

Abstract
Major applications of RNA-seq data include studies of how the transcriptome is modulated at the levels of gene expression and RNA processing, and how these events are related to cellular identity, environmental condition, and/or disease status. While many excellent tools have been developed to analyze RNA-seq data, these generally have limited efficacy for annotating 3′ UTRs. Existing assembly strategies often fragment long 3′ UTRs, and importantly, none of the algorithms in popular use can apportion data into tandem 3′ UTR isoforms, which are frequently generated by alternative cleavage and polyadenylation (APA). Consequently, it is often not possible to identify patterns of differential APA using existing assembly tools. To address these limitations, we present a new method for transcript assembly, Isoform Structural Change Model (IsoSCM) that incorporates change-point analysis to improve the 3′ UTR annotation process. Through evaluation on simulated and genuine data sets, we demonstrate that IsoSCM annotates 3′ termini with higher sensitivity and specificity than can be achieved with existing methods. We highlight the utility of IsoSCM by demonstrating its ability to recover known patterns of tissue-regulated APA. IsoSCM will facilitate future efforts for 3′ UTR annotation and genome-wide studies of the breadth, regulation, and roles of APA leveraging RNA-seq data. The IsoSCM software and source code are available from our website https://github.com/shenkers/isoscm.
Keywords: 3′ UTR, APA, RNA-seq, IsoSCM
INTRODUCTION
The recent astonishing advances in RNA-sequencing (RNA-seq) technologies have spurred the development of numerous methods to exploit these data for diverse applications, such as inferring transcript structures, differential gene expression, and alternative RNA processing. These analyses require models of transcript structure that serve as a foundation for transcriptome analysis, and to quantify differences in read mapping between conditions (Martin and Wang 2011). While transcript structures were historically inferred from full-length cDNA sequences (Adams et al. 1995), the wealth of data from RNA-seq experiments has greatly expanded transcriptome annotation pipelines more recently (Djebali et al. 2012; Brown et al. 2014).
Strategies for transcript assembly can be categorized based on their dependence on a reference genome sequence. Genome-independent (de novo) approaches typically construct a De Brujin graph representing reads sharing compatible subsequences, then use heuristics to decompose this graph to recover full-length transcript sequences (Martin and Wang 2011). Genome-dependent approaches decompose the transcript assembly problem into smaller subproblems by first mapping the reads to the reference sequence, and then constructing gene models that are consistent with the aligned reads (Denoeud et al. 2008; Mortazavi et al. 2008; Yassour et al. 2009; Guttman et al. 2010; Trapnell et al. 2010). Since these strategies operate in the absence of any reference gene models, they are termed ab initio transcript assembly methods.
In addition to transcript assembly, a separate class of methods attempts to quantify the relative abundance of transcripts and isoforms using RNA-seq data (Feng et al. 2011; Li et al. 2011a,b; Behr et al. 2013; Hiller and Wong 2013). Since alternatively processed gene products are frequently encoded in overlapping genomic locales, and RNA-seq reads typically cannot be unambiguously assigned to overlapping transcripts, these methods attempt to construct parsimonious models for transcript abundance that are consistent with observed read counts. Importantly, these approaches cannot be applied if a set of reference models is not available, and may provide inaccurate quantification if these models are not complete.
While the problem of full-length transcript assembly from short reads is in general difficult and underdetermined (Behr et al. 2013), current methods have particular difficulties in annotating 3′ terminal exons correctly. In contrast to the boundaries of internal exons, which can be identified precisely by virtue of reads that span splice junctions, terminal exon boundaries only evince themselves in RNA-seq data as the position at which read coverage decreases. To identify terminal boundaries the popular transcriptome assembly tool Cufflinks constructs a minimum path cover from compatible RNA-seq reads, resulting in a single terminal exon annotation that extends from the last splice acceptor site to the position where there is zero read coverage (Trapnell et al. 2010). Since low-frequency polyadenylation site read-through events are captured in RNA-seq experiments, this strategy will often result in the terminal 3′ exon boundary being extended beyond the dominantly used polyadenylation site. Cufflinks implements a post hoc trimming process to mitigate this problem. Another well-utilized transcriptome assembly tool, Scripture, identifies transcribed segments of the genome using scan statistics. Here, the terminal exon boundary is determined as the location where the statistic calculated within a window overlapping the terminal exon drops below the genome-wide significance threshold (Guttman et al. 2010).
Neither of these strategies is specifically designed to identify the set of positions at which the level of RNA-seq coverage transitions from high-to-low coverage, and neither is capable of generating more than one terminal exon annotation. Consequently, the terminal exon annotations built from RNA-seq data can be inaccurate and incomplete. This point was emphasized during a recent comparative assessment of 14 different algorithms for transcript assembly and exon identification, conducted as part of the RNA-seq Genome Annotation Assessment Project (RGASP) (Steijger et al. 2013). In particular, the outputs for transcript termini from all of the algorithms tested were sufficiently inaccurate that a relaxed criteria exon correctness was used that evaluated only on the 5′ boundary of the 3′ terminal exon (Steijger et al. 2013). This highlighted the need for improved methods to identify transcript termini from RNA-seq data.
The 3′ untranslated region (3′ UTR) is an important location of post-transcriptional regulation, and deregulation of 3′ end formation has medical relevance (Elkon et al. 2013). In recent years, there has been increasing appreciation that most genes are subject to alternative cleavage and polyadenylation (APA) to yield multiple 3′ UTR isoforms, and that APA is frequently modulated in tissue-specific, state-specific, or environmentally responsive manner (Miura et al. 2014). Although various specialized techniques have been developed to sequence the 3′ ends of transcripts (Elkon et al. 2013), conventional RNA-seq methods remain the methodology of choice for most laboratories, and the amount of existing RNA-seq data is far greater than for 3′-seq data. Thus, it would be highly desirable to improve the accuracy with which we can infer 3′ UTR boundaries and alternative isoform expression from RNA-seq data.
Recently, we used tissue-specific RNA-seq data to refine terminal exon models in the human and mouse genomes (Miura et al. 2013). We encountered unique challenges when refining terminal exons, which motivated the implementation of a specialized annotation process for 3′ terminal exons. For example, while transposable element insertions are strongly selected against in the coding exons of a gene, UTRs are less constrained and harbor thousands of repetitive elements genome wide (Chen et al. 2009). Nonuniform read coverage arising from sequence specific (Hansen et al. 2010; Lahens et al. 2014) as well as positional biases (Bohnert and Rätsch 2010), and uncertain allocation of multimapping reads (Mortazavi et al. 2008) can cause artificial local gaps in RNA-seq coverage, making it difficult to annotate full-length 3′ UTR models using next-generation sequencing data alone (Fig. 1A). We previously developed an ad hoc procedure to bridge short gaps in RNA-seq coverage when known sources of sequencing bias could be identified. This enabled us to extend thousands of 3′ UTR models in the extensively annotated mouse and human genomes. A substantial proportion of these extensions show tissue-specific expression patterns, and identify a previously unappreciated potential for post-transcriptional regulation in the extended genes (Miura et al. 2013).
FIGURE 1.

Challenges and proposed solutions for 3′ UTR annotation from RNA-seq data. (A) Genome browser view of Kif3a illustrates how regions of low-coverage RNA-seq can result in fragmented 3′ UTR assemblies reported by Cufflinks and Scripture. There is an annotated repetitive element coinciding with the position of the gap, which could explain the observed decrease in coverage at this location, due to ambiguous read mappability. (B) Hdlbp illustrates a gene with tandem polyadenylation sites that is not completely annotated using either Cufflinks or Scripture. The Ensembl 73 annotation contains transcript models with alternative short/long 3′ UTR isoforms, and the relative abundance of these isoforms is reflected by the pattern of RNA-seq depth. However, neither Cufflinks nor Scripture is able to assemble the short isoform; both exclusively report the long isoform. (C) The maximum marginal likelihood segmentation is computed using a dynamic programming algorithm that recursively computes Q(t), the likelihood of the optimal segmentation of the subsequence of observations yt:n, given that there is a change point at position t − 1. This graphic illustrates how the value of Q(t) at each position in the sequence is decomposed into a component representing the likelihood of sequences of the data starting at position t (red) and the maximum likelihood segmentation of the remainder of the data (blue), and a component representing the likelihood that there are no change points in the sequence yt:n (green). RNA-seq data are shown for the Ict1 gene, which utilizes tandem alternative 3′ ends, both of which are supported by 3′-seq data. (D) Toy data are used to illustrate the effect of constraints on the segmentation process. Above, the scatter plot displays simulated coverage data, with a local region of low coverage indicated by the bracket at top. Below, vertical lines indicate location of change points identified using the standard and constrained formulations of the change-point inference algorithm, where red lines indicate locations of decreased coverage and green lines indicate the locations of increased coverage. The unconstrained solution identifies both the local dip and the most distal change point, while the constrained solution reports the most likely configuration of change points that conforms to the requirement for monotonically decreasing coverage in sequential segments.
We formalize this procedure here, extending our previous work for terminal exon annotation, using a segmentation approach that integrates long-range patterns of RNA-seq coverage to identify polyadenylation sites with greater sensitivity and specificity than existing methods. More importantly, we demonstrate its utility for identifying complex patterns of tandem polyadenylation site usage that are inaccessible with conventional annotation strategies. We implement our approach as the stand-alone program Isoform Structural Change Model (IsoSCM), which is available from our website (https://github.com/shenkers/isoscm).
RESULTS
Transitions in coverage depth identify 3′ UTR boundaries
RNA-seq protocols sample reads from across transcript bodies, approximately uniformly, although with certain biases (Mortazavi et al. 2008). Existing approaches use minimum path coverage (Trapnell et al. 2010), or a scan statistic (Guttman et al. 2010), to identify transcribed segments, and annotate at most one 3′ boundary for each terminal exon, typically the longest isoform compatible with the reads. Since the longest isoform will not in general reflect the dominant 3′ UTR isoform used by a gene, Cufflinks uses a heuristic post-assembly processing step to trim terminal exon annotations to a prespecified fraction of the average level of coverage. While such a strategy will identify high abundance short isoforms at a subset of loci, a single trimming parameter will not result in optimal annotations genome wide. Moreover, these strategies tend to generate incomplete 3′ UTR assemblies because they cannot capture tandem terminal exon isoforms that are coexpressed in a given sample, as illustrated in Figure 1B.
Given the unique challenges associated with transcript assembly within 3′ UTRs, and to address the limitations of existing tools, we developed a more expressive framework for transcript assembly that incorporates information from the patterns of read coverage into the process of UTR boundary definition. If we assume that sequenced reads are distributed approximately uniformly across the transcript, the boundaries of transcription will be marked by a change in the level of coverage. In instances where a shorter exon is nested within a longer exon, there can still be a significant number of reads aligning downstream from the shorter isoform, creating a “step-like” pattern of coverage at the boundary of the nested exon model. For example, RNA-seq data for the Hdlbp and Ict1 genes show such drop-offs within their 3′ UTRs, indicative of tandem APA events (Fig. 1B,C).
To identify terminal exon boundaries, we thus seek critical points (“change points”) that mark transitions in RNA-seq coverage. Previously, segmentation approaches were used to identify transcript boundaries from tiling microarray probe intensities (Huber et al. 2006), and while these change points have been described in RNA-seq data (Nagalakshmi et al. 2008), no existing RNA-seq ab initio transcript assembly tool fully leverages this information to annotate 3′ UTR boundaries. To fill this gap, we adapt multiple change-point inference to the problem of 3′ UTR isoform identification.
Inference for multiple change-point problems
To implement change-point inference, we made use of a Bayesian framework for change-point inference established previously (Fearnhead 2006). For a sequence of n observations y1:n = y1,…,yn representing the level of coverage at sequential genomic positions, we consider all possible combinations of m change points τ1,…,τm where 0 < τi < n and τi < τi+1, and 0 ≤ m < n, such that the jth segment pertains to the observed level of coverage between two successive change points. We assume that the level of coverage observed at each position within a segment are independent samples from a common probability distribution f(x|θ), parameterized by θ, with prior distribution π(θ). To model the expected length of a segment, we define a probability mass function g(t) for the length t of the genomic segment between two successive change points, with a cumulative mass function . Given this probability model, the goal of change-point inference is to identify the set of change points that maximizes the marginal likelihood of the data, i.e., the set of change points that “best explain” the observed pattern of read coverage.
To achieve this, we implemented a dynamic programming algorithm that recursively calculates the maximum marginal likelihood solution for nested subsequences of the observations. To begin, for all indices in a subsequences yt:s, such that 0 ≤ t ≤ s ≤ n we calculate a marginal likelihood P(t,s) that the observations yt:s were sampled from a common distribution as the integral of the joint data likelihood over the possible parameter values within that segment:
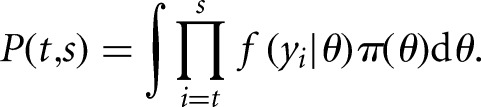 |
(1) |
The most likely segmentation using this model is defined recursively in terms of the likelihood of the current segment (Fig. 1C, red bracket), and the likelihood of the remainder of the data (Fig. 1C, blue bracket), and alternately, the likelihood of the remainder of the data coming from a single segment (Fig. 1C, green bracket). This requires the construction of two tables Q(t) and R(t) of length n, indexed by t, the location of the start of the current segment. Q(t) stores the maximum marginal likelihood of a segmentation of the subsequence of observations from yt:n, given a change point at t − 1, while R(t) stores the index of the next change point in the maximum marginal likelihood segmentation, given a change point at t − 1. These tables can be recursively computed in O(n2) operations using the formulas:
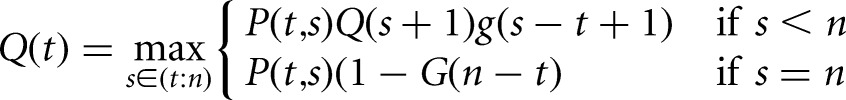 |
(2) |
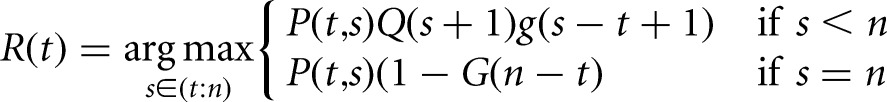 |
(3) |
The sequence of change points can be recovered by performing a trace-back through table R(t); the location of the first change point τ1 is given by R(1), and subsequent change points are given by τj + 1 = R(τj), while τj < n. A more detailed derivation of these equations along with proofs is given previously (Fearnhead 2006).
Constraining the location of change points
Although the change point model is able to tolerate a degree of variation within each segment, real RNA-seq data contain sequencing biases that can cause the sampling of reads across the transcript body to deviate from a uniform distribution. These biases typically cause short segments of the transcript to be sequenced at a lower frequency, resulting in a local drop in the level of coverage. The conventional (unconstrained) implementation of the change-point detection procedure identifies these local in-homogeneities in coverage as a segment with a distinct coverage distribution. However, in the context of annotating terminal exon boundaries we wish to disregard these local aberrations as they do not correspond to terminal exon boundaries. In order to minimize the effect of local biases on transcript model inference we introduce additional constraints to the change-point identification procedure.
To distinguish local changes from change points that mark a sustained decrease in the level of coverage, we restrict the set of identified change points to conform to a pattern of monotonically decreasing coverage over sequential segments. Solutions satisfying this requirement are achieved by discarding any configurations of change points in which the fold change in the level of coverage between two neighboring segments is less than a specified fold-ϕ.
This constraint is implemented by modifying the recursions in Equations 2 and 3. To do so, we introduce the functions M(t,s) to represent a point estimator of the level of coverage for observations yt:s, and C(s) to represent the maximum estimated coverage of the most likely segmentation of observations ys:n, respectively. We enforce this constraint by assigning zero probability to all configurations in which the estimated coverage of a downstream segment exceeds the estimated coverage of an upstream segment. Formally, this requires replacing the unconstrained recursions (Equations 2 and 3), with constrained versions (Equations 4 and 5).
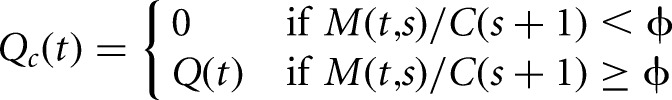 |
(4) |
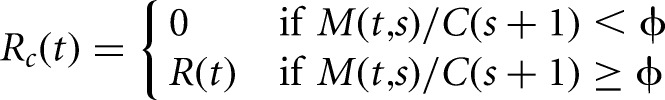 |
(5) |
Here, a parameterization ϕ = 1 corresponds to a requirement that the level of coverage of sequential segments are strictly decreasing, while ϕ = 2 the level of coverage of sequential segments drops at least twofold at each change point. The consequences of including these constraints are illustrated for a toy example in Figure 1D. In this scenario, a hypothetical 3′ UTR has relatively uniform coverage, except for a local region (Fig. 1D, bracket), where read coverage is underrepresented. When the unconstrained implementation of change-point detection procedure is applied to identify polyadenylation sites within the last exon, it detects two locations where the coverage drops, one upstream of the underrepresented segment, and the other at the polyadenylation site of the hypothetical exon. While this accurately reflects the observed pattern of coverage, for the purpose of polyadenylation site annotation, we are only interested in the location of the second change point.
Application of the constrained segmentation procedure has the desired effect; a change point is reported at the position of the polyadenylation site, while the change point caused by the local drop in coverage is omitted. While this limits the ability to identify arbitrary combinations of change points, reads sampled from tandem terminal exon isoforms are expected to produce a “step-like” coverage pattern. Thus, this constraint is intended to promote sensible inference of change-point location in situations where read distributions deviate from a theoretically uniform sampling across the transcript body.
Implementing change-point detection for 3′ UTR annotation
In order to implement the described change-point inference algorithm, one needs to specify the distributions f(yi|θ), π(θ), and g(t). While the depth of sequencing coverage is a discrete counting process and would typically be modeled using a Poisson distribution, it has been suggested that read counts from sequencing data are overdispersed, and are more appropriately modeled by a Negative Binomial distribution (Anders and Huber 2010). Therefore, we model f(yi|θ) ∼ NB(p,r), and use an uninformative conjugate prior π(p) ∼ β(1,1), which allows for analytical integration of Equation 1.
While the procedure described above efficiently identifies optimal sets of change points, these do not by themselves provide meaningful transcript models. For this reason the change points need to be interpreted in the context of the exon that they occur. For example, a transition from high-to-low coverage (over increasing genomic coordinates) would suggest the presence of a polyadenylation site on the plus strand, and vice versa for a gene on the minus strand. To enable application of change-point detection in an ab initio setting, prior to segmentation, covered genomic segments and spliced reads are used to identify regions that will be searched for change points. The output, including inferred overlapping terminal exons models, is reported in a compact splice graph. This graph identifies exon boundaries and connections between exons supported by spliced reads. The stages of the IsoSCM annotation algorithm are illustrated in Figure 2.
FIGURE 2.
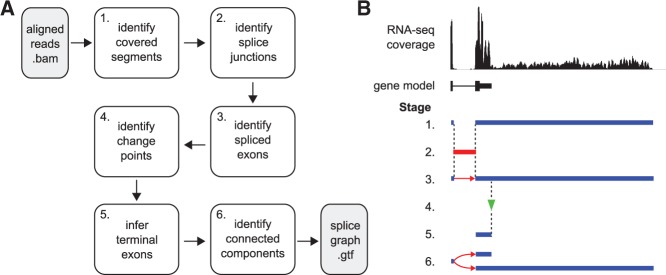
Implementation of the IsoSCM analysis pipeline. (A) The sequence of steps IsoSCM uses to build a splice graph are illustrated as a flow chart, starting from a set of reads that have been mapped to a genome using splice-aware alignment software. (B) The assembly operations from the flow chart at left are illustrated for a prototypical gene with tandem 3′ ends. These steps are (1) identification of segments of the genome where continuous read coverage is observed; if paired end reads information is available mate-pairs will be used to scaffold segments together. Locations with expected low coverage (i.e., repetitive elements or low-complexity sequence) can also be used to scaffold segments separated by gaps. (2) The location of splice junctions is recovered from the mapped reads; (3) the boundaries of spliced exons are inferred by intersecting continuously transcribed regions with the positions of splice junctions; (4) change points in the level of coverage are identified, using the constrained segmentation procedure; (5) terminal exon structure is inferred from change-point location; (6) the assembled splice-graph is reported, where exons are labeled as elements of a common transcription unit if they are connected either by spliced reads, or occupy overlapping genomic segments.
Method evaluation: simulated data
To assess the benefit of incorporating coverage-based segmentation for terminal exon annotation, we compared the performance of IsoSCM with two widely used ab initio reference based annotation tools: Cufflinks (Trapnell et al. 2010) and Scripture (Guttman et al. 2010). We prepared a test set using simulated data, where the transcript structures underlying the sequencing data are known a priori (illustrated in Supplemental Fig. 1A). This test set was comprised of 14,263 nonoverlapping genes, based on transcript models obtained from the mouse Ensembl 73 release. For each gene, we selected a single-transcript isoform, and generated a new isoform by randomly truncating the terminal exon at a random position that was at least 150 nt from both the 5′ and 3′ boundaries of the original exon.
As we expect the accuracy of assembled models to relate to sequencing depth, we measured the performance of each method over a range of coverage levels. For each pair of isoforms we simulated reads sampled uniformly across the body of the transcript, such that the aggregate density of reads over exonic segments was 2000 reads per kilo base (kb). This library was recursively subsampled such that evaluations spanned a range of read densities from 5 to 2000 reads/kb. For each simulated data set, we used IsoSCM, Cufflinks, and Scripture to assemble a set of predicted transcript models. For each assembly we assessed the correctness of 3′ UTR predictions by classifying assembled terminal exons as true positives (TP), true negatives (TN), false positives (FP), and false negatives (FN) with respect to the reference ground truth transcript set.
Since it is difficult to predict nested terminal exon boundaries with nucleotide-level accuracy from RNA-seq data, and there is value in an annotation that is “close” to the true transcript model, we use a classification scheme that allows a degree of flexibility. In contrast to the relatively generous assessment scheme used by the RGASP project (Steijger et al. 2013), which required only the spliced 5′ boundary of the terminal exon to match the reference and disregards the predicted 3′ end, we require both 5′ and 3′ boundaries to be similar to reference models. We classified predictions that are within a small (100 nt) distance of a reference model to be correct by dividing the interval spanning the terminal exon into segments that are positive or negative for polyadenylation, such that assemblies can be judged for their consistency with these segments (Supplemental Fig. 1C). The genomic segments within 100 nt of a reference 3′ ends were counted as TPs if an assembled 3′ end fell within that segment, and were otherwise counted as FN. Likewise, segments >100 nt from a reference end were counted as FP if an assembled 3′ end fell within that segment, and TN otherwise. Using these counts we calculated the PPV = TP/TP + FP, TPR = TP/TP + FN, FPR = FP/FP + TN, NPV = TN/TN+ FN, for each assembly.
These metrics are plotted as a function of sequencing depth in Figure 3A,B and Supplemental Figure 2. In these simulations, IsoSCM identifies the terminal exon boundaries with a PPV comparable to Cufflinks and Scripture at all sequencing depths, indicating that the change-point inference procedure does not inappropriately increase the number of false positives compared with methods that attempt to assemble only a single 3′ UTR isoform. In contrast, comparisons of the TPR achieved by each method highlight the fact that the models built by Cufflinks and Scripture were unable to annotate tandem terminal exons. Even as the number of available reads is increased toward infinity, the maximum TPR achieved by these methods was 0.5. A priori, we expect that Cufflinks and Scripture cannot exceed this level of sensitivity since the gene models they build are not expressive enough to represent tandem overlapping terminal exons, and one out of every two 3′ terminal exons in the test set is nested within a longer exon. In contrast, given sufficient data to statistically identify changes in the level of coverage, IsoSCM is able to achieve perfect TPR for the test set (Fig. 3A).
FIGURE 3.

Comparison of IsoSCM with existing transcript assembly methods. (A,B) Simulated RNA-seq data were used to assess predictive positive value (A) and true positive rate (B) of IsoSCM, Cufflinks, and Scripture outputs for 3′ end prediction. We generated a set of 14,263 nonoverlapping gene models that contain transcripts with nested 3′ terminal exons, and used these as a reference set of “true” transcripts. These metrics were calculated for simulated sequencing depths ranging from 5 to 2000 reads/kb. (C,D) We assessed the positional accuracy of IsoSCM outputs across a range of change-point magnitudes. We partitioned these events into quintiles (with n = 3005 termini in each group), corresponding to bins of <4x, 4–10x, 10–40x, 40–175x, and ≥175x drop-offs in read coverage. For each group the fraction of predicted termini with either canonical polyadenylation signals (PAS, AATAAA, or ATTAAA) or 3′-seq tags within 20 nt are shown at each position relative to the predicted boundary. Based on signals for appropriate positional enrichment, we utilized the top four cutoffs for running IsoSCM. (E) Proximity of IsoSCM, Cufflinks, and Scripture terminal outputs relative to Ensembl 3′ end annotations. The cumulative number of annotations at each distance to the closest Ensembl 3′ end is plotted. Based on apparent inflection points (dashed lines), we categorize annotations within 20 nt of Ensembl as precise annotations, and ones between 20 and 100 nt as imprecise matches to reference models. (F) Validation of IsoSCM, Cufflinks, and Scripture terminal outputs within ±20-nt windows of various types of supporting evidence. Ends were initially assigned, if possible, to Ensembl models, and then checked for proximity to PAS and/or 3′-seq tags. Of the remaining termini with “no evidence,” many would be validated using a relaxed 100-nt window. As the largest numbers of these correspond to “imprecise” calls of Ensembl ends (see E), we marked their numbers as sub-bars in the “no evidence” category. Regardless of the type or types of evidence considered, IsoSCM yields the largest numbers of validated termini without inflating numbers of unvalidated predictions.
We explored TPR, PPV, NPV, FPR measurements further, by subdividing the aggregate performance on the entire test set into component corresponding to proximal and distal isoforms exclusively (Supplemental Fig. 2). The methods performed comparably for the task of distal isoform identification, and the performance gain achieved by IsoSCM corresponds to an increased number of correct annotations of nested isoforms. To explore the relationship between sequencing depth and annotation accuracy we repeated the evaluation, requiring that predictions be within 10 nt of the reference transcript to be classified as TP. Under these conditions, we observed qualitatively the same pattern; only IsoSCM is able to reconstruct the nested isoforms correctly (Supplemental Fig. 3). However, while a sequencing depth of 200 reads/kb was sufficient to achieve a TPR = 0.9 and FPR = 0.1 with 100-nt resolution, this level of performance required over 1000 reads/kb at the 10-nt resolution. We note that all methods fail to reliably assemble low expressed transcripts, and that the advantages of IsoSCM are more fully realized when there are sufficient data to distinguish overlapping isoforms. Nevertheless, IsoSCM performs equally or better to Cufflinks and Scripture over a range of sequencing depths.
Method evaluation: real RNA-seq data
Of course, we do not expect experimentally generated RNA-seq data to be as well-behaved as simulated data. To verify that the benefits observed in our simulation experiment are achieved when IsoSCM is applied to real RNA-seq data, we utilized published RNA-seq data comprised of nine mouse tissues analyzed in triplicates (Merkin et al. 2012). In contrast to the simulation experiment, the ground truth set of transcripts expressed in each sample is not known a priori. As a first measure of model correctness, we compared the terminal boundaries of constructed transcript models with those in the Ensembl 73 reference annotation. However, as existing 3′ UTR reference annotations are incomplete (Miura et al. 2013), we expect that sole comparisons to the Ensembl reference to inflate FPR estimates. Therefore, we sought additional orthogonal sources of evidence to support predicted polyadenylation sites. Directed 3′-sequencing (3′-seq) protocols permit experimental mapping of polyadenylation sites, and we used an atlas of 3′-seq data from multiple mouse tissues (Derti et al. 2012). As well, cleavage and polyadenylation sites have long been known to be associated with a polyadenylation signal (PAS) hexamer (Proudfoot and Brownlee 1976), most frequently either AATAAA or ATTAAA, located ∼21 upstream of the cleavage site. While such motifs are not sufficient to specify 3′ end formation, we could assay PAS enrichment in the vicinity of novel 3′ end annotations as another measure of their quality.
We first sought to evaluate change-point cutoffs that delivered appropriate accuracy for genuine 3′ termini. We tested this using mouse brain (SRR594393), and partitioned predicted ends into quintiles by the magnitude of the change point. We then evaluated the positional enrichment of PAS and 3′-seq data supporting these groups, to examine the genomic precision with which IsoSCM called 3′ termini. We observed robust positional enrichments of PAS and 3′-seq tags in the appropriate locations relative to IsoSCM annotations (Figure 3C,D), demonstrating that these sources of evidence could be used to evaluate de novo predictions. As expected, annotated 3′ termini associated with the sharpest drops in coverage identified genuine polyadenylation sites with greatest accuracy, while positional support for PAS and 3′-seq tags was distributed more broadly for weaker change points. We focused on models for which the coverage drops at least fourfold in order to balance sensitivity and specificity of the predictions.
We then compared the performance of the different transcript assembly methods on the mouse brain RNA-seq reads, from which 11,000 3′ terminal exon models were constructed by IsoSCM, 9722 by Cufflinks, and 6282 by Scripture. Upon examining the distribution of the distance to the closest Ensembl 3′ end for each method (Fig. 3E) we observed that all three methods generate two populations of predictions distinguished by the precision with which they identify Ensembl 3′ ends. The inflection point at ∼20 nt identifies a population of predictions that capture annotated termini quite precisely, while a second inflection point at ∼100 nt identifies a set of predictions that are localized in a wider window around annotated 3′ ends (Fig. 3E, “imprecise”). For many types of genome-wide analysis, i.e., when assessing general trends of transcript isoform expression, nucleotide-precise annotations of 3′ termini are not necessary. Nevertheless, as the narrow 20-nt window enables the merits of predicted models to be evaluated stringently, we assessed how frequently each method produced 3′ termini that were validated by polyadenylation site features at a distance of ±20 nt (Fig. 3F).
Of the total set of predictions, IsoSCM made 4785 annotations that were ±20 nt of Ensembl 3′ ends, compared with 2450 by Cufflinks and 1827 by Scripture. In addition, each method annotated thousands of termini not supported by reference models. Among these, IsoSCM identified 2718 novel termini supported by 3′-seq tags, PAS, or both features, within 20 nt, compared with 1914 identified by Cufflinks and 941 identified by Scripture (Fig. 3F). These evaluations demonstrate the strong performance of IsoSCM on real RNA-seq data using a stringent cutoff. Finally, a substantial portion of the remaining termini annotated by each method (“no evidence”) could be considered validated if one utilized a broader window, e.g., 100 nt. Since the majority of these events can be defined as being “imprecise” captures of Ensembl ends, we have noted their numbers within the “no evidence” category of Fig. 3F, and using the relaxed 100-nt criteria in Supplemental Figure 4A. A full accounting of the termini annotated by the three algorithms using both 20- and 100-nt windows is given in Supplemental Table 1. Overall, we find that IsoSCM correctly annotates more 3′ UTRs while simultaneously reporting the fewest unvalidated annotations, regardless of the window size used for model evaluation.
Positional enrichments of functional features in predicted 3′ termini
To gain further insight into the resolution of the predictions made by each method, we investigated positional enrichments of PAS signals, 3′-seq reads, and conservation profiles around predicted 3′ termini. Considering all predictions of each method in aggregate, we see PAS signals enriched ∼21-nt upstream, 3′-seq tags at position 0, and a conservation profile characteristic of polyadenylation sites for all three methods (Fig. 4A–C). However, when we compare the relative frequency of evidence at these positions, we see that ends predicted by IsoSCM are supported 22.6%–23.1% more frequently by 3′-seq (Fig. 4A) and 19.9%–20.4% more frequently by a PAS (Fig. 4B) than either Cufflinks or Scripture. The 3′ ends annotated by IsoSCM also exhibited greater overall conservation, as assessed by PhastCons (Fig. 4C).
FIGURE 4.

Evaluating spatial accuracy of 3′ end predictions. Analysis of all 3′ ends reported by each method shows the frequency of 3′-seq evidence (A), canonical PAS (B), and genomic conservation (C) relative to predicted termini. The fraction of predicted 3′ end sites with the indicated type of support within 20 nt is shown at each position relative to the predicted boundary. The peak position of the PAS and 3′-seq tags are at their characteristic locations, about −21 and 0, respectively, while average PhastCons show characteristic peaked conservation at −21. All of these features are most robust for IsoSCM termini. (D–F) Positional enrichment of features examined for all method-specific ends, defined as termini that are >100 nt away from those generated by the other methods. Here, IsoSCM is the only method with substantial enrichment of 3′-seq evidence (D) and PAS (E) in the appropriate locations. The IsoSCM-specific annotations also exhibit the most robust conservation signature at these termini (F). (G–I) Positional enrichment of features examined for all method-specific ends that are “substantially novel,” defined as being located >250 nt from Ensembl termini and >100 nt away from other method predictions. Again, IsoSCM is the only method that displays the expected enrichment of polyadenylation site features for this subset of predicted 3′ ends.
Close inspection of these analyses reveal differential biases of the methods. In particular, consideration of the tail of “imprecise” PAS and 3′-seq predictions showed that they are biased to be upstream of annotations provided by Scripture, while they are more likely to be downstream from Cufflinks and IsoSCM predictions (Fig. 4A,B). This suggests that Scripture has some tendency to overextend transcript models, whereas IsoSCM and Cufflinks are slightly more likely to truncate 3′ UTRs. The overextension bias observed for Scripture is a known consequence of the scan statistic used by this method to define the boundaries of transcribed regions (Huber et al. 2006). We might expect a bias toward slight truncation using IsoSCM and Cufflinks if the regions directly upstream of the polyadenylation site are underrepresented in input reads, perhaps due to a cloning, sequencing, or mapping bias. Nevertheless, the robust peaks at positions −21 and 0, for PAS and 3′-seq evidence, respectively, indicate that IsoSCM accurately localizes polyadenylation sites in the majority of its predictions, and that its performance is substantially higher than Cufflinks or Scripture.
We next judged the qualities of the substantial populations of 3′ end annotations that were specific to each method. Of the 11,000 annotations of 3′ ends, IsoSCM identified 4115 termini that were >100 nt away from annotations made by either Cufflinks or Scripture, and these annotations exhibit enrichment for polyadenylation site features that is comparable to the aggregate set of predictions for the three methods (Fig. 4D–F). In contrast, analysis of 3′ end annotations specific to Cufflinks (n = 3717) or Scripture (n = 2962) outputs show little or no positional enrichment of 3′-seq tags (Fig. 4D) or PAS (Fig. 4E) at the appropriate locations. Therefore, IsoSCM identifies thousands of sites that in aggregate exhibit the expected features of 3′ ends, and are not reported by either Cufflinks or Scripture. Reciprocally, the ends that are uniquely provided by the other methods are not supported by similar evidence.
Finally, we were interested to assess the quality of substantially novel, method-specific termini. As mentioned, all methods report a population of ends that localize near Ensembl termini, but do not identify cleavage sites precisely (Fig. 3C). We found that positional assessments of bulk novel ends was compromised by inclusion of these “Ensembl-imprecise” termini, which create offset peaks of evidence at the boundary of the window utilized (Supplemental Fig. 4B–D). To provide clarity to these comparisons, we differentiated the population of “substantially novel” ends from imprecise annotations, by investigating predictions located >250 nt from Ensembl annotations. At this distance, IsoSCM identified 2472 termini, compared with 3513 by Cufflinks and 2367 by Scripture (Supplemental Fig. 4D). To highlight the differences between methods, we focused on “substantially novel” annotations that were specific to each method. This yielded sets of IsoSCM-specific (n = 1340), Cufflinks-specific (n = 2485), and Scripture-specific (n = 1969) ends that were >100 nt from each other. Analysis of these novel 3′ termini showed that only the IsoSCM-specific ends were enriched for 3′-seq tags (Fig. 4G) and PAS (Fig. 4H), whereas the other sets of method-specific ends were not distinguishable from background. As well, the IsoSCM-specific novel ends exhibited the highest level of genomic conservation (Fig. 4I).
Robust performance of IsoSCM across data sets
To assess if the performance differences we observed are representative, we used the framework for evaluating transcript assemblies from the simulation experiment to compare the performance of each method for 26 other mouse tissue RNA-seq data sets (Merkin et al. 2012). To provide a compact visualization of the evaluation, we used evidence of Ensembl terminal exons, 3′-seq, and PAS within ±20 nt to define sets of true and false predicted 3′ ends and plotted the differences in TPR and PPV obtained using IsoSCM and Cufflinks or Scripture in Figure 5A.
FIGURE 5.
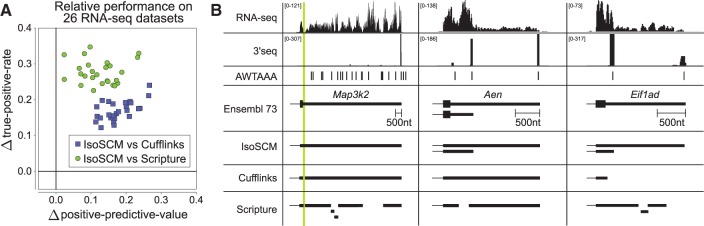
Relative performance of IsoSCM, Cufflinks, and Scripture on multiple data sets. (A) We used 26 RNA-seq data sets from nine mouse tissues to assess the ability of each method to correctly annotate 3′ terminal exons. For this analysis, we estimated the true positive rate and positive predictive value of each method by its ability to recapitulate Ensembl reference termini, or otherwise be supported by 3′-seq and/or PAS (as described in Fig. 3). The sensitivity and specificity of Cufflinks and Scripture was compared with IsoSCM by subtracting the value of each metric from the value obtained using IsoSCM, such that each point represents the relative performance of a pair of methods on a single sample. Each comparison involved evaluation of 9237–22,955 3′ exon annotations (median 18,737), depending on the number expressed in each sample. For every data set comparison, the TPR and PPV of IsoSCM annotations exceed those of Cufflinks or Scripture. (B) The genes Map3k2, Aen, and Eif1ad illustrate scenarios where Cufflinks and Scripture assemble gene models that are less optimal than those reported by IsoSCM. Map3k2 is a gene where a gap in RNA-seq coverage fragments the 3′ UTR of models reported by Cufflinks and Scripture, due to a repetitive element (green bar). IsoSCM is able to scaffold the complete 3′ UTR together. Aen and Eif1ad are examples of genes whose tandem polyadenylation sites are missed by Cufflinks and Scripture, but are captured by IsoSCM. IsoSCM correctly annotates the short and long 3′ UTR isoforms of both genes by identifying a change point in the level of RNA-seq coverage. In these examples, Cufflinks and Scripture annotate at most one isoform correctly, and sometimes neither. AWTAAA track indicates genomic matches to the two most common PAS, AATAAA and ATTAAA; note that many such instances are not actually functional PAS.
While there is sample-to-sample variability in these estimated performance metrics, IsoSCM consistently achieved a TPR that is at least 12.2% higher than the other methods, without compromising the PPV. Additional metrics of predictive performance for each method, including evaluations at both the 20-nt and 100-nt resolution, are provided in Supplemental Table 2. Concrete examples illustrating how IsoSCM reduces the number of incorrectly truncated 3′ UTRs and identifies additional 3′ UTR isoforms that are missed by Cufflinks and Scripture are given in Figure 5B.
Application of IsoSCM to identify tissue-differential tandem APA events
To illustrate a practical application, we next applied change-point inference to examine differential usage of 3′ UTR isoforms between different tissues. In the previous section, we demonstrate that the IsoSCM segmentation strategy is effective for annotating terminal transcript boundaries within an individual sample; however, the quantification of differential isoform usage requires an accounting of all transcripts expressed among the samples being compared. When generating a single annotation that is representative of two samples, one could attempt to merge annotations assembled independently from each sample. However, it is not obvious what the optimal strategy is for combining annotations from separate samples, while preserving alternative events. For example, the cuffmerge program, available as part of the Cufflinks suite, merges transcript models that share overlapping and compatible chains of introns, and during this process discards shorter terminal exon annotations, reporting only the longest 3′ exon. While such an approach could be used to identify the longest 3′ UTR isoform, it discards potential alternative events, and is thus inappropriate for assessing tissue-differential tandem APA events.
Conveniently, the change-point detection framework described above can be extended naturally to the assembly of tandem 3′ UTR boundaries from multiple samples simultaneously. To do this, we define Pjoint, the joint likelihood of k independent samples,
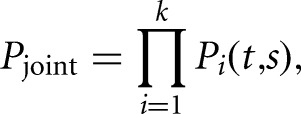 |
(6) |
where Pi is the marginal likelihood of the ith sample. Thus, by replacing P in Equations 2 and 3 with Pjoint, the segmentation procedure will identify the configuration of change points that maximize the joint marginal likelihood of all the samples simultaneously. Importantly, this modification enables identification of polyadenylation sites that are specific to one condition, in addition to sites that are common among the samples being compared.
Previously, we and others observed that the nervous system accumulates many transcripts with longer 3′ UTR isoforms than in other tissues, whereas the testis expresses many transcripts with relatively shorter 3′ UTR isoforms (Zhang et al. 2005; Jan et al. 2011; Derti et al. 2012; Smibert et al. 2012; Ulitsky et al. 2012; Miura et al. 2013). Using the IsoSCM framework, we reexamined these observations by analyzing differential polyadenylation site usage in nine mouse tissues. By performing the joint segmentation for each pair of tissues, we identified a set of change points representing tandem polyadenylation events. For each of these events, we estimated a polyadenylation-site-usage index in each condition, representing the relative frequency with which a particular polyadenylation site is used. By calculating the difference in this index between a pair of conditions, we identified polyadenylation events with differential usage patterns among tissues. St8sia3 and Sap30l typify transcripts IsoSCM identifies as being differentially polyadenylated between a pair of tissues, and are illustrated in Figure 6A. By identifying global differences in the distribution of the polyadenylation-site-usage statistic at proximal sites between tissue pairs, we assessed whether there were systematic patterns of altered 3′ UTR length. Indeed, we observed clear signatures in which the termini annotated by IsoSCM were broadly extended in brain relative to all other tissues, whereas they were generally shorter in testis relative to all other tissues (Fig. 6B).
FIGURE 6.

Analysis of tissue-differential APA using IsoSCM. (A) Instances of tissue-differential APA identified by IsoSCM are illustrated St8sia3 and Sap30l. IsoSCM models generated by the joint segmentation procedure are compared with models assembled by Cufflinks and Scripture analyzing each tissue independently. In these examples, Cufflinks is able to recover switches in tandem polyadenylation site usage for St8sia3 when comparing independent assemblies of the data sets, but does not detect the more nuanced difference with Sap30l as the short was not assembled. Scripture does not generate alternative models in either case. Since IsoSCM uses change-point detection for 3′ UTR boundary annotation, IsoSCM assembles both isoforms correctly, and is able to consistently capture these patterns. The AWTAAA track indicates genomic matches to the two most common PAS, AATAAA and ATTAAA; note that many such instances are not actually functional PAS. (B) Exon models built using IsoSCM were used to identify global patterns in differential 3′ UTR usage between nine mouse tissues. Events arising from overlapping tandem 3′ exons were used to estimate the relative usage of 3′ ends in each tissue. For all genes that are expressed in a pair of tissues, the difference in relative usage of a polyadenylation site is computed. To assess whether there are global trends toward 3′ UTR lengthening (or shortening) between tissues, the observed distribution of differences in 3′ end usage is compared with a null model that lengthening (or shortening) events are equally likely to occur in either sample. The 36 pairwise comparisons are represented in a matrix, with the tissues being compared labeled at the left of each row, and the bottom of each column. A cell shaded blue indicates that the tissue labeled at the left of the row tends to have lower usage of proximal 3′ ends than the tissue labeled at the bottom of the column, while red shading indicates the opposite pattern. Here, we see that the brain tends to use polyadenylation sites that are more distal, while the testis tends to use more proximal polyadenylation sites. (C) Neural APA event detected for Shank2; the alternative 3′ UTRs annotated by IsoSCM are supported by 3′-seq data. Probes were designed against a universal region present in both isoforms (green), and a region exclusive to the extended isoform (red). (D) Sequential Northern blotting shows that the universal probe hybridizes to two bands with estimated lengths of 8.2 and 9.7 kb, while the extension probe hybridizes only with the 9.7-kb band. These bands are consistent with the 3′ exon annotations assembled by IsoSCM, and likely correspond to the transcript ENSMUST00000105900 (8169-nt long) as well as an isoform bearing a 1.5-kb 3′ UTR extension.
To emphasize the importance of using accurate terminal exon annotations when quantifying tandem terminal exon expression patterns, we repeated the analysis using the transcript assemblies of Cufflinks and Scripture in place of the models generated by IsoSCM. As these methods do not provide a means to assemble transcript models considering two samples simultaneously, we used the union of terminal exon models from tissues being compared to define the set of tandem polyadenylation events identified by these methods. As shown in Figure 6B, there is only modest agreement between patterns identified using models built by Cufflinks and Scripture and IsoSCM. Enhanced usage of proximal polyadenylation sites in the testis was reliably recovered using Cufflinks. This is likely because the abrupt truncation events observed in testis can be captured by the Cufflinks assembly procedure, as is the case for St8sia3 (Fig. 6A). However, neither Cufflinks nor Scripture were able to assemble models that consistently captured the pattern of increased abundance of extended 3′ UTRs when comparing brain with other tissues (Fig. 6B).
Cufflinks and Scripture seek only to annotate the longest terminal isoform that is consistent with the reads, and in brain we observe many cases where both isoforms are expressed and only their relative usage changes between tissues. In such cases, it is expected that the short 3′ UTR isoform will not be captured by these methods. This is illustrated by the gene Sap30l in Figure 6A, for which both a short and long isoform are expressed in brain and liver, but there is a higher proportion of isoforms bearing the extended 3′ UTR in the brain. In this case, using Cufflinks or Scripture this alternative event is not identified, since neither method annotates the short isoform correctly.
We previously provided extensive molecular validation of neural 3′ UTR lengthening events of a similar quality to those identified by the IsoSCM pipeline, although those APA events were previously cataloged by a combination of visual annotation and partially automated procedures (Smibert et al. 2012; Miura et al. 2013). Along these lines, we tested a 3′ UTR extension for Shank2 identified by IsoSCM (Fig. 6C) by Northern blot. We designed double stranded DNA probes that either hybridize to the region common to both transcripts (“uni”), or that exclusively recognizes the extended 3′ UTR isoform (“ext”). Based on the predicted size of the full-length transcript differing only in the 3′ boundary of the terminal exon, we expect full-length transcripts of length 8.2 and 9.7 kb. Northern blotting showed that the common probe hybridize to two dominant bands that are consistent with these lengths, while extension-specific probe hybridizes exclusively to the longer RNA species (Fig. 6D), confirming our inference of tandem terminal exon boundaries from stepped patterns of RNA-seq coverage. Overall, these analyses are consistent with previously described patterns of tissue-specific APA, and suggest that the methodology encapsulated by IsoSCM can be readily applied in other settings to gain new insights into alternative 3′ UTR isoform regulation.
CONCLUSIONS
The inference of transcript structures from short RNA-seq reads is a complex problem, particularly for the identification of 3′ UTR boundaries. As we have illustrated, confounding factors such as the presence of repetitive sequences, overlapping coexpressed isoforms, and nonuniform coverage patterns make the identification of 3′ UTR boundaries from RNA-seq data a nontrivial problem. These issues are exacerbated by the absence of a framework to incorporate observed patterns of read depth into the transcript assembly process. To address these challenges and the limitations of existing tools, we developed IsoSCM. By benchmarking IsoSCM against state of the art methods for transcript assembly on simulated and experimentally generated RNA-seq data sets, we have demonstrated by several measures that the performance of IsoSCM is superior to existing tools for 3′ UTR reconstruction. Although evaluations indicate substantial benefits of change-point inference, we reiterate that the advantages of IsoSCM are manifest where sufficient levels of isoform expression exist, and that it does not correctly identify 3′ UTR isoforms if the level of coverage over exonic segments is not proportional to isoform abundance. These are indeed general challenges for ab initio transcript assemblers. Nevertheless, we show that IsoSCM consistently identifies thousands of functionally supported 3′ termini in excess of Cufflinks and Scripture, and reduces the number of false predictions reported.
The improvement in sensitivity and accuracy of reconstructed transcript models gained by using IsoSCM has implications beyond transcriptome annotation. A major application of RNA-seq technologies is for the inference of alternative isoform regulation, and these inferences require accurate gene models. By analyzing a panel of RNA-seq samples from different tissues, we demonstrate how previously observed patterns of APA are faithfully recapitulated by IsoSCM. Moreover, by extending the constrained change-point detection procedure we developed for single-sample annotation to the joint analysis of two samples, we obtain an effective method to detect and annotate differential APA in an ab initio setting. Notably, these patterns are not recovered using existing tools for transcript reconstruction, because these methods are by design not expressive enough to annotate tandem 3′ UTR isoforms.
While this work was in revision, Wagner and colleagues reported their algorithm DaPars and used it to identify genes with altered polyadenylation patterns following CFIm25 knockdown and in glioblastoma (Masamha et al. 2014). While their work also exploits change points in RNA-seq data, it provides only a subset of the functionalities of IsoSCM, making direct comparisons of the methods difficult. A significant advantage of IsoSCM is that it operates in an ab initio setting, and its accuracy does not depend on the quality and completeness of a reference annotation. Even for the well-annotated mouse transcriptome, dominant isoforms are missing (i.e., the extended isoform of Sap30l) (Fig. 6A), and could only be analyzed using ab initio methods such as IsoSCM. For many species, especially nonmodel organisms, 3′ UTR models are incomplete or absent, precluding the application of DaPars to these transcriptomes. The generality of IsoSCM will enable change-point analysis to be applied to APA in a wider scope of problems.
While IsoSCM utilizes only RNA-seq to identify 3′ ends, there also exist specialized protocols for high-throughput mapping of 3′ UTR boundaries by direct cloning and sequencing of 3′ ends. Clearly such methods provide greater and more direct information on 3′ ends. However, as these strategies are technically more complicated than conventional RNA-seq protocols, the available 3′-seq data are only a small fraction of the aggregate RNA-seq data that are available for diverse organisms. We present IsoSCM as a methodological advance for ab initio 3′ end identification that can take advantage of existing RNA-seq data and extend its analysis beyond current algorithms. The refinement of 3′ UTR boundaries provided by IsoSCM provides greater accuracy and more systematic accounting of 3′ end processing, and enables diverse studies of differential APA using the wealth of RNA-seq data now available.
MATERIALS AND METHODS
RNA-seq simulations
A graphic overview of the simulation framework is given in Supplemental Figure 1A. To generate a set of reference transcript models with tandem polyadenylation events we selected 14,263 nonoverlapping transcript models that contain a terminal exon at least 500-nt long from the Ensembl 73 annotation for RNA-seq simulations. Within the last exon of each transcript model, a truncated isoform was generated by uniformly sampling a position at least 150 nt from both 5′ and 3′ exon boundaries, and generating a new model sharing the complete upstream exon structure, while bearing a terminal exon truncated to this sampled position. Unstranded 50-nt single end reads were simulated by sampling alignment start positions uniformly across the body of the transcript until a target depth of 2000 reads/kb of exonic sequence (94,962,730 reads total) was reached. This library was recursively subsampled to generate a test set spanning the range of depths (5, 7.5, 10, 20, 30, 40, 50, 100, 200, 400, 600, 800, 1000, 2000) reads/kb (Supplemental Fig. 1B).
Evaluation of transcript models
For each method, we assessed the quality of 3′ terminal exon assemblies of annotated protein coding genes. After identifying assembled 3′ terminal exon models for each method that shared a 5′ splice boundary with the terminal coding exon of an expressed gene from the Ensembl 73 annotation, we assessed the extent to which that annotation is supported by various types of evidence. For each predicted 3′ end we assessed whether an Ensembl 73 annotation, 3′-seq tag, or polyadenylation signal were detected within a short distance of the predicted boundary of the 3′ UTR. We assessed evidence supporting assembled exon model using both a stringent (20 nt) and relaxed (100 nt) windows. Annotations from one method that were within 100 nt of an annotation made by another method were defined to be common between those methods, while annotations that were >100 nt from an annotation made by any other method were considered to be method-specific ends. The number of distinct 3′ ends was counted, such that a 3′ end that was common between two terminal exon models was only counted once.
To calculate the positional enrichment of polyadenylation signals and 3′-seq data, all annotations of terminal exons of known protein coding genes were aligned with their 3′ boundary at position 0. At each position in a 200-nt window upstream of and downstream from each annotation, we calculated the fraction of sequences that have either a PAS or 3′-seq evidence within 20 nt of that position. For the PAS we considered the two most frequent motifs, AATAAA and ATTAAA, and at the terminally aligned position of a 3′-seq read was used as the position for 3′-seq evidence.
Transcript assembly
Cufflinks 2.2.0 was downloaded from http://cufflinks.cbcb.umd.edu/downloads/cufflinks-2.2.0.Linux_x86_64.tar.gz, and was run with the default parameters, except that --library-type fr-firststrand was provided to indicate the strandedness of the sequencing data, and the --overlap-radius was set to 100 bp.
The scripture-beta2.jar was downloaded from ftp://ftp.broadinstitute.org/pub/papers/lincRNA/scripture-beta2.jar. Scripture was run on each chromosome independently, with the default parameters except that 48 GB of memory was allocated, as smaller allocations resulted in “out of memory” errors. The resulting bed files from assemblies for each chromosome were concatenated to form the final assembly for each sample.
Data sets analyzed
Sequencing fastq files with RNA-seq (GSE41637) (Merkin et al. 2012) and 3′-seq GSE30198 (Derti et al. 2012) were downloaded from GEO. Reference genome sequences were downloaded from Ensembl, ftp://ftp.ensembl.org/pub/release-73/fasta/. RNA-seq was mapped to the genome using Tophat2 with default parameters, except that an Ensembl 73 reference annotation was provided with the -G option, and the --segment-length was set to 20. Bowtie was used to map the 3′-seq data, with the default parameters.
Identification of tissue differential APA events
For each pair of tissues, a joint segmentation of the two samples was computed. For each change point identified between a pair of conditions, we calculated the magnitude of change in the level of coverage in each condition. From these values a usage score (U) was calculated as U = 1 − (covdn/covup), where covup and covdn are point estimates of the level of read density in the segments upstream of and downstream from the change point, respectively. The differential usage between conditions A and B was calculated as the difference in the estimated usage at that change point, UΔAB = UA − UB.
Northern analysis
We used PCR to amplify universal and proximal regions of the predicted isoforms of Shank2 using these primer sets.
shank2_uni; GGACCTCTTTGGCTTGAACC
shank2_uni; CTATGGCAGCCTCTGAGACC
chr7:151606740-151607287
shank2_ext2; GGGAGCAGAAGACTGAGTGG
shank2_ext2; CAGCATCATCAGGACAGTGG
chr7:151610801-151611426
Random primed radiolabeled probes were generated using these templates. RNA was isolated from mouse brains, and sequentially hybridized with extension and universal probes as described previously (Miura et al. 2013).
Software availability
IsoSCM transcript assembly is implemented as a standalone Java program, available at https://github.com/shenkers/isoscm. Using the “assemble” keyword IsoSCM will assemble the mapped reads in a BAM file into a splice graph, identify nested terminal exons boundaries using the constrained segmentation procedure, and report the resulting models in GTF format. Functionality to enumerate splice isoforms from the assembled splice graph can be accessed by the keyword “enumerate.” Pairwise comparison of tandem isoform usage can be performed using the “compare” keyword, which reports the relative usage of change points in each sample in a tabular format. Complete documentation is available from the IsoSCM website (https://github.com/shenkers/isoscm).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
S.S. was supported by the Tri-Institutional Training Program in Computational Biology and Medicine. P.M. was supported by a fellowship from the Canadian Institutes of Health Research. Work in E.C.L.’s group was supported by the Burroughs Wellcome Fund and the National Institute of General Medical Sciences of the National Institutes of Health (National Institute of Neurological Disorders and Stroke: R01-NS074037 and R01-NS083833).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.046037.114.
REFERENCES
- Adams MD, Kerlavage AR, Fleischmann RD, Fuldner RA, Bult CJ, Lee NH, Kirkness EF, Weinstock KG, Gocayne JD, White O, et al. 1995. Initial assessment of human gene diversity and expression patterns based upon 83 million nucleotides of cDNA sequence. Nature 377: 3–174. [PubMed] [Google Scholar]
- Anders S, Huber W 2010. Differential expression analysis for sequence count data. Genome Biol 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behr J, Kahles A, Zhong Y, Sreedharan VT, Drewe P, Rätsch G 2013. MITIE: simultaneous RNA-Seq-based transcript identification and quantification in multiple samples. Bioinformatics 29: 2529–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnert R, Rätsch G 2010. rQuant.web: a tool for RNA-Seq-based transcript quantitation. Nucleic Acids Res 38: W348–W351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JB, Boley N, Eisman R, May G, Stoiber M, Duff MO, Booth BW, Wen J, Park S, Suzuki AM, et al. 2014. Diversity and dynamics of the Drosophila transcriptome. Nature 512: 393–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Ara T, Gautheret D 2009. Using Alu elements as polyadenylation sites: a case of retroposon exaptation. Mol Biol Evol 26: 327–334. [DOI] [PubMed] [Google Scholar]
- Denoeud F, Aury JM, Da Silva C, Noel B, Rogier O, Delledonne M, Morgante M, Valle G, Wincker P, Scarpelli C, et al. 2008. Annotating genomes with massive-scale RNA sequencing. Genome Biol 9: R175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derti A, Garrett-Engele P, Macisaac KD, Stevens RC, Sriram S, Chen R, Rohl CA, Johnson JM, Babak T 2012. A quantitative atlas of polyadenylation in five mammals. Genome Res 22: 1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. 2012. Landscape of transcription in human cells. Nature 489: 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkon R, Ugalde AP, Agami R 2013. Alternative cleavage and polyadenylation: extent, regulation and function. Nat Rev Genet 14: 496–506. [DOI] [PubMed] [Google Scholar]
- Fearnhead P 2006. Exact and efficient Bayesian inference for multiple changepoint problems. Stat Comput 16: 203–213. [Google Scholar]
- Feng J, Li W, Jiang T 2011. Inference of isoforms from short sequence reads. J Comput Biol 18: 305–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, Adiconis X, Fan L, Koziol MJ, Gnirke A, Nusbaum C, et al. 2010. Ab initio reconstruction of cell type–specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol 28: 503–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KD, Brenner SE, Dudoit S 2010. Biases in Illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Res 38: e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller D, Wong WH 2013. Simultaneous isoform discovery and quantification from RNA-seq. Stat Biosci 5: 100–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber W, Toedling J, Steinmetz LM 2006. Transcript mapping with high-density oligonucleotide tiling arrays. Bioinformatics 22: 1963–1970. [DOI] [PubMed] [Google Scholar]
- Jan CH, Friedman RC, Ruby JG, Bartel DP 2011. Formation, regulation and evolution of Caenorhabditis elegans 3′UTRs. Nature 469: 97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahens NF, Kavakli IH, Zhang R, Hayer K, Black MB, Dueck H, Pizarro A, Kim J, Irizarry R, Thomas RS, et al. 2014. IVT-seq reveals extreme bias in RNA sequencing. Genome Biol 15: R86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JJ, Jiang CR, Brown JB, Huang H, Bickel PJ 2011a. Sparse linear modeling of next-generation mRNA sequencing (RNA-Seq) data for isoform discovery and abundance estimation. Proc Natl Acad Sci 108: 19867–19872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Feng J, Jiang T 2011b. IsoLasso: a LASSO regression approach to RNA-Seq based transcriptome assembly. J Comput Biol 18: 1693–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JA, Wang Z 2011. Next-generation transcriptome assembly. Nat Rev Genet 12: 671–682. [DOI] [PubMed] [Google Scholar]
- Masamha CP, Xia Z, Yang J, Albrecht TR, Li M, Shyu AB, Li W, Wagner EJ 2014. CFIm25 links alternative polyadenylation to glioblastoma tumour suppression. Nature 510: 412–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkin J, Russell C, Chen P, Burge CB 2012. Evolutionary dynamics of gene and isoform regulation in mammalian tissues. Science 338: 1593–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura P, Shenker S, Andreu-Agullo C, Westholm JO, Lai EC 2013. Widespread and extensive lengthening of 3′ UTRs in the mammalian brain. Genome Res 23: 812–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura P, Sanfilippo P, Shenker S, Lai EC 2014. Alternative polyadenylation in the nervous system: To what lengths will 3′ UTR extensions take us? Bioessays 36: 766–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5: 621–628. [DOI] [PubMed] [Google Scholar]
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M 2008. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320: 1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proudfoot NJ, Brownlee GG 1976. 3′ Non-coding region sequences in eukaryotic messenger RNA. Nature 263: 211–214. [DOI] [PubMed] [Google Scholar]
- Smibert P, Miura P, Westholm JO, Shenker S, May G, Duff MO, Zhang D, Eads BD, Carlson J, Brown JB, et al. 2012. Global patterns of tissue-specific alternative polyadenylation in Drosophila. Cell Rep 1: 277–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steijger T, Abril JF, Engstrom PG, Kokocinski F, Hubbard TJ, Guigó R, Harrow J, Bertone P; RGASP Consortium. 2013. Assessment of transcript reconstruction methods for RNA-seq. Nat Methods 10: 1177–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I, Shkumatava A, Jan CH, Subtelny AO, Koppstein D, Bell GW, Sive H, Bartel DP 2012. Extensive alternative polyadenylation during zebrafish development. Genome Res 22: 2054–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yassour M, Kaplan T, Fraser HB, Levin JZ, Pfiffner J, Adiconis X, Schroth G, Luo S, Khrebtukova I, Gnirke A, et al. 2009. Ab initio construction of a eukaryotic transcriptome by massively parallel mRNA sequencing. Proc Natl Acad Sci 106: 3264–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Lee JY, Tian B 2005. Biased alternative polyadenylation in human tissues. Genome Biol 6: R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.