

Abstract
mRNA capping enzymes are directed to nascent RNA polymerase II (Pol2) transcripts via interactions with the carboxy-terminal domains (CTDs) of Pol2 and transcription elongation factor Spt5. Fission yeast RNA triphosphatase binds to the Spt5 CTD, comprising a tandem repeat of nonapeptide motif TPAWNSGSK. Here we report the crystal structure of a Pct1·Spt5-CTD complex, which revealed two CTD docking sites on the Pct1 homodimer that engage TPAWN segments of the motif. Each Spt5 CTD interface, composed of elements from both subunits of the homodimer, is dominated by van der Waals contacts from Pct1 to the tryptophan of the CTD. The bound CTD adopts a distinctive conformation in which the peptide backbone makes a tight U-turn so that the proline stacks over the tryptophan. We show that Pct1 binding to Spt5 CTD is antagonized by threonine phosphorylation. Our results fortify an emerging concept of an “Spt5 CTD code” in which (i) the Spt5 CTD is structurally plastic and can adopt different conformations that are templated by particular cellular Spt5 CTD receptor proteins; and (ii) threonine phosphorylation of the Spt5 CTD repeat inscribes a binary on–off switch that is read by diverse CTD receptors, each in its own distinctive manner.
Keywords: transcription, elongation, mRNA processing, mRNA capping, X-ray crystallography
INTRODUCTION
The m7GpppN cap of eukaryal mRNA is formed by three enzymatic reactions: (i) RNA triphosphatase hydrolyzes the RNA 5′ triphosphate end (pppRNA) to a diphosphate (ppRNA); (ii) GTP:RNA guanylyltransferase converts ppRNA to GpppRNA via a covalent enzyme-(lysyl-Nζ)-GMP intermediate; and (iii) AdoMet:RNA(guanine-N7)-methyltransferase converts GpppRNA to m7GpppRNA (Ghosh and Lima 2010). Capping is targeted to nascent RNA polymerase II (Pol2) transcripts via physical interactions of one or more of the capping enzymes with the carboxy-terminal domain (CTD) of the Pol2 Rpb1 subunit. The Pol2 CTD consists of tandemly repeated heptapeptides of consensus sequence Y1S2P3T4S5P6S7. Phosphorylation and dephosphorylation of the Tyr1, Ser2, Thr4, Ser5, and Ser7 residues and cis–trans isomerization of Pro3 and Pro6 inscribe a complex “CTD code” read by diverse receptor proteins that control transcription, modify chromatin structure, and catalyze or regulate mRNA capping, splicing, and polyadenylation (Buratowski 2009; Corden 2013; Eick and Geyer 2013; Jeronimo et al. 2013).
The Ser5-PO4 “letter” of the CTD code plays a vital role in recruiting RNA guanylyltransferase (GTase) to the Pol2 elongation complex (Ho and Shuman 1999; Pei et al. 2001a; Schwer and Shuman 2011; Schwer et al. 2012). Indeed, the direct binding of GTase to the Ser5-PO4 form of Pol2 CTD is a conserved theme among diverse eukaryal taxa (Ho and Shuman 1999; Pei et al. 2001a; Fabrega et al. 2003; Ghosh et al. 2011). Comparisons of the crystal structures of Candida albicans GTase (Cgt1), mammalian GTase (Mce1), and Schizosaccharomyces pombe GTase (Pce1) bound to Ser5-phosphorylated Pol2-CTD ligands illuminated how eukarya take divergent structural routes to achieve GTase·CTD interaction (Fabrega et al. 2003; Ghosh et al. 2011; Doamekpor et al. 2014). Whereas the CTD docking sites are dominated in each case by interactions of GTase with Ser5-PO4 and Tyr1, the CTD segments bound to the GTases differ in their length and in the phase of the heptad sequence bound. For example, fission yeast GTase recognizes an 8-amino acid CTD segment (S5PPSYSPTS5P) bracketed by two Ser5-PO4 marks; mammalian GTase captures a 6-amino acid segment (S5PPSYSP); and Candida GTase has two distinct CTD-docking sites that recognize Ser5-PO4 containing heptads in differently phased registers (TS5PPSYSP and PSYSPTS5PP). Moreover, the atomic contacts to CTD differ among the three GTases and few of the CTD-interacting side chains are conserved. Thus, capping enzymes from different taxa have evolved unique strategies to read the same Pol2 CTD code.
The RNA triphosphatase (TPase) components of the mammalian and budding yeast capping apparatus are recruited passively to the Pol2 CTD, by virtue of their physical association with the GTase: in cis as a covalently fused TPase-GTase enzyme Mce1 in mammals (Ho and Shuman 1999) or in trans as separately encoded subunits of a TPase·GTase complex in budding yeast (Ho et al. 1999; Gu et al. 2010). The fission yeast S. pombe has a distinctive strategy for targeting cap formation to Pol2 transcripts, whereby the TPase (Pct1) and GTase (Pce1) enzymes are not associated physically, but instead bind independently to the Ser5-phosphorylated Pol2 CTD (Pei et al. 2001a; Doamekpor et al. 2014).
Mammalian and fungal capping enzymes can also access nascent Pol2 transcripts via physical interactions with transcription elongation factor Spt5 (Wen and Shatkin 1999; Pei and Shuman 2002; Doamekpor et al. 2014). Spt5 is a large polypeptide, composed of multiple domain modules, that associates with the Pol2 transcription complex shortly after initiation and can exert negative and positive effects on transcription elongation (Hartzog and Fu 2013). Fission yeast Spt5 has a distinctive C-terminal repeat domain (the “Spt5 CTD”), composed of 18 repeats of a nonapeptide motif (T1P2A3W4N5S6G7S8K9), that (i) binds the RNA capping enzymes Pct1 and Pce1 (Pei and Shuman 2002) and (ii) is targeted for threonine phosphorylation by the Cdk9 kinase (Pei and Shuman 2002, 2003; Viladevall et al. 2009). The CTDs of fission yeast Pol2 and Spt5 play overlapping roles in recruiting the capping enzymes in vivo (Schneider et al. 2010). Unlike the capping enzyme·Pol2-CTD interactions, which stringently depend on the Ser5-PO4 mark, Thr1 phosphorylation of the Spt5 CTD is not required for binding to fission yeast Pct1 or Pce1 (Pei et al. 2001a; Pei and Shuman 2002) and Pce1 binding to a Spt5 CTD is antagonized by Thr1 phosphorylation (Doamekpor et al. 2014).
Key insights to the interaction of the capping apparatus with Spt5 were gained via the crystal structure of a fission yeast Pce1·Spt5-CTD complex, which revealed a docking site in the OB domain of the GTase enzyme that captures the Trp4 residue of the fission yeast Spt5 nonamer repeat (Doamekpor et al. 2014). We found that a disruptive mutation in the Spt5-CTD binding site of Pce1 was synthetically lethal with mutations in the Pol2-CTD binding site (Doamekpor et al. 2014), signifying that the Spt5 and Pol2 CTDs cooperate to recruit guanylyltransferase to the transcription elongation complex in vivo.
Here we analyze the fission yeast RNA triphosphatase Pct1 and its interactions with the Spt5 CTD. We report crystal structures of the Pct1 apoenzyme and a Pct1·Spt5-CTD complex and show that Pct1 binding to Spt5 CTD is antagonized by threonine phosphorylation. We establish by structure-guided mutagenesis the relevance of the CTD interface to CTD binding in vitro and Pct1 function in vivo. Our results fortify an emerging definition of an “Spt5 CTD code.”
RESULTS
Crystallization of Pct1 and a Pct1·Spt5-CTD complex
Crystallization trials were conducted with S. pombe RNA triphosphatase Pct1 that had been premixed with (i) manganese and the high-affinity competitive inhibitor tripolyphosphate (Pei et al. 2001b), in an effort to mimic a substrate complex, or (ii) an 18-mer Spt5-CTD peptide TPAWNSGSRTPAWNSGSK composed of two nonamer repeats corresponding to Spt5-(845–862). Crystals of the Pct1, tripolyphosphate, manganese mixture were grown by vapor diffusion against a precipitant solution containing 2.8 M sodium chloride, 10% PEG6000. Crystals were in space group P21. The structure was solved by molecular replacement, using the S. cerevisiae Cet1 homodimer (pdb 1D8I) as the search model, and ultimately refined at 2.6 Å resolution with R/Rfree of 0.199/0.245 (Supplemental Table S1). No electron density was observed for tripolyphosphate or manganese; therefore we refer to this structure as Pct1 apoenzyme. The Pct1 apoenzyme crystal contained four protomers in the asymmetric unit organized as two homodimers, A/B and C/D. Ordered amino acids in A/B include 40–161, 165–303/40–161, 166–303 and in C/D include 40–108, 112–160, 166–303/40–108, 112–160, 165–303.
Crystals of the mixture of Pct1 and Spt5-CTD peptide were grown by vapor diffusion against 3 M sodium formate as the precipitant. The Pct1·Spt5-CTD crystals were in space group P21212. The structure was solved by molecular replacement using the Pct1 apoenzyme homodimer as the search model. Electron density corresponding to a Spt5 CTD peptide fragment was evident. The structure was refined at 2.8 Å resolution with R/Rfree of 0.191/0.248 (Supplemental Table S1). Ordered amino acids in A/B include 43–163, 166–303/39–161, 166–303 and in C/D include 43–247, 253–303/40–161, 164–246, 254–303. Each homodimer was bound to two copies of an internal 5-amino acid CTD peptide T1P2A3W4N5 (Fig. 1A).
FIGURE 1.
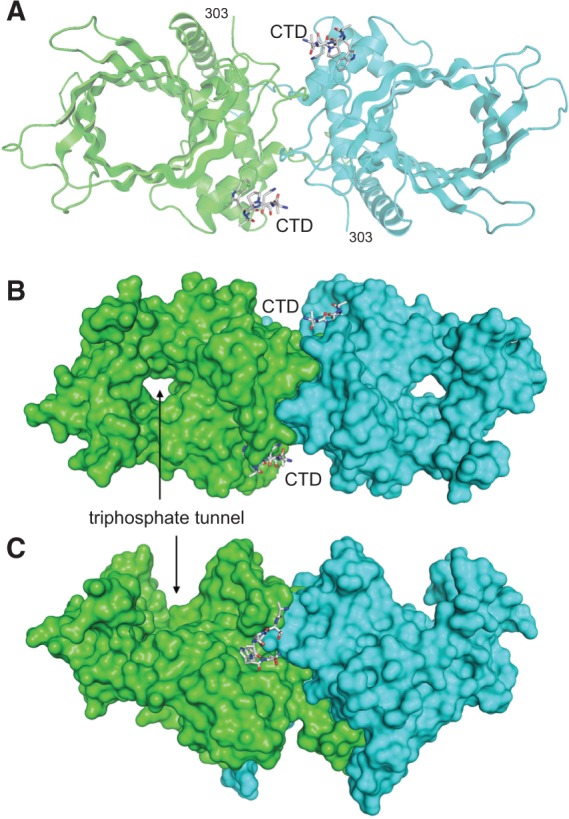
Structure of the Pct1·Spt5 complex. (A) The structure of the Pct1 homodimer is depicted as a cartoon model with the A and B protomers colored green and cyan, respectively. The two Spt5 CTD petides are rendered as stick models with gray carbons. (B) Surface view of the Pct1 homodimer in the same orientation as in panel A, highlighting the through-and-through aperture of the triphosphate tunnel. (C) Surface view of the homodimer rotated 90° about the x-axis.
Overview of the Pct1 structure
The fold of Pct1 is shown in Figure 1A, with the A and B protomers colored green and cyan, respectively; the two Spt5 CTD peptides are depicted as stick models. The Pct1 homodimer consists of parallel triphosphate tunnels, each comprising a topologically closed 8-strand antiparallel β-barrel, resting on a predominantly α-helical globular pedestal. The pedestal forms the dimer interface at a central twofold symmetry axis, running perpendicular to the page in the view in Figure 1A. A surface model of the homodimer in Figure 1B, in the same orientation, highlights the through-and-through tunnel aperture. Rotating the surface model 90° around the x-axis in Figure 1C underscores the concave entrance to the triphosphate tunnel.
A DALI search of the Protein Database with the Pct1 structure recovered S. cerevisiae RNA triphosphatase Cet1 (pdb 1D8I; Z score 26.8; 2.1 Å RMSD at 247 Cα positions) and the RNA triphosphatase domain of mimivirus capping enzyme (pdb 2QY2; Z score 14.0; 3.8 Å RMSD at 176 Cα positions) as the most closely related homologs (Lima et al. 1999; Benarroch et al. 2008). These enzymes belong to, and define, the triphosphate tunnel metalloenzyme superfamily (Gong et al. 2006; Keppetipola et al. 2007). They share a tunnel architecture and a mechanism of metal-dependent hydrolysis of the γ phosphate of nucleoside triphosphates and RNA 5′-triphosphate ends.
A horizontally offset superposition of the Pct1 and Cet1 protomers is shown in Supplemental Figure S1A. The loop segment from amino acids 43 to 54 preceding the first helix of the pedestal appears to be dangling free; however, as shown in Figure 1C, this segment actually extends across the twofold axis to interdigitate with the pedestal domain of the partner protomer. This feature of the structure accords with an earlier finding (Hausmann et al. 2003) that Pct1 homodimerization depends on the peptide segment 41VPKIEMNFLN50. Deletion of this peptide converts Pct1 into a catalytically active monomer, which is defective in vivo in S. pombe and hypersensitive to thermal inactivation in vitro. In contrast, deleting the N-terminal 40 amino acids of Pct1, for which there was no interpretable electron density in our Pct1 crystal structures, had no effect on Pct1 homodimerization, activity in vivo, or thermal stability in vitro (Hausmann et al. 2003). The structure of the homologous budding yeast Cet1 protein extends from amino acids 265 to 539. The Pct1 structure includes two interstrand loops that were disordered in the Cet1 structure (Supplemental Fig. S1A). The N-terminal 240 amino acids of Cet1 are dispensable for its RNA triphosphatase function in vivo and in vitro (Lehman et al. 1999). The Cet1 segment from amino acid 245–261 is essential for Cet1 function in vivo, but not for TPase catalytic activity in vitro, because this segment mediates tight association of Cet1 with the budding yeast GTase Ceg1 (Ho et al. 1999; Lehman et al. 1999; Gu et al. 2010). S. pombe Pct1 lacks a counterpart of this GTase-binding motif, consistent with the fact that Pct1 does not interact with the fission yeast GTase Pce1 (Pei et al. 2001a).
Pct1 active site
The TPase active site within the tunnel aperture is shown in stereo in Supplemental Figure S1B. The aligned Cet1 active site, which is depicted in Supplemental Figure S1C, features a manganese ion in an octahedral coordination complex that includes a sulfate anion as a putative mimetic of the substrate γ phosphate (Lima et al. 1999). Virtually all of the catalytic residues of the Cet1 active site are conserved in Pct1 (Supplemental Fig. S1B,C). From the superposition, we infer that Pct1 acidic side chains Glu78, Glu80, Asp240, Glu260, and Glu262—projecting up from the tunnel floor—bind the essential metal cofactor; this inference is consistent with prior findings that mutating Glu78, Glu80, or Glu260 to alanine abolished Pct1 triphosphatase activity (Pei et al. 2001b). An ensemble of basic side chains emanating from the lateral walls and roof of the Pct1 tunnel (Arg169, Lys185, Lys227, Arg229) is implicated, by analogy to Cet1, in binding the γ phosphate and stabilizing the transition state of the phosphohydrolase reaction.
Pct1·Spt5-CTD complex
An Spt5 CTD peptide T1P2A3W4N5 is docked on the lateral portion of pedestal domain of each protomer of the Pct1 homodimer (Fig. 1A; Supplemental Fig. S1A). (The main chain and Cβ atoms of the Arg9 residue of the preceding nonamer of the input 18-mer CTD peptide were placed into electron density, but the rest of the Arg side chain was disordered.) The N-termini of the two Spt5 CTD peptides are oriented toward each other and are separated by ∼40 Å across the protein surface, underscoring that each CTD docking site on the homodimer is occupied by a different copy of the 18-mer CTD. We estimate that 11–12 amino acids would be required to bridge the 40 Å gap between TPAWN segments. As such, a Spt5 CTD composed of TPAWN-SGSKTPAWNSGSK-TPAWN with one intervening repeat could be sufficient to engage both sites. While it is not known if both binding sites are required for function in vivo, a S. pombe mutant strain with a Spt5 CTD containing just three repeats was sufficient for normal growth (Schneider et al. 2010). The surface view in Figure 1C highlights the proximity of the Spt5 CTD to the homodimer interface. The close-up stereo view of the CTD docking site (Fig. 2A) affirms that elements of both Pct1 protomers contribute to the Spt5 CTD binding site.
FIGURE 2.
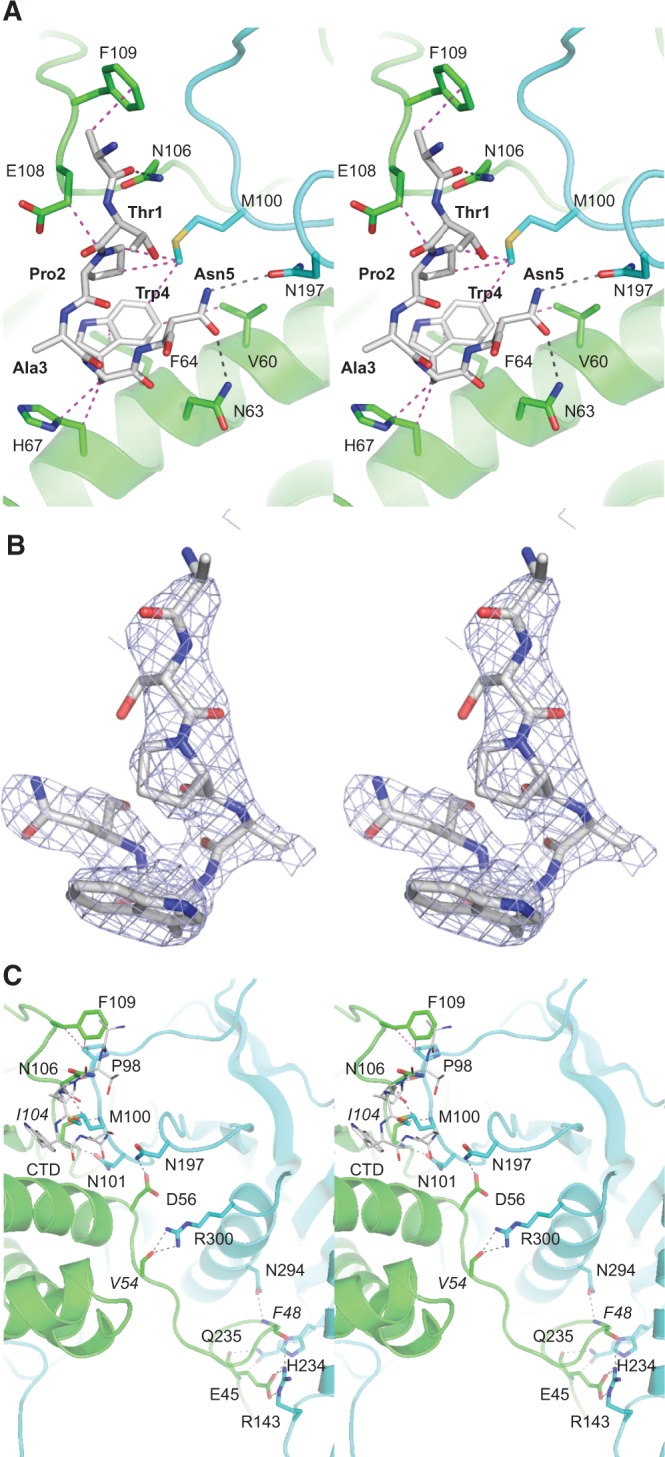
Architecture of the Pct1·Spt5-CTD and homodimer interfaces. (A) Stereo view of the Pct1·Spt5-CTD interface. The tertiary structures and side chain carbons of the A and B protomers are colored green and cyan, respectively. The CTD peptide is rendered as a stick model with gray carbons. Hydrogen bonds are denoted by black dashed lines; van der Waals contacts are indicated by magenta dashed lines. (B) Stereo view of the Spt5 peptide rotated ∼180° from the view in A. Blue mesh represents a simulated annealing 2fo-fc omit map contoured at 1.0 σ. (C) Stereo view of the Pct1 homodimer interface colored as in panel A. The CTD peptide is depicted as a thin stick model.
The atomic contacts of the Spt5 CTD amino acids with Pct1, proceeding from N to C termini of the bound peptide, are as follows: Arg9-Cβ makes van der Waals contacts with Phe109; the Arg9 main chain carbonyl is within hydrogen bonding distance of Asn106-Nδ; Pro2 makes van der Waals contact with Glu108 and with Met100 of the other protomer; Trp4 makes van der Waals contacts with Val60, Phe64, and His67, and with Met100 of the other protomer; Asn5-Oδ is within hydrogen bonding distance of Asn63-Nδ, while Asn5-Nδ is within hydrogen bonding distance of Asn197-Oδ of the other protomer (Fig. 2A). A noteworthy feature of the CTD conformation is that the peptide backbone makes a tight U-turn about Trp4 so that Pro2 stacks over the Trp4 indole (Fig. 2A,B). The centrality of Trp4 to the crystallographic Pct1·Spt5-CTD interface rationalizes previous findings that Pct1 failed to bind in vitro to an Spt5 CTD nonamer repeat array in which every Trp4 was replaced by alanine (Schneider et al. 2010). The Spt5 CTD Thr1 side chain makes no contacts with Pct1 in the crystal (Fig. 2A), consistent with an earlier report that mutating every Thr1 to alanine did not affect Pct1 binding to an Spt5 nonamer array (Schneider et al. 2010).
The fission yeast Pct1 106NPEF109 motif that engages the Spt5 CTD is not conserved in budding yeast Cet1; indeed, the PEF109 tripeptide is an inserted element at the tip of an interstrand loop (denoted by the arrow in Supplemental Fig. S1A, left panel) that is not even present in Cet1, where the corresponding shorter loop is displaced compared with its position in Pct1 (see arrow in Supplemental Fig. S1A, right panel). This motif acquisition in Pct1 likely contributes to its distinctive interactions with the Spt5 CTD.
Alignment of the structures of the Pct1·Spt5-CTD complex and the Pct1 apoenzyme reveals an Spt5-induced local conformational change, depicted in Figure 3, where the apoenzyme fold and pertinent side chain carbons are colored beige and the Spt5-bound Pct1 fold and side chain carbons are colored green. The positions of most of the Spt5-interacting amino acids (e.g., Val60, Asn63, Phe64, His67, Met100, Asn106, and Asn197) are unchanged in the two structures. In contrast, residues 108 to 112 undergo a significant rearrangement. In the apoenzyme, this segment adopts a 310 helix secondary structure. However, when Spt5 CTD is bound, the segment refolds to a loop. This transition is accompanied by an 8 Å movement of the Phe109 aromatic ring, bringing it to the position formerly occupied by Glu108 in the apoenzyme, after which it contacts the Spt5 CTD. Glu108 is displaced by 7 Å to a position where it, too, contacts the CTD. The flanking Leu111 side chain undergoes an 11 Å shift in apoenzyme versus Spt5 CTD complex, to occupy the position originally taken by Phe109 (Fig. 3). Otherwise, the folds of the two structures are highly similar.
FIGURE 3.
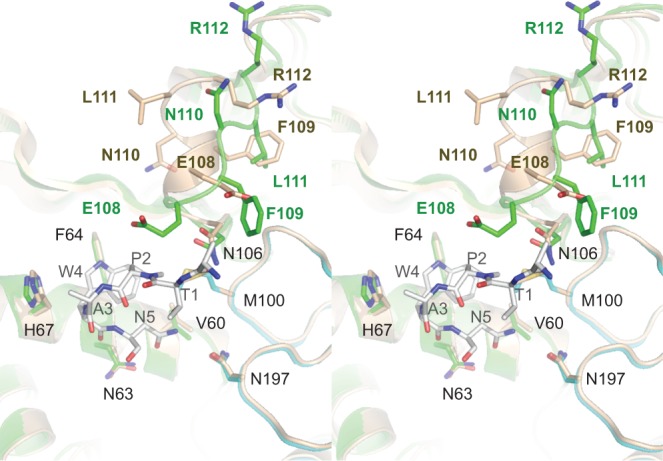
Spt5 CTD binding elicits a local conformational change in Pct1. Stereo view of the superimposed structures of the Pct1·Spt5-CTD complex (with the Pct1 fold and side chain carbons colored green and the Spt5 CTD stick model carbons colored gray) and the Pct1 apoenzyme (colored beige) reveals an Spt5-induced local conformational change involving residues 108–112. In the apoenzyme, this segment adopts a 310 helix secondary structure. However, when Spt5 CTD is bound, the segment unfolds to a loop.
Pct1 homodimer interface
The homodimer is stabilized by two distinct Pct1·Pct1 interfaces, each repeated twice across the twofold axis. The pertinent cross-protomer contacts are depicted in Figure 2C. The first interface is between the N-terminal leader peptide and the pedestal of the other protomer. Specifically, the Glu45 carboxylate makes a bidentate salt bridge to Arg143 and is within hydrogen bonding distance of His234; the Glu45 main chain carbonyl and amide nitrogen hydrogen bond with Gln235 Nɛ and Oɛ; the Phe48 main chain carbonyl and amide nitrogen hydrogen bond with the Arg143 and Asn294 side chains, respectively; and the Val54 main chain carbonyl receives hydrogen bonds from the Arg300 guanidinium nitrogens (Fig. 2C). The second dimer interface abuts the Spt5 CTD (shown as a thin stick model in Fig. 2C) and involves several of the amino acids that contact the CTD. This dimer interface entails the following: van der Waals interaction of Asn106 with Met100 and of Phe109 with Pro98; hydrogen bonds between Ile104 and Asn106 main chain atoms in one protomer and the Asn101 side chain and Met100 and Pro98 main chain atoms in the other protomer; and a hydrogen bond between Asp56 and Asn197 side chains (Fig. 2C). Many of the charged and polar amino acids at the Pct1 homodimer interface are conserved in Cet1. For example, Pct1 Asp56 is the counterpart of Cet1 Asp280, mutation of which to alanine converts the budding yeast triphosphatase into a catalytically active monomer that is unable to function in vivo (Hausmann et al. 2003).
Effect of threonine phosphorylation of the Spt5 CTD on interaction with Pct1
We implemented a fluorescence polarization assay to study the binding of Pct1 to purified recombinant Spt5-(801–898) protein, comprising 10 nonamer repeats (Pei and Shuman 2002), that was labeled at the N-terminus with BODIPY-FL. Fluorescence anisotropy increased with Pct1 concentration, with an apparent Kd of 0.68 ± 0.10 µM (Fig. 4A). We also gauged Pct1 binding to a fluorescein-labeled 22-amino acid synthetic Spt5 CTD peptide SGSKTPAWNSGSKTPAWNSGSK that contained two complete nonamer repeats. The apparent Kd of 124 ± 10 µM (Fig. 4B) was 180-fold higher than the Kd of Pct1 for the longer Spt5-(801–898) ligand. This acute dependence on the number of nonamer repeats for high-affinity binding of Pct1 to Spt5 CTD in the fluorescence assay accords with results obtained using a yeast two-hybrid interaction assay, which showed that Pct1 binding to the Spt5 CTD in vivo required a long tandem nonamer repeat array (Pei and Shuman 2002). The Pct1·Spt5-CTD structure suggests how a longer nonamer array would have high avidity, based on simultaneous occupancy of both CTD docking sites on the Pct1 homodimer.
FIGURE 4.

Threonine phosphorylation of the Spt5 CTD weakens interaction with Pct1. (A) Pct1 binding to a fluorescent-tagged Spt5-CTD-(801–898) ligand was assayed by a change in anisotropy as described under Materials and Methods. Anisotropy is plotted as a function of input Pct1 polypeptide. Each datum is the average of three independent experiments ± SEM. (B) Pct1 binding to a fluorescent-tagged 22-amino acid Spt5 CTD peptide (primary structure as shown, with the N-terminal tag denoted by *) was assayed by a change in anisotropy as described under Materials and Methods. Anisotropy is plotted as a function of input Pct1 polypeptide. Each datum is the average of three independent experiments ± SEM. (C) Effect of Thr1 phosphorylation on competition for the Spt5 binding site. Pct1 was preincubated with increasing concentrations of untagged Spt5 CTD peptide or untagged Spt5-Thr1-PO4 peptide prior to addition of fluorescein-Spt5-CTD peptide. Relative anisotropy is plotted as a function of competitor concentration. Each datum is the average of three independent experiments ± SEM.
Threonine-1 of the S. pombe Spt5 nonamer motif is targeted for phosphorylation by Cdk9. Here and in previous studies, we found that Pct1 binds to the unphosphorylated Spt5 CTD. However, it is unclear whether and how the Pct1·Spt5 interaction is affected by phosphorylation. Although the Thr1 side chain does not contact the Pct1 interface in our cocrystal structure of Pct1 bound to the peptide TPAWN (Fig. 2A), the addition of a phosphate group at Thr1-Oγ could clash with the tight hairpin turn conformation of the Pct1-bound CTD peptide if the Thr1 side chain rotamer is maintained after phosphorylation. While a clash could be alleviated if the Thr1 side chain adopted an alternative rotamer, the structure engenders a prediction that Thr1 phosphorylation might be inimical to Spt5 CTD binding to Pct1.
To evaluate the impact of Thr1 phosphorylation, we tested the ability of unphosphorylated and Thr1-phosphorylated 22-amino acid synthetic peptides SGSKTPAWNSGSKTPAWNSGSK and SGSK(Tp)PAWNSGSK(Tp)PAWNSGSK to compete with a fluorescein-tagged 22-mer Spt5 CTD peptide for binding to Pct1 in a fluorescence polarization assay. Whereas unphosphorylated 22-mer CTD displaced the fluorescent CTD with a Ki of 79 ± 9 µM, the Thr1-phosphorylated CTD did so with a Ki of 2600 ± 280 µM (Fig. 4C). Thus, Thr1 phosphorylation elicited a 33-fold decrement in affinity of the fission yeast TPase for the Spt5 CTD.
Functional probing of the Pct1·Spt5-CTD interface
Mutations of Pct1 amino acids comprising the Spt5 CTD docking site, and a three amino acid internal deletion of the Pct1-specific loop segment at the docking site (▵106NPE108), were introduced by marked allelic replacement into one chromosomal pct1 locus of a diploid S. pombe strain. The diploids were sporulated and viable haploids were obtained and genotyped to verify the allelic replacement. Serial dilutions of the wild-type and mutant strains containing equal numbers of cells were spot tested for growth on YES agar medium at 20°C, 25°C, 30°C, 34°C and 37°C. The M100A, N197A, and N197R mutants grew as well as wild-type cells at all temperatures (as gauged by colony size). The F64A, H67A, and ▵106NPE108 mutants thrived at 20°C–25°C, but displayed severe temperature-sensitive (ts) growth defects at 30°C–37°C. The M100R strain grew slowly at 20°C–30°C and failed to thrive at 37°C (Fig. 5A). Because of the overlap between the Spt5 CTD and homodimer interfaces of Pct1, and prior studies indicating that weakening the homodimer interface of Cet1 elicits a ts growth defect and renders the protein thermolabile in vitro (Hausmann et al. 2003), we surveyed the mutant strains for their steady-state levels of Pct1 protein by Western blotting of whole-cell extracts of equal numbers of cells grown in liquid medium at 30°C. Probing the blot with anti-Spt5 antibody verified comparable sample loading. However, probing with anti-Pct1 antibody showed that the steady-state levels of Pct1 were decreased in F64A, H67A, and ▵106NPE108 cells, likely accounting for the growth defects of these strains (Fig. 5B). In contrast, the Pct1 level in M100A and M100R cells were similar to that of wild-type cells, implying that the slow growth of M100R cells at 30°C was not caused by depletion of the Pct1 pool. Moreover, the kinetics of Mn2+-dependent ATP hydrolysis by purified recombinant M100A and M100R proteins were similar to the rate of ATP hydrolysis by recombinant wild-type Pct1, arguing against a catalytic defect as the cause of the M100R growth phenotype (Fig. 5C).
FIGURE 5.

Functional analysis of the Pct1·Spt5-CTD interface. (A) Growth of S. pombe strains with the indicated chromosomal pct1 alleles in which amino acids at the Spt5 CTD interface were mutated or deleted. (B) Western blots of whole-cell extracts prepared from equal numbers of cells with pct1 alleles as specified. The blots were probed with antibodies to fission yeast Spt5 and Pct1. The positions and sizes (kDa) of polypeptide size markers are denoted on the left. (C) Purified recombinant wild-type Pct1 and mutants M100A and M100R were assayed for manganese-dependent ATP hydrolysis as described under Materials and Methods. The extents of phosphate release (from 1 nmol of input ATP) are plotted as a function of reaction time. (D) Mutations that disrupt binding to the Spt5 CTD do not affect binding to the phosphorylated Pol2 CTD. Wild-type Pct1 and the indicated mutants were assayed for binding to BODIPY-FL-Spt5 CTD-(801–898) and BODIPY-FL-Pol2 CTD-Ser5-PO4 ligands. The Kd values are shown.
We proceeded to test the binding of recombinant M100A and M100R proteins to the BODIPY-FL-Spt5-(801–898) ligand by Pct1 titration in the fluorescence polarization assay. The Kd values were 1.4 ± 0.15 µM for M100A and 21.4 ± 2.4 µM for M100R, versus 0.68 ± 0.10 µM for wild-type Pct1. Thus, M100A, which had no apparent effect on fission yeast growth, had a twofold effect on Spt5 CTD binding (which presumably reflects loss of the van der Waals contact of Met100 with the CTD), whereas M100R, which compromised yeast growth, elicited a 31-fold decrement in Spt5 CTD binding, likely via steric hindrance at the CTD docking site (Fig. 2A). To confirm that defects associated with M100 mutations were specific to Spt5 we proceeded to test the binding of Pct1 to a phosphorylated 28 amino acid Pol2 CTD ligand containing four YSPTSPS consensus motifs. Pct1 bound to BODIPY-Pol2-CTD-PO4 with a Kd value of 90.2 ± 5.2 (Supplemental Fig. S2A), interactions that were competed effectively with an unlabeled CTD ligand (Supplemental Fig. S2B). Interaction with BODIPY-Pol2-CTD-PO4 was also dependent on phosphorylation as interactions with the same ligand were decreased by at least an order of magnitude after treatment with calf-intestinal alkaline phosphatase (Supplemental Fig. S2A). No defects were observed in binding for recombinant M100A and M100R proteins to BODIPY-Pol2-CTD-PO4 (Fig. 5D). We infer that the M100R allele reflects the contribution of Spt5 CTD interaction to Pct1 activity in vivo.
DISCUSSION
Structural plasticity of the Spt5 CTD
The fission yeast Spt5 CTD has a relatively uniform primary structure consisting of tandem repeats of a nonapeptide motif that extend to the very C-terminus of the 990-amino acid Spt5 protein (Pei and Shuman 2002). Metazoan Spt5 proteins have an analogous repeat domain, often referred to as the CTR (C-terminal repeat), located near but not at the C-terminus (Hartzog and Fu 2013). Whereas the repeated elements of fission yeast Spt5 CTD and metazoan Spt5 CTRs characteristically include a Thr-Pro or Ser-Pro dipeptide (targeted for threonine or serine phosphorylation by a cyclin-dependent kinase), the spacer intervals between the dipeptide repeats differ among metazoan species, and they also differ from the regular spacing in S. pombe Spt5. The metazoan repeats do not adhere to the S. pombe consensus sequence TPAWNSGSK. In particular, whereas the residue located two positions downstream from the Thr-Pro dipeptide in S. pombe Spt5 is typically a tryptophan, it is never a tryptophan in the human, nematode, or zebrafish proteins; instead, this position is usually occupied by tyrosine or histidine (Pei and Shuman 2002). The consensus view is that the Spt5 CTD/CTR functions to recruit cellular proteins to elongating Pol2 complexes with which Spt5 is engaged.
The present structural snapshot of the RNA capping enzyme Pct1 bound to Spt5 CTD sequence TPAWN extends insights from the recently reported crystal structure of a human Spt5 CTR binding protein (Wier et al. 2013): the Plus3 domain of Rtf1 bound to a phosphorylated CTR peptide SGSRTPPMYGSQ. Figure 6 shows an alignment of the Spt5 ligands from the two structures, superimposed at their respective Thr-Pro dipeptides. The proline is in the trans conformation in both structures. In the case of Rtf1, the Spt5 interface is dominated by hydrogen bonding contacts to the phosphate group of the Spt5 ligand, consistent with the fact that threonine phosphorylation is required for Rtf1 binding to Spt5-CTR (Wier et al. 2013). In contrast, Pct1 makes no contacts with the Spt5 threonine side chain and its binding to Spt5 is antagonized by threonine phosphorylation. The phosphothreonine in the Rtf1 complex adopts a different rotamer configuration than does threonine in the Pct1 complex. Although our structure did not visualize the SGSR peptide segment preceding Thr1, it is noteworthy that the amino acid sequence of this segment of S. pombe Spt5 is identical to the threonine-flanking peptide segment present in the Rtf1 structure (Fig. 6). The two Spt5 CTD conformations diverge sharply after the Cα of the amino acid following the proline. In Rtf1, the bound Spt5 segment YGSQ adopts a 310 helix-like conformation that bears no resemblance to the tight hairpin turn made by the WN dipeptide of the Pct1-bound Spt5. Indeed, the positions of the aromatic tyrosine and tryptophan side chains of the human and S. pombe Spt5 are different. Whereas the tyrosine is a minor contributor to the interface with Rtf1 (i.e., compared with phosphothreonine), the fission yeast tryptophan is the dominant player at the interface with Pct1 (Fig. 2A). The comparison of the fission yeast and human Spt5 CTD/CTR complexes highlights the theme that the Spt5 CTD is structurally plastic and can adopt different conformations that are templated by particular cellular Spt5 CTD receptor proteins. In these respects, the Spt5 CTD resembles the Pol2 CTD.
FIGURE 6.
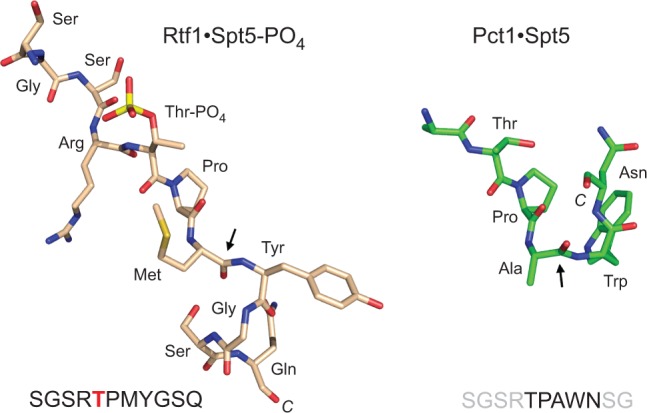
Spt5 CTD structure is phosphorylation-specific and templated by its receptor protein. The Spt5 ligands from the human Rtf1·Spt5-PO4 (pdb 4L1U) and fission yeast Pct1·Spt5 structures were aligned and then offset horizontally. The sites at which the trajectory of the CTD diverges in the two structures are highlighted by arrows.
An Spt5 CTD code
Spt5 binds to and travels with elongating Pol2, helping to coordinate elongation kinetics with RNA processing and chromatin modification (Hartzog and Fu 2013). Several of the key effector functions of Spt5 rely on its CTD/CTR as a platform to recruit cellular proteins to the Pol2 elongation complex. Available evidence suggests that threonine phosphorylation of Spt5 CTD repeats inscribes a binary code (on–off) that is read by diverse CTD receptors, each in its own distinctive manner. Whereas the Paf1 complex subunit Rtf1 recognizes only the phosphorylated Spt5 CTR (Wier et al. 2013), the RNA guanylyltransferase Pce1 (Doamekpor et al. 2014) and the RNA triphosphatase Pct1 (present study) specifically bind to the unmodified form of the CTD. Moreover, the positive elongation function of Spt5 depends on phosphorylation of the CTD/CTR by Cdk9 (Yamada et al. 2006).
The organization of the Spt5 CTD/CTR as a tandem array of Thr/Ser-Pro-containing motifs allows (in principle) for simultaneous engagement of multiple cellular CTD receptors that respond to different CTD cues. Taking into account just the on–off phosphorylation state of the repeated Thr-Pro and Ser-Pro motifs, the fission yeast Spt5 CTD can adopt up to 262,144 different primary structures. Cdk9 is a principal Spt5 CTD kinase in fission yeast (Pei and Shuman 2003; Viladevall et al. 2009), but it is conceivable that other kinases may also contribute to the writing of an Spt5 CTD code. How the code is edited by Spt5 CTD phosphatases is still uncharted territory, at least in fission yeast. Finally, by analogy to the Pol2 CTD, it is conceivable that the complexity of the Spt5 CTD code is amplified by cis–trans isomerization of the defining proline residue of the repeated Spt5 motif.
Two key themes highlighted by structural, biochemical, and genetic analyses of the reading of the Pol2 and Spt5 CTD codes by the fission yeast capping apparatus are (i) that CTD phosphorylation has opposite effects on the interaction of fission yeast guanylyltransferase and triphosphatase with Pol2 (Ser5-PO4 is required for binding) versus Spt5 (Thr1-PO4 inhibits binding) and (ii) the two CTD codes function in parallel to aid cotranscriptional capping.
MATERIALS AND METHODS
Purification of S. pombe Pct1
The pct1 open reading frame was inserted into pET-based bacterial expression plasmid pSMT3 so as to encode Pct1 fused to an N-terminal His6Smt3 domain. Mutations were introduced into the pSMT3-Pct1 plasmids with a QuickChange kit (Stratagene). The mutated pct1 ORFs were sequenced to verify the absence of unwanted coding changes. The expression plasmids were transformed into E. coli BL21(DE3) codon plus RIL (Stratagene). Cultures of E. coli containing the expression plasmids were grown in super broth in baffled flasks at 37°C to an A600 of 1.5, after which the cells were cold-shocked by placing the flasks on ice for 30 min. The cultures were adjusted to 0.5 mM isopropyl-β-D-thiogalactoside (IPTG) and then incubated overnight at 18°C with constant shaking. Cells were harvested by centrifugation at 16,000g and resuspended in buffer containing 50 mM Tris–HCl, pH 8.0, and 20% (w/v) sucrose. All subsequent procedures were performed at 4°C.
For purification of the His6Smt3-tagged Pct1 proteins, cells were resuspended in lysis buffer containing 20 mM Tris–HCl, pH 8.0, 500 mM NaCl, 20 mM imidazole, 1 mM β-mercaptoethanol (BME), 0.1% IGEPAL (Fluka), 1 mM phenylmethanesulfonylfluoride (PMSF) and then disrupted by sonication. Lysates were clarified by centrifugation at 45,000g and the resulting supernatants were applied to a Ni-NTA superflow resin (Qiagen) that had been equilibrated in buffer A (20 mM Tris-HCl, pH 8.0, 350 mM NaCl, 1 mM BME) containing 20 mM imidazole. Bound proteins were eluted with 250 mM imidazole in buffer A. The His6Smt3 tags were removed by incubating the 250 mM imidazole eluate fractions with Ulp1, a Smt3-specific protease (Mossessova and Lima 2000), at a protein:Ulp1 ratio of 1000:1 (w/w) for 6–18 h at 4°C. The tag-free Pct1 proteins were separated from His6Smt3 by gel-filtration through a Superdex 200 column equilibrated in buffer A. The peak Pct1 fractions were pooled, dialyzed against buffer B (20 mM Tris–HCl, pH 8.0, 50 mM NaCl, 1 mM BME), and concentrated by centrifugal ultrafiltration. For crystallography experiments, wild-type Pct1 was further purified by cation exchange chromatography. The Superdex fraction was adsorbed to a MonoS 10/10 column (GE Healthcare) and eluted with a 20-column-volume linear gradient of 50 mM to 1 M NaCl in 20 mM Tris–HCl, pH 8.0, 1 mM BME. Pct1 was then concentrated to 10 mg/mL in 20 mM Tris–HCl, pH 8.0, 50 mM NaCl, 1 mM TCEP.
Pct1 crystallizations and structure determinations
Pct1 apoenzyme
Aliquots of a mixture of 140 µM Pct1, 2 mM tripolyphosphate, 5 mM manganese chloride were mixed on a cover slip with an equal volume of precipitant solution containing 2.8 M sodium chloride, 10% (w/v) PEG6000. Crystals were grown at 18°C by hanging drop vapor diffusion against the precipitant solution, then flash-frozen in liquid nitrogen. Diffraction data from a single crystal to 2.6 Å resolution were collected on a Rigaku RAXIS-4 image plate detector with CuKα radiation from a Rigaku RU200 rotating anode generator. The data were indexed, integrated and scaled using HKL2000 (Otwinowski and Minor 1997). The crystal was in space group P21. Initial phases were obtained by molecular replacement in PHASER (McCoy et al. 2007) using the coordinates of the S. cerevisiae Cet1 homodimer (pdb 1D8I) as the search model. Iterative rounds of refinement and adjustment in CNS (Brunger et al. 1998), PHENIX (Adams et al. 2010), and COOT (Emsley and Cowtan 2004) yielded a 2.6 Å Pct1 model with R/Rfree of 0.199/0.245 and good geometry (97.9%, 99.9%, and 0.1% in favored, allowed, and disallowed regions of Ramachandran space, respectively) (Chen et al. 2010). The model contained four Pct1 protomers in the asymmetric unit organized as two homodimers (A/B and C/D). No electron density was observed for tripolyphosphate or manganese.
Pct1·Spt5-CTD complex
A mixture of 140 µM Pct1 and 340 µM 18-mer Spt5-CTD peptide T1P2A3W4N5S6G7S8R9T1P2A3W4N5S6G7S8K9 was incubated for 1 h at 4°C, then mixed on a cover slip with an equal volume of 3 M sodium formate. Crystals were grown at 18°C by hanging drop vapor diffusion against 3 M sodium formate and cryoprotected with 3.2 M sodium formate, 20% (v/v) ethylene glycol. Diffraction data from a single crystal to 2.8 Å resolution were collected at APS NE-CAT beamline 24-IDE equipped with an ADSC Quantum 315 detector. Data were indexed, integrated and scaled using HKL2000. The crystal was in space group P21212. The structure was solved by molecular replacement in PHASER using the coordinates of the Pct1 apoenzyme homodimer as the search model. After a single round of refinement in CNS, electron density corresponding to a Spt5-CTD peptide fragment was evident. Model refinement and rebuilding in CNS, PHENIX, and COOT yielded a final 2.8 Å model with R/Rfree of 0.191/0.248 and good geometry (96.0%, 100%, and 0% in most favored, allowed, and disallowed Ramachandran regions, respectively). The model contained four Pct1 protomers in the asymmetric unit organized as two homodimers (A/B and C/D). Each homodimer bound two copies of a Spt5-CTD peptide, of which five amino acids corresponding to TPAWN were ordered in electron density.
Assays of Pct1·CTD interactions by fluorescence polarization
Recombinant N(Cys)-Spt5-(801–898) protein was produced in E. coli, purified, and labeled with BODIPY-FL at the N-terminus via maleimide chemistry as described previously (Doamekpor et al. 2014). A fluorescein-labeled 22-amino acid synthetic Spt5 CTD peptide SGSKTPAWNSGSKTPAWNSGSK was purchased from New England Peptides. A 28-amino acid synthetic Pol2 CTD-Ser5-PO4 peptide (YSPTS5pPSYSPTS5pPSYSPTS5pPSYSPTS5pPS) was labeled with BODIPY-FL at the N terminus via N-hydroxy succinimidyl ester chemistry as previously described (Doamekpor et al. 2014).
Binding reaction mixtures (18 µL) containing 20 mM Tris–HCl, pH 8.0, 50 mM NaCl, 4 mM dithiothreitol (DTT), either 300 nM BODIPY-FL-N(Cys)-Spt5-(801–898), or 25 nM fluorescein-labeled 22-amino acid synthetic Spt5 CTD peptide and increasing concentrations of Pct1 polypeptide as specified were incubated on ice for 15 min. For binding reactions of Pct1 with the Pol2-CTD, 400 nM untreated labeled BODIPY-FL-Pol2 CTD-Ser5-PO4 or labeled BODIPY-FL-Pol2 CTD-Ser5-PO4 which had previously been incubated with 0.5 units alkaline phosphatase (CIP, New England Biolabs) at 37°C for 2 h were used. Aliquots (15 µL) were transferred to wells of a 384-well microplate and fluorescence polarization was measured at 23°C with a SpectraMax M5 microplate reader (Molecular Devices) using an excitation wavelength of 485 nm, emission wavelength of 538 nm, and a cutoff of 530 nm for BODIPY-FL labeled peptides. Fluorescence polarization using fluorescein-labeled peptide was measured using an excitation wavelength of 485 nm, emission wavelength of 525 nm, and a cutoff of 515 nm. Experiments were performed in triplicate except for the reaction involving the CIP treated Pol2-CTD which was performed in duplicate. For the experiments using a 22-amino acid Spt5-CTD ligand or a 28-amini acid Pol2-CTD, in which the Pct1 concentration that achieved 50% saturation was at least three times greater than the concentration of the labeled ligand, the Kd value was derived by fitting the data in Prism to a single-site binding model (Lundblad et al. 1996). For experiments with Spt5-(801–898), in which the Pct1 concentration that achieved 50% saturation was similar to the concentration of labeled ligand, the Kd value was derived by fitting the data in Prism to a single-site binding model that accounts for receptor depletion (Lundblad et al. 1996). For competition experiments, Pct1 was incubated with serial dilutions of the unlabeled competitor for 15 min on ice. Fluorescent-labeled Spt5-CTD or Pol2-CTD-PO4 was then added and incubation was continued for another 15 min on ice. Aliquots (15 µL) were transferred to the wells of a 384-well microplate, and measurements were obtained as described above. In assays with the 22-mer Spt5-CTD, the fluorescent ligand and Pct1 concentrations were 25 nM 22-mer fluorescein-Spt5-CTD and 120 µM Pct1 polypeptide. In the assays with the Pol2-CTD, the fluorescent ligand and Pct1 concentrations were 400 nM BODIPY-FL-Pol2 CTD-Ser5-PO4 and 40 μM Pct1 polypeptide. Experiments were performed in triplicate, and data were fit in Prism to a one-site binding model for competitor.
Effects of CTD interfacial mutations on Pct1 function in vivo
Allelic replacement at chromosomal pct1+ locus was performed as follows. We constructed series of integration cassette plasmids containing, in the 5′–3′ direction, (i) a 1.4-kbp segment encompassing the pct1 cDNA (wild-type or mutated) and 484-bp of 5′ flanking chromosomal DNA; (ii) a 268-bp DNA segment harboring the nmt1+ transcription termination signal; (iii) a hygMX cassette conferring resistance to hygromycin; and (iv) a 498-bp segment of 3′ flanking pct1 chromosomal DNA. The 5′-pct1–hygMX-pct1-3′ integration cassettes were excised from the plasmids and transfected into S. pombe diploids. Transformants selected for growth on YES agar containing 0.3 mg/mL hygromycin were genotyped by Southern blotting to confirm correct integration of hygMX at one of the pct1 loci. The pct1–hygMX allele was then PCR-amplified and sequenced to confirm the presence of the desired mutation(s). The heterozygous pct1+ pct1–hygMX strains were sporulated. A random population of haploid progeny (∼2500, as gauged by plating on YES agar) was plated on YES-hygromycin agar. Viable pct1–hygMX haploid strains formed colonies on selective hygromycin agar at frequencies consistent with random segregation. To gauge the effect of the pct1 mutations on vegetative growth, cultures of haploid S. pombe pct1–hygMX strains were grown in liquid medium at 30°C until A600 reached 0.6–0.9. The cultures were adjusted to a final A600 of 0.1 and aliquots (3 µL) of serial fivefold dilutions were spotted on YES agar. The plates were incubated at 20°C, 25°C, 30°C, 34°C, and 37°C.
ATPase activity
Reaction mixtures (100 µL) containing 20 mM Tris–HCl, pH 8.0, 50 mM NaCl, 0.5 mM DTT, 2 mM MnCl2, 0.2 mM ATP, and 40 nM of the Superdex preparations of wild-type or mutant Pct1 polypeptide were incubated at 30°C. At the times specified, aliquots (5 µL) were removed and mixed with 100 µL Biomol green reagent (Enzo) for 30 min at 22°C before measuring the A620. The molar amount of phosphate released was determined by interpolation to a phosphate standard curve.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank NE-CAT beamlines (Advanced Photon Source) supported by RR-15301 (NIH NCRR). APS is supported by the US Department of Energy, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award numbers GM061906 (C.D.L.) and GM052470 (B.S. and S.S.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. S.S. is an American Cancer Society Research Professor. C.D.L. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.048181.114.
REFERENCES
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Cryst D Biol Crystallogr 66(Pt 2):213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benarroch D, Smith P, Shuman S 2008. Characterization of a trifunctional mimivirus mRNA capping enzyme and crystal structure of the RNA triphosphatase domain. Structure 16: 501–512. [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges N, Pannu NS, Read RJ, et al. 1998. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr 54(Pt 5):905–921. [DOI] [PubMed] [Google Scholar]
- Buratowski S 2009. Progression through the RNA polymerase II CTD cycle. Mol Cell 36: 541–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arrendall WB 3rd, Headd JJ, Keady DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Cryst D Biol Crystallogr 66(Pt 1):12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corden JL 2013. RNA polymerase II C-terminal domain: tethering transcription to transcript and template. Chem Rev 113: 8423–8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doamekpor SK, Sanchez AM, Schwer B, Shuman S, Lima CD 2014. How an mRNA capping enzyme reads distinct RNA polymerase II and Spt5 CTD phosphorylation codes. Genes Dev 28: 1323–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eick D, Geyer M 2013. The RNA polymerase II carboxy-terminal domain (CTD) code. Chem Rev 113: 8456–8490. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60(Pt 12 Pt 1):2126–2132. [DOI] [PubMed] [Google Scholar]
- Fabrega C, Shen V, Shuman S, Lima CD 2003. Structure of an mRNA capping enzyme bound to the phosphorylated carboxy-terminal domain of RNA polymerase II. Mol Cell 11: 1549–1561. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Lima CD 2010. Enzymology of RNA cap synthesis. Wiley Interdiscip Rev RNA 1: 152–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Shuman S, Lima CD 2011. Structural insights to how mammalian capping enzyme reads the CTD code. Mol Cell 43: 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong C, Smith P, Shuman S 2006. Structure–function analysis of Plasmodium RNA triphosphatase and description of a triphosphate tunnel metalloenzyme superfamily that includes Cet1-like RNA triphosphatases and CYTH proteins. RNA 12: 1468–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu M, Rajashankar KR, Lima CD 2010. Structure of the Saccharomyces cerevisiae Cet1-Ceg1 mRNA capping apparatus. Structure 18: 216–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartzog GA, Fu J 2013. The Spt4-Spt5 complex: a multi-faceted regulator of transcription elongation. Biochim Biophys Acta 1829: 105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausmann S, Pei Y, Shuman S 2003. Homodimeric quaternary structure is required for the in vivo function and thermal stability of Saccharomyces cerevisiae and Schizosaccharomyces pombe RNA triphosphatases. J Biol Chem 278: 30487–30496. [DOI] [PubMed] [Google Scholar]
- Ho CK, Shuman S 1999. Distinct roles for CTD Ser-2 and Ser-5 phosphorylation in the recruitment and allosteric activation of mammalian capping enzyme. Mol Cell 3: 405–411. [DOI] [PubMed] [Google Scholar]
- Ho CK, Lehman K, Shuman S 1999. An essential surface motif (WAQKW) of yeast RNA triphosphatase mediates formation of the mRNA capping enzyme complex with RNA guanylyltransferase. Nucleic Acids Res 27: 4671–4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeronimo C, Bataille AR, Robert F 2013. The writers, readers, and functions of the RNA polymerase II C-terminal domain code. Chem Rev 113: 8491–8522. [DOI] [PubMed] [Google Scholar]
- Keppetipola N, Jain R, Shuman S 2007. Novel triphosphate phosphohydrolase activity of Clostridium thermocellum TTM, a member of the triphosphate tunnel metalloenzyme superfamily. J Biol Chem 282: 11941–11949. [DOI] [PubMed] [Google Scholar]
- Lehman K, Schwer B, Ho CK, Rouzankina I, Shuman S 1999. A conserved domain of yeast RNA triphosphatase flanking the catalytic core regulates self-association and interaction with the guanylyltransferase component of the mRNA capping apparatus. J Biol Chem 274: 22668–22678. [DOI] [PubMed] [Google Scholar]
- Lima CD, Wang LK, Shuman S 1999. Structure and mechanism of yeast RNA triphosphatase: an essential component of the mRNA capping apparatus. Cell 99: 533–543. [DOI] [PubMed] [Google Scholar]
- Lundblad JR, Laurance M, Goodman RH 1996. Fluorescence polarization analysis of protein–DNA and protein–protein interactions. Mol Endocrinol 10: 607–612. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ 2007. Phaser crystallographic software. J Appl Crystallogr 40(Pt 4):658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossessova E, Lima CD 2000. Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Mol Cell 5: 865–876. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Pei Y, Shuman S 2002. Interactions between fission yeast mRNA capping enzymes and elongation factor Spt5. J Biol Chem 277: 19639–19648. [DOI] [PubMed] [Google Scholar]
- Pei Y, Shuman S 2003. Characterization of the Schizosaccharomyces pombe Cdk9/Pch1 protein kinase: Spt5 phosphorylation, autophosphorylation and mutational analysis. J Biol Chem 278: 43346–43356. [DOI] [PubMed] [Google Scholar]
- Pei Y, Hausmann S, Ho CK, Schwer B, Shuman S 2001a. The length, phosphorylation state, and primary structure of the RNA polymerase II carboxyl-terminal domain dictate interactions with mRNA capping enzymes. J Biol Chem 276: 28075–28082. [DOI] [PubMed] [Google Scholar]
- Pei Y, Schwer B, Hausmann S, Shuman S 2001b. Characterization of Schizosaccharomyces pombe RNA triphosphatase. Nucleic Acids Res 29: 387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider S, Pei Y, Shuman S, Schwer B 2010. Separable functions of the fission yeast Spt5 carboxyl-terminal domain (CTD) in capping enzyme binding and transcription elongation overlap with those of the RNA polymerase II CTD. Mol Cell Biol 30: 2353–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Shuman S 2011. Deciphering the RNA polymerase II CTD code in fission yeast. Mol Cell 43: 311–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Sanchez AM, Shuman S 2012. Puncuation and syntax of the RNA polymerase II CTD code in fission yeast. Proc Nat Acad Sci 109: 18024–18029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viladevall L, St Amour CV, Rosebrock A, Schneider S, Zhang C, Allen JJ, Shokar KM, Schwer B, Leatherwood JK, Fisher RP 2009. TFIIH and P-TEFb coordinate transcription with capping enzyme recruitment at specific genes in fission yeast. Mol Cell 33: 738–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y, Shatkin AJ 1999. Transcription elongation factor hSPT5 stimulates mRNA capping. Genes Dev 13: 1774–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wier AD, Mayekar MK, Héroux A, Arndt KM, VanDemark AP 2013. Structural basis for Spt5-mediated recruitment of the Paf1 complex to chromatin. Proc Natl Acad Sci 110: 17290–17295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Yamaguchi Y, Inukai N, Okamoto S, Mura T, Handa H 2006. P-TEFb-mediated phosphorylation of hSpt5 C-terminal repeats is critical for processive transcription elongation. Mol Cell 21: 227–237. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.