

Abstract
Blastic plasmacytoid dendritic neoplasm is an exceedingly rare tumor that has undergone several changes in nomenclature over the last two decades, largely because of confusion regarding its cell of origin. It does, however, have distinctive clinical features with a particularly aggressive clinical course and no standard treatment. Overall, prognosis is poor and relapse is routine after initial response to chemotherapy. In this report, we describe a typical patient with this disease and reconcile the available literature and its evolution. We emphasize the leukemic nature of this tumor’s behavior, with extensive central nervous system and skin involvement, and describe for the first time a potential role for maintenance chemotherapy in its treatment.
Key words: blastic plasmacytoid dendritic cell neoplasm, skin, central nervous system, leukemia
Case Report
A 69 year old Caucasian man from Texas (USA) presented with several months history of a diffuse skin rash. He also reported fatigue and a 10 pound weight loss over the same period of time. The rash initially involved the trunk, but later progressively extended to his extremities and face, and was only associated with minimal pruritus. His past medical history was remarkable for coronary artery disease, congestive heart failure, and mild chronic renal insufficiency. On physical examination, he had a diffuse, pigmented, plaque-like, bruised rash over his trunk, extremities, and face (Figure 1). He was also noted to have a low grade temperature of 100 degrees Fahrenheit (37.8°C) and mildly enlarged, non-tender epitrochlear and cervical lymph nodes. Laboratory evaluation was remarkable for a white blood cell count of 32,000 with 50% blasts. Hemoglobin was 12.5 g/dL, platelets 357,000/dL, and lactate dehydrogenase (LDH) was elevated at 473 (normal range 100-190). Review of his peripheral blood smear revealed the presence of large basophilic cells with agranular cytoplasm, cleaved nuclei with a high nuclear cytoplasmic ratio, and prominent discrete nucleoli (Figure 2A). Because of the leukemic nature of his disease, cerebrospinal fluid (CSF) analysis was done and revealed the presence of immature appearing blasts; similar to the peripheral blood findings (Figure 2B). Biopsy of a one of the skin lesions showed diffuse perivascular and peri-adnexal infiltration by a monomorphic population of medium to large malignant-appearing cells, with convoluted nuclei and prominent nucleoli, similar in appearance to the cells present in the circulation and CSF (Figure 3). Bone marrow biopsy and aspirate specimens were markedly hypercellular with diffuse infiltration of immature blasts similar to the skin and peripheral blood (not shown). On flow cytometry of peripheral blood the malignant cells were positive for CD4, CD7, CD45, CD56, and HLA-DR. The same cell population was negative for CD2, CD3, CD5, CD8, CD10, CD13, CD14, CD19, CD22, CD23, CD33, CD34, FMC7, and for both kappa and lambda light chains. Terminal deoxynucleotidyl transferase (TdT) was diffusely positive by immunohistochemistry in both the skin and bone marrow. CD45 was also diffusely positive, with moderate to strong staining intensity. Further molecular studies of the malignant cells showed no clonal T cell receptor (TCR) re-arrangement present and Ebstein Barr Virus (EBV) early RNA elements (EBER) were negative. Testing for serum antibodies for HTLV-I and HTLV-II was negative. Cytogenetic analysis of the bone marrow aspirate showed normal male karyotype. Imaging studies, including brain and sinus magnetic resonance imaging (MRI), were all negative except for the presence of a mildly enlarged spleen.
Figure 1.
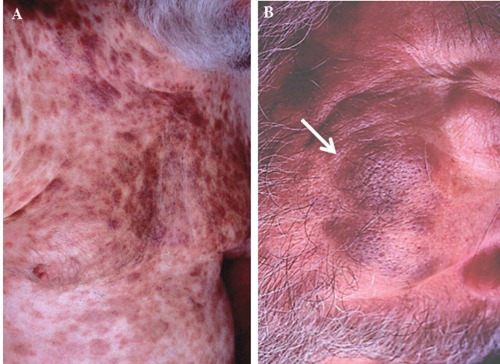
A plaque-like pigmented bruised rash over the patient’s trunk (A) and face (B).
Figure 3.
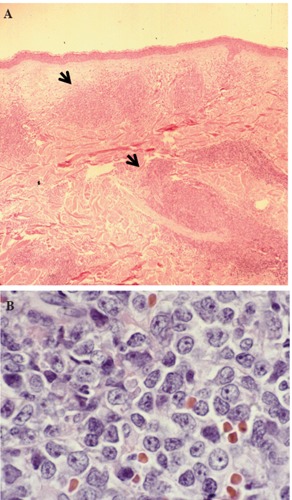
Hematoxylin & Eosin stain of skin lesion biopsy. Low power view of leukemic infiltrate corresponding to the raised plaque (A, black arrows) and high power view of the malignant cells in the skin infiltrate (B).
Based on the clinical presentation and the profile of the malignant cells, the patient was diagnosed with acute Natural Killer Cell lymphoblastic Leukemia (NK-ALL) (based on the prevailing diagnostic nomenclature, later renamed blastic plasmacytoid dendritic cell neoplasm). The absence of myeloid (CD13, CD33), T cell (CD2, CD3, TCR rearrangement) and B cell (CD19, CD10, CD22, immunoglobulin light chains) markers essentially restricted the differential to monocytic and BPDC neoplasms. Human monocytes express dim CD4 and monocytic leukemias have the tendency to infiltrate extrahematopoietic tissues. In his case, the size of the cells, the absence of cytoplasmic azurophilic granules and the morphology of the nuclei and the absence of CD14 as well as the positivity for TdT and the presence of CD7, essentially excluded the possibility of monocytic/monoblastic leukemia. The patient was started on COP (cyclophosphamide 600 mg/m2 IV on day 1, vincristine 2 mg IV on day 1, and prednisone 100 mg per mouth daily on days 1-5) repeated every 21 days, along with weekly intrathecal methotrexate. The schedule was selected because of his borderline performance status and multiple significant comorbidities. Doxorubicin was not administered because of known congestive heart failure. There was a rapid clinical response to treatment, with complete disappearance of leukemic blasts from the blood and CSF, and fading of the skin lesions. No tumor lysis was observed and treatment was well-tolerated with no significant toxicities. After 3 cycles of intravenous COP chemotherapy and 6 intrathecal methotrexate treatments, repeat biopsy of an old skin lesion site and bone marrow biopsy were both negative for malignant cells. Leukemic blasts also disappeared from the CSF after the 1st intrathecal methotrexate with no later reappearance during subsequent follow up. Repeat flow cytometry of the bone marrow showed complete disappearance of the malignant clone and the patient was therefore declared to be in complete remission. Because of the known markedly increased risk of relapse reported with this type of malignancy and because of its clinical behavior resembling that of acute lymphoblastic leukemia, the patient was placed on maintenance chemotherapy with monthly POMP (prednisone 100 mg po (per os) day 1-5, vincristine 2 mg IV on day 1, methotrexate 20 mg po once a week, and 6-mercaptopurine 100 mg po daily) along with monthly intrathecal methotrexate. This regimen was administered for 8 months altogether, during which time repeat assessments showed continued complete remission, but was subsequently discon-tinued because of recurrent pancytopenia and decreased patient tolerance. Unfortunately, two months after POMP was discontinued, the patient experienced widespread recurrence of his leukemia and succumbed to his illness.
Discussion and Conclusions
Blastic plasmacytoid dendritic cell neoplasm (BPDCN) is an exceedingly rate neoplasm that was reported under multiple different names in the literature, reflecting a mix of clinical descriptive terminology and some confusion about the cell of origin. The most recent nomenclature of BPDCN is reported in the updated World Health Organization classification and conforms to the proposed cell of origin.1 Originally, it was named blastic natural killer cell leukemia because of suspected origin at that time and later changed to CD4+/CD56+ hematodermotropic tumor (HDT),2 before the most current classification. Blastic plasmacytoid dendritic cell neoplasm primarily affects middle-aged or elderly individuals, with male predominance, and a peculiar predilection for skin involvement, where it forms nodules, plaques and patches of variable sizes. Bone marrow, peripheral blood, lymph nodes and soft tissues are commonly involved, but CNS involvement is more unusual and reported in only a few cases.3,4 The term blastic was initially adopted because the tumor is composed of medium to large-sized cells with fine chromatin and scanty cytoplasm resembling lymphoblasts, or in some cases, myeloblasts,5 and may on occasion exhibit sub-membranous cytoplasmic vacuolations surrounding the nucleus. The leukemic cells are usually agranular, and angiodestructive or necrotic lesions are rare. EBV is negative in all cases and TCR genes are typically in germline configuration with no rearrangement seen. Tumor cells are invariably CD4+ and CD56+, and usually HLA-DR and CD45RA are positive as well. CD2 and CD34 are usually negative; and expression of TdT, CD7 and cytoplasmic CD3 is variable. Although in earlier reports, this tumor was originally referred to as blastic NK lymphoma, or cutaneous CD-4 positive lymphoma as noted earlier,5-9 the natural killer reference in the nomenclature was subsequently removed because of the absence of EBV genomic material or antigens in the tumor cells in which the expression of CD4 which is not considered a NK cell marker. The absence of cytotoxic molecules, like TIA-1, granzyme B and perforin, raised more questions regarding the origin of this tumor, and it was later found that these CD4+/CD56+ tumors actually express IL-3 receptor alpha (CD123) and the T-cell leukemia-1 (TCL1) molecule, both considered plasmacytoid dendritic cell (pDC) markers, along with other related pDC markers; the blood dendritic cell antigen 2 (BDCA-2) and interferon.10 Most authorities, therefore, agree that BPDCN is a pDC precursor-related and not a true NK neoplasm. It is worthwhile noting that there is no pathognomonic cytogenetic abnormality, but deletion in 5q- is the most frequent associated cytogenetic change.7
Clinically, patients with BPDCN have a poor prognosis and usually survive on average a little more than one year after the diagnosis is made. Typically, initial response to treatment is dramatic (70% complete response and 10% partial response), but later tumor cells become resistant. CHOP or COP-like regimens have been mainly used, as well as regimens for acute leukemia, but almost all patients relapse after initial response and eventually succumb to the disease. Maintenance chemotherapy, such as what was used in this case, may be critical in long term control of this leukemia, similar to other non-myelogenic leukemias, but has not been well studied. We believe that maintenance chemotherapy may be critical in preventing the inevitable rapid relapse of this tumor after initial therapy and could be a reasonable consideration to help maintain prolonged freedom from disease relapse.
Figure 2.
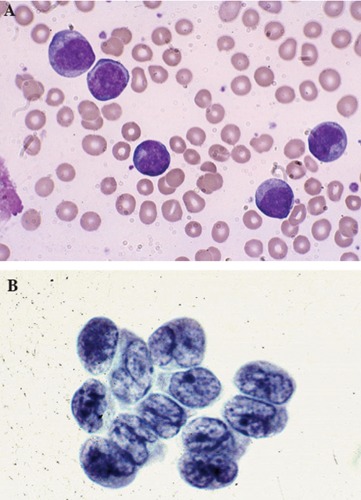
Large malignant-appearing cells, with agranular cytoplasm, cleaved nuclei and prominent neocleoli on peripheral blood smear using Wright stain (A) and similar blast cells present in cerebrospinal fluid (B).
References
- 1.Campo E, Swerdlow SH, Harris NL, et al. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood 2011; 117:5019-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood 2005;105:3768-85. [DOI] [PubMed] [Google Scholar]
- 3.Bekkenk MW, Jansen PM, Meijer CJ, Willemze R. CD56+ hematological neoplasms presenting in the skin: a retrospective analysis of 23 new cases and 130 cases from the literature. Ann Oncol 2004;15: 1097-108. [DOI] [PubMed] [Google Scholar]
- 4.Eros N, Marschalko M, Balassa K, et al. Central nervous system involvement in CD4+/CD56+ hematodermic neoplasm: a report of two cases. J Neurooncol 2010;97: 301-4. [DOI] [PubMed] [Google Scholar]
- 5.DiGiuseppe JA, Louie DC, Williams JE, et al. Blastic natural killer cell leukemia/lymphoma: a clinicopathologic study. Am J Surg Pathol 1997;21:1223-30. [DOI] [PubMed] [Google Scholar]
- 6.Feuillard J, Jacob MC, Valensi F, et al. Clinical and biologic features of CD4+CD56+ malignancies. Blood 2002;99: 1556-63. [DOI] [PubMed] [Google Scholar]
- 7.Leroux D, Mugneret F, Callanan M, et al. CD4(+), CD56(+) DC2 acute leukemia is characterized by recurrent clonal chromosomal changes affecting 6 major targets: a study of 21 cases by the Groupe Francais de Cytogenetique Hematologique. Blood 2002;99:4154-9. [DOI] [PubMed] [Google Scholar]
- 8.Petrella T, Bagot M, Willemze R, et al. Blastic NK-Cell Lymphomas (Agranular CD4+CD56+ Hematodermic Neoplasms). Am J Clin Pathol 2005;123:662-75. [PubMed] [Google Scholar]
- 9.Reichard KK, Burks EJ, Foucar MK, et al. CD4(+) CD56(+) lineage-negative malignancies are rare tumors of plasmacytoid dendritic cells. Am J Surg Pathol 2005;29: 1274-83. [DOI] [PubMed] [Google Scholar]
- 10.Herling M, Teitell MA, Shen RR, et al. TCL1 expression in plasmacytoid dendritic cells (DC2s) and the related CD4+ CD56+ blastic tumors of skin. Blood 2003;101:5007-9. [DOI] [PubMed] [Google Scholar]