

Abstract
Introduction
Giant cell arteritis (GCA) is characterized by intimal hyperplasia leading to ischaemic manifestations that involve large vessels. Neurotrophins (NTs) and their receptors (NTRs) are protein factors for growth, differentiation and survival of neurons. They are also involved in the migration of vascular smooth muscle cells (VSMCs). Our aim was to investigate whether NTs and NTRs are involved in vascular remodelling of GCA.
Methods
We included consecutive patients who underwent a temporal artery biopsy for suspected GCA. We developed an enzymatic digestion method to obtain VSMCs from smooth muscle cells in GCA patients and controls. Neurotrophin protein and gene expression and functional assays were studied from these VSMCs. Neurotrophin expression was also analysed by immunohistochemistry in GCA patients and controls.
Results
Whereas temporal arteries of both GCA patients (n = 22) and controls (n = 21) expressed nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), tropomyosin receptor kinase B (TrkB) and sortilin, immunostaining was more intense in GCA patients, especially in the media and intima, while neurotrophin-3 (NT-3) and P75 receptor (P75NTR) were only detected in TA from GCA patients. Expression of TrkB, a BDNF receptor, was higher in GCA patients with ischaemic complications. Serum NGF was significantly higher in GCA patients (n = 28) vs. controls (n = 48), whereas no significant difference was found for BDNF and NT-3. NGF and BDNF enhanced GCA-derived temporal artery VSMC proliferation and BDNF facilitated migration of temporal artery VSMCs in patients with GCA compared to controls.
Conclusions
Our results suggest that NTs and NTRs are involved in vascular remodelling of GCA. In GCA-derived temporal artery VSMC, NGF promoted proliferation and BDNF enhanced migration by binding to TrkB and p75NTR receptors. Further experiments are needed on a larger number of VSMC samples to confirm these results.
Electronic supplementary material
The online version of this article (doi:10.1186/s13075-014-0487-z) contains supplementary material, which is available to authorized users.
Introduction
Giant cell arteritis (GCA) is a primary vasculitis involving aorta and its extra- and intracranial branches. Histopathological findings show cellular infiltrates, internal elastic lamina disruption and intimal hyperplasia, leading to luminal stenosis. Macrophages and giant cells produce platelet-derived growth factor (PDGF), which stimulates migration of vascular smooth muscle cells (VSMCs) from the media to the intima to initiate intimal hyperplasia. However, several mechanisms involved in vascular remodelling of GCA still remain unclear [1].
Neurotrophins (NTs) are growth factors initially described in the nervous system and now include vascular cells [2]. Three NTs and their receptors (NTRs) are well investigated. They include the nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF) and neurotrophin-3 (NT-3), the selective tropomyosin receptor kinase (Trk), TrkA for NGF, TrkB for BDNF and TrkC for NT-3, a non-selective receptor p75NTR and a co-receptor, which is usually associated with p75NTR, called sortilin. In the cardiovascular system, NTs and NTRs are involved in cardiovascular development, blood vessel growth, VSMCs and cardiomyocyte functions [2]. NTs and NTRs are detected in vivo and in vitro in endothelial cells and human aortic VSMC donors and in rats [3]. In addition, NGF promotes similar VSMC migration as PDGFs, but not their proliferation [3,4]. Moreover, NGF and BDNF and their two specific Trk receptors are involved in aortic intimal hyperplasia induced by balloon angioplasty in rats [3], while activation of p75NTR by NGF, NT-3 and, to a lesser extent, BDNF, induce VSMC apoptosis [5].
Using temporal artery VSMCs (TASMCs) obtained prospectively from suspected GCA patients, we hypothesized that NTs and NTRs are involved in the pathogenesis of GCA, especially in the vascular remodelling stage.
Methods
Patients
We prospectively enrolled patients who had undergone a temporal artery biopsy (TAB) for suspected GCA at Limoges and Paris (Cochin and Hotel-Dieu) university hospitals, since January 2010. GCA diagnosis was established according to American College of Rheumatology (ACR) criteria and biopsy-proven GCA lesions [6]. Patients with a negative TAB without clinical diagnosis of arteritis were defined as controls. All subjects gave their informed consent prior to participation and the study was approved by the Ethics Committee of the Cochin University of Paris (collection dc-2010-1079). Clinical and biological data are shown in Table 1.
Table 1.
Clinical and biological findings in the study cohort patients with GCA
| Clinical characteristics | GCA patients (Plasma study) N (%) | GCA patients (IHC study) N (%) | GCA patients (Cohort study) N (%) |
|---|---|---|---|
| Number of patients | 30 | 22 | 42 |
| General characteristics | |||
| Age in years, median (range) | 77 (60-92) | 80.7 (73-89) | 76.3 (60-92) |
| Sex, women/men | 23/7 | 14/8 | 30/12 |
| Systemic manifestations | 15 (50) | 17 (77) | 33 (78.5) |
| Fever | 9 (30) | 4 (18) | 13 (31) |
| Weight loss | 15 (50) | 12 (54.5) | 20 (47.6) |
| Cranial symptoms | 21 (70) | 14 (63.6) | 31 (73.8) |
| Headache | 21 (70) | 14 (63.6) | 31 (73.8) |
| Jaw claudication | 15 (50) | 11 (50) | 19 (45.2) |
| Scalp tenderness | 14 (46.6) | 12 (54.5) | 21 (50) |
| Abnormal temporal arteries* | 13 (43) | 7 (32) | 7 (16.6) |
| Cranial ischaemic events | 9 (30) | 8 (36) | 11(26) |
| Permanent visual loss | 1 (3) | 3 (13.6) | 2 (4.7) |
| Amaurosis fugax | 6 (20) | 8 (36) | 11 (26) |
| Stroke | 2 (6.6) | 1 (4.5) | 3 (7) |
| Polymyalgia rheumatica | 7 (23) | 7 (32) | 10 (24) |
| Biological findings | Mean ± SD | Mean ± SD | Mean ± SD |
| Erythrocyte sedimentation rate (mm/h) | 85.8+/-25 | 84+/-28 | 77.8+/-26 |
| C-reactive protein (mg/L) | 102.2+/-70.3 | 92.6+/-66 | 86.2+/-78 |
| Platelet count (x 109/L) | 476+/-157 | 484+/-162 | 448+/-150 |
| Haemoglobin (g/L) | 11.5+/-1.6 | 12.2+/-1.4 | 11.8+/-1.7 |
*Abnormal temporal arteries at physical examination (painful, swollen, indurated and/or with decreased or absent pulsation). GCA, giant cell arteritis; IHC, immunohistochemistry; SD, standard deviation.
Cell culture
TASMCs were isolated from segments of 5 to 10 mm of temporal artery (TA) after removal of adventitia and enzymatic digestion of media with 500 U/mL of type I collagenase in 5 mL of Dulbecco’s modified Eagle’s medium (DMEM) Glutamax II (Gibco, Grand Island, NY, USA). Cultures were performed, as previously described [7], in Smooth Muscle Cell Basal Medium (PromoCell, Heidelberg, Germany) supplemented with foetal calf serum (FCS) (5%), insulin (5 μg/mL), fibroblast growth factor (FGF-2) (2 ng/mL), epidermal growth factor (0.5 ng/ml) and streptomycin/penicillin (1%) at 37°C in 5% CO2. Functional assay cells were growth factor-starved and maintained in DMEM-free medium for 24 h before stimulation.
Cultures contained 90% VSMC cells expressing α-smooth-muscle actin (Sigma-Aldrich, St Louis, MO, USA), without CD-90 fibroblastic staining (Dianova, Hamburg, Germany) (Additional file 1). TASMCs were used for the experiments between passages 3 and 8. The cells were studied for NTs and NTRs expression at protein and transcriptional levels and in proliferation and migration assays. GCA-derived TASMCs used for western blotting, real-time polymerase chain reaction (PCR) analysis and functional assays were obtained from patients with positive TAB for GCA.
Immunohistochemistry
Immunohistochemistry was performed on paraffin-embedded sections of TA deparaffinised in 22 biopsy-proven GCA patients according to previous described criteria [8] and 21 controls with rabbit anti-NGF, -BDNF, -NT-3, -TrkA, -TrkB, -TrkC, -p75NTR and goat anti-sortilin antibodies (Ab) (Santa Cruz, Heidelberg, Germany) at 1:100 and anti-CD3 Ab for T lymphocytes (1/400; Dako Glostrup, Denmark). Sections of TA were rehydrated and subjected to steam-heat antigen retrieval in citrate buffer in a microwave oven (750 W). Endogenous peroxidases were inhibited with 5% H2O2 in methanol and non-specific sites were blocked with phosphate-buffered saline (PBS)-3% bovine serum albumin. The primary Ab as above were incubated at a 1/100 dilution overnight at 4°C and revealed with the anti-rabbit HRP Envision™ (Dako). After counterstaining, the slides were studied with a Leica microscope (×200 magnification) (Leica, Wetzlar, Germany). Immunostaining was scored using a staining index based on the percentage of positive cells. Positive controls were obtained from neuroblastoma and breast cancer tissues. Negative controls were done with isotypic immunoglobulin controls (data not shown). To perform semi-quantitative measurement of immunostaining, we chose three representative area of temporal artery (×200 magnification) to count staining cells that expressed myofibroblast or VSMC phenotype (that is, elongated cells). The mean of positive cell percentage of these three measurements for each patient or control was used to determine immunostaining intensity. The results were expressed as the mean of three independent quantifications made by two different observers.
Western blotting
Western blotting was performed as previously described [9] with rabbit anti-NGF, -BDNF, -NT-3, -TrkA, -TrkB, -p75NTR and goat anti-sortilin Ab (Santa Cruz) or anti-TrkC (R&D Systems, Minneapolis, MN, USA) at 1/200 dilution. Signalling activation was studied by using rabbit anti-protein kinase B (Akt) and -phospho-Akt Ab and -phospho-extracellular signal-regulated kinase (Erk)1/2 and mouse anti-Erk1/2 Ab (at 1/1000 dilution; R&D Systems). Proteins (50 μg/lane) obtained from whole-cell lysates of TASMCs using lysis buffer [9] were separated on 10 to 12% SDS-polyacrylamide gels (Invitrogen, Carlsbad, CA, USA) and transferred onto PVDF membranes (Merck Millipore, Billerica, MA, USA). After saturation (5% nonfat dry milk, 0.1% Tween in PBS), Ab were incubated overnight at 4°C, and revealed after 1 h incubation at room temperature (RT) with anti-rabbit or -goat Ig-HRP-conjugated Ab (Santa Cruz, dilution 1/1000) by chemiluminescence (ECL reagent; Amersham, Little Chalfont, UK). Protein-loading control was performed with anti-actin Ab (Santa Cruz, 1/10,000). Western blots were scanned using a bioimaging system (Genesnap; Syngene, Cambridge, UK).
RNA extraction, reverse transcription and real-time PCR assays
Total RNAs were extracted with Qiagen RNeasy Isolation Kit (Qiagen, Venlo, Netherlands), treated with RNase-free DNase I (Qiagen), and quantified by NanoDrop (ND-1000) spectrophotometer (NanoDrop, Wilmington, DE, USA). The RNA quality was evaluated on an Agilent 2100 bioanalyzer using the RNA 6000 Labchip kit (Agilent Technologies, Palo Alto, CA, USA). cDNA synthesis was performed with a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA, USA), as recommended by the manufacturer. Real-time PCR was performed by predesigned primer/probe TaqMan gene expression assays (Applied Biosystems) and normalized to HPRT housekeeping gene (Additional file 2). Experiments were assessed in triplicate at least using TaqMan Fast-Univ Master Mix (Applied Biosystems) using an Applied Biosystems StepOnePlus Real-Time PCR System and analysed by StepOne Software v2.2.2 with a two-step PCR protocol as previously described [9]. The comparative ΔCt method was used for relative quantification of gene expression on duplicate of each reaction.
Cell viability, proliferation and chemoinvasion assays
Cell viability, proliferation and migration assays were studied using exogenous NGF (Alomone Labs, Jerusalem, Israel), recombinant human BDNF (Promega, Madison, WI, USA), or PDGF-AB (R&D Systems, Minneapolis, Minnesota, USA), alone or both with K252a, a pharmacological Trk inhibitor (Alomone), ANA-12 (100 μM), a specific TrkB inhibitor, acting without altering TrkA and TrkC functions (Tocris, Bristol, UK) [10] or an anti-p75NTR antagonist Ab (Alomone).
Cell viability of TASMCs was assessed using the colorimetric XTT assay (Cell Proliferation Kit II, XTT, Roche, Basel, Switzerland) according to the manufacturer’s instructions. A range of concentrations was evaluated for each of the conditions studied (Additional file 3). Efficient concentrations were defined as follows: NGF (10 ng/mL), BDNF (200 ng/mL), K252a Trk inhibitor (100 ng/mL), ANA-12 (100 μM), anti-p75NTR antagonist Ab (10 mg/mL) and PDGF (40 ng/mL), which was used as a positive control [11].
Cell proliferation was determined by incorporation of BrdU (Cell Signaling, Danvers, MA, USA) according to the manufacturer’s instructions. Assays were studied using recombinant human NGF (10 ng/mL), recombinant human BDNF (200 ng/mL), or recombinant human PDGF-AB (40 ng/mL), alone or both with K252a Trk inhibitor (100 ng/mL), or ANA-12 specific TrkB inhibitor (100 μM) or an anti-p75NTR antagonist Ab (10 mg/mL). Efficient concentrations of exogenous NTs, PDGF and NT receptor inhibitors were previously determined with XTT assay. Cells were seeded in 96-well plates (5,000 cells/well) and maintained in DMEM-free serum medium during 48 h before stimulation. Proliferation assays were performed after one, three and four days of TASMC incubation with NT or NTR inhibitors, or both.
Chemoinvasion assay
The Boyden chamber method was performed in a BD Biocoat Matrigel Invasion Chambers (BD Biosciences, Franklin Lakes, NJ, USA) with an 8-μm pore-size membrane for the chemoinvasion assay. Experiments were performed in duplicate in at least two independent experiments. Serum-starved TASMCs were seeded (1 × 105 cells/well) in 500 μL of serum-free DMEM into the upper chamber of the membrane-embedded insert during 48 h before stimulation. DMEM containing either 10% FCS alone or NGF, BDNF or PDGF at the concentrations defined above were placed in the lower chamber and the cells were incubated for 24 or 48 h at 37°C/5% CO2. After incubation, the lower chamber was treated with 50 μM Calcein AM fluorescent dye for 30 min at 37°C/5% CO2. Non-invading cells in the upper part of the insert were carefully removed. Then, 8 μl of Calcein AM fluorescent dye (BD Biosciences) was added for 15 min to the lower chamber. Non-invading cells in the upper part of the insert were carefully removed. The migration of TASMCs to the lower part was quantified by fluorescent counting spectrophotometry at 488 nm (Berthold, Bad Wildbad, Germany) and images were obtained using a fluorescence microscope M2FLIII (Leica).
Measurement of sera and supernatant neurotrophin secretion
Cell-culture medium was collected and centrifuged for 30 min at 3,000 g in Vivaspin columns (Merck Millipore). Concentrations of NGF, BDNF and NT-3 in TASMC supernatant and 1:100 diluted sera from GCA patients and controls were measured using enzyme-linked immunosorbent assay (ELISA) kits according to the manufacturer’s instructions (NGF or BDNF or NT-3 EmaxH ELISA, Promega).
Statistical analysis
Results were expressed as their means ± standard errors of the mean (SEM) of at least three independent experiments. Statistical analyses were performed using Student’s t test; a P value <0.05 was considered statistically significant. Asterisks in figures indicate statistically significant differences: *P <0.05, **P < 0.01, ***P <0.001.
Results
Neurotrophins are overexpressed in temporal arteries from patients with GCA compared to controls
TAs from 22 GCA patients and 21 controls expressed NGF, BDNF, TrkB and sortilin, whereas TrkA and TrkC were not detected in any studied TAs (Figure 1A). However, significant differences in the intensity and location of staining were observed between GCA patients and controls. Indeed, NGF expression was more intense in the adventitia (76.2 +/- 19 vs. 35.4 +/- 22%, P = 0.002) and the media (59.6 +/- 25% vs. 26 +/- 22%, P = 0.004), respectively. BDNF was markedly expressed in the media (82 +/- 21 vs. 26.5 +/- 27%, P <0.0001) and the intima (78 +/- 20 vs. 36 ± 27%, P = 0.0007), respectively. Concerning sortilin, expression was prominent in the adventitia (31 ± 28 vs. 2 ± 4%, P = 0.003) and the intima (41 ± 14 vs. 27 ± 14%, P = 0.02), respectively (Figure 1B). Interestingly, only GCA arteries strongly expressed NT-3 and P75NTR in all artery layers (Figure 1A).
Figure 1.
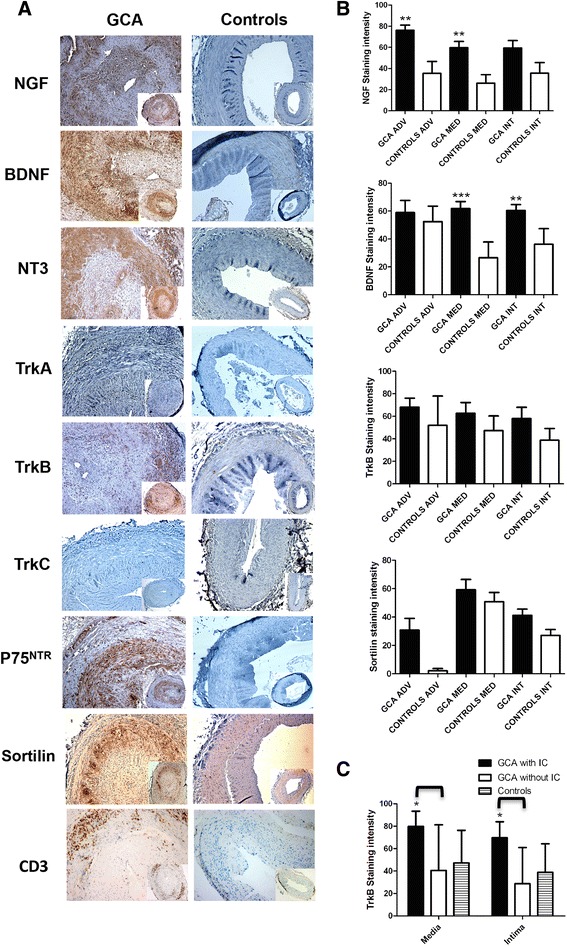
Immunostaining of NTs and their receptors and T lymphocytes (CD3+) in temporal arteries of patients and controls. (A) Panels are representative examples of immunohistological images (x50 and x200) of temporal arteries from GCA patients (n = 22) compared to controls (n = 21). (B). Panels are measurements of significant NT immunostaining intensities. Results are shown as mean values and standard errors of the mean (SEM). (C) TrkB immunostaining in GCA patients with or without ischaemic complications. Immunoreactivity to TrkB in the media and intima of TAs in GCA patients with (n = 9) or without (n = 13) ischaemic complications. GCA, giant cell arteritis; NT, neurotrophin; TA, temporal artery; Trk, tropomyosin receptor kinase.
By contrast, T lymphocytes (anti-CD3+) staining was localized in the media intima junction.
To search for a relationship between these expressions and the course of the disease, we evaluated the staining of NTs and NTRs in association with the clinical parameters of the 22 GCA patients (Table 1).
TrkB is overexpressed in temporal arteries of GCA patients with ischaemic complications
Strikingly, only GCA patients with ischaemic events (stroke, amaurosis or permanent visual loss) (n = 9) had significant overexpression of TrkB in the media (P = 0.04) and intima (P = 0.015), compared to GCA patients without ischaemia (n = 13) (Figure 1C). No difference in expression of NTs and NTRs was found in the TAs of GCA patients regarding other clinical characteristics or inflammatory biomarkers. Moreover, we did not find any difference in immunostaining of NTs and NTRs between GCA patients who received corticosteroids before a TAB (n = 14) and untreated patients (n = 8).
Considering these differences in the expression of NTs and NTRs on ex vivo TAs in GCA patients, we achieved primary cultures for six TAs from biopsy-proven GCA patients and from 10 suspected GCA patients who had normal histological TAs as controls.
Expression of neurotrophins and receptors in TASMCs from the temporal arteries of GCA patients and controls
Protein expression was assessed by western blotting in proteins extracted from the TASMCs of GCA patients and controls (Figure 2A). Whereas NGF, BDNF, TrkB, TrkC, p75NTR and sortilin were detected in TASMCs from patients and controls, proteins for TrkA receptor were not detected in cells from either of these groups. In contrast, NT-3 protein was only detected in GCA and not in control cells. Exploration of signalling pathways at baseline showed that phospho-Akt is predominantly detected in GCA-derived TASMCs than control. The ratio of phospho-Akt/ Akt/actin is significantly higher in GCA cells patients in comparison to patients controls (P = 0.033) (Figure 2B) whereas no difference was found for ERK 1/2 and phospho-ERK 1/2. In parallel, we have confirmed by RT-PCR that the corresponding mRNAs for NGF, BDNF, TrkB (truncated 95 length) and p75NTR were present but not those for TrkA, as expected (Figure 2B). However, some discrepancies were noted between protein and mRNA expressions. This was evidenced for NT-3 mRNA, which was detected in all sampled cells from patients and controls, and feebly for the TrkC transcript. Therefore, a quantitative RT-PCR was performed to determine the differences between transcript expression of NT and NTR genes in cells from patients and controls.
Figure 2.
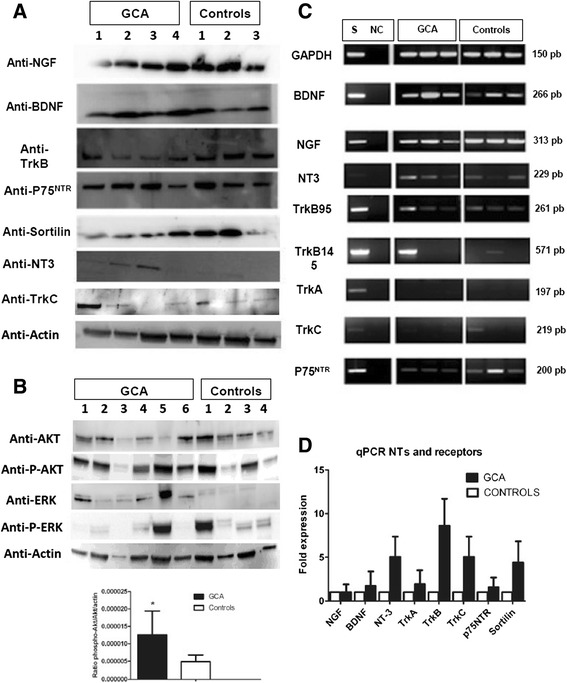
Western blot (WB) analysis of GCA patients’ and controls’ TASMC lysates. WB analysis of GCA patients’ (n = 6) and controls’ (n = 4) TASMC lysates for NTs and NT receptors (A) and for Akt, phospho-Akt, ERK 1/2 and phospho-ERK 1/2 and histograms for phospho-Akt /Akt/ actin ratio (B). Panels are representative examples of WB analysis. (C) Transcription of NTs and NT receptors by TASMCs from GCA patients (n = 6) and controls (n = 6) cultured with 10% FCS. The neuroblastoma cell line SH-SY-5Y (S) was used as a positive control. Constitutively expressed GAPDH is a control of PCR efficiency. Panels are representative examples of the transcription assay. NC: negative control. (D) qRT-PCR analysis of NT and NT receptor expression in TASMC cultures from GCA patients (n = 6) and controls (n = 6), normalized to HPRT gene expression. Akt, protein kinase B; ERK, extracellular signal-regulated kinase FCS, foetal calf serum; GCA, giant-cell arteritis; NGF, nerve growth factor; NT, neurotrophin; PCR, polymerase chain reaction; TASMC, temporal artery VSMC; VSMC, vascular smooth muscle cell.
While BDNF, NGF and NT-3 mRNA were expressed, those of NT-3, TrkB, TrkC and sortilin were overexpressed (increase by fivefold) in TASMCs from GCA patients compared to controls, but the difference was not significant (Figure 2C). Thus, because NGF and BDNF and their receptors were detected in TASMCs from GCA and control patients, the functional effects of these two NTs were evaluated in cultured cells from TAs from the two groups and compared with PDGF as a positive control.
Proliferation effects of NT and NT receptor blockers in GCA patients
NGF promoted proliferation of TASMCs from GCA patients compared to controls (P = 0.025) similarly to FCS (P = 0.012) and PGDF (P = 0.023) by day 1. BDNF tended to enhance cell proliferation by day 1 especially in TASMCs from GCA patients (P = 0.052) (Figure 3A). Proliferation effects with NTs, FCS 10% and PDGF tended to disappear by day 3 and day 4 with no difference between the two groups (Additional file 4). Addition of K252a, ANA-12 or anti-p75NTR Ab prior NTs did not induce a significant difference between the two groups. However, ANA-12 significantly decreased proliferation of TASMCs from GCA in the presence of BDNF (P = 0.010), demonstrating that BDNF-induced proliferation was depending on TrkB activation (Figure 3B).
Figure 3.
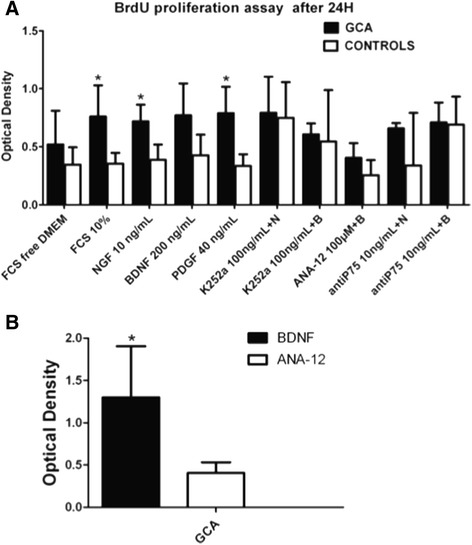
Effects of NTs and the inhibitors of Trk (K252a), TrkB (ANA-12) and p75 NTR (anti-p75) on TASMC proliferation in GCA patients and controls. Proliferation assay performed with a BrdU assay on day 1 (A) in serum-starved TASMC incubated with NT or NT receptor inhibitors, or a combination of NT and NT receptor inhibitors, in GCA patients (n = 6) and controls (n = 10). In all figures, bars represent the mean of three experiments with their SEMs. K252a: specific Trk receptor inhibitor; ANA-12: a specific TrkB receptor inhibitor; anti-p75: p75NTR Ab inhibitor; N (NGF) or B (BDNF) + K (K252a) or anti-p75NTR: NGF or BDNF are added at the same time with the specific inhibitor. (B) Effect of ANA-12 on GCA-derived TASMCs proliferation. Three independent assays were performed and cells were seeded in triplicate for each cell line. BDNF, brain-derived neurotrophic factor; GCA, giant-cell arteritis; NGF, nerve growth factor; NT, neurotrophin; SEM, standard errors of the mean; TASMC, temporal artery VSMC; Trk, tropomyosin receptor kinase; VSMC, vascular smooth muscle cell.
Given that NGF and, to a lesser extent, BDNF induced proliferation in TASMCs from GCA patients compared to controls, we searched for the potential effect of NTs in TASMC invasion properties in Matrigel chambers to determine whether NTs are involved in intimal hyperplasia as shown by immunohistochemistry.
BDNF activates TASMC migration in GCA patients but not in controls
Comparison of TASMC invasion in GCA and control patients demonstrated that TASMCs from GCA patients were significantly able to migrate in the presence of exogenous BDNF compared to those from controls (P = 0.012) after a 24-h exposure (Figure 4). In contrast, no effect was shown for exogenous NGF whatever the status of TASMCs in patients or controls. Of note, only cells from GCA patients were able to significantly invade Matrigel chambers in the presence of 10% FCS (P = 0.0175, compared to cells from controls) as was also observed for exogenous PDGF (P <0.0001 vs. cell migration in controls). Strikingly, neither BDNF nor PDGF induced migration of TASMCs in controls. Migration of TASMCs in GCA patients was only detected after a 24-h exposure and disappeared after 48 h of incubation (data not shown).
Figure 4.
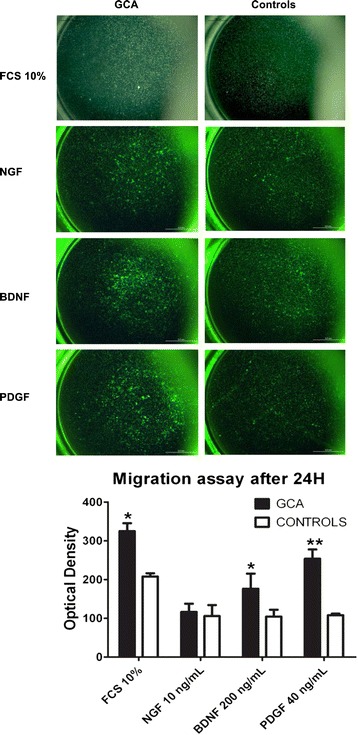
Migration assay of TASMC with NGF and BDNF. Migration assays were performed with TASMCs from GCA patients (n = 6) and controls (n = 6) incubated in the upper chamber with serum-free DMEM during 48 h whereas NGF (10 ng/mL), BDNF (200 ng/mL), PDGF (40 ng/mL) or 10% FCS were added in the lower chamber during 24 h. Top: representative images of the lower chamber after migration and fluorescent staining. Bottom: data analysis obtained from two independent assays performed with cells seeded in duplicate for each cell line. BDNF, brain-derived neurotrophic factor; DMEM, Dulbecco’s modified Eagle’s medium; FCS, foetal calf serum; GCA, giant cell arteritis; NGF, nerve growth factor; PDGF, platelet-derived growth factor; TASMC, temporal artery VSMC; VSMC, vascular smooth muscle cell.
Because NTs are expressed in TAs and in VSMCs of GCA patients, we looked for secretions by TASMC cells in cultures and the sera.
NGF concentration was higher in the sera and in TASMC culture-supernatants from GCA patients
NGF sera concentrations were significantly higher in GCA patients compared to controls (177 +/- 67.6 vs. 145.5 +/- 66 pg/mL, P = 0.04) (Figure 5A) whereas no difference was found for BDNF (Figure 5A) or NT-3 (data not shown). There was no correlation between NGF, BDNF or NT-3 level and ischaemic events or inflammatory syndrome. NGF and BDNF secretion was measured in cell supernatants. NGF levels tended to be higher in TASMCs from GCA patients (P = 0.06) (Figure 5B) whereas no difference was detected for BDNF and NT-3 supernatants (data not shown).
Figure 5.
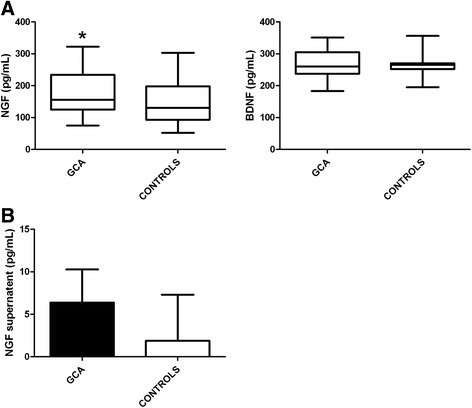
Sera and supernatant concentrations of NGF and BDNF from GCA patients and controls. (A) NGF and BDNF sera concentrations from patients with GCA (n = 30) compared to controls (n = 48) with a sera dilution of 1:100. (B) NGF TASMC-related supernatant concentration from GCA patients (n = 8) compared to controls (n = 8). Supernatants were collected when VSMC reached 80% confluence, between passages 3 and 5. Experiments were performed in duplicate, with three independent ELISA assays and data are presented as pg/mL. BDNF, brain-derived neurotrophic factor; ELISA, enzyme-linked immunosorbent assay; GCA, giant cell arteritis; NGF, nerve growth factor; TASMC, temporal artery VSMC; VSMC, vascular smooth muscle cell.
Discussion
Despite substantial improvement in understanding the pathophysiologic features of GCA, some mechanisms still remain unclear, especially in relation to vascular remodelling. Herein, we report on the expression and functions of NTs and their receptors, which are potentially involved in vascular remodelling in GCA. It is worth noting that NGF, BDNF and sortilin are significantly overexpressed in different histological layers of TAs in GCA patients compared to those of controls. In addition, TrkB staining was significantly higher in the TAs of GCA patients with a cranial ischaemic event. TrkA and TrkC were not detected in patients or controls, whereas NT-3 and p75NTR were enhanced only in GCA patients. These data dealt with mRNA and protein expression of NTs and NTRs in TASMCs from GCA patients and controls. Enhanced TrkB transcription was observed in GCA cells whereas those of TrkA and TrkC were feebly detected.
The expressions of NGF, BDNF and sortilin in the TAs of GCA patients could be related to other inflammatory disorders associated with localized expression of NTs. Indeed, NGF is expressed in endothelial cells and keratinocytes in patients with psoriasis [12], and NGF and BDNF are present in the inflamed joints of patients with rheumatoid arthritis [13,14]. Of note, constitutive NGF expression and release in both human and mouse articular chondrocytes is enhanced by interleukin (IL)-1β [15]. Likewise, NTs are detected in other bronchial smooth muscle cells during asthma [16] or sarcoidosis [17] and their relationship with inflammatory cytokines, IL-1β and interferon (IFN)-γ has been established by the induction of NGF, and BDNF mRNA expression and secretion in cell-culture supernatants [18,19]. Hence, the overexpression of NTs and their receptors in TAs from GCA patients could be, in part, related to the presence of proinflammatory cytokines in all layers of the inflamed arterial wall. Moreover, it has been demonstrated that ischaemia contributes to the overexpression of some NTs and their related receptors, as we have shown with the increased TrkB expression in GCA patients with a cranial ischaemic event. Indeed, in experimental arterial injury that leads to ischaemia, enhanced expression of NGF, BDNF and their specific receptors has been described [3]. Furthermore, in a murine experimental model of atherosclerosis, the TrkB receptor is overexpressed in neointimal VSMC cells whereas decreased TrkB expression reduces atherosclerotic lesions [20].
Moreover, sortilin expression is enhanced in the adventitia and intima of TAs from GCA patients. This data could be correlated with intimal TrkB overexpression, as sortilin acts as an intracellular protein transporter for immature NTs [21] and controls the regulation of BDNF trafficking and release [22]. Such a function could be related to BDNF and sortilin overexpression in TAs from GCA patients. In addition, sortilin is also a co-receptor for p75NTR, which was only observed in the TAs of GCA patients. p75NTR is mostly absent in healthy controls (HCs): its expression is enhanced by pathologic conditions such as diabetes or atherosclerosis [3,5,23].
Serum NGF levels are significantly enhanced in GCA patients, but not BDNF or NT-3. Such elevated NGF levels have been previously reported in inflammatory diseases such as Sjögren syndrome [24].
The proliferation effects of NTs are well described in smooth muscle cells other than VSMCs, especially for NGF and BDNF [25-28]. Given that isolation and expansion in the culture of smooth muscle cells from temporal arteries was previously reported [29], functional studies of TASMCs isolated from GCA and controls were performed in our study. Indeed, NGF promoted proliferation in GCA-derived TASMCs similarly to PDGF. Because TrkA was not detected in TASMCs, this effect probably took place through p75NTR activation as already described in brain tumor-initiating cells [30]. BDNF induced GCA-derived TASMCs proliferation depending on TrkB as demonstrated by ANA-12, a specific inhibitor of TrkB [10]. This effect, well reported in smooth muscle cells in pulmonary hypertension [27] is not significant probably because of our small number samples. In GCA patients we have demonstrated that BDNF promotes TASMCs migration rather than their proliferation. Because both TrkB and BDNF were prominently expressed in the same areas of GCA temporal arteries, it is probable that BDNF activates TASMCs migration from TAs of patients through its interaction with TrkB. Our data are consistent with previous studies on the effects of VSMCs on NTs [3,4,31]. Indeed, BDNF-related migration was comparable to that observed with PDGF, as described previously, [4] and was more consistent than that observed with NGF.
Exploration of signalling pathways in our study showed that PI3/Akt pathway seemed to be more activated than ERK1/2 pathway in GCA patients compared to controls. Though PI3/Akt signalling pathway can be activated by TNF-α in human smooth muscle cells [32] or IL-1β in rat brain astrocytes [33], our results are consistent with BDNF-induced migration in human chondrosarcoma [34] through a transducing signal involving TrkB.
Conclusions
Neurotrophins inducing proliferation and migration of VSMC are potentially implicated in vascular remodelling in GCA.
Taken together, our results provide new insights regarding the involvement of NTs and NTRs in the vascular remodelling of GCA pathogenesis. Their overexpression in TAs from GCA patients could facilitate VSMC proliferation and migration from media to intima, thus contributing to intimal hyperplasia. Particularly, a fine-tuning role of the BDNF/TrkB axis is implicated in proliferation and migration of VSMC from GCA patients (and not from controls) as demonstrated in vitro (Figure 6). However, further studies are needed to elucidate the signalling pathways that allow VSMC migration in order to define new therapeutic approaches for patients with GCA.
Figure 6.
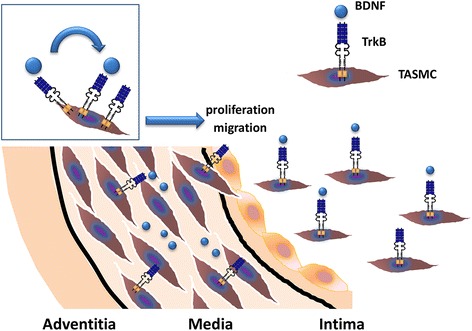
Fine-tuning role of BDNF and TrkB in proliferation and migration of TASMC. BDNF, brain-derived neurotrophic factor; TrkB, tropomyosin receptor kinase B; TASMC, temporal artery vascular smooth muscle cell.
Acknowledgements
AR received financial support from the Direction Régionale et Départementale de Champagne-Ardennes et de la Marne and the Société Nationale Française de Médecine Interne (SNFMI) and SS from Conseil Régional du Limousin.
Abbreviations
- Ab
antibody
- ACR
American College of Rheumatology
- Akt
protein kinase B
- BDNF
brain-derived neurotrophic factor
- DMEM
Dulbecco’s modified Eagle’s medium
- ELISA
enzyme-linked immunosorbent assay
- ERK
extracellular signal-regulated kinase
- FCS
foetal calf serum
- FGF
fibroblast growth factor
- GCA
giant cell arteritis
- HC
healthy control
- IL
interleukin
- INF
interferon
- NGF
nerve growth factor
- NT
neurotrophin
- NT-3
neurotrophin-3
- NTR
neurotrophin receptor
- PCR
polymerase chain reaction
- PDGF
platelet-derived growth factor
- RT
reverse transcriptase
- SEM
standard errors of the mean
- TA
temporal artery
- TAB
temporal artery biopsy
- TASMC
temporal artery VSMC
- TNF-α
tumour necrosis factor alpha
- Trk
tropomyosin receptor kinase
- VSMC
vascular smooth muscle cell
Additional files
VSMC phenotype in immunocytochemistry assay.
Primers used in qPCR and RT-PCR studies.
Dose-response of NT and NT receptor inhibitors.
Proliferation assays on days 3 and 4.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
KHL carried out the design of the study, cell culture, immunohistochemistry, western blotting, molecular, ELISA, proliferation and migration assays, analysed the results and drafted the manuscript. AR participated to cell culture, immunoassays and drafted the manuscript. EM participated in immunohistochemistry and molecular experiments and edited the manuscript. SS participated in molecular experiments, statistical analysis and edited the manuscript. PS participated in immunohistochemistry analysis, statistical analysis and drafted the manuscript. ALF provided sera patients, analysed the results and drafted the manuscript. CLJ, VWS, PB and EV provided sera patients, participated in the acquisition of clinical and biological data and helped to revise the manuscript. AB, PYR and JLB provided temporal arteries, clinical data and helped to revise the manuscript. FL participated in the acquisition, analysis of immunohistochemistry data and drafted the manuscript. LM provided sera, participated in the analysis of clinical data and the drafting of the manuscript. MOJ carried out the design of the study with KHL, designed the experiments, analysed the results and drafted the manuscript. All authors read and approved the final manuscript.
Contributor Information
Kim Heang Ly, Email: kim.ly@chu-limoges.fr.
Alexis Régent, Email: alexisregent@hotmail.com.
Elsa Molina, Email: elsa13_molina@yahoo.fr.
Sofiane Saada, Email: sofiane.saada@etu.unilim.fr.
Philippe Sindou, Email: philippe.sindou@unilim.fr.
Claire Le-Jeunne, Email: claire.le-jeunne@htd.aphp.fr.
Antoine Brézin, Email: antoine.brezin@cch.aphp.fr.
Véronique Witko-Sarsat, Email: veronique.witko@inserm.fr.
François Labrousse, Email: francois.labrousse@unilim.fr.
Pierre-Yves Robert, Email: pierre-yves.robert@unilim.fr.
Philippe Bertin, Email: Philippe.Bertin@chu-limoges.fr.
Jean-Louis Bourges, Email: jean-louis.bourges@htd.aphp.fr.
Anne-Laure Fauchais, Email: anne-laure.fauchais@unilim.fr.
Elisabeth Vidal, Email: elisabeth.vidal@unilim.fr.
Luc Mouthon, Email: luc.mouthon@cch.aphp.fr.
Marie-Odile Jauberteau, Email: jauberte@unilim.fr.
References
- 1.Ly KH, Regent A, Tamby MC, Mouthon L. Pathogenesis of giant cell arteritis: More than just an inflammatory condition? Autoimmun Rev. 2010;9:635–645. doi: 10.1016/j.autrev.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 2.Caporali A, Emanueli C. Cardiovascular actions of neurotrophins. Physiol Rev. 2009;89:279–308. doi: 10.1152/physrev.00007.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Donovan MJ, Miranda RC, Kraemer R, McCaffrey TA, Tessarollo L, Mahadeo D, Sharif S, Kaplan DR, Tsoulfas P, Parada L, Toran-Allerand CD, Hajar DP, Hempstead BL. Neurotrophin and neurotrophin receptors in vascular smooth muscle cells. Regulation of expression in response to injury. Am J Pathol. 1995;147:309–324. [PMC free article] [PubMed] [Google Scholar]
- 4.Kraemer R, Nguyen H, March KL, Hempstead B. NGF activates similar intracellular signaling pathways in vascular smooth muscle cells as PDGF-BB but elicits different biological responses. Arterioscler Thromb Vasc Biol. 1999;19:1041–1050. doi: 10.1161/01.ATV.19.4.1041. [DOI] [PubMed] [Google Scholar]
- 5.Wang S, Bray P, McCaffrey T, March K, Hempstead BL, Kraemer R. p75(NTR) mediates neurotrophin-induced apoptosis of vascular smooth muscle cells. Am J Pathol. 2000;157:1247–1258. doi: 10.1016/S0002-9440(10)64640-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hunder GG, Bloch DA, Michel BA, Stevens MB, Arend WP, Calabrese LH, Edworthy SM, Fauci AS, Leavitt RY, Lie JT, Lightfoot RW, Jr, Masi AT, McShane DJ, Mills JA, Wallace SL, Zvaifler NJ. The American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum. 1990;33:1122–1128. doi: 10.1002/art.1780330810. [DOI] [PubMed] [Google Scholar]
- 7.Regent A, Dib H, Ly KH, Agard C, Tamby MC, Tamas N, Weksler B, Federici C, Broussard C, Guillevin L, Mouthon L. Identification of target antigens of anti-endothelial cell and anti-vascular smooth muscle cell antibodies in patients with giant cell arteritis: a proteomic approach. Arthritis Res Ther. 2011;13:R107. doi: 10.1186/ar3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Emilie D, Liozon E, Crevon MC, Lavignac C, Portier A, Liozon F, Galanaud P. Production of interleukin 6 by granulomas of giant cell arteritis. Hum Immunol. 1994;39:17–24. doi: 10.1016/0198-8859(94)90096-5. [DOI] [PubMed] [Google Scholar]
- 9.Saada S, Marget P, Fauchais AL, Lise MC, Chemin G, Sindou P, Martel C, Delpy L, Vidal E, Jaccard A, Troutaud D, Lalloue F, Jauberteau MO. Differential expression of neurotensin and specific receptors, NTSR1 and NTSR2, in normal and malignant human B lymphocytes. J Immunol. 2012;189:5293–5303. doi: 10.4049/jimmunol.1102937. [DOI] [PubMed] [Google Scholar]
- 10.Cazorla M, Premont J, Mann A, Girard N, Kellendonk C, Rognan D. Identification of a low-molecular weight TrkB antagonist with anxiolytic and antidepressant activity in mice. J Clin Invest. 2011;121:1846–1857. doi: 10.1172/JCI43992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benjamin CW, Jones DA. Platelet-derived growth factor stimulates growth factor receptor binding protein-2 association with Shc in vascular smooth muscle cells. J Biol Chem. 1994;269:30911–30916. [PubMed] [Google Scholar]
- 12.Raychaudhuri SP, Raychaudhuri SK. Role of NGF and neurogenic inflammation in the pathogenesis of psoriasis. Prog Brain Res. 2004;146:433–437. doi: 10.1016/S0079-6123(03)46027-5. [DOI] [PubMed] [Google Scholar]
- 13.Barthel C, Yeremenko N, Jacobs R, Schmidt RE, Bernateck M, Zeidler H, Tak PP, Baeten D, Rihl M. Nerve growth factor and receptor expression in rheumatoid arthritis and spondyloarthritis. Arthritis Res Ther. 2009;11:R82. doi: 10.1186/ar2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klein K, Aeschlimann A, Jordan S, Gay R, Gay S, Sprott H. ATP induced brain-derived neurotrophic factor expression and release from osteoarthritis synovial fibroblasts is mediated by purinergic receptor P2X4. PLoS One. 2012;7:e36693. doi: 10.1371/journal.pone.0036693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pecchi E, Priam S, Gosset M, Pigenet A, Sudre L, Laiguillon MC, Berenbaum F, Houard X. Induction of nerve growth factor expression and release by mechanical and inflammatory stimuli in chondrocytes: possible involvement in osteoarthritis pain. Arthritis Res Ther. 2014;16:R16. doi: 10.1186/ar4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Braun A, Lommatzsch M, Lewin GR, Virchow JC, Renz H. Neurotrophins: a link between airway inflammation and airway smooth muscle contractility in asthma? Int Arch Allergy Immunol. 1999;118:163–165. doi: 10.1159/000024056. [DOI] [PubMed] [Google Scholar]
- 17.Dagnell C, Grunewald J, Kramar M, Haugom-Olsen H, Elmberger GP, Eklund A, Olgart Hoglund C. Neurotrophins and neurotrophin receptors in pulmonary sarcoidosis - granulomas as a source of expression. Respir Res. 2010;11:156. doi: 10.1186/1465-9921-11-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kemi C, Grunewald J, Eklund A, Hoglund CO. Differential regulation of neurotrophin expression in human bronchial smooth muscle cells. Respir Res. 2006;7:18. doi: 10.1186/1465-9921-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Freund V, Pons F, Joly V, Mathieu E, Martinet N, Frossard N. Upregulation of nerve growth factor expression by human airway smooth muscle cells in inflammatory conditions. Eur Respir J. 2002;20:458–463. doi: 10.1183/09031936.02.00269202. [DOI] [PubMed] [Google Scholar]
- 20.Kraemer R, Baker PJ, Kent KC, Ye Y, Han JJ, Tejada R, Silane M, Upmacis R, Deeb R, Chen Y, Levine DM, Hempstead B. Decreased neurotrophin TrkB receptor expression reduces lesion size in the apolipoprotein E-null mutant mouse. Circulation. 2005;112:3644–3653. doi: 10.1161/CIRCULATIONAHA.105.587980. [DOI] [PubMed] [Google Scholar]
- 21.Nykjaer A, Willnow TE. Sortilin: a receptor to regulate neuronal viability and function. Trends Neurosci. 2012;35:261–270. doi: 10.1016/j.tins.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 22.Evans SF, Irmady K, Ostrow K, Kim T, Nykjaer A, Saftig P, Blobel C, Hempstead BL. Neuronal brain-derived neurotrophic factor is synthesized in excess, with levels regulated by sortilin-mediated trafficking and lysosomal degradation. J Biol Chem. 2011;286:29556–29567. doi: 10.1074/jbc.M111.219675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salis MB, Graiani G, Desortes E, Caldwell RB, Madeddu P, Emanueli C. Nerve growth factor supplementation reverses the impairment, induced by Type 1 diabetes, of hindlimb post-ischaemic recovery in mice. Diabetologia. 2004;47:1055–1063. doi: 10.1007/s00125-004-1424-5. [DOI] [PubMed] [Google Scholar]
- 24.Fauchais AL, Boumediene A, Lalloue F, Gondran G, Loustaud-Ratti V, Vidal E, Jauberteau MO. Brain-derived neurotrophic factor and nerve growth factor correlate with T-cell activation in primary Sjogren's syndrome. Scand J Rheumatol. 2009;38:50–57. doi: 10.1080/03009740802378832. [DOI] [PubMed] [Google Scholar]
- 25.Freund-Michel V, Bertrand C, Frossard N. TrkA signalling pathways in human airway smooth muscle cell proliferation. Cell Signal. 2006;18:621–627. doi: 10.1016/j.cellsig.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 26.Stawowy P, Marcinkiewicz J, Graf K, Seidah N, Chretien M, Fleck E, Marcinkiewicz M. Selective expression of the proprotein convertases furin, pc5, and pc7 in proliferating vascular smooth muscle cells of the rat aorta in vitro. J Histochem Cytochem. 2001;49:323–332. doi: 10.1177/002215540104900306. [DOI] [PubMed] [Google Scholar]
- 27.Kwapiszewska G, Chwalek K, Marsh LM, Wygrecka M, Wilhelm J, Best J, Egemnazarov B, Weisel FC, Osswald SL, Schermuly RT, Olschewski A, Seeger W, Weissmann N, Eickelberg O, Fink L. BDNF/TrkB signaling augments smooth muscle cell proliferation in pulmonary hypertension. Am J Pathol. 2012;181:2018–2029. doi: 10.1016/j.ajpath.2012.08.028. [DOI] [PubMed] [Google Scholar]
- 28.Aravamudan B, Thompson M, Pabelick C, Prakash YS. Brain-derived neurotrophic factor induces proliferation of human airway smooth muscle cells. J Cell Mol Med. 2012;16:812–823. doi: 10.1111/j.1582-4934.2011.01356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lozano E, Segarra M, Garcia-Martinez A, Hernandez-Rodriguez J, Cid MC. Imatinib mesylate inhibits in vitro and ex vivo biological responses related to vascular occlusion in giant cell arteritis. Ann Rheum Dis. 2008;67:1581–1588. doi: 10.1136/ard.2007.070805. [DOI] [PubMed] [Google Scholar]
- 30.Forsyth PA, Krishna N, Lawn S, Valadez JG, Qu X, Fenstermacher DA, Fournier M, Potthast L, Chinnaiyan P, Gibney GT, Zeinieh M, Barker PA, Carter BD, Cooper MK, Kenchappa RS. p75 neurotrophin receptor cleavage by alpha- and gamma-secretases is required for neurotrophin-mediated proliferation of brain tumor-initiating cells. J Biol Chem. 2014;289:8067–8085. doi: 10.1074/jbc.M113.513762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Urban D, Lorenz J, Meyborg H, Ghosh S, Kintscher U, Kaufmann J, Fleck E, Kappert K, Stawowy P. Proprotein convertase furin enhances survival and migration of vascular smooth muscle cells via processing of pro-nerve growth factor. J Biochem. 2013;153:197–207. doi: 10.1093/jb/mvs137. [DOI] [PubMed] [Google Scholar]
- 32.Lee CW, Lin CC, Lin WN, Liang KC, Luo SF, Wu CB, Wang SW, Yang CM. TNF-alpha induces MMP-9 expression via activation of Src/EGFR, PDGFR/PI3K/Akt cascade and promotion of NF-kappaB/p300 binding in human tracheal smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2007;292:L799–L812. doi: 10.1152/ajplung.00311.2006. [DOI] [PubMed] [Google Scholar]
- 33.Wu CY, Hsieh HL, Sun CC, Tseng CP, Yang CM. IL-1 beta induces proMMP-9 expression via c-Src-dependent PDGFR/PI3K/Akt/p300 cascade in rat brain astrocytes. J Neurochem. 2008;105:1499–1512. doi: 10.1111/j.1471-4159.2008.05318.x. [DOI] [PubMed] [Google Scholar]
- 34.Lin CY, Chen HJ, Li TM, Fong YC, Liu SC, Chen PC, Tang CH. beta5 integrin up-regulation in brain-derived neurotrophic factor promotes cell motility in human chondrosarcoma. PLoS One. 2013;8:e67990. doi: 10.1371/journal.pone.0067990. [DOI] [PMC free article] [PubMed] [Google Scholar]