed by extraction with phenol and then chloroform. The RNA was dissolved in 200 μl of DEPC-treated water and stored at −70°C.
Reverse transcription was carried out with Superscript II (Invitrogen) in a 20-μl reaction mixture containing 4 μg of total RNA and 3 μg of random primers according to the manufacturer's instructions. In control tubes, Superscript II was omitted from the reaction. Following reverse transcription, the mixture was diluted fourfold in DEPC-treated water, and 4 μl was used for PCR. The primers used for the amplification of ftf were SL184 (GCAGATGAAGCCAATTC) and SL185 (ACAAGATAGCGATCTCC), and those used for the amplification of recA were SL194 (GGGCCGGAATCTTCTG) and SL195 (GTACCAAGCTCCAGCTT). The SL184-SL185 primer pair was designed to amplify a 1-kb internal fragment of ftf, while the SL194-SL195 primer pair was designed to amplify a 0.6-kb internal fragment of recA. The PCR products were analyzed on 1% agarose gels.
SDS-PAGE and Western immunoblotting.
Sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) was performed with 7.5% gels as described by Laemmli (16). Western immunoblotting was performed by the method of Towbin et al. (27). Nitrocellulose blots were probed with a mouse polyclonal antibody raised against the 66-kDa trypsin fragment of FTF (1/5,000; see below), anti-antigen P1 monoclonal antibody 4-10A (1/7,000; courtesy of A. S. Bleiweis, University of Florida, Gainesville), or a rabbit polyclonal anti-FTF antibody (1/100; courtesy of R. R. B. Russell, University of Newcastle, Newcastle upon Tyne, United Kingdom), followed by a goat anti-mouse or anti-rabbit immunoglobulin G-alkaline phosphatase conjugate (Sigma-Aldrich).
Trypsin digestion and N-terminal sequencing.
Cells from late-exponential-phase cultures grown in FMC medium (5 ml) were washed with phosphate-buffered saline and resuspended to the same cell density in 200 μl of 50 mM potassium phosphate buffer (pH 7.4) containing 100 μg of sequencing-grade trypsin (Boehringer Mannheim Canada, Laval, Quebec, Canada)/ml. The cell suspension was incubated with gentle rotation at 37°C for 1 h, at which time the cells were pelleted by centrifugation (10 min, 10,000 × g). The clear supernatant was saved, and 20-μl aliquots were analyzed by SDS-PAGE. The 66-kDa protein band from the tryptic digest of covR cells was transferred to polyvinylidene difluoride membranes, and its N-terminal sequence was determined by microsequencing (National Research Council of Canada, Montreal, Quebec, Canada).
FTF assay.
The FTF activity of the 66-kDa protein was assayed as described by Aduse-Opoku et al. (1). Briefly, following SDS-PAGE, the protein was renatured by washing with five changes of Tris buffer (pH 8.0) for 1 h and incubated with 1% raffinose-1% Triton X-100-0.01% dextran T10 (Sigma-Aldrich)-50 mM sodium phosphate (pH 6.5) for 8 h. The fructan synthesized appeared as an opaque band.
Generation of an antibody against the tryptic digest containing the 66-kDa protein.
Six-week-old BALB/c mice (n = 6) were each immunized intraperitoneally with 40 μg of protein from the tryptic digest of covR cells in Freund's complete adjuvant. The animals were boosted with the same amount of antigen in incomplete adjuvant 14 days later. At 21 days, the animals were euthanatized, and serum was collected following heart puncture. The antiserum obtained was referred to as the anti-66-kDa FTF antibody.
Preparation and analysis of extracellular carbohydrates.
Late-exponential-phase cultures (50 ml) of S. mutans grown in FMC-glucose medium were centrifuged. Four volumes of ice-cold acetone were added to the culture supernatant, and carbohydrates were precipitated at 4°C for 24 h. The carbohydrates were recovered by centrifugation, dissolved in 1 ml of distilled water, and reprecipitated with acetone as described above. The pellets obtained from the second precipitation were washed three times with cold acetone, air dried, and dissolved in distilled water. The cells were stirred in 1 M KOH at 4°C for 2 h (10), and the extracted carbohydrates were recovered by centrifugation and dialyzed against water. The carbohydrates were freeze-dried and stored at −20°C. The materials recovered from the culture supernatant and cells were designated extracellular and cell-bound carbohydrates, respectively. Preliminary results showed that the amounts of carbohydrates recovered from the cells were very low and that there were no major differences in the amounts of cell-bound carbohydrates between the mutants and the wild type. Therefore, subsequent experiments focused on extracellular carbohydrates.
Total carbohydrate and uronic acid contents in nonhydrolyzed samples were measured with phenol-sulfuric acid (5) and m-hydroxydiphenyl (3) and with glucose and glucuronic acid as the standards, respectively. To analyze sugar compositions, aliquots (2 mg) of extracellular carbohydrates were hydrolyzed in 2 N trifluoroacetic acid at 121°C for 1 h. The hydrolysates were dried by using a rotary evaporator under reduced pressure at 40°C. The residues obtained were dissolved in 0.5 ml of water and analyzed by ascending paper chromatography on Whatman no. 1 chromatography paper with the solvent system pyrindine-ethyl acetate-water (4:10:3). Glucose, fructose, galactose, rhamnose, glucuronic acid, and N-acetylglucosamine were used as the standards. Sugars were revealed by staining the paper with alkaline silver oxide reagents (21). Fructose in hydrolyzed samples was also measured enzymatically by using a fructose assay kit (Sigma-Aldrich).
Analysis of band intensities.
The relative intensities of bands in Western blots and DNA gels were analyzed by using NIH Image software (National Institutes of Health, Bethesda, Md.). For estimation of the ftf transcripts, the bands were normalized to the recA bands.
Nucleotide sequence accession number.
The sequence described in this study can be obtained at GenBank accession number AF393849.
RESULTS
covRSX operon.
The cov operon was identified by BLAST searching of the S. mutans UA159 genome at the University of Oklahoma site by using the S. pyogenes covR gene as the query sequence. The cov operon was cloned as a 3,120-bp fragment amplified by PCR from the genome of S. mutans NG8. Sequence analysis showed that the S. mutans NG8 cov operon consisted of three genes, covR, covS, and covX (Fig. 1A). Analysis of the deduced amino acid sequences of the three open reading frames suggested that CovR (235 amino aids) and CovS (450 amino acids) were the responder and sensor proteins, respectively, of a two-component regulatory system. CovX (274 amino acids) was an unknown protein. The proteins encoded by the three genes were highly homologous to that encoded by the covRSX operon of S. pyogenes. A single putative promoter was found upstream of covR, each open reading frame was preceded by a Shine-Dalgarno sequence, and a putative Rho-independent terminator was identified approximately 100 bp downstream of the stop codon of covX. These features suggest that the three genes form an operon. At the promoter-operator region of the operon was a pair of imperfect inverted repeats; the second repeat overlapped the −10 region (Fig. 1C).
Construction of a covR mutant.
A covR mutant of S. mutans NG8 was constructed by insertion of pVA981 through homologous recombination with pCovR/981. Following transformation of S. mutans NG8 with pCovR/981, five tetracycline-resistant transformants were obtained. The five transformants showed autoaggregation during growth in broth cultures, and one of them was selected for further study.
Southern analysis showed that the pVA981 probe hybridized to 7.5- and 2.6-kb HindIII fragments from the covR mutant DNA, while no signal was obtained from the NG8 DNA (Fig. 2A). When the DNA was hybridized with the covS probe, a 10.5-kb SalI DNA fragment was obtained from the covR mutant DNA and a high-molecular-weight band was observed for the NG8 DNA (Fig. 2B). Computer analysis of the S. mutans genome showed that the closest SalI site upstream of that found in covS is 54 kb away. Therefore, the high-molecular-weight NG8 DNA band represented the expected 54-kb SalI fragment. In contrast, the integration of pVA981 into covR introduced a new SalI site (from the pVA981 sequence) upstream of covS, resulting in the appearance of the 10.5-kb band. In addition, when the DNA was hybridized with the pCovR/18 probe, the expected 2.6-kb HindIII fragment was observed for NG8, while three fragments of the expected sizes (7.5, 2.6, and 0.9 kb) were observed for the covR mutant (Fig. 2C). These hybridization patterns further confirmed the insertion of pVA981 into the covR gene of S. mutans (Fig. 1B).
FIG. 2.

Southern hybridization of S. mutans NG8 DNA (lanes 1) and covR mutant DNA (lanes 2). (A) DNAs were digested with HindIII and probed with the pVA981 probe. (B) DNAs were digested with SalI and probed with an internal covS fragment. (C) HindIII-digested DNAs were probed with a 400-bp internal covR fragment carried on pUC18. The location of the 1-kb DNA ladder is indicated.
Characteristics of the covR mutant.
The covR mutant displayed a growth rate and an extent of growth (optical density at 600 nm [OD600] after 24 h, 1.74) similar to those of the wild type at a neutral pH. The covR mutant grew to an OD600 of 0.696 at pH 6 but did not grow at pH 5.5. In contrast, NG8 grew to OD600s of 1.095 and 0.380 at pHs 6 and 5.5, respectively. At a neutral pH, the mutant autoaggregated extensively and settled on the bottom of the culture tube, leaving the medium almost clear. In contrast, the wild type grew as a cloudy suspension. A covRSX-complemented mutant, obtained by transforming pCovRSX/276 into the covR mutant, also grew as a cloudy suspension. Under a light microscope, the individual aggregated mutant cells showed the same shape and size as wild-type cells.
The extensive aggregation displayed by the mutant suggested changes on the cell surface. Therefore, the cells were subjected to trypsinization. However, trypsinization of the mutant cells did not dissociate the aggregates. Interestingly, when the tryptic digests were analyzed by SDS-PAGE, a 66-kDa protein was present at a higher quantity in the mutant digest than in the wild-type digest (Fig. 3). The N-terminal amino acid sequence of this 66-kDa protein fragment was determined to be NTIDEYGLTEQARKIAT; this sequence was identical to that of the S. mutans GS5 FTF enzyme (GenBank accession number M18954) from amino acid residues 148 to 164. The amino acid at residue 147 of the GS5 FTF is a lysine, indicating a trypsin cleavage site. The 66-kDa trypsin fragment produced an opaque band following incubation with raffinose in the FTF assay (data not shown).
FIG. 3.
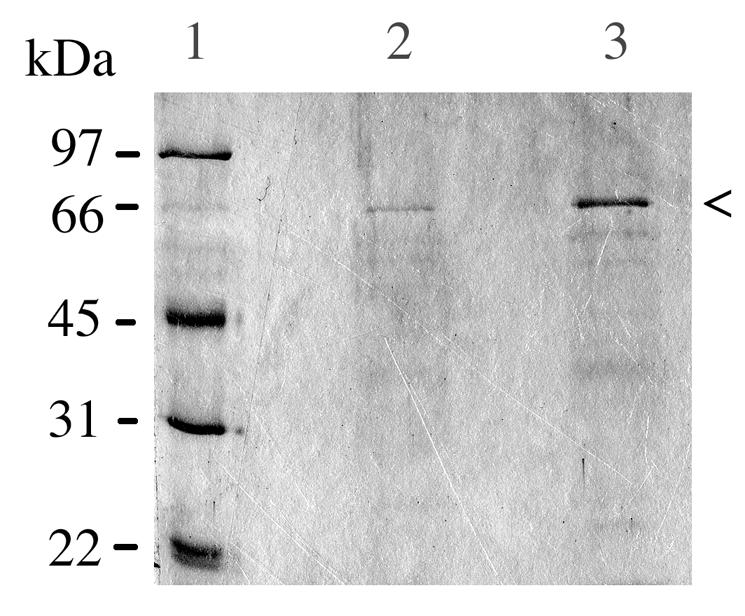
Coomassie blue-stained SDS-polyacrylamide gel of proteins released by trypsin treatment from S. mutans NG8 (lane 2) and covR (lane 3) cells. Each of those lanes contained 20 μl of supernatant from tryptic digests of equivalent numbers of NG8 and covR cells. Lane 1 contained low-molecular-weight markers. The arrowhead indicates the 66-kDa FTF fragment.
A polyclonal antibody raised against the 66-kDa trypsin fragment recognized a 94-kDa protein from S. mutans NG8 and covR cultures, as shown by Western blotting (Fig. 4). This 94-kDa protein and the 66-kDa trypsin fragment were recognized by a rabbit anti-FTF antibody (data not shown), confirming their identity as FTF. Collectively, these results indicate that the 66-kDa protein is a fragment of full-length 94-kDa FTF.
FIG. 4.

Western immunoblots of FTF (A) and antigen P1 (B) produced by S. mutans NG8, the covR mutant, and the covRSX-complemented mutant. Lanes 1 and 2, noninduced and induced NG8; lanes 3 and 4, noninduced and induced covR mutant; lanes 5 and 6, noninduced and induced covRSX-complemented mutant. Each lane contained 20 μl of a late-exponential-phase culture with an OD600 of 1.3. Equal amounts of samples were loaded for FTF and antigen P1. Noninduced cultures were those grown in FMC; induced cultures were those grown in FMC supplemented with 0.5% (wt/vol) sucrose. For FTF, the probe was mouse anti-66-kDa FTF antibody; for antigen P1, the probe was anti-P1 monoclonal antibody 4-10A. The FTF band in panel A was estimated to be 94 kDa. In panel B, the top band represented the 185-kDa antigen P1, and the two lower bands (165 and 155 kDa) were the commonly observed degradation products of P1 (13).
Regulation of ftf expression by covRSX.
The higher level of the 66-kDa FTF fragment in the tryptic digest of the covR cells suggests that FTF may be overexpressed by the mutant. When we used the anti-66-kDa FTF antibody as a probe, the covR mutant showed a threefold increase in the level of FTF relative to the wild type under noninducing conditions (Fig. 4A, lanes 1 and 3). Interestingly, the levels of FTF produced by the mutant were similar in the presence and in the absence of sucrose (Fig. 4A, lanes 3 and 4). In contrast, wild-type FTF production was increased about three times when sucrose was present (Fig. 4A, lanes 1 and 2).
A covRSX-complemented mutant was obtained by transforming pCovRSX/276 into the covR mutant. The production of FTF by the covRSX-complemented mutant was inducible by sucrose, as in the wild type (Fig. 4A, lanes 5 and 6). Under noninducing conditions, the level of FTF production by the covRSX-complemented mutant was about one-third the level of FTF production by the wild type, while under sucrose induction, the level of FTF produced by the mutant was similar to that produced by the wild type. In contrast, the levels of a constitutive protein, major surface protein antigen P1, produced by the three strains remained the same regardless of whether sucrose was present or absent (Fig. 4B).
The regulation of ftf expression by covRSX was further demonstrated by analysis of the levels of ftf transcripts produced by the wild type, the covR mutant, and the covRSX-complemented mutant. As shown in Fig. 5, the level of ftf mRNA produced by the covR mutant was about three times higher than that produced by the wild type, while that produced by the covRSX-complemented mutant was barely detectable, presumably due to the effect of multiple copies of covRS.
FIG. 5.

Transcription of ftf by S. mutans NG8 and mutants. ftf transcripts were obtained by PCR with cDNA synthesized by reverse transcription. Lane 1, 1-kb DNA ladder; lanes 2, 3, and 4, ftf transcripts from wild-type NG8, covR mutant, and covRSX-complemented mutant, respectively; lanes 5, 6, and 8, recA transcripts from NG8, covR mutant, and covRSX-complemented mutant; lane 7, similar to lane 3 but with the reverse transcription step omitted.
Overexpression of extracellular carbohydrate by the covR mutant.
Initial observations of thin sections of covR cells under transmission electron microscopy revealed the presence of amorphous material loosely associated with the cells, suggesting that extracellular carbohydrate might be overproduced by the mutant. These observations were confirmed by the total carbohydrate assay, which showed that the amounts of extracellular carbohydrates (mean and standard deviation [SD]) produced by the covR mutant, NG8, and the covRSX-complemented mutant were 1.97 ± 0.39, 0.54 ± 0.16, and 0.90 ± 0.04 mg of glucose per 109 cells, respectively. The results of the fructose assay and paper chromatography showed that fructose was not present in the trifluoroacetic acid-hydrolyzed carbohydrate, indicating that the carbohydrate was not fructan. Interestingly, on the chromatogram, the samples from the covR mutant and the complemented mutant showed two spots with Rf values identical to those of glucose and glucuronic acid, while the NG8 sample showed only the glucose spot. The presence of glucuronic acid was confirmed by the uronic acid assay, which showed that the covR and complemented mutant samples contained 494 ± 114 (mean and SD) and 125 ± 19 μg of glucuronic acid per mg of extracellular carbohydrate, respectively. The NG8 sample contained no detectable glucuronic acid. The amounts of glucose present in the covR mutant, complemented mutant, and NG8 samples were 103 ± 11 (mean and SD), 30 ± 8, and 44 ± 8 μg per mg of extracellular carbohydrate, respectively. N-Acetylglucosamine was not detected in the carbohydrates from all three strains.
DISCUSSION
In the present study, a two-component regulatory system encoded by the covRSX operon was cloned from S. mutans NG8. Sequence analysis suggested that covR and covS code for the responder and sensor kinases, respectively, while covX codes for an unknown protein. The genetic organization of the covRSX operon resembles that in S. pyogenes. The results showed that FTF expression in S. mutans NG8 is regulated either directly or indirectly by the two-component regulatory covRSX operon. Inactivation of this operon resulted in constitutive overexpression of FTF, and covRSX operon complementation restored the sucrose-inducible expression of FTF. These findings suggest that covRSX negatively regulates FTF expression. The results from the ftf transcript analysis suggest that the regulation occurs at the transcriptional level. Several research laboratories (14, 15, 18, 29) have shown that sucrose is an inducer of FTF expression in S. mutans, but the molecular basis of the induction is not known. Our results provide some insight into the induction mechanism. We hypothesize that S. mutans senses the level of sucrose (signal) in the environment, leading to the derepression of ftf transcription. This process is likely achieved through the commonly accepted mechanism of phosphorelay transfer between the sensor and the responder (CovR). The sensor molecule may be CovS, but further experimentation is required to prove this notion. Phosphorylated CovR then causes an increase in the transcription of the ftf gene, either directly or indirectly. This mechanism of regulation apparently is not responsible for the sucrose-inducible expression of GTFs (14, 18, 29) because the level of GTFs in the covR mutant remains the same as that in the wild type (G. Spatafora, personal communication).
One most interesting and novel finding of this work is that the covR mutant produces an increased amount of extracellular carbohydrate, which apparently contains glucose and glucuronic acid but not fructose as constituent sugars. This finding indicates that the extracellular carbohydrate is not fructan, although FTF is overexpressed. The extracellular carbohydrate contained no detectable N-acetylglucosamine, suggesting that it is not hyaluronic acid. The type 3 capsule of Streptococcus pneumoniae consists of repeating units of glucose and glucuronic acid (4). The presence of an unequal molar ratio of glucose and glucuronic acid suggests that the S. mutans extracellular carbohydrate is not identical to the type 3 capsule. Furthermore, searching of the S. mutans genome did not reveal any sequence homology to the S. pneumoniae cps3DSU operon, which is responsible for capsule synthesis. It is not clear at this time whether this new extracellular carbohydrate is made solely of glucuronic acid or a mixture of glucose and glucuronic acid. The ability of S. mutans to produce a glucose- and glucuronic acid-containing extracellular carbohydrate was not previously described, possibly because this novel extracellular carbohydrate is not expressed under laboratory conditions. The biological importance of the glucose- and glucuronic acid-containing extracellular carbohydrate produced by S. mutans is not clear at this time.
The expression of the glucose- and glucuronic acid-containing extracellular carbohydrate is also under the negative control of covRS, as is evident from the results of the inactivation and complementation studies. This finding is consistent with the situation for S. pyogenes, in that the capsular hyaluronic acid is also negatively regulated by covRS (7, 17). It is interesting that the covRS-complemented mutant produced an intermediate level of extracellular carbohydrate, indicating that complementation in trans did not restore the wild-type phenotype fully. In contrast, full restoration of FTF expression was observed in the covRS-complemented mutant. These results suggest that although ftf and the extracellular carbohydrate synthesis gene are both regulated by covRS, there are differences in the form of the regulation.
Two other findings reported in this study deserve mention. One is the autoaggregation phenotype of the covR mutant. This phenotype is likely due to the overproduction of the extracellular carbohydrate, which imparted to the cell surface a highly negative charge that promoted cell clumping, presumably in the presence of divalent cations. covRS complementation reduces extracellular carbohydrate production and returns the cells to the nonaggregation phenotype. The second finding is that the covR mutant is more sensitive to an acidic pH than the wild type. Li et al. (19) recently reported a two-component regulatory system, hk11/rr11, that regulates genes required for acid resistance and biofilm formation. They showed that a histidine kinase mutant (hk11) but not a responder mutant (rr11) of S. mutans was sensitive to an acidic pH. These researchers suggested that the gene product of hk11 acts as a pH sensor and cross talks with other response regulators to confer acid resistance in S. mutans. The fact that covRS and hk11/rr11 are two different loci suggests that S. mutans has more than one two-component system to respond to an acidic pH. It is not clear at this time whether CovR is the putative responder that cross talks with Hk11 or whether covRS is similar to hk11/rr11 in that CovS is the pH sensor and cross talks with other responders to confer acid resistance.
In conclusion, we have identified a two-component regulatory system that regulates virulence traits in S. mutans, including FTF expression and acid resistance. The system also regulates the synthesis of a novel extracellular carbohydrate. These findings suggest that CovR is a global regulator in S. mutans, similar to its counterpart in S. pyogenes.
Acknowledgments
We thank Roy Russell for providing the rabbit anti-FTF antibody and Le Tao for technical assistance.
This study was supported by NSERC.
Editor: V. J. DiRita
REFERENCES
- 1.Aduse-Opoku, J., M. L. Gilpin, and R. R. B. Russell. 1989. Genetic and antigenic comparison of Streptococcus mutans fructosyltransferase and glucan-binding protein. FEMS Microbiol. Lett. 59:279-282. [DOI] [PubMed] [Google Scholar]
- 2.Biswas, I., and J. R. Scott. 2003. Identification of rocA, a positive regulator of covR expression in the group A streptococcus. J. Bacteriol. 185:3081-3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blumenkrantz, N., and G. Asboe-Hansen. 1973. New method for quantitative determination of uronic acids. Anal. Biochem. 54:484-489. [DOI] [PubMed] [Google Scholar]
- 4.Dillard, J. P., M. W. Vandersea, and J. Yother. 1995. Characterization of the cassette containing genes for the type 3 capsular polysaccahride biosynthesis in Streptococcus pneumoniae. J. Exp. Med. 181:973-983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dubois, M., K. A. Gillies, J. K. Hamilton, P. A. Rebers, and F. Smith. 1956. Colorimetric method for determination of sugars and related substances. Anal. Chem. 28:350-356. [Google Scholar]
- 6.Dunny, G. M., L. N. Lee, and D. J. LeBlanc. 1991. Improved electroporation and cloning vector system for gram-positive bacteria. Appl. Environ. Microbiol. 57:1194-1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Federle, M. J., K. S. McIver, and J. S. Scott. 1999. A response regulator that represses transcription of several virulence operons in the group A streptococcus. Infect. Immun. 181:3649-3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Federle, M. J., and J. S. Scott. 2002. Identification of binding sites for the group A streptococcal global regulator CovR. Mol. Microbiol. 43:1161-1172. [DOI] [PubMed] [Google Scholar]
- 9.Graham, M. R., L. M. Smoot, C. A. Lux Migliaccio, K. Virtaneva, D. E. Sturdevant, S. F. Porcella, M. J. Federle, G. J. Adams, J. R. Scott, and J. M. Musser. 2002. Virulence control in group A Streptococcus by a two-component gene regulatory system: global expression profiling and in vivo infection modeling. Proc. Natl. Acad. Sci. USA 99:13855-13860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guggenheim, B., and H. E. Schroeder. 1967. Biochemical and morphological aspects of extracellular polysaccharides produced by cariogenic streptococci. Helv. Odontol. Acta 11:131-152. [PubMed] [Google Scholar]
- 11.Hamada, S., and H. D. Slade. 1980. Biology, immunology, and cariogenicity of Streptococcus mutans. Microbiol. Rev. 44:331-384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoch, J. A., and T. J. Silhavy (ed.). 1995. Two-component signal transduction. ASM Press, Washington, D.C.
- 13.Homonylo-McGavin, M. K., and S. F. Lee. 1996. Role of the C terminus in antigen P1 surface localization in Streptococcus mutans and two related cocci. J. Bacteriol. 178:801-807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hudson, M. C., and R. Curtiss III. 1990. Regulation of expression of Streptococcus mutans genes important to virulence. Infect. Immun. 58:464-470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kiska, D. L., and F. L. Macrina. 1994. Genetic regulation of fructosyltransferase in Streptococcus mutans. Infect. Immun. 62:1241-1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 17.Levin, J. C., and M. R. Wessels. 1998. Identification of csrR/csrS, a genetic locus that regulates hyaluronic acid capsule synthesis in group A Streptococcus. Mol. Microbiol. 30:209-219. [DOI] [PubMed] [Google Scholar]
- 18.Li, Y., and R. A. Burne. 2001. Regulation of the gtfBC and ftf genes of Streptococcus mutans in biofilms in response to pH and carbohydrate. Microbiology 147:2841-2848. [DOI] [PubMed] [Google Scholar]
- 19.Li, Y. H., P. C. Y. Lau, N. Tang, G. Svensater, R. P. Ellen, and D. G. Cvitkovitch. 2002. Novel two-component system involved in biofilm formation and acid resistance in Streptococcus mutans. J. Bacteriol. 184:6333-6342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loesche, W. J. 1986. Role of Streptococcus mutans in human dental decay. Microbiol. Rev. 50:353-380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Menzies, I. S., and J. W. T. Seakins. 1969. Sugars, p. 310-329. In I. Smith (ed.), Chromatographic and electrophoretic techniques, 3rd ed., vol. 1. Pitman Press, Bath, England. [Google Scholar]
- 22.Miller, A. A., N. C. Engleberg, and V. J. DiRita. 2001. Repression of virulence genes by phosphorylation-dependent oligomerization of CsrR at target promoters in S. pyogenes. Mol. Microbiol. 40:976-990. [DOI] [PubMed] [Google Scholar]
- 23.Munro, C., S. M. Michalak, and F. L. Macrina. 1991. Cariogenicity of Streptococcus mutans V403 glucosyltransferase and fructosyltransferase mutants constructed by allelic exchange. Infect. Immun. 59:2316-2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schroeder, V. A., S. M. Michalak, and F. L. Macrina. 1989. Biochemical characterization and evaluation of virulence of a fructosyltransferase-deficient mutant of Streptococcus mutans V403. Infect. Immun. 57:3560-3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Terleckyj, B., N. P. Willet, and G. D. Shickman. 1975. Growth of several cariogenic strains of oral streptococci in a chemically defined medium. Infect. Immun. 11:649-655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tobian, J. A., M. L. Cline, and F. L. Macrina. 1984. Characterization and expression of a cloned tetracycline resistance determinant from the chromosome of Streptococcus mutans. J. Bacteriol. 160:553-563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Towbin, H., T. Staehelin, and J. Gordon. 1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. USA 76:4350-4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vats, N., and S. F. Lee. 2001. Characterization of a copper transport copYAZ operon from Streptococcus mutans. Microbiology 147:39-43. [DOI] [PubMed] [Google Scholar]
- 29.Wexler, D. L., M. C. Hudson, and R. A. Burne. 1993. Streptococcus mutans fructosyltransferase (ftf) and glucosyltrasferase (gtfBC) operon fusion strains in continuous culture. Infect. Immun. 61:1259-1267. [DOI] [PMC free article] [PubMed] [Google Scholar]