

Abstract
Recently, atmospheric-pressure non-thermal plasma-jets (APPJ) are being for the cancer treatment. However, APPJ still has drawbacks such as efficiency and rise in temperature after treatment. So, in this work, a synergetic agent D2O vapour is attached to APPJ which not only increase the efficiency of plasma source against cancer treatment, but also controlled the temperature during the treatment. OD generated by the combination of D2O + N2 plasma helped in enhancing the efficiency of APPJ. We observed OD induced apoptosis on melanocytes G361 cancer cells through DNA damage signalling cascade. Additionally, we observed that plasma induces ROS, which activated MAPK p38 and inhibits p42/p44 MAPK, leading to cancer cell death. We have also studied DNA oxidation by extracting DNA from treated cancer cell and then analysed the effects of OD/OH/D2O2/H2O2 on protein modification and oxidation. Additionally, we attempted molecular docking approaches to check the action of D2O2 on the apoptosis related genes. Further, we confirmed the formation of OD/OH simultaneously in the solution using optical emission spectroscopy. Moreover, the simultaneous generation of D2O2/H2O2 was detected by the use of confocal Raman spectroscopy and density measurements.
The induction of apoptosis or programmed cell death, in cancer cells is the fundamental step towards success for the cancer treatment. Most of the techniques applied for the control of cancer is depend on the use of anticancer agents, which unfortunately provide restricted effects. Hence, development of a new methodology for the cancer treatment is a hot topic for research. Recently, atmospheric-pressure non-thermal plasma-jet (APPJ) has been an emerging field which has been actively applied to resolve various medical problems1 such as treatment of skin diseases2,3,4, dental cavities5,6, control the contamination of various surfaces7, promote cell proliferation8, enhance cell transfection9, wound healing10, and ablate the cultured cancer cells11. APPJ has been well known for the generation of charged particles (electrons, ions), electronically excited atoms, and reactive oxygen species (ROS) including the hydroxyl radical (OH), nitric oxide (NO) and superoxide radicals12,13,14. Since, the growing demands of APPJ in clinical utility needs further innovation to increase its wide area of application in the various therapies. However, still in plasma source, efficiency and abruptly temperature rise during treatment are the main problems that need attention. Hence, there is an urgent need to develop or modify the plasma source.
Herein, we are proposing a synergetic agent D2O vapour along with nitrogen gas (N2) which can overcome the heat problem during exposure and enhance more efficacies through the propagation of high amount of free radicals when compared with other plasma jet source alone. Deuterium oxide or heavy water (D2O) is known for exhibiting various kinds of different biological activities15,16,17. Since there have been several papers reported for the incubation of tumor cells with varying concentrations of D2O, leading to inhibition of cell proliferation. Additionally, they have found to be an active in various cancer cell line in-vitro and in-vivo15,16,17,18,19. However, very little evidence is available for investigation of the cellular consequences of D2O. Somlyai and co-workers stated that naturally occurring deuterium might be essential for normal cell growth; in addition, they showed that the proliferation of tumor cells can be suppressed by deuterium-depleted water20. Recently, it has been reported that D2O causes mitotic arrest and inhibited DNA and protein synthesis in several cell lines21,22,23. However, there is no study reported for the combined action of plasma with the D2O for utility in cancer therapy. Therefore, to investigate their biological applications, we have studied the action of our new technology (combined action of plasma and deuterium oxide radical (OD)) on the anti-cancer activity of melanocytes G361 cancer cells. In this work, we have studied the simultaneous generation of OD/OH using optical emission spectroscopy. However, the instantaneous formation of deuterium dioxide (D2O2)/hydrogen peroxide (H2O2) was measured by confocal Raman spectroscopy and density meter measurements after D2O + N2 plasma treatment. Furthermore, we have studied the oxidative stress related protein signalling such as mitogen-activated protein kinases (MAPK p38) and inhibit p42/p44 MAPK (extracellular signal-regulated kinases (Erk2)). Moreover, DNA oxidation of extracting cell DNA was analysed using circular dichroism (CD), 8-OHdG measurements and Agrose electrophoresis. In addition to this, we have also described the structural changes of proteins [Myoglobin (Mb) and Hemoglobin (Hb)] using the CD, protein oxidation and gel electrophoresis. Further, we have used molecular docking approach to investigate the action of D2O2/H2O2 on the apoptosis related genes such as ataxia telangiectasia mutated (ATM), p53 and cysteine-aspartic acid protease (caspase-8).
Results
Physical and chemical parameter estimation during D2O plasma exposure in media
Since plasma is responsible for altering the physical parameters of the solution24, as well as in most of the cases gradually enhanced the temperature25. However, the combination of D2O vapour with plasma has a significant effect to overcome the heat problems and provides the synergetic effect of OD with N2 plasma. In order to reveal the plasma constituent, we apply D2O along with N2 gas and examine the emission spectra of plasma to find out the OD and OH peaks. Figure 1D shows the emission spectrum, which reveals the presence of the highest OD peak by applying 600 sccm of D2O vapour. We applied D2O to enhance the high amount of OD radicals, which was consequently converted into D2O2. The richness of these species may make this high efficiency of APPJ compression with other sources. After treatment with the D2O + N2 plasma jet at different time interval of 0, 1, 3, 5 and 10 min, we measured the pH and temperature of RPMI (being used as main media in the experiments). There was no significance change in pH of media with or without D2O + N2 plasma; this could probably be due to buffering capacity of media, as shown in Figure 2A. Although, the temperature of D2O + N2 plasma doesn't show any change over the treatment time. Whereas, in case of N2 plasma alone (without D2O vapour) temperature of the solutions continuously increased with increase in treatment time and reached ~ 55°C after 5 min treatment (Figure 2B).
Figure 1.

Schematic diagram of D2O + N2 plasma jet device and its OES spectra (A) A plasma jet system consisting mainly of a high-voltage power supply, electrodes and dielectrics; (B) Shows vacuum chamber consists of vacuum pump which used to take out atmospheric gas from inside the chamber and then chamber was filled with nitrogen gas only; (C) D2O + N2 plasma jet device was used in this experiment. The distance between the cell media and outer electrode is kept as 3 to 4 mm during exposure; (D) Measurement optical emission spectra during treatment with plasma.
Figure 2.

pH, temperature and ROS/RNS measurements after plasma exposure in media (A) Measurement of pH, (B) Temperature in 1 ml of RPMI media were exposed to N2 plasma and D2O + N2 plasma for different time interval 1, 3, 5 and 10 min; (C) Determination of OH concentration; (D) Determination of H2O2 concentration; (E) NO in 1 ml media after N2 and D2O + N2 plasma exposure for 1, 3, and 5 min only. All values are expressed as ± SD in triplicates. Students' t-test was performed to control (* denotes P<0.05 and ** denotes P<0.01).
It may be possible that the interaction between environment surrounding and plasma affected the level and composition of ROS and RNS during treatment24. Therefore, we have performed the plasma treatment in the vacuum conditions (before plasma treatment we removed out the atmospheric gas and then filled with nitrogen gas only), in order to generate the ROS without interference of environmental conditions. In this environment, we treated 1 ml RPMI media on different time intervals. In case of D2O + N2 plasma, the relative levels of OH, H2O2 and NO radical were higher in comparison to N2 plasma (Figures 2C and D), the low levels of NO species were observed in both conditions of plasma treatment (Figure 2E). To measure the OH radicals, we used the standard protocol to detect the OH concentration. Hence, we are not sure that these protocols can differentiate between the OH and OD radicals. Therefore, the detected concentration of the OH in Figure 2C, might be the combined effect of both OH and OD radicals in D2O + N2 plasma. Similarly, the Kit provided to detect the H2O2 concentration after the treatment cannot distinguish between the H2O2 and D2O2, i.e exhibits the total concentration of H2O2 or D2O2 present in solution. During the treatment, the generation of nitrogen atom concentration may be low that results in the insignificant generation of NO in D2O + N2 plasma (Figure 2E).
In order to check the generation of the D2O2 and H2O2 in water solution, we have studied the confocal Raman spectroscopy as displayed in Figure 3A, B. We added the titanyl ion that reacts with D2O2 and H2O2 in solution to form different complexes which were then detected on a different wavelength26. The peaks for Ti-OD and Ti-OH appeared at 1450 cm−1 and 1600 cm−1, which clearly is an evidence that our device can generate the both D2O2 and H2O2 in solution at the same time. Whereas, in the D2O + N2 plasma we have not observed the peak at 2530 cm−1(present in the D2O control sample) which being related to the ν(O-D) of D2O. This, further proves that in our experimental setup there were no free D2O vapours, all D2O vapours were converted either in the D2O2 or OD from the plasma source. On the other hand, we observed the peak at 3400 cm−1 in D2O + N2 plasma, attributed to the ν(O-H) of H2O, similar to control H2O sample (without treatment). On comparing the peak height and the area, we found that the peak height and area decreased in the D2O + N2 plasma as compared to the control H2O sample. This reveals that during the treatment few water molecules are converted into H2O2. Figure 3C shows the concentration of the H2O2 in the solution using a chemical kit after the 3 min and 5 min treatment. We observed that as the time of treatment increased, the concentration of H2O2 also increased. Additionally, we have also checked the density of water solution after treatment with D2O + N2 plasma for 3 min. We observed that after the treatment of the D2O + N2 plasma, the density of water was found to be 1.012358 g cm−3, whereas the density of water before treatment was 0.998204 g cm−3 and density of D2O 1.107000 g cm−3. This also indicated that after the treatment there is a generation of H2O2 and D2O2 that resulted in the increased solution density. This increase in density after the treatment clearly evidences the generation of D2O2 and H2O2 in the solution. Therefore, OH, H2O2, OD and D2O2 could be the main radicals generated during the treatment which increase the efficiency of our plasma source against cancer cells.
Figure 3.

(a) Raman spectra of the H2O (black), D2O (red), D2O + N2 plasma (blue) after binding with titanyl ion after 5 min; (b) Raman spectra of the H2O (black), D2O (red), D2O + N2 plasma (blue) after binding with titanyl ion to check the conformation of D2O2; (c) Determination of H2O2 concentration in 1 ml water solution after D2O + N2plasma exposure for 3 and 5 min. All values are expressed as ± SD in triplicates. Students' t-test was performed to control (* denotes P<0.05 and ** denotes P<0.01).
Plasma induced apoptosis study on different time intervals
To find the plasma treatment time and dose at sublethal level, cell viability was examined at different plasma doses. For determination of cell viability and toxicity of D2O + N2 plasma on normal cells, we performed an MTT assay. After treating with D2O + N2 plasma and N2 plasma for 0, 1, 3, and 5 min, G361cancer cells showed significant decrease in viability after 3 and 5 min treatment for D2O + N2 plasma, whereas the decrease was insignificant for the N2 plasma, during incubation time 24, 48, and 72 h (Figures 4A and B). Although D2O + N2 plasma effects on normal-human dermal-fibroblast (NHDF) cells showed no significant decrease in viability in comparison to the control (without treatment, 0 min) and after 5 min treatment, respectively, during incubation time 24, 48, and 72 h (Figure S1). On the other hand, after treatment with D2O + N2 gas for 0, 1, 3, and 5 min on G361 cells, no effects on viability were observed during incubation time 24, 48, and 72 h (Figure S2). Additionally, we also checked the G361 cell viability by applying deionised water vapour (H2O) instead of D2O. The same condition and treatment time were kept same as used in D2O experiment. After treating with H2O + N2 plasma, G361cancer cells showed about 55–65 % viability of cells on 5 min treatment (Figure S3). Although, after exposure to D2O + N2 plasma for 5 min, only 20–30% viability of cells was observed. No significant effect on cell viability was observed on 5 min N2 plasma exposure until 72 h incubation (Figures 4A and B). This indicates that D2O + N2 plasma is more effective than H2O + N2 plasma. MTT assay data shows that D2O + N2 plasma has an inhibitory effect after 3 min treatment on the growth of G361 cancer cells, but no toxic effects on the growth NHDF cells upto 5 min exposure. Further, we determined the plasma sublethal dose by treating the cells on different time intervals (0, 1, 3, and 5 min) and also checked the viability on every 3 h interval or incubation. Figure 4C, reveals that cells become apoptotic after 3 min plasma treatment and 12 h incubation.
Figure 4.

Viability of G361 cells treated with (A) D2O + N2 plasma; (B) N2 plasma (C) cell viability during different time intervals after plasma treatment with D2O + N2 plasma. Viability of plasma treated cells was measured after 3, 6, 9, 12 and 24 h incubation by MTT assay; (D) Early and late apoptosis analysis through flow cytometry after Annexin V-FITC/PI staining of plasma treated G361cells; (E) The normalized cell number distribution to Annexin V-FITC/PI intensity of G361 cells was plotted by plasma exposure time. Actinomycin D was used to make positive control. All values are expressed as ± SD in triplicates. Students' t-test was performed to control (* denotes P<0.05 and ** denotes P<0.01).
To obtain direct evidence that supports a causative link between over production of ROS and apoptosis in G361 cancer cells, a comparative study using actinomycine D was conducted. Apoptotic activity was assayed using annexin V-FITC/PI staining (annexin V-FITC/PI described in method section) for control cells (without treatment), treated cells (3 min treatment of D2O + N2 plasma) and in the presence of 1 µM of actinomycin D treatment, in Figures 4D and E. Plasma treated and actinomycin D treated cells induced apoptosis of ~52 % and 24 % of the total cells, respectively. However, ~21 % early apoptosis and ~31 % late apoptosis were observed for 3 min plasma treatment, whereas, in the presence of actinomycin D ~13 % early and ~11 % post apoptosis were observed (Figure 4D). These results strongly suggest that over production of ROS generated by D2O + N2 plasma could be the only factor responsible for the induced apoptosis in G361 cancer cells.
Plasma stimulated Intracellular ROS effect on apoptosis related gene study
Intracellular ROS levels dramatically increased over the D2O + N2 plasma-treated cells, and it was significantly higher after 3 min treatment in comparison with 0 min (Figures 5A and B). Although in the presence of trolox with 3 min treatment, there was no significant enhancement in ROS due to scavenging activity of trolox (Figure 5A). Therefore, the presence of trolox proves the enhancement of intracellular ROS through extracellular ROS, generated by D2O + N2 plasma. Oxidative stress inside the cell modulates the several kinds of protein signalling such as MAPKs family which take part in the regulation of cell survival and apoptosis related gene expression27. In mammalian cells, basically there are least 3 different groups of MAPKs, including p38 MAPK, p42/p44 MAPK (Erk2), and c- jun N-terminal kinase (JNK)27. Among the MAPKs family, MAPK p38 are known as highly responsive to oxidant stress inside the cell28. In this study, we showed the mechanistically induced oxidative stress by reactive oxygen species ROS with 3 min exposure of D2O + N2 plasma and 20 µM concentration of H2O2, which activated phosphorylation of MAPK p38 and inhibited the phosphorylation of p42/p44 MAPK (Erk2) respectively, triggering cell death (Figure S4).
Figure 5.
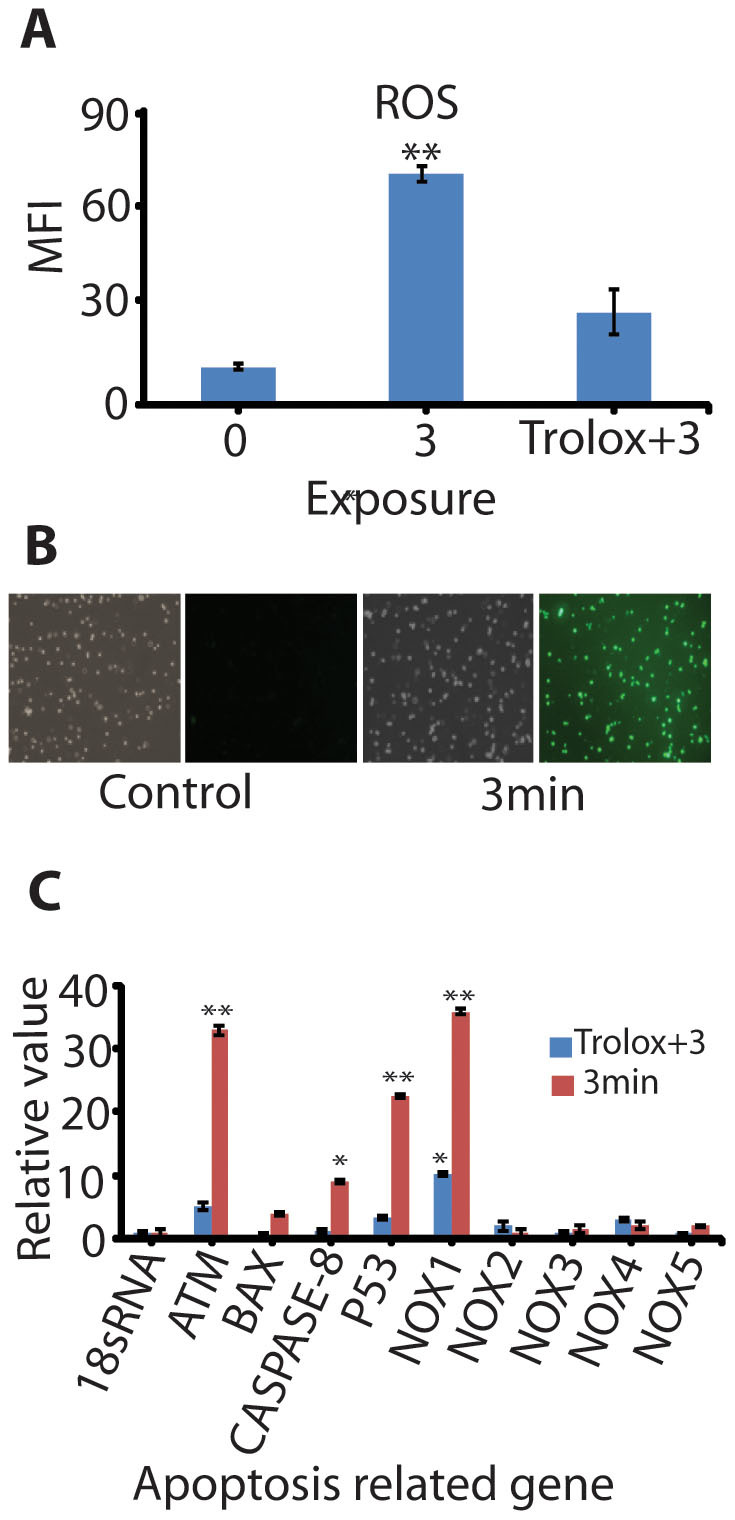
Intracellular ROS estimation and apoptotic related gene expression analyses (A) The level of intracellular ROS after D2O + N2 plasma exposure for 0 min (control), 3 min and ROS scavenger trolox + 3 min treatment. All values are expressed as mean fluorescence intensity (MFI) and ± SD in triplicates; (B) Qualitative analysis of the intracellular ROS level after plasma treatment through fluorescence microscopy using fluorescent probes H2DCFDA; (C) The relative value of mRNA expression of nine apoptosis and related genes of G361melanocytes cancer cells after plasma exposure for 3 min with and without presence of trolox. The relative mRNA expression of p53, Caspase-8, ATM, Bax and NOX(1-5) subfamily were measured by real-time RT-PCR on day 1 and 18s rRNA was used as a reference gene. All values are expressed as (MFI) and ± SD in triplicates. Students' t-test was performed to control (* denotes P<0.05 and ** denotes P<0.01).
Further, to establish that role of ROS in cancer cell death induced by D2O plasma, we examined the mRNA expression of five oxidative stress related gene (NOX 1-5) and four apoptosis (ATM, BAX, caspase-8, p53) related genes. The generation of ROS by nicotinamide adenine dinucleotide phosphate-oxidase (NOX) is essential for signal transduction. NOX generate ROS in a variety of cells and tissues in response to apoptosis related signalling29,30. While, ATM has belonged to one of the largest protein kinase family, that is responsible for regulating cellular responses and activated H2AX and p53 in the DNA damage signalling cascade31. This study illustrate that ROS induction after treatment of cells with D2O + N2 plasma increased the levels of intracellular ROS that might be mediated through NOX-1 and p53/ATM genes over expression. In our result (Figure 5C), cancer cells exposed to D2O + N2 plasma for 3 min without the presence of trolox showed high expression of NOX-1, ATM and p53 genes. This proves that the over expression of these genes leads to DNA damage in G361 cancer cells. Moreover, slightly enhanced expression of Caspase-8 gene can activate apoptosis and lead to induction of cell death through the oxidative DNA damage. Although in the presence of trolox with 3 min treatment, showed no significant enhancement in gene expression due to scavenging activity of trolox. Therefore, ROS play a key role in initiating cell death in response to oxidative DNA damage.
Effects of ROS on DNA oxidation of G361 cells
High levels of ROS generated through the NTP (non thermal plasma) in tumour cells elevated the levels of oxidative stress or oxidative DNA damage in human malignancies relative to normal cells31. In order to check the effect of plasma on DNA oxidation, we have studied the CD spectra (CD is described in method section) of DNA. The ellipticity (θ) measurement has been widely used to supervise the conformational changes of DNA32. The changes in the CD spectra of DNA are triggered by the certain factors such as stacking interactions, hydrogen bonding between the electrostatic repulsion of the negatively charged phosphate groups and complementary bases33. The obtained changes in ellipticity as a function of wavelength before and after plasma, in the presence of actinomycine D drug and 1M urea (added the 1M urea in cancer cell DNA, to check the extent of DNA denaturation in cancer cell DNA) are shown in Figure 6A. Before the plasma treatment, (control) G361 cancer cell DNA exhibited a negative peak at 209 nm and positive peak at 243 nm. After 3 min treatment of D2O + N2 plasma, the negative peak appeared at 212 nm and positive peak at 252 nm. While cancer cell DNA treated with the actinomycine D drug revealed the negative peak at 211 nm and positive peak at 249 nm. In order to investigate DNA damage by plasma and urea, we have extracted the DNA of cancer cells and then treated with 1M urea. We obtained the positive peak at 244 nm and negative peak at 210 nm. This indicates that the deformation occurring in the DNA varies with the type of treatment. These deformations in the DNA structure could be due to DNA oxidation34. From the Figure 6B, we observed that the maximum 8-OHdG was formed after 3 min D2O + N2 plasma treatment and the concentration of 8-OHdG formation was less for the actinomycine D. This also boosts the fact that in the change in conformation of DNA was found to be different in the presence of different type of treatment (urea, plasma and actinomycine D). For further validation, we did 1% agarose gel analysis, which showed that DNA oxidation increased significantly at 3 min treatment when compared with control and actinomycine D (Figure 6C). Gel bandwidth became thicker in 3 min D2O + N2 plasma treatment as compared to actinomycine D. This reveals that the formation of DNA oxidation was dependent on the presence of D2O2/H2O2 and OD/OH radicals produced by D2O + N2 plasma. Further, we observed the action of D2O + N2 plasma on the Mb and Hb as standard proteins.
Figure 6. DNA oxidation and genomic DNA modification analyses.

Melanocytes G361 cancer cells were exposed to D2O + N2 plasma for 3 min and used for the analyses of DNA oxidation and genomic DNA modification through (A) Circular dichroism; (B) Production of 8-OHdG measured. Each value is the average of three technical replicates. Students' t-test was performed to control (*denotes P<0.05 and **denotes P<0.01); (C) 1%Agarose gel electrophoresis analysis of genomic DNA extracted from the treated G361cells.
Estimation of protein modification
In order to obtain a detailed understanding of the extent of modification by D2O + N2 plasma on Mb and Hb, we further performed CD experiments. CD spectrum (190–240 nm wavelength range) was used to determine the variation in the secondary structure. The far-UV CD spectra of Hb and Mb under treatment are shown in Figures 7A and B. As seen from Figures 7A and B, the far-UV CD spectra of proteins indicate that the deformation of secondary structure of proteins. The CD spectrum for a polypeptide chain which is usually organized in α-helix conformation with two well-labelled minima at ≈ 210 and ≈ 222 nm for proteins in water35,36. The results in the Table 1 reveal that in case of both proteins (without treatment) have 51 % and 50 % of α-helical structure for Mb and Hb respectively. Whereas, after 1 min D2O + N2 plasma treatment Mb ≈ 41 % and Hb ≈ 49 %, while, after 3 min treatment Mb ≈ 38 % and Hb ≈ 31 % of α-helical structure. However, 5 min treatment of D2O + N2 plasma leads to the Mb ≈ 30 % and Hb ≈ 25 % of α-helical structure. The α-helical structure of Mb ≈ 26 % and Hb ≈ 12 % decreased in 30% (v/v) of H2O2 treatment. Therefore, the denaturation was very different in H2O2 treatment as compared to D2O + N2 plasma because in the D2O + N2 plasma there is generation of both H2O2 and D2O2 which may result to different extent of secondary structural changes as compared to control (without treatment). Further, to see more clearly the action of D2O + N2 plasma treatment on proteins, we have studied gel electrophoresis. From gel electrophoresis, we predicted the change of molecular weight of the Mb and Hb after the D2O + N2 plasma treatment. The results summarized in Figure 7C reveal that the intensity of gel bands increases more in case of Mb and Hb after D2O + N2 plasma, as compared to 30% (v/v) H2O2 treatment. The increase in bandwidth is due to protein oxidation or protein carbonylation. Protein oxidation is widely used as an indicator of oxidative stress. In order to gain insight into these problems, we have studied the protein carbonyl test in the presence of 3 min, 5 min plasma treatment and H2O2, as displayed in Figure 7D. This shows that percentage of protein oxidation after 5 min treatment is maximum, as compared to 3 min and H2O2. So, in order to check the attacking position of D2O2/H2O2 with cancer cell proteins, we also explored the molecular docking studies.
Figure 7.

Protein oxidation and extracellular protein modification analyses (A) Mb; (B) Hb proteins were exposed to D2O + N2 plasma for different time intervals through circular dichroism; (C) SDS gel electrophoresis of treated Mb and Hb proteins for 5 min and 30% (v/v) H2O2 treatment; (D) Production of carbonyl content measured the treated protein with D2O + Nitrogen plasma for without treatment (control), 3 min, 5 min and 50 μM H2O2.Each value is the average of three technical replicates. Students' t-test was performed to control (* denotes P<0.05 and ** denotes P<0.01).
Table 1. Secondary structure changes of Mb and Hb proteins, determined from CD spectra in different condition at 25°C determined.
| Samples | α- sheet (%) | β-sheet (%) | Turn (%) | Random coil (%) |
|---|---|---|---|---|
| Mb control | 51 | 22 | 1 | 26 |
| 1 min | 41 | 20 | 15 | 23 |
| 3 min | 31 | 23 | 22 | 24 |
| 5 min | 25 | 29 | 19 | 26 |
| H2O2 | 26 | 38 | 0 | 35 |
| Hb control | 50 | 22 | 10 | 18 |
| 1 min | 49 | 20 | 10 | 20 |
| 3 min | 38 | 33 | 6 | 23 |
| 5 min | 30 | 31 | 7 | 31 |
| H2O2 | 12 | 49 | 0.4 | 38 |
Exploration of binding site interacting residues through molecular docking
The propose of the molecular docking analysis was to elucidate whether D2O2/H2O2 and actinomycin D modulate the anticancer target or not. However, this study was used to identify the binding site pocket against the long-familiar anticancer molecular targets Erk2 (Figure 8A), p53 (Figure 8B) and caspase-8 (Figure 8C) protein receptor. The result of the molecular docking reveals that D2O2/H2O2 binds well to the Erk2, p53 and caspase-8 proteins. This is theoretically validated by computational simulation process i.e., molecular docking. Docking procedure is used to show the molecular insight of interacting compound with that of target protein. This method is used to show the molecular interaction and the interaction can be measured in terms of docking score or docking energy. At the same time, we get the information related to docked binding site and interacting residues. Molecular docking experiments mimic the biological process under standard simulated parameters. This is most commonly used approach to see the molecular interaction and interlinked the experimental results very well matched with our data. Through molecular docking simulations, we successfully explored the binding affinities and orientations (in terms of docking score refer here as ‘total score’) of H2O2 against the anticancer targets Erk2 (Figure 8D), p53 (Figure 8E) and caspase-8 (Figure 8F) protein receptors.
Figure 8.

Backbone ribbon diagram of the Erk2 (A), p53 (B) and caspase-8 (C) heterodimer. Inhibitors are shown as in magnatacolor. Electrostatic potential mapped onto molecular surface for substrate binding-site (active pocket) of Erk2 (D), p53 (E) and caspase-8 (F). Inhibitors are shown as in green color in binding pocket site. In silico molecular docking studies elucidating the interaction of actinomycin D in the binding site of Erk2 (G), p53 (H) and Caspase-8 (I) protein. H2O2/D2O2 docked on target with total docking score 3.4751 (J), 3.7161 (K) and 2.6876 (L) show two H-bond of length 1.9 and 1.9 Å to binding pocket residue ASP-106 and MET-108, with Erk2, four H-bond of length 1.8,1.8, 1.9 and 2.0 Å to binding pocket residue GLY-154, THR-155, CYS-220 and GLU-221 with p53 and two H-bond of 2.1 Å to binding pocket residue GLU-396 with caspase-8 within selection radius of 4Å from bound substrate revealing the binding site pocket of active conformation.
The molecular docking procedure reliability was corroborated by using the known crystallographic X-ray structure of target protein Erk 2 complex with known anticancer (against skin cancer) inhibitor norathyriol37. The co-crystallized anticancer inhibitor norathyriol was re-docked into the binding site and the docked conformation with the highest total score of 4.4761 was selected as the most probable binding conformation. The low root mean-square deviation (RMSD) of 0.5628Å between the docked (computational) and the experimental structure conformations37, indicates the high reliability of Surflex-dock software (Sybyl-X, Certara, USA) in reproducing the experimentally observed binding site for this inhibitor. The RMSD measure is normally used to see the statistical deviation between two superimposed structures within the same binding (active) site of the target protein. Before running any docking experiment on unknown compounds, in general, mostly docked compound/inhibitor structure is compared with the actual co-crystallazed bound inhibitor structure by using the superimposition method of molecular docking and deviation from the original conformation crystallographic structure37 is measured in terms of RMSD. We set the standard cut-off of RMSD not more than 0.6. Re-docked inhibitor norathyriol was almost in the same active site as shown in Figure S5. The experimental X-ray crystallography data37 showed that the polar acidic (negative charged) residue aspartic acid (Asp-106) and nonpolar hydrophobic residue methionine (Met-108) were the “gatekeeper” residues, an important determinant of inhibitory functional specificity in the Erk2 binding site.
For comparison of binding affinity and docking score, additional known anticancer drug actinomycin D and its known anticancer targets Erk2, p53 and caspase-8 were selected for docking simulations37,38,39. The docking results of actinomycin D with the known anticancer targets Erk2, p53 and caspase-8 showed significant binding affinity, as indicated by docking score (i.e., total score) of 1.0890 (Figure 8G), 3.4505 (Figure 8H) and 3.3725 (Figure 8I), respectively, and displayed in Table 2. These docking results were considered as positive control data and later used to compare the significance level on the basis of binding affinity (docking score) of H2O2. Although structural diversity resulted in the variation in binding site residues and H-bonds formation. On comparing with actinomycin D, the docking results for H2O2/D2O2 against target Erk2 showed high binding affinity docking score as indicated by total score of 3.4751 and also revealed the formation of two H-bonds of length 1.95Å and 1.90Å with the polar acidic residue aspartic acid (Asp-106) and nonpolar hydrophobic residue methionine (Met-108) similar to inhibitor norathyriol37. In this docking pose, other binding site residues within a selection radius of 4Å from bound substrate were polar uncharged residue glutamine (Gln-106), polar acidic (negative charged) residue aspartic acid (Asp-106), nonpolar hydrophobic residues e.g., alanine (Ala-52), leucine (Leu-107, Leu-156), methionine (Met-108) and isoleucine (Ile-31, Ile-84), as a result, the bound substrate depicted a strong interaction with Erk2, thus contributing to more inhibitory activity and stability with H2O2 (Figure 8J).
Table 2. Comparison of binding affinity of D2O2/H2O2, Actinomycin D and Norathyriol and against Erk2, p53 and Caspase-8 receptor protein.
| Inhibitor/Compound | Target protein/PDB ID | Total Score | Amino acid involved in active pocket in 4 Ă | Involved group of Amino Acid | Length of H-bond Ă | No. of Hydrogen Bond |
|---|---|---|---|---|---|---|
| H2O2 (Test substrate) | Erk2/3SA0 | 3.4751 | ILE-31, ALA-52, ILE-84, GLN-106, ASP-106, LEU-107, MET-108, LEU-156 | ASP-106 MET-108 | 1.951.90 | 2 |
| p53/3ZME | 3.7161 | PRO-153, GYS-154, THR-155, ARG-156, ARG-202, PRO-219, CYS-220, GLU-221 | GLY-154THR-155CYS-220GLU-221 | 1.861.881.992.00 | 4 | |
| Caspase-8/3KJQ | 2.6876 | GLU-396, PHE-399, THR-469 | GLU-396 | 2.132.15 | 2 | |
| Actinomycin (positive control) | Erk2/3SA0 | 1.0890 | TYR-30, ILE-31, GLY-32, GLU-33, GLY-34, ALA-35, VAL-39, GLU-109, THR-110, ASP-111, TYR-113, LYS-114, LEU-115, LYS-117, LEU-156 | - | - | - |
| p53/3ZME | 3.4505 | THR-150, PRO-153, ARG-202, GLU-221, PRO-222, PRO-223, GLU-224, VAL-225 | - | - | ||
| Caspase-8/3KJQ | 3.3725 | PRO-322, TYR-334, GLU-335, SER-338, GLN-339, THR-341, LYS-344, GLU-396, THR-467, THR-469 | LYS-344 | 1.99 | 1 | |
| Norathyriol37 | Erk2/3SA0 | 4.4761 | GLY-34, VAL-39, ALA-52, LYS-54, ILE-84, GLN-105, ASP-106, LEU-107, MET-108, ASP-111, LYS-114, LEU-156 | MET-108ASP-106GLN-105 | 1.832.031.96 | 3 |
| QC538 | p53/3ZME | 4.6541 | LEU-145, VAL-147, ASP-148, THR-150, PRO-151, PRO-152, PRO-153, CYS-220, GLU-221, PRO-222, THR-230 | - | - | - |
| B9439 | Caspase-8/3KJQ | 3.8974 | TYR-334, THR-337, SER-338, GLU-396, PHE-399, THR-467, PHE-468, THR-469 | - | - | - |
Although, H2O2/D2O2 docking outcomes against the anticancer target protein p53 showed a high binding affinity with total docking score of 3.7161 and simultaneously revealed the formation of four H-bonds of length 1.86 Å, 1.88 Å, 1.99 Å and 2.00 Å to the polar uncharged residues e.g., glycine (Gly-154), threonine (Thr-155), cysteine (Cys-220) and polar acidic residue glutamine acid (Glu-221) similar to inhibitor QC5 (2-(4-(4-fluorophenyl)-5-(1H-pyrrol-1-yl)-1H-pyrazol-1-yl)-N,N-dimethylethanamine)38. In this docking pose of the H2O2-p53complex, the chemical nature of binding site residues within a radius of 4Å was polar basic due to arginine (Arg-156, Arg-202) and nonpolar hydrophobic due to proline (Pro-153, Pro-219) similar to Thr-150, PRO-153, Glu-221 and Pro-222, as a result, the bound substrate showed a strong interaction with p53 protein (Figure 8K).
Contrary to this, docking results for H2O2/D2O2 against anticancer target caspase-8 showed low binding affinity score as indicated by total score of 2.6876 and simultaneously revealed the formation of two H-bonds of length 2.13Å and 2.15Å to the polar acidic residue glutamine acid (Glu-396) similar to inhibitor B94 ((3S)-3-({[(5S,8R)-2-(3-carboxypropyl)-8-(2-{[(4-chlorophenyl)acetyl]amino}ethyl)-1,3-dioxo-2,3,5,8-tetrahydro-1H-[1,2,4]triazolo[1,2-a] pyridazin-5-yl]carbonyl}amino)-4-oxopentanoic acid)39. Moreover, in this docking pose, the chemical nature of other binding site residues within a radius of 4Åwas nonpolar hydrophobic due to amino acid residue phenylalanine (Phe-399) and polar uncharged due to residue threonine (Thr-469) similar to GLU-396, PHE-399, THR-469, as a result, the bound substrate showed weak molecular interaction with caspase-8, thus leading to less stability and inhibitory activity with H2O2/D2O2 (Figure 8L).
Discussion
Combination of D2O and N2 plasma is the advance technology which not only controls the temperature of the plasma source but also increases the efficiency for anti-cancer activity. D2O vapour is such a synergetic agent that helps in increasing the efficiency of APPJ. However, by applying D2O vapour, there was no significant change in pH and temperature. This indicates that D2O + N2 plasma during the treatment has no direct effect of pH and temperature on cell viability. Plasma generated from the D2O + N2 plasma contained radicals, mainly OD and nitrogen atom. Nitrogen atoms dissociated from nitrogen molecules may form nitrogen monoxide by the reaction of N + OD → NO + D in D2O + N2 plasma. But, electrons require relatively-high energy for dissociation of nitrogen molecules by electron impact because of a high dissociation-energy of 10 eV. Hence the OD radicals react with other OD radicals to form D2O2 as the main component in the solution (OD + OD → D2O2), rather than NO. Generation of OD/D2O2 is very well interpreted using the optical emission spectroscopy and Raman spectroscopy analysis. Further, the increase in density solution after the treatment clearly revealed the generation of D2O2. Therefore, the direct generation of OD and D2O2 in culture media by plasma enhanced the intracellular ROS level. In response to oxidative stress induced by the ROS, p38 MAPK are generally phosphorylated40. In Figure S4, up regulation of phosphorylation of p38 MAPK and inhibition of p42/p44 MAPK (Erk2) signalling after D2O+ N2 plasma exposure, which may upregulate proapoptotic genes through distinct phosphorylation events, resulting in stress stimulus DNA damage40. These results are supported by Chen et al.40 They suggested that enhanced ROS activated p38 MAPK phosphorylation and inhibits Erk2 phosphorylation, results in tumor cell death.
In our experiment, the OD and D2O2 generated by the D2O + N2 plasma produce oxidative potential that is partially mediated through NOX-1 and p53/ATM over expression. Over expression of these genes clearly indicates that ROS dependent DNA damage-triggered signalling leading to cell death. So, in order to check the attaching position of H2O2/D2O2 with a binding site pocket of the well-known anticancer molecular target Erk2, p53 and caspase-8 protein receptor we studied the docking studies. These results reveal that the binding affinity of D2O2/H2O2 is very high than actinomycin D, hence it supports our experimental data that activity of combined action of OD and plasma is more as compared to actinomycin D. Additionally, we observed the DNA damage or oxidation on treatment with D2O + N2 plasma using CD, DNA oxidation and gel electrophoresis. Further, molecular docking approach to show that D2O2 strongly binds to the active site of apoptosis related genes of Erk2, p53 and caspase-8 as compared to actinomycin D. Hence, the presence of the OD/OH radicals, or D2O2/H2O2 that generated from D2O + N2 plasma resulted in the oxidative stress and finally leads to DNA oxidation. Later, we have studied the far-UV CD spectra of Hb and Mb after D2O + N2 plasma treatment. It leads to different extent of secondary structural changes in both proteins. Moreover, we observed that D2O + N2 plasma can produce oxidants that can generate carbonyls into proteins can result in the structural changes.
However, DNA damage through the D2O2/H2O2 generation, the detailed mechanism remains to be uncovered and still need to be investigated. Our results show that D2O + N2 plasma source is a useful therapy against human skin carcinoma cells in vitro. It is the fact that makes it more potential candidate for the treatment of skin tumors. However, in vivo studies are needed to determine the value of D2O for the treatment of these, mostly fatal, tumors. Nevertheless, uses of D2O + N2 plasma can be one of the cheap and effective alternative methodology for cancer therapy. Hence, this approach makes a remarkable milestone to increase the utility of APPJ for the plasma medicines.
Methods
Plasma properties
The nonthermal plasma jet system used in our study (Figure 1A) mainly consists of a high voltage power supply, electrodes and dielectrics41. The vacuum chamber is fabricated to study the activity in a closed environment as shown in Figure 1B. A commercial transformer for the neon light as a commercial transformer is used for the high-voltage power supply. The inner electrode is an injection needle made of stainless steel with thickness of 0.2 mm and inner diameter of 1.2 mm. Quartz tube as a tight cover with an outer diameter of 7 mm. The outer electrode is also fabricated from stainless steel and centrally perforated with a hole of mm, through which the plasma jet is ejected into the surrounding ambient air. Porous alumina with a diameter of 12 mm and a length of 17 mm is machined to be in tight contact with the inner and outer electrodes (Figure 1C). The discharge gap distance is adjusted to be 2 mm between the inner and outer electrodes. Gas is injected into the injection needle and then ejected through the 1 mm hole in the outer electrode via the porous alumina, which is approximately 30 % (vol.) porosity with an average pore diameter of about 300 μm. Nitrogen with D2O (heavy water) vapor 600 sccm (standard cubic centimeter per minute) is used as the feeding gas, and the flow rate is controlled to be 1 lpm (liter per minute) by mass flow as well as flow meter controller. These flow meters are used for mixing D2O vapour and nitrogen gas, once the nitrogen gas flow to move through the heavy water bottle to evaporate heavy water and mix the vapour molecules with nitrogen gas. The actual voltage and current are distorted sinusoidal waveforms due to micro-discharges through porous alumina41. A typical rms values of current and voltage are approximately 11 mA and 2 kV, respectively. In order to know the plasma constituent, we apply D2O along with nitrogen used as feeding gas and examined the emission spectra of plasma to find out the OD and OH peak (Figure 1D).
OES spectra measurement of NPJ
Spectra of the NPJ emission are recorded by the use of HR4000CG-UV-NIR (Ocean Optics, FL, USA) over a wide wavelength range 200–1100 nm and cover the whole experimental conditions explored in the study under electrical power of about 7 W (2.2 kV, 11 mA with a phase angle of 60° between the current and voltage) with a 60 V input voltage at 60 Hz frequency and we used nitrogen gas flow of 1lpm with a combination of 600 sccm of D2O vapours.
pH & Temperature measurement
RPMI media in a 48 well plate (1 ml per well) were exposed to D2O + N2 plasma for 0, 1, 3, 5 and 10 min. After exposure, the pH and temperature of the media was measured using a pH meter (Eutech Instruments, Singapore) and Infrared (IR) camera (Fluke Ti100 Series Thermal Imaging Cameras, UK). All measurements were carried out in triplicate.
Cell lines used in the study
Melanocytes G361 cell lines were used for the effect of plasma on cell viability and Normal human dermal fibroblast (NHDF) cell lines were used for toxic effect on normal cells effect after treatment with D2O + N2 plasma. Both cells were maintained in RPMI and DMEM supplemented with 10 % fetal bovine serum, 1 % nonessential amino acids, 1 % glutamine, 1 % penicillin (100 IU/ml) and streptomycin (100 mg/ml) (all from Hyclone, USA). All cultures were maintained at 37°C, 95 % relative humidity and 5 % CO2. The cells allowed to grow in 75 cm2 tissue culture flasks until confluence were then sub-cultured for experimentation.
MTT assay
Cells were seeded in culture plates at a concentration of 2~4 × 105 cells/plate in 1 ml of complete media and incubated for 12 h at 37°C in a 5% CO2 atmosphere to allow for cell adhesion. Cells were treated with D2O + N2 plasma for 0, 1, 3 and 5 min. A control (without treatment) was included in each assay. After incubation times of 24, 48 and 72 h, each plate was carefully rinsed with 1 ml phosphate buffered saline (PBS). Viability was assessed using MTT (3-[4,5-dimethylthiazol-2yl]-2,5-diphenyltetrazolium bromide). 50 μl MTT solutions (5 mg/ml in PBS) were added to each well in the plate and the plates were incubated for 3 h. Following incubation, the medium was removed and the purple formazan precipitates in each well were released in the presence of 1 ml DMSO (Dimethyl sulphoxide). All assays were performed as three independent sets of tests. Absorbance was measured using a micro plate reader (Biotek, Techan, USA) at 540 nm and the cell viabilities were assessed as the ratio of absorbance between plasma treated and control cells which, was directly proportional to the metabolically active cell number42,43. Percentage (%) viability was calculated as equation 1:
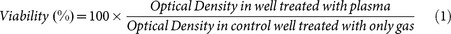 |
Measurement of ROS and RNS inside the Solution and Cells
For the comparative study between N2 and D2O + N2 plasma, ROS and RNS levels in Media (RPMI) were analysed after N2 Plasma and D2O + N2 plasma exposure for 0, 3, 5 min and 3 min+Trolox/cPITO (1 µM Trolox used for OH, H2O2 scavenger and 0.5 mM cPITO used for NO scavenger), and then the concentrations of OH, H2O2 and NO radical was measured in the solutions RPMI media, we have used the previously reported method from our group24. To study the total ROS inside the cell, we have used the H2DCFDA. Attached G361 melanocytes cells in RPMI were treated with D2O+ N2 plasma for 0, 3 min and 3 min + Trolox then, transferred to a microcentrifuge tube. The cells were washed with PBS and 500 ml of 10 mM H2DCFDA was added. After 30°C incubation for 1 h, the cells were washed twice with PBS. Then, the cells were recovered in again with PBS at 30°C for 30 min and read at 495/515 (ex./em.) nm using a microplate reader. Mean fluorescence intensity is calculated between plasma and control (no treated) cells. MFI (%) = 100 × (fluorescent value at an excitation wavelength of 485 nm and an emission at 535 nm of plasma treated cells – Control)/Control.
Detection of apoptosis
Apoptosis in human skin melanocytes G361 cancer cell lines were treated with D2O + N2 plasma for 0 (control) and 3 min detected using the EzWay AnnexinV-FITC apoptosis detection kit, Komabiotech Inc, Korea. (This kit can identify apoptosis at earlier stage as compared to DNA based assay. During apoptosis, phosphatidylserine (PS) exposed to extracellular environment binds to annexin V. After 12 h incubation, cells were trypsinized and washed with 0.5 ml of cold 1x binding buffer. Then, 1.25 µl of Annexin V-FITC (0.5 µg/ml) were added and incubated for 15 min at room temperature in the dark environment. Cells were washed and stained with 10 µl PI (0.3 µg/µl) and observed by flow cytometry (BD FACS Verse, BD Biosciences).
DNA extraction for 8-OHdG detection and agrose gel analysis
Genomic DNA was extracted after plasma exposure and media in a 48 well plate (1 ml per well) was exposed to D2O + N2 plasma for 0 (control), 3 min and 0.5 µM of actinomycin D for OH-dG detection, 1 % agrose gel and CD analysis. Treated cells were subjected to genomic DNA extraction kit. Genomic DNA was extracted following a standard molecular biology protocol and re-suspended in 50 µl water24. The same amount of genomic DNA (2 mg) extracted from cells was used for the detection of 8-OH-dG level using a oxidative DNA damage ELISA kit (Cell Biolabs, inc. USA), same amount of DNA was loaded on a 1 % agarose gel and runned for 1 h. Then, the DNA bands were photographed, after staining with ethidium bromide.
RNA extraction for quantitative real time PCR
We measured mRNA expression of nine apoptosis related genes, including p53, caspase-8, ATM, BAX and NOX1-5. The total cellular RNA was isolated from cancer cell line after 1 day (3 min and 3 min + Trolox) D2O + N2 plasma exposure. Primers designing, cDNA synthesis and quantified every gene, according to the previously reported method from our group44. Quantitative real-time PCR was performed by using following forward and reverse primer sequences were listed in Table 3.
Table 3. Primer sequences used for evaluation of the mRNA expression.
| Genes | Forward primers [5-3] | Reverse primers [5-3] |
|---|---|---|
| 18S rRNA | AACGAGACTCTGGCATGCTAACTA | CGCCACTTGTCCCTCTAAGAA |
| p53 | CACATGACGGAGGTTGTGAG | ACACGCAAATTTCCTTCCAC |
| Caspase8 | TTGAACCCAAGAGGTCAAGG | ACGGGGTCTTGTTCTGTCAC |
| ATM | GGGCAACATGGTGAAACTCT | CCTTAACCTCCAGGGCTCAG |
| Bax | AACATGGAGCTGCAGAGGAT | CAGTTGAAGTTGCCGTCAGA |
| NOX1 | CCACTGTAGGCGCCCTAAGTT | ATGACCGGTGCAAGGATCC |
| NOX2 | GCCCAAAGGTGTCCAAGC | TCCCCAACGATGCGGATAT |
| NOX3 | CCTTCTGTAAAGACCGCTATGCA | GACCACAGGGCCTAAAATCCA |
| NOX4 | ACCCTGTTGGATGACTGGAA | ACCAACGGAAAGGACTGGATA |
| NOX5 | CAGGCACCAGGAAAAGAAAGCAT | TGTTGATCCAGATAAAGTCCACCTT |
Western blotting
The whole cell protein extracts from control and treated cells were prepared as described previously45. Protein of 50 mg were resolved on 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis gels and latter blotted onto nitrocellulose membranes as described in previous paper24. We have used p38 MAP kinase, p42/p44 MAPK (Cell Signaling Technology, Danvers, MA, USA), phospho-p38 MAP kinase, phospho- p42/p44 MAPK (Cell Signaling Technology), and actin (Abcam) antibodies. The analysis were measured using SuperSignal West Pico Chemiluminescent substrate (Pierce, Rockford, IL, USA) and imaged using a Vilver imaging system (Vilver, Upland, CA, USA).
Circular dichroism Spectroscopy (CD,) confocal Raman spectroscopy and density meter measurement analysis
CD spectroscopic studies were performed using a J-815 spectrophotometer (Jasco, Japan) equipped with a Peltier system for controlling the temperature. CD was calibrated using the previous reported protocols46,47. Far UV CD spectra of proteins reveals the important characteristics of the secondary structure of proteins. and also provides the conformational properties of DNA. The secondary structure of Mb and Hb were monitored by using 1.0 cm path length cuvette. The concentration of secondary structures of proteins was 0.1 mg/ml, each spectrum being an average of six spectra. However, for CD DNA the concentration was 0.2 mg/ml. Each sample spectrum was obtained by subtracting appropriate blank media without proteins and DNA. The percentages of secondary structures of proteins were calculated by using Jasco inbuilt software.
A confocal Raman microscope (WITec, Alpha 300 R) with a 632.8 nm of He-Ne laser was utilized to measure the Raman spectra at room temperature. The incident laser beam was focused onto the sample using a microscope objective (100×) in order to obtain the Raman spectra for the titanyl ion and N2 + D2O plasma treated mixtures. The scattered light was collected by same objective lens and then dispersed by a grating. And, it was detected with the help of a charge-coupled-device array detector.
The density (ρ) measurement was performed with an Anton-Paar DSA 5000 with an accuracy of temperature of ± 0.01 K. The uncertainties in the density measurement was ± 0.00005 g cm−3. Prior to measurements, the instrument was calibrated with deionized water and dry air as standards at 293.15 K.
In silico molecular modelling and docking parameters
To find the possible bioactive conformations of D2O2/H2O2 and actinomycin D, the Sybyl X 2.0 interfaced with Surflex-Dock module was used for molecular docking. The program automatically docks ligand into the binding pocket of an enzyme/receptor protein using a protomol-based algorithm and empirically produced scoring function. The X-ray crystallographic structures of skin cancer receptor protein targeting Erk2 (PDB:3SA0)37, p53 (PDB:3ZME)38 and caspase-8 (PDB:3KJQ)39 protein receptor was taken from the protein data bank (PDB) and modified for docking calculations48,49,50,51, the detail explanation is given in supporting information.
Statistical analysis
All values are represented by the mean ± S.D of the indicated number of replicates. Statistical analyses of the data were performed using student's t-test to establish significance between data points, and significant differences have been based on the P<0.05 or P<0.01.
Author Contributions
N.K. and P.A. conceived and designed the experiments, performed research, analyzed the data. P.A. wrote the manuscript. D.K.Y. attempted insilico drug design approaches. J.C. provided assistance with Raman experiments. P.A. performed the protein experiment and CD spectroscopy. H.S.U and E.H.C supervised the study/project, provided assistance in design the plasma setup and provides the OES spectra measurement characterization.
Supplementary Material
Supporting information
Acknowledgments
This work was financially supported by the SRC program (Grant # 2010-0029421) and by Basic Science Research Program of the National Research Foundation (NRF), and was also partially supported by CBDRC program in Defense Ministry. This work was also supported by R & D Program of ‘Plasma Advanced Technology for Agriculture and Food (Plasma Farming)’ through the National Fusion Research Institute of Korea (NFRI) funded by the Government funds and Kwangwoon University 2014.
References
- Winter T. et al. Characterization of the global impact of low temperature gas plasma on vegetative microorganisms. Proteomics 11, 3518–3530 (2011). [DOI] [PubMed] [Google Scholar]
- Fridman G. et al. Applied plasma medicine. Plasma Processes Polym. 5, 503–533 (2008). [Google Scholar]
- Daeschlein G. et al. Antimicrobial activity of an atmospheric pressure plasma jet again relevant wound pathogens in vitro on a simulated wound environment. Plasma Processes Polym. 7, 224–230 (2010). [Google Scholar]
- Ehlbeck J. et al. Low temperature atmospheric pressure plasma sources for microbial decontamination. J. Phys. D: Appl Phys. 44, 013002 (2011). [Google Scholar]
- Fridman G. et al. Blood coagulation and living tissue sterilization by floating electrode dielectric barrier discharge in air. Plasma Chem. Plasma Process 26, 425–442 (2006). [Google Scholar]
- Shekhter A. B. et al. Beneficial effect of gaseous nitric oxide on the healing of skin wounds. Nitric Oxide 12, 210–219 (2005). [DOI] [PubMed] [Google Scholar]
- Sladek R. E. J. et al. Plasma treatment of dental cavities: a feasibility study. IEEE Trans. Plasma Sc 32, 1540–1543 (2004). [Google Scholar]
- Bussiahn R. et al. The hairline plasma: An intermittent negative DC-corona discharge at atmospheric pressure for plasma medical applications. Appl. Phys. Lett. 96, 14370 (2010). [Google Scholar]
- Gadri R. B. et al. Sterilization and plasma processing of room temperature surfaces with a one atmosphere uniform glow discharge plasma (OAUGDP). Surf. Coat. Technol. 131, 528–542 (2000). [Google Scholar]
- Isbary G. et al. Cold atmospheric argon plasma treatment may accelerate wound healing in chronic wounds: Results of an open retrospective randomized controlled study in vivo. Clinical Plasma Medicine 1, 25–30 (2013). [Google Scholar]
- Leszczynski D. et al. Non-thermal activation of the hsp27/p38MAPK stress pathway by mobile phone radiation in human endothelial cells: Molecular mechanism for cancer- and blood-brain barrier-related effects. Differentiation 70, 120–129 (2002). [DOI] [PubMed] [Google Scholar]
- Kim C. H. et al. Effects of atmospheric nonthermal plasma on invasion of colorectal cancer cells. Appl. Phys. Lett 96, 24370 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X. et al. The Effect of Tuning Cold Plasma Composition on Glioblastoma Cell Viability. PLOS ONE 9, e98652 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar N. et al. Enhancement of glucose uptake in skeletal muscle L6 cells and insulin secretion in pancreatic hamster-insulinoma-transfected cells by application of non-thermal plasma jet. Appl. Phys. Lett 103, 203701 (2013). [Google Scholar]
- Takeda H. et al. Mechanisms of cytotoxic effects of heavy water (deuterium oxide: D2O) on cancer cells. Anticancer Drugs 9, 715–725 (1998). [DOI] [PubMed] [Google Scholar]
- Uemura T. et al. Experimental validation of deuterium oxide mediated antitumoral activity as it relates to apoptosis in murine malignant astrocytoma cells. J. Neurosurg 96, 900–908 (2002). [DOI] [PubMed] [Google Scholar]
- Hartmann J. et al. Effects of Heavy Water (D2O) on Human Pancreatic Tumor Cells. Anticancer Res 25, 3407–3412 (2005). [PubMed] [Google Scholar]
- Altermatt H. J. et al. Heavy water delays growth of human carcinoma in nude mice. Cancer 62, 462–466 (1988). [PubMed] [Google Scholar]
- Laissue J. A. et al. Survival of tumor bearing mice exposed to heavy water or heavy water plus methotrexate. Cancer Res. 42, 1125–1129 (1982). [PubMed] [Google Scholar]
- Somlyai G. et al. Naturally occurring deuterium is essential for the normal growth rate of cells. FEBS Lett. 317, 1–4 (1993). [DOI] [PubMed] [Google Scholar]
- Lamprecht J. et al. Disorganization of mitosis in HeLa cells by deuterium oxide. Eur. J. Cell Biol. 50, 360–369 (1989). [PubMed] [Google Scholar]
- Schroeter D. et al. Deuterium oxide (heavy water) arrests the cell cycle of PtK2 cells during interphase. Eur. J. Cell Biol. 58, 365–370 (1992). [PubMed] [Google Scholar]
- Uemura T. et al. Experimental validation of deuterium oxide mediated antitumoral activity as it relates to apoptosis in murine malignant astrocytoma cells. J. Neurosurg. 96, 900–908 (2002). [DOI] [PubMed] [Google Scholar]
- Ryu Y. H. et al. Effects of Background Fluid on the Efficiency of Inactivating Yeast with Non-Thermal Atmospheric Pressure Plasma. PLoS ONE 8, e66231 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirpour S. et al. The Selective Characterization of NonthermalAtmospheric Pressure Plasma Jet on Treatment of Human Breast Cancer and Normal Cells. IEEE Trans. Plasma Sc. 42, 315–322 (2014). [Google Scholar]
- Mededovic S. et al. Electrical Discharges in Mixtures of Light and Heavy Water. Plasma Process Polym. 5, 76–83 (2008). [Google Scholar]
- Kim E. K. et al. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 1802, 396–405 (2010). [DOI] [PubMed] [Google Scholar]
- Wagner E. F. et al. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 9, 537–49 (2009). [DOI] [PubMed] [Google Scholar]
- Bedard K. & Krause K. H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol Rev. 87, 245–313 (2007). [DOI] [PubMed] [Google Scholar]
- Yamaura M. et al. NADPH Oxidase 4 Contributes to Transformation Phenotype of Melanoma Cells by Regulating G2-M Cell Cycle Progression. Cancer Res. 69, 2647–2654 (2009). [DOI] [PubMed] [Google Scholar]
- Kang J. et al. Functional Interaction of H2AX, NBS1, and p53 in ATM-Dependent DNA Damage Responses and Tumor Suppression. Mol.Cell. Biol 25, 661–670 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maheswari P. U. & Palaniandavar M. J. Circular Dichroism: Principles and Applications, 2nd ed, Wiley-VCH: New York. Inorg. Biochem 98, 219–230 (2004). [Google Scholar]
- Nowicka A. M. et al. Oxidation of DNA Followed by Conformational Change after OH Radical Attack. Anal. Chem. 85, 355–361 (2013). [DOI] [PubMed] [Google Scholar]
- Gao D. et al. Benzo[a]pyrene and its metabolites combined with ultraviolet A synergistically induce 8-hydroxy-2V-deoxyguanosine via reactive oxygen species. Free Radic. Biol. Med 39, 1177–1183 (2005). [DOI] [PubMed] [Google Scholar]
- Donne I. D. et al. Protein carbonylation, cellular dysfunction, and disease progression. J. Cell. Mol. Med. 10, 389–406 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimsrud P. A. et al. Oxidative Stress and Covalent Modification of Protein with Bioactive Aldehydes. J. Biol. Chem. 283, 21837–21841 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. et al. Norathyriol Suppresses Skin Cancers Induced by Solar Ultraviolet Radiation by Targeting ERK Kinases. Cancer Res. 72, 260–270 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X. et al. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res. 41, 6034–44 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z. et al. Kinetic and structural characterization of caspase-3 and caspase-8 inhibition by a novel class of irreversible inhibitors. Biochim. Biophys. Acta. 1804, 1817–31 (2010). [DOI] [PubMed] [Google Scholar]
- Wenxing C. et al. Cryptotanshinone Activates p38/JNK and Inhibits Erk1/2 Leading to Caspase-Independent Cell Death in Tumor Cells. Cancer Prev. Res. 5, 778–787 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Y. C. et al. Atmospheric pressure air-plasma jet evolved from microdischarges: Eradication of E. coli with the jet. Phys. Plasmas 16, 123502 (2009). [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65, 55–63 (1983). [DOI] [PubMed] [Google Scholar]
- Kaushik N. K. et al. Effect of jet plasma on T98G human brain cancer cells. Curr. Appl. Phys. 13, 176–180 (2013). [Google Scholar]
- Panngom K. et al. Differential responses of cancer cell lines to non-thermal plasma from dielectric barrier discharge. Curr. Appl. Phys. 13, S6–S11 (2013). [Google Scholar]
- Chen X. L. et al. Role of p38 mitogen-activated protein kinase in Kupffer cell secretion of the proinflammatory cytokines after burn trauma. Burns. 29, 533–539 (2003). [DOI] [PubMed] [Google Scholar]
- Attri P. et al. Water and a Protic Ionic Liquid Acted as Refolding Additives for Chemically Denatured Enzymes. Org. Biomol. Chem. 10, 7475–7478 (2012). [DOI] [PubMed] [Google Scholar]
- Attri P. & Choi E. H. Influence of Reactive Oxygen Species on the Enzyme Stability and Activity in the Presence of Ionic Liquids. PloS One 8, e75096 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav D. K. et al. Design, Synthesis and in Vitro Evaluation of 18β-Glycyrrhetinic Acid Derivatives for Anticancer Activity Against Human Breast Cancer Cell Line MCF-7. Curr. Med. Chem. 21, 1160–1170 (2014). [DOI] [PubMed] [Google Scholar]
- Yadav D. K. et al. QSAR and docking studies on Chalcone derivatives for anti-tubercular activity against M. tuberculosis H37Rv. J. Chemometrics 21, 1160–70 (2014). [Google Scholar]
- Yadav D. K. et al. QSAR and docking based semi-synthesis and in vitro evaluation of 18β-glycyrrhetinic acid derivatives against human lung cancer cell line A-549. Med. Chem. 9, 1073–1084 (2013). [DOI] [PubMed] [Google Scholar]
- Yadav D. K. et al. QSAR and docking based semi-synthesis and in vivo evaluation of artemisinin derivatives for antimalarial activity. Current Drug Target 15, 753–61 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information