

Abstract
RNA–protein interactions are central to biological regulation. Cross-linking immunoprecipitation (CLIP)-seq is a powerful tool for genome-wide interrogation of RNA–protein interactomes, but current CLIP methods are limited by challenging biochemical steps and fail to detect many classes of noncoding and nonhuman RNAs. Here we present FAST-iCLIP, an integrated pipeline with improved CLIP biochemistry and an automated informatic pipeline for comprehensive analysis across protein coding, noncoding, repetitive, retroviral, and nonhuman transcriptomes. FAST-iCLIP of Poly-C binding protein 2 (PCBP2) showed that PCBP2-bound CU-rich motifs in different topologies to recognize mRNAs and noncoding RNAs with distinct biological functions. FAST-iCLIP of PCBP2 in hepatitis C virus-infected cells enabled a joint analysis of the PCBP2 interactome with host and viral RNAs and their interplay. These results show that FAST-iCLIP can be used to rapidly discover and decipher mechanisms of RNA–protein recognition across the diversity of human and pathogen RNAs.
Keywords: genomics, noncoding RNA, RNA–protein interactions, virology
INTRODUCTION
Across the increasingly diverse repertoire of RNA species, RNA–protein interaction is a nearly ubiquitous function (Rinn and Ule 2014). While RNA–protein complexes such as the ribosome and spliceosome, central regulators of cell biology, are well-studied, a vast and enigmatic repertoire of noncoding and nonhuman RNA–protein interactomes await further characterization (Rinn and Ule 2014). Considering the diverse reach of RNA-binding proteins (RBPs) in cell biology, substantial effort has been focused on methods for genome-wide interrogation of RNA–protein interactions using next-generation sequencing (NGS) (König et al. 2012). By stabilizing direct interactions in vivo combined with stringent purification steps, UV cross-linking immunoprecipitation and sequencing (CLIP-seq) enables specific isolation of an RBP's target RNA-binding sites for NGS analysis. Following NGS, the topological profile of the RBP can be informatically extracted using numerous computational tools, including peak-calling algorithms (Corcoran et al. 2011; Lovci et al. 2013), to isolate the salient features of the RBP of interest.
Much of the pioneering work has been focused on well-studied proteins and the protein-coding transcriptome, leading to numerous important advances, on both methodological (PAR-CLIP, iCLIP, and BrdU-CLIP) and computational fronts (Hafner et al. 2010; König et al. 2010; Zhang and Darnell 2011; Weyn-Vanhentenryck et al. 2014). These results have spurred broader interest in CLIP, particularly with respect to the interactome of noncoding RNAs (Guttman and Rinn 2012) or across the diversity of viruses and microbes that impinge on human health (Papenfort and Vogel 2010). Yet, extending CLIP across many RBPs is challenging for at least two reasons: (1) The sample preparation protocol is inefficient and time consuming and (2) informatic methods are not easily implemented or generally applicable to any RBP, particularly if RBP targets are not obvious a priori, or if the RBP binds various classes of noncoding transcripts as well as nonhuman transcriptomes.
To put these challenges in context, the CLIP workflow can be thought of as a stack of tasks—starting with NGS biochemistry, followed by informatic transformations of the resulting data, and finally protein-specific questions or analyses. Because the specificity of work increases as one moves across the stack, we sought to address common challenges to any CLIP investigation by improving the efficiency of sample preparation, extending the intermediate analysis to include a diverse set of user-definable transcriptomes (protein coding, noncoding, repetitive, retroviral, and nonhuman), and also standardizing data format output such that comparisons between RBPs are straightforward (Fig. 1A,B). We applied this Fully Automated and Standardized iCLIP (FAST-iCLIP) pipeline to a published iCLIP data set of the RBP hnRNP-C, recapitulating the published biology of this protein. We then applied FAST-iCLIP to the human and viral interactomes of Poly-C binding protein 2 (PCBP2), a KH domain (hnRNP-K homology) (Lunde et al. 2007) containing RBP that has a preference for C/U-rich motifs (Choi et al. 2009) as well as pathological associations in cancer (Eiring et al. 2010; Han et al. 2013) and infectious disease (Gamarnik and Andino 2000; Herold and Andino 2001; Shetty et al. 2013). The unbiased characterization of the PCBP2 protein revealed novel RNA–protein interactions and provided supporting evidence for previously known cellular and viral roles for PCBP2.
FIGURE 1.

FAST-iCLIP incorporates experimental improvements and standardized experimental interface to enable iCLIP analysis. (A) Biochemical improvements to the standard iCLIP procedure reduce experimental time by half. (B) A standard interface for analysis iCLIP data increases analysis efficiency and dissects many known sources of RNA transcripts including both the repetitive and nonrepetitive human genome as well as nonhuman genomes. (C) Histogram of the types and number of genes identified by FAST-iCLIP analysis of publically available hnRNP-C iCLIP data. (D) Percentage of all hnRNP-C iCLIP reads mapping to mRNA loci subdivided by functional domain. (E) Average histogram of hnRNP-C iCLIP reads along a normalized mRNA transcript. Each gene's functional regions (5′ UTR, CDS, and 3′ UTR) are binned into 200 units plotted along the same axis. Intronic reads are not visualized in this plot. (F) Logo visualization of the HOMER motif output from all hnRNP-C iCLIP reads with the fraction of iCLIP target regions containing that motif. hnRNP-C crosslink sites and region-shuffle control are shown.
RESULTS
Well established CLIP methods require a combination of multiple single-stranded RNA ligations, RNA precipitations, and gel size selections on low abundance material, all leading to complex and time-consuming protocols. We reasoned that replacing the 3′-blocking moiety (often dideoxyC [ddC] or puromycin) on the 3′ RNA ligation adaptor with a biotin would allow for purification of CLIP'ed material throughout the library preparation procedure without the need for preamplification gel selection or precipitations. By exchanging the 3′ddC blocker from the standard iCLIP 3′ adaptor with a 3′-biotin, we reduced the time required to perform iCLIP by 50% relative to conventional protocols (Fig. 1A). Additionally, we modified the circularizing cDNA synthesis primer to avoid cytidine at the 5′-end as CircLigase has significant biases against this nucleotide (A Mele and R Darnell, unpubl., pers. comm.). Combining these improvements and other innovations (Huppertz et al. 2014; Vanharanta et al. 2014; Weyn-Vanhentenryck et al. 2014), our methods reduced the sample preparation time from 4 to 2 d, utilizing the high affinity biotin–streptavidin interaction for rapid and specific biochemical purification (Fig. 1A).
We next sought to address challenges associated with analysis of iCLIP data across RNA classes and transcriptomes (Fig. 1B). While each RBP subjected to CLIP is likely to harbor unique binding features, we reasoned that a standardized set of analyses and visualizations could be used to rapidly identify these features independent of the protein of interest, and allow direct comparison across RBPs. We therefore implemented an automated informatics pipeline, termed Fully Automated and Standardized (FAST-iCLIP). The pipeline is synergistic with existing tools, as its aim is to bridge between the diversity of available algorithms and the biological questions that underpin any CLIP experiment (Fig. 1B). Specifically, we focused on analyses related to four broad RNA classes that CLIP data sets are well-positioned to interrogate, including (1) nonrepetitive, protein-coding regions of the genome, (2) noncoding RNAs (ncRNAs), (3) highly abundant, repetitive RNAs such as ribosomal RNAs (rRNA), small nuclear RNAs (snRNAs), endogenous retroviral, and transposable elements that are often ignored in current CLIP analyses, and (4) nonhuman genomes, such as RNA viruses.
Highlighting its synergy with existing algorithms built for analysis of CLIP data, we integrated FAST-iCLIP with the CLIPper peak-calling algorithm (Lovci et al. 2013) in order to isolate high confidence binding regions. After mapping, isolating reverse-transcription (RT) stop positions that are conserved across biological replicates, and extracting high confidence reads using CLIPper, the FAST-iCLIP pipeline partitions reads and applies different analyses based on the gene type. Protein-coding RT stops are further partitioned to 5′ untranslated region (UTR), coding sequence (CDS), 3′ UTR, and intronic regions. The binding topology across these mRNA regions is determined and gene lists with unique regional binding profiles are extracted. Similarly, the binding topology across each class of ENSEMBL-defined ncRNA, including long ncRNAs, is determined and genes lists for each RNA class are extracted. A similar strategy is applied to repetitive RNA genes using a custom index that includes several short repeat RNAs, including splicing RNAs, SRP, 7SK, Y RNAs, and the 5S rRNA, as well as the full ribosomal DNA locus. Binding to endogenous retroviral and transposable elements is evaluated using a custom index of over 1000 distinct elements enabling rapid identification of highly bound elements along with binding topology. Collectively, these analyses provide a comprehensive snapshot of binding topology that informs follow-up protein-specific analyses.
We tested the performance of FAST-iCLIP on hnRNP-C, a well-characterized RBP that regulates splicing in a binding-site specific manner. Importantly, hnRNP-C has been the focus of two iCLIP studies and thus provided a well-studied RNA–protein interactome on which to validate the performance of FAST-iCLIP (König et al. 2010; Zarnack et al. 2013). We obtained raw data for biological replicates from a prior study (Zarnack et al. 2013) and processed them with FAST-iCLIP, revealing a strong preference for protein-coding targets (Fig. 1C) and a binding preference for intronic regions (∼67.6% of the reads) (Fig. 1D) relative to 5′ UTR and CDS regions (Fig. 1D,E). FAST-iCLIP also uses the HOMER algorithm (Heinz et al. 2010) to search for de novo primary sequence motifs in CLIPper called RT stops, uncovering a strong poly-uridine (polyU) track binding motif (Fig. 1F). Additionally, we identify at least two previously unreported features in the published data set, but which are easily extracted from the FAST-iCLIP output. First, hnRNP-C binds to multiple places across the rRNA in a unique pattern (more below). Second, we find ∼21.6% of the iCLIP reads fall within 3′ UTRs, representing 1829 mRNAs with hnRNP-C binding exclusively within the 3′ UTR, suggesting there are additional cellular roles hnRNP-C may be playing. Together, this example demonstrates that FAST-iCLIP can accurately identify known features as well as quickly provide binding information of novel transcripts bound by a well-studied RBP.
We next focused on applying the experimental and computational workflow of FAST-iCLIP to Poly(rC) binding protein 2 (PCBP2), a KH domain protein with important roles in translational control and stabilization of cellular and viral RNAs. PCBP2 has a preference for C-rich motifs (Choi et al. 2009) and notable association with infectious disease (Han et al. 2013). PCBP2 was a favorable target, as features of its biology are well understood, but no CLIP-seq study has been reported to date (Waggoner and Liebhaber 2003; Han et al. 2013). We performed FAST-iCLIP in the human Huh-7 (human hepatoma) cells and processed the data, revealing that the majority (2116) of targets were mRNAs with 68% of reads found within 3′ UTRs (Fig. 2A,B). Motif search of PCBP2-bound regions revealed polyC/U-rich binding sites, as expected based upon prior functional studies (Luo 1999) and biochemistry (Fig. 2C; Du et al. 2005). Based upon global analysis of PCBP2 binding preference across mRNA genes, we found a preference for 3′-UTR binding (Fig. 2D), but also identified 48, 246, and 1113 mRNAs to which PCBP2 cross-links exclusively in 5′ UTR, CDS, and 3′ UTR regions, respectively. We used DAVID (Huang et al. 2009) in order to identify enriched Gene Ontology (GO) terms within each gene set (Fig. 2E). Notably, we found that PCBP2 interacts with mRNAs with distinct and coherent biological functions based on binding in distinct RNA regions. PCBP2 interacted with mRNAs encoding proteins involved in “cellular response to stress” at their 5′ ends, those associated with “translation and translational elongation” throughout the CDS, and those associated with “protein localization,” “transport and RNA splicing” within their 3′ UTRs. The diversification in functions of RNA elements within different mRNA regions, provides new insights into the diverse biological functions attributed to PCBP2.
FIGURE 2.
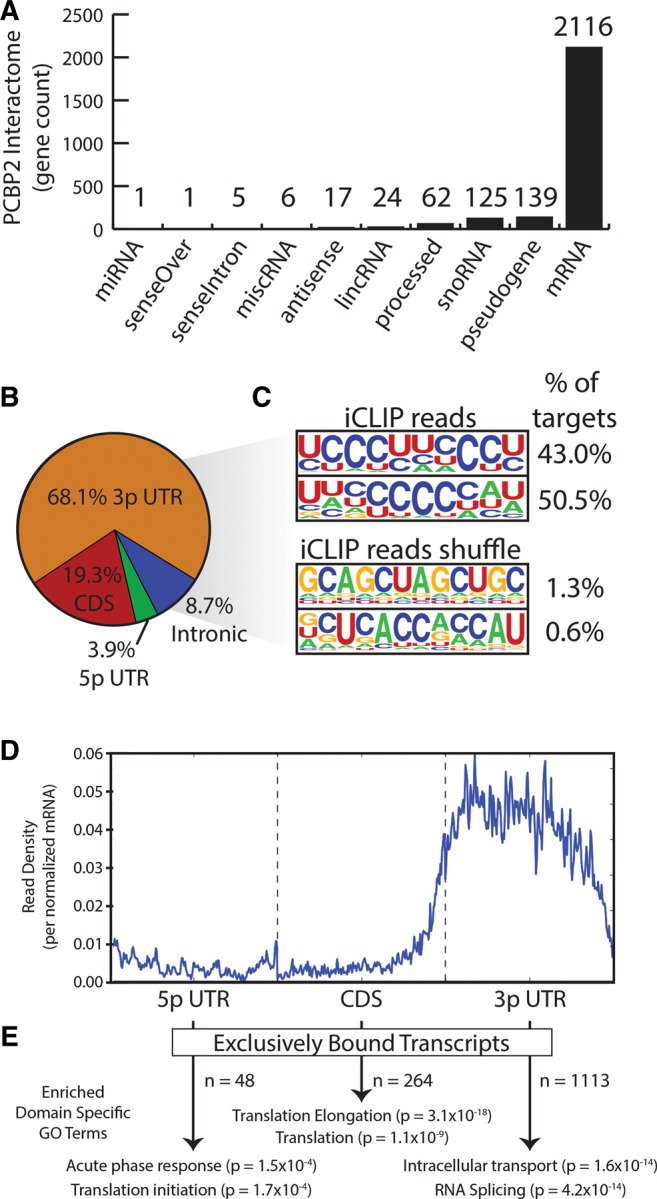
PCBP2 is a major mRNA-binding protein that exhibits distinct binding modes. (A) Histogram of the types and number of genes identified by FAST-iCLIP bound to PCBP2. (B) Percentage of all PCBP2 iCLIP reads mapping to mRNA loci subdivided by functional domain. (C) Logo visualization of the top two HOMER motif output generated from all PCBP2 iCLIP reads. The fraction of target regions with each motif and PCBP2 or region-shuffle results are displayed. (D) Average histogram of PCBP2 iCLIP reads along a normalized mRNA transcript. Each gene's functional regions (5′ UTR, CDS, and 3′ UTR) are binned into 200 units plotted along the same axis. (E) For each functional region in D, DAVID was run to obtain enriched GO terms.
After extracting distinct binding modes across the protein-coding transcriptome for PCBP2, we used additional features of FAST-iCLIP in order to uncover its binding topology across the noncoding transcriptome. Without a priori knowledge of an RBP's function it is important to explore all possible binding events, including coding and noncoding transcripts as well as RNAs that are multicopy (repetitive) in the genome. Though often discarded from CLIP workflows, repetitive sequences include some of the most abundant and important structural noncoding RNAs as well as pseudogenes that may have functional activity (Rapicavoli et al. 2013). We constructed a custom annotation of 18 short repetitive ncRNAs and the full 42 kilobase (kb) ribosomal DNA (rDNA) locus, allowing us to map iCLIP reads to these RNA transcripts. We examined crosslinked sites of PCBP2 across the transcribed region of the rDNA locus revealing a preference for the 18S rRNA (Fig. 3A). To determine if iCLIP was nonspecifically cross-linking to the rRNA, we compared the PCBP2 and hnRNP-C data sets. Analysis of the hnRNP-C iCLIP revealed a different binding profile, enriched for cross-linking in the intergenic spacers (Fig. 3B), suggesting that even analyzing highly abundant noncoding RNAs such as rRNA can provide protein-specific results. Leveraging the exhaustive set of noncoding analyses provided by FAST-iCLIP, we identified 125 PCBP2-bound snoRNAs with a slight preference for the C/D box type (Fig. 3C). By plotting the distribution of iCLIP cross-linking sites across the C/D and H/ACA box subtype we find PCBP2 globally binds snoRNAs in clustered regions (Fig. 3C). Similarly, we identified PCBP2 binding to poly-pyrimidine residues of two repetitive RNAs (Y RNA 1 and Y RNA 3) that belong to a small class of ncRNAs implicated in modulating the immune response and are known to have poly-pyrimidine tracks (Fig. 3D; Perreault et al. 2007; Verhagen and Pruijn 2011). In silico folding of these two noncoding RNAs predicts the iCLIP sites to be largely single stranded (Fig. 3D), compatible with the RNA-binding properties of PCBP2 (Du et al. 2005). Based upon its noncoding binding topology, PCBP2 appears to have broad regulatory scope through interaction with 18S rRNA as well as serving as a translational regulator in both cancer (Eiring et al. 2010; Han et al. 2013) and infectious disease (Gamarnik and Andino 2000; Herold and Andino 2001; Shetty et al. 2013).
FIGURE 3.

Noncoding and repeat RNA analysis reveals novel PCBP2–RNA interactions. (A,B) Coverage histogram of PCBP2 or hnRNP-C (respectively) iCLIP reads mapping to the 42-kb human rDNA locus. Total reads mapping to the rDNA were used to calculate a fraction of total per RT stop and binding sites are reported as such. The mature sequences of the 18S, 5.8S, and 28S rRNAs are highlighted in gray. (C) PCBP2 iCLIP reads mapping to snoRNA loci were tallied and plotted as percentages mapping to the three snoRNA classes; C/D box, H/ACA box, and scaRNAs (center). Reads mapping to C/D box or H/ACA box snoRNAs were extracted and plotted across a normalized snoRNA transcript of 100 units long and read percentages were calculated as in left and right bar plots, respectively. (D) PCBP2 iCLIP reads mapping to the Y RNA 1 (left) and Y RNA 3 (right) transcripts are plotted as in (A). Secondary structure predictions of each RNA were produced with mFold and nucleotides identified as having PCBP2 iCLIP RT stops are highlighted in red.
To further elucidate the connection between PCBP2, translational regulation, and disease, we next performed FAST-iCLIP in Huh-7 cells infected with the JFH-1 strain of Hepatitis C virus (HCV) (Fig. 4A). Though PCBP2 is required for HCV replication (Wang et al. 2011), the molecular details are poorly understood. Several studies focused on the HCV 5′ UTR have led to the suggestion that a complex between PCBP2 and SL1 of the 5′ UTR as well as an undefined region of the 3′ UTR of the viral RNA may be formed that facilitates viral circularization (Wang et al. 2011). Both SL1 and stem–loop structures in the 3′ UTR of the viral genome are required for viral RNA replication (Tellinghuisen et al. 2007). In addition, the proximity of SL1 to the conserved miR-122 sites in the HCV genome suggests that PCBP2 may coordinate with miR-122 in protection of the uncapped 5′ end of the viral RNA from degradation and/or the switch between viral translation and RNA replication (Machlin et al. 2011; Li et al. 2013a). Because FAST-iCLIP can accommodate any viral or custom genome, we generated coverage histograms of iCLIP RT stops across the HCV genome for two biological replicates, observing favorable concordance and global preference for binding U/C-rich regions of the genome (Fig. 4B, r2 = 0.93). The 5′ UTR of the HCV RNA genome has well-annotated (Fraser and Doudna 2006) structural elements that play critical roles in the viral life cycle (Fig. 4C; Tellinghuisen et al. 2007). Consistent with prior studies (Shetty et al. 2013), we observed a strong binding peak at SL1, but also detected PCBP2 occupancy that extends from SLI through the two miR-122 binding sites to the base of SL2. Surprisingly, we also detected strong binding around the translation start codon within SLVI of the internal ribosome entry site (IRES) (Fig. 4D).
FIGURE 4.

Systematic mapping of PCBP2 iCLIP data to the JFH-1 HCV genome. (A) Experimental design to identify in vivo PCBP2 interaction sites across the JFH-1 HCV genome. (B) Genomic structural features within both UTRs with functional annotation. (C) Scatter plot of individual RT stops mapping to the JFH-1 genome comparing the two iCLIP biological replicates. (D,E) Coverage histogram across the 5′ UTR revealing peaks at SL1, across the SL1-SLII junction, and around the start codon (D) and across the 3′ UTR revealing peaks on hairpins involved in the kissing interaction as well as the poly-U/C tract (E). Both coverage histograms include bin size of 5 bp.
PCBP2's interaction with viral 3′ UTR was significantly stronger than with the well-studied 5′ UTR (Fig. 4D,E [cf. y-axis scales]). PCBP2 binding to the 3′ UTR occurred primarily in the single-stranded regions between stem–loops 5BSL3.2 and the variable region, a domain that includes the viral stop codon and that is implicated in both stimulation of translation and replication (Fig. 4C,E; Song et al. 2006; Tellinghuisen et al. 2007). Not surprisingly, PCBP2 also bound to the poly(U)/UC region of the viral genome, consistent with binding to single-stranded poly(U)/C regions (Fig. 4C,E). In addition to the UTRs, we observe multiple robust peaks of PCBP2 occupancy across the full viral gene body, which includes previously unreported binding across the coding region of the virus (data not shown). Comparative analysis of the identified PCBP2-bound host transcripts between control and HCV-infected Huh-7 cells indicate that HCV interactions with PCBP2 do not significantly alter the cellular PCBP2 binding repertoire (r2 = 0.87 between tag count across protein-coding target genes in HCV infected relative to uninfected cells) (data not shown), consistent with the relatively low copy number of the HCV RNA genome (∼1000–3000 copies per cell (Wakita et al. 2005; Miyamoto et al. 2006; Steinmann and Pietschmann 2013). Together, our FAST-iCLIP analysis revealed PCBP2's interactions with the viral genome in a distinct and reproducible pattern, consistent with both the reported PCBP2 activity and previously identified PCBP2-binding sites. Additionally, we identified binding regions in the 3′ UTR and novel binding to regions of the genome important to the viral life cycle (Fig. 4C–E). Our data suggest that PCBP2 may play a role in both viral translation and replication as distinct binding to regions of the genome implicated in regulation of both these activities were found. Further mutational and functional analyses will be required to reveal the precise mechanism of PCBP2 regulation of the HCV life cycle.
DISCUSSION
FAST-iCLIP incorporates a protocol that reduces experimental time by ∼50% with a computational pipeline that produces standardized data sets across protein coding, noncoding, and user-definable nonhuman transcriptomes. As sequencing continues to reveal novel noncoding RNA (Rinn and Chang 2012) classes and further characterize microbial biodiversity, FAST-iCLIP can scale beyond the human- and protein-centric scope of CLIP investigation. Highlighting the importance of careful analysis of the noncoding interactome, PCBP2 binds to the rRNA, supporting its known role in translation and identifying novel interaction sites. Moreover, we took advantage of FAST-iCLIP's modularity to incorporate analysis of nonhuman genomes to examine the PCBP2-HCV interactome in both infected and uninfected cells.
The global PCBP2 binding topology uncovered by FAST-iCLIP is consistent with known functional roles for the protein, including 5′-UTR binding to modulate translation of cellular transcripts (Eiring et al. 2010) as well as 3′-UTR binding to modulate transcript stability through occlusion of endonuclease cleavage sites (Weiss and Liebhaber 1995). Yet, our data extend the scope of this regulation considerably and indicates novel occupancy in the coding region of cellular mRNAs. Our application of FAST-iCLIP to HCV suggests that these regulatory functions of PCBP may be co-opted by the virus, as we also observe PCBP2 binding to the viral 5′ UTR, coding region, and 3′ UTR. We observe a peak of PCBP2 around the SL1/miR-122 binding site junction in the HCV genome, suggesting that PCBP2 may act in concert with miR-122 to restrict viral degradation from the 5′ UTR by cellular exonucleases such as Xrn2 (Li et al. 2013b). PCBP2 also strongly bound to the translational start codon and the 3′ UTR of the HCV genome including the viral stop codon and conserved stem–loop structures required for viral RNA replication, a mode of binding that is topologically similar to that observed in poliovirus where PCBP2 plays a critical role in the viral life cycle (Gamarnik and Andino 2000). In the context of a poliovirus infection, PCBP2 mediates cross-talk between the viral 5′ and 3′ UTRs in order to regulate the switch between viral translation and RNA replication (Gamarnik and Andino 2000; Walter et al. 2002). Our data are consistent with a symmetrical role for PCBP2 in the context of HCV infection and overlays in vivo biophysical detail from prior reports showing PCBP2-mediates circularization of the HCV genome (Wang et al. 2011) in vitro. Thus, our application of FAST-iCLIP reveals a common binding topology of PCBP2 across the human transcriptome as well as the HCV genome. In both cases, a 3′ UTR bias is evident and suggests that PCBP2 regulatory functions may be co-opted by the virus. Informed by this in vivo biophysical data, targeted functional studies can be designed to enrich mechanistic understanding.
Collectively, FAST-iCLIP can be used to comprehensively survey the repertoire of protein-coding and noncoding human transcript classes, revealing novel hypotheses about function. The extensibility of FAST-iCLIP to novel genomes is well positioned for future study on pathogen and microbiome interactomes. Additionally, by utilizing the established framework within FAST-iCLIP, novel analytic modules can be easily built and implemented to analyze even more features of iCLIP data. Finally, the standardized data output format eases comparative analysis. Continued efforts to standardize analyses and share workflows, along with novel technologies for high-throughput RNA–protein biochemistry to validate hypothesis on the same scale as their generation (Martin et al. 2012), will hasten the adoption of CLIP and shed greater light on vastly unexplored human and nonhuman protein–RNA interactomes.
MATERIALS AND METHODS
Cell culture
Huh-7 cells were maintained in DMEM and supplemented with 10% FBS, 1% nonessential amino acids and 200 μM L-glutamine as described previously (Machlin et al. 2011). Huh-7 cells were infected at 37°C with the JFH-1 isolate of HCV (Wakita et al. 2005) at a multiplicity of infection (MOI) of 0.01. Five hours after infection, cells were trypsinized, and replated in duplicate tissue culture plates. Infected cells were cultured for 3 d post-infection before harvesting. Uninfected and JFH-1-infected Huh-7 cells were UV-C crosslinked to a total of 0.3 J/cm2.
FAST-iCLIP
The FAST-iCLIP method is based on the published iCLIP protocol with the following modifications. Crosslinked cells were scraped and pelleted and whole cell lysates were generated in CLIP Lysis Buffer (50 mM HEPES, 200 mM NaCl, 1 mM EDTA, 10% Glycerol, 0.1% NP-40, 0.2% TritonX-100, 0.5% N-lauroylsarcosine). Typically, one 15-cm plate of crosslinked cells were lysed in 1 mL of CLIP Lysis Buffer and briefly sonicated using a probe-tip Branson sonicator to solubilize chromatin. Lysates were then clarified by centrifugation and the supernatants were subjected to partial RNaseA (Affymetrix) digestion (ranging from 0.1 to 2.0 ng of RNaseA per 1 mL of lysate) for 10 min at 37°C and quenched on ice. Dynabeads Protein-A (Life Technology) were conjugated overnight at 4°C to antiPCBP2 antibody (Sigma, HPA038356). Typically, 40 μL of Protein-A Dynabeads (Life Technologies) and 4 μg of antiPCBP2 antibody were used and added 1 mL of digested lysate for 4 h at 4°C for imunoprecipitation. Samples were washed sequentially in 1 mL for 5 min each at 4°C: 1× high stringency buffer (15 mM Tris–HCl, pH 7.5, 5 mM EDTA, 2.5 mM EGTA, 1% TritonX-100, 1% Na-deoxycholate, 120 mM NaCl, 25 mM KCl), 1× high salt buffer (15 mM Tris–HCl pH 7.5, 5 mM EDTA, 2.5 mM EGTA, 1% TritonX-100, 1% Na-deoxycholate, 1 M NaCl), 1× NT2 buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1 mM MgCl2, 0.05% NP-40). Samples were processed as previously described for 3′-end RNA dephosphorylation, 3′ end ssRNA ligation, 5′ labeling, SDS-PAGE separation and transfer, autoradiograph, RNP isolation, ProteinaseK treatment, and overnight RNA precipitation took place as previously described (Huppertz et al. 2014). The 3′-ssRNA ligation adaptor was modified to contain a 3′-biotin moiety as a blocking agent.
FAST-iCLIP library construction
FAST-iCLIP uses the standard iCLIP procedures isolate RBP protected RNA fragments for deep sequencing, however a 3′-end biotin blocked adaptor is used in place of the traditionally 3′-end ddC blocked adaptor (L3_Biotin: /5rApp/AGATCGGAAGAGCGGTTCAG/3Biotin/; 5rApp = 5′ pre-adenylation. After overnight precipitation of the ProteinaseK treated CLIP'ed RNA at −20°C the samples were pelleted at 15,000 rpm at 4°C for 1 h. RNA pellets were washed once in ice-cold 80% ethanol and air-dried. The RNA was resuspended in 6.25 μL of water, 1 μL of 1 μM RT primer (iCLIP_ddRT_BC1: /5phos/DDDNNAACCNNNNAGATCGGAAGAGCGTCGTGAT/iSp18/GG ATCC/iSp18/TACTGAACCGC; /5phos/ = 5′ phosphorylation, /iSp18/ = Carbon-18 spacer), and 0.5 μL of 10 mM dNTPs. Samples were heated to 70°C for 5 min and cooled to 25°C. To each sample 0.25 μL of SuperScriptIII, 2 μL of 5× first Strand Synthesis Buffer and 0.5 μL of 100 mM DTT was added and RT proceeded under the following program: 5′ at 25°C, 20′ at 42°C, 40 min at 52°C, 4°C forever. Note, after this point do not elevate the temperature past 37°C until after cDNA circularization. Next, 1 μL of RNase Cocktail (Life Technologies) and 1 μL of RNaseH (Enzymatics) was added to each sample and incubated at 37°C for 15 min. During this time 5 μL, per sample, of Dynabeads MyOne Streptavidin C1 (C1) beads were washed twice 200 μL of StrepBead Wash Buffer (100 mM Tris–HCl, pH 7, 1 M NaCl, 10 mM EDTA, 0.1% Tween-20). Each 5 μL volume of beads were finally resuspended in 40 μL of Strep Bead Wash Buffer. After RNase digestion 40 μL of prewashed C1 beads were added to each sample and incubated at 25°C for 30-min rotation. After incubating the samples were applied to a 96-well magnet for 2 min and the buffer was discarded. Each sample was washed with 4 × 100 μL with MyOne Wash Buffer (100 mM Tris–HCl pH 7, 4 M NaCl, 10 mM EDTA, 0.2% Tween-20) and 2 × 100 μL in NT2 Buffer. The purified cDNA was then circularized by added 20 μL of CircLigase Reaction Mix (2 μL 10× CircLigaseII 10× Reaction Buffer, 1 μL 50 mM MnCl2, 1 μL CircLigaseII ssDNA Ligase (100 U/μL), 16 μL ddH2O) to each sample and incubated for 1 h at 60°C, 10 min at 80°C, and 4°C forever. Samples were then applied to the 96-well magnet, the reaction buffer removed and saved, and 20 μL of Elution Buffer (10 mM Tris pH 7.5 and 5 μM P3 Solexa Short oligo) added and heated to 95°C for 2 min. Samples were placed on the magnet for 30 sec, elution buffer removed and added to the initial 20 μL and the elution was repeated twice for a final volume of 60 μL. MiniElute columns were used with Buffer PNI (Qiagen) as per the manufacturer's instructions to clean up small DNA samples and the circularized cDNA was eluted twice in 12 μL of Elution Buffer (Qiagen) (final of 22 μL). Short-arm qPCR was then performed on the entire sample by added 28.5 μL of Phusion PCR Mix (25 μL 2× Phusion HF Master Mix (NEB), 2.5 μL of 10 μM P3/P5 Solexa Short (P3 Solexa Short: 5′-CTGAACCGCTCTTCCGATCT-3′; P5 Solexa Short: 5′-ACACGACGCTCTTCCGATCT-3′), 1 μL of 15× SybrGreenI (Life Technologies) to each sample. Samples were amplified using the iCLIP PCR program (98°C, 30 sec; repeat as needed [98°C 15 sec, 61°C 30 sec, 72°C 45 sec]; image for Sybr at the end of the 45-sec extension) and individual PCR reactions were stopped at the end of the extension step once reaching a fluorescence value of 8000 units on a Mx3000 qPCR System (Agilent). PCR reactions were cleaned up as before using MiniElute columns and the resulting DNA was size-selected on home-made 6% 7 M Urea PAGE gels. Amplified DNA was visualized with SybrGold (Life Technologies) and iCLIP inserts with sizes between 30 and 70 nt were isolated. The PAGE gel was crushed and the DNA was eluted in 400 μL of Crush-Soak Buffer (500 mM NaCl, 1 mM EDTA) at 50°C on rotation overnight. The eluted DNA was purified over a SpinX column (Corning), precipitated for 1 h at −80°C and the pelleted at 4°C for 1 h at 15,000 rpm. The samples were then reamplified using long arm PCR (P3 Solexa: 5′-CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT-3′; P5 Solexa: 5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT-3′) primers with the same PCR program without SybrGreenI and for only three cycles each. The final PCR was purified with AMPure XP beads (Beckman) as per the manufacture's conditions and eluted in 20 μL of water. One microliter of each sample was used for quantification on the BioAnalyzer High Sensitivity DNA chip and then sent for deep sequencing on the Illumina HiSeq 2500 machine for 1 × 75-bp cycle run.
Publically available data
Human hnRNP-C iCLIP data were downloaded from the iCount server (http://icount.biolab.si/) and processed with the FAST-iCLIP pipeline. The link of all code is https://github.com/ChangLab/FASTCLIP
ACKNOWLEDGMENTS
We thank A. Mele and R. Darnell for advice with library preparation and G. Pratt, M. Lovci, and G. Yeo for support implementing the CLIPper peak-calling algorithm. This work is supported by grants from National Defense Science & Engineering Graduate Fellowship (L.M.), National Institutes of Health (NIH) Training Grant PHS NRSA 5T32-CA09302 (R.A.F.), NIH R01-AI069000 (P.S.), The International Life Sciences Institute Research Foundation (S.M.S.), Operating grant from the Canadian Institutes of Health Research (S.M.S.), and NIH R01-HG004361 (H.Y.C.). H.Y.C. and S.R.Q. are funded by the Howard Hughes Medical Institute.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.047803.114.
Freely available online through the RNA Open Access option.
REFERENCES
- Choi HS, Hwang CK, Song KY, Law PY, Wei LN, Loh HH 2009. Poly(C)-binding proteins as transcriptional regulators of gene expression. Biochem Biophys Res Commun 380: 431–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran DL, Georgiev S, Mukherjee N, Gottwein E, Skalsky RL, Keene JD, Ohler U 2011. PARalyzer: definition of RNA binding sites from PAR-CLIP short-read sequence data. Genome Biol 12: R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z, Lee JK, Tjhen R, Li S, Pan H, Stroud RM, James TL 2005. Crystal structure of the first KH domain of human poly(C)-binding protein-2 in complex with a C-rich strand of human telomeric DNA at 1.7 Å. J Biol Chem 280: 38823–38830. [DOI] [PubMed] [Google Scholar]
- Eiring AM, Harb JG, Neviani P, Garton C, Oaks JJ, Spizzo R, Liu S, Schwind S, Santhanam R, Hickey CJ, et al. 2010. miR-328 functions as an RNA decoy to modulate hnRNP E2 regulation of mRNA translation in leukemic blasts. Cell 140: 652–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser CS, Doudna JA 2006. Structural and mechanistic insights into hepatitis C viral translation initiation. Nat Rev Microbiol 5: 29–38. [DOI] [PubMed] [Google Scholar]
- Gamarnik AV, Andino R 2000. Interactions of viral protein 3CD and poly (rC) binding protein with the 5′ untranslated region of the poliovirus genome. J Virol 74: 2219–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Rinn JL 2012. Modular regulatory principles of large non-coding RNAs. Nature 482: 339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr, Jungkamp AC, Munschauer M, et al. 2010. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141: 129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W, Xin Z, Zhao Z, Bao W, Lin X, Yin B, Zhao J, Yuan J, Qiang B, Peng X 2013. RNA-binding protein PCBP2 modulates glioma growth by regulating FHL3. J Clin Invest 123: 2103–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold J, Andino R 2001. Poliovirus RNA replication requires genome circularization through a protein–protein bridge. Mol Cell 7: 581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA 2009. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppertz I, Attig J, D'Ambrogio A, Easton LE, Sibley CR, Sugimoto Y, Tajnik M, König J, Ule J 2014. iCLIP: protein–RNA interactions at nucleotide resolution. Methods 65: 274–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- König J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B, Turner DJ, Luscombe NM, Ule J 2010. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol 17: 909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- König J, Zarnack K, Luscombe NM, Ule J 2012. Protein–RNA interactions: new genomic technologies and perspectives. Nat Rev Genet 13: 77–83. [DOI] [PubMed] [Google Scholar]
- Li Y, Masaki T, Lemon SM 2013a. miR-122 and the Hepatitis C RNA genome: more than just stability. RNA Biol 10: 919–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Masaki T, Yamane D, McGivern DR, Lemon SM 2013b. Competing and noncompeting activities of miR-122 and the 5′ exonuclease Xrn1 in regulation of hepatitis C virus replication. Proc Natl Acad Sci 110: 1881–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovci MT, Ghanem D, Marr H, Arnold J, Gee S, Parra M, Liang TY, Stark TJ, Gehman LT, Hoon S, et al. 2013. Rbfox proteins regulate alternative mRNA splicing through evolutionarily conserved RNA bridges. Nat Struct Mol Biol 20: 1434–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunde BM, Moore C, Varani G 2007. RNA-binding proteins: modular design for efficient function. Nat Rev Mol Cell Biol 8: 479–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo G 1999. Cellular proteins bind to the poly (U) tract of the 3′ untranslated region of hepatitis C virus RNA genome. Virology 256: 105–118. [DOI] [PubMed] [Google Scholar]
- Machlin ES, Sarnow P, Sagan SM 2011. Masking the 5′ terminal nucleotides of the hepatitis C virus genome by an unconventional microRNA-target RNA complex. Proc Natl Acad Sci 108: 3193–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin L, Meier M, Lyons SM, Sit RV, Marzluff WF, Quake SR, Chang HY 2012. Systematic reconstruction of RNA functional motifs with high-throughput microfluidics. Nature Methods 9: 1192–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto M, Kato T, Date T, Mizokami M, Wakita T 2006. Comparison between subgenomic replicons of hepatitis C virus genotypes 2a (JFH-1) and 1b (Con1 NK5.1). Intervirology 49: 37–43. [DOI] [PubMed] [Google Scholar]
- Papenfort K, Vogel J 2010. Regulatory RNA in bacterial pathogens. Cell Host Microbe 8: 116–127. [DOI] [PubMed] [Google Scholar]
- Perreault J, Perreault JP, Boire G 2007. Ro-associated Y RNAs in metazoans: evolution and diversification. Mol Biol Evol 24: 1678–1689. [DOI] [PubMed] [Google Scholar]
- Rapicavoli NA, Qu K, Zhang J, Mikhail M, Laberge RM, Chang HY 2013. A mammalian pseudogene lncRNA at the interface of inflammation and anti-inflammatory therapeutics. Elife 2: e00762–e00762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn JL, Chang HY 2012. Genome regulation by long noncoding RNAs. Annu Rev Biochem 81: 145–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn JL, Ule J 2014. ‘Oming in on RNA–protein interactions. Genome Biol 15: 401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty S, Stefanovic S, Mihailescu MR 2013. Hepatitis C virus RNA: molecular switches mediated by long-range RNA–RNA interactions? Nucleic Acids Res 41: 2526–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Friebe P, Tzima E, Junemann C, Bartenschlager R, Niepmann M 2006. The hepatitis C virus RNA 3′-untranslated region strongly enhances translation directed by the internal ribosome entry site. J Virol 80: 11579–11588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmann E, Pietschmann T 2013. Cell culture systems for hepatitis C virus. Curr Top Microbiol Immunol 369: 17–48. [DOI] [PubMed] [Google Scholar]
- Tellinghuisen TL, Evans MJ, von Hahn T, You S, Rice CM 2007. Studying hepatitis C virus: making the best of a bad virus. J Virol 81: 8853–8867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanharanta S, Marney CB, Shu W, Valiente M, Zou Y, Mele A, Darnell RB, Massague J 2014. Loss of the multifunctional RNA-binding protein RBM47 as a source of selectable metastatic traits in breast cancer. Elife 3: e02734–e02734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhagen AP, Pruijn GJ 2011. Are the Ro RNP-associated Y RNAs concealing microRNAs? Y RNA-derived miRNAs may be involved in autoimmunity. Bioessays 33: 674–682. [DOI] [PubMed] [Google Scholar]
- Waggoner SA, Liebhaber SA 2003. Identification of mRNAs associated with αCP2-containing RNP complexes. Mol Cell Biol 23: 7055–7067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Kräusslich HG, Mizokami M, et al. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11: 791–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter BL, Parsley TB, Ehrenfeld E, Semler BL 2002. Distinct poly(rC) binding protein KH domain determinants for poliovirus translation initiation and viral RNA replication. J Virol 76: 12008–12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Jeng KS, Lai MM 2011. Poly(C)-binding protein 2 interacts with sequences required for viral replication in the hepatitis C virus (HCV) 5′ untranslated region and directs HCV RNA replication through circularizing the viral genome. J Virol 85: 7954–7964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss IM, Liebhaber SA 1995. Erythroid cell-specific mRNA stability elements in the α2-globin 3′nontranslated region. Mol Cell Biol 15: 2457–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyn-Vanhentenryck SM, Mele A, Yan Q, Sun S, Farny N, Zhang Z, Xue C, Herre M, Silver PA, Zhang MQ, et al. 2014. HITS-CLIP and integrative modeling define the Rbfox splicing-regulatory network linked to brain development and autism. Cell Rep 6: 1139–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarnack K, König J, Tajnik M, Martincorena I, Eustermann S, Stévant I, Reyes A, Anders S, Luscombe NM, Ule J 2013. Direct competition between hnRNP C and U2AF65 protects the transcriptome from the exonization of Alu elements. Cell 152: 453–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Darnell RB 2011. Mapping in vivo protein–RNA interactions at single-nucleotide resolution from HITS-CLIP data. Nat Biotechnol 29: 607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]