

Abstract
The sequencing of the human genome and the advent of low-cost genome-wide assays that generate millions of observations of individual genomes in a matter of hours constitute a disruptive innovation for social science. Many public-use social science datasets have or will soon add genome-wide genetic data. With these new data come technical challenges, but also new possibilities. Among these, the lowest hanging fruit and the most potentially disruptive to existing research programs is the ability to measure previously invisible contours of health and disease risk within populations. In this article, we outline why now is the time for social scientists to bring genetics into their research programs. We discuss how to select genetic variants to study. We explain how the polygenic architecture of complex traits and the low penetrance of individual genetic loci pose challenges to research integrating genetics and social science. We introduce genetic risk scores as a method of addressing these challenges and provide guidance on how genetic risk scores can be constructed. We conclude by outlining research questions that are ripe for social science inquiry.
The sequencing of the human genome and the advent of low-cost genome-wide assays that generate millions of observations of individual genomes in a matter of hours constitute a disruptive innovation for social science. For a few hundred dollars, any tissue collected from a subject in a research study can be transformed into an inventory of disease risks and behavioral proclivities that is continuously updated by an ever-growing army of scientists from around the world. Many of the datasets that fuel social science research have or will soon add genome-wide genetic data. With these new data come technical challenges, but also new possibilities. Among these, the lowest hanging fruit and the most potentially disruptive to existing research programs is the ability to measure previously invisible contours of health and disease risk within populations.
Much of social science is concerned with uncovering sources of heterogeneity within and between populations in the processes that determine life courses. Non-DNA biomarkers—including observations of physical and cognitive function, clinical indexes of frailty, and measured constituents of blood and other tissues—are now in wide use for this purpose. (Crimmins, Kim, and Vasunilashorn 2010; Carey and Vaupel 2005) Genome-wide genetic data are something different. They are different because of their primacy; DNA sequence is fixed at conception, before the individual is even born. They are different because of their stability; with rare exception (Forsberg, Absher, and Dumanski 2013), DNA sequence is the same across tissues and there are no circadian, seasonal, or age- or morbidity-related changes. (Reverse causality—a central consideration in most other biomarker research—is not a primary concern in genetics.) They are different because of their scope; DNA sequence variation is related to virtually any behavioral or disease phenotype that can be imagined. In these three ways – primacy, stability, and scope of influence – the DNA sequence stands apart from other biomarkers.
The remainder of this article is divided into four sections. We begin by outlining why now is the time for social scientists to bring genetics into their research programs. We next discuss how to select genetic variants to study. We introduce genetic risk scores as a method of adapting genetic information into social science research designs. Finally, we outline research questions that are ripe for social science inquiry. This article is not intended as a comprehensive survey of all the things that can be done with genetic information. Instead, we aim to suggest some initial forays into the integration of social science and genetics that represent what we view as the low hanging fruit and that are appropriate to newcomers and veterans alike.
Three reasons now is the time to integrate genetics and social science
First, data are available. Decline in costs of measuring genomes enabled several large social surveys to add genome-wide genetic information to their databases. The Health and Retirement Study (HRS) has completed this process. The Wisconsin Longitudinal Study, and the National Longitudinal Study of Adolescent Health (Add Health) will soon join the HRS. In addition to these traditional social science datasets, the National Heart Lung and Blood Institute’s Atherosclerosis Risk in Communities (ARIC), Multiethnic Study of Atherosclerosis (MESA), Framingham Heart Study, Coronary Artery Risk Development in Young Adults (CARDIA) and various other resources bring together genome-wide data, longitudinal follow-up tracking health, disease, and mortality, and varying degrees of social contextual information on large population-based samples. Importantly, all of these data are now or will be publicly available through the NIH dbGaP (http://www.ncbi.nlm.nih.gov/gap).
Second, data are analytically tractable. Analyzing genome-wide data has been compared to drinking from a fire hose (Hunter and Kraft 2007). The high dimensionality of the datasets—usually a million or more observations per individual—makes business as usual data analysis impossible. But tools and methods for handling genome-wide data are now well established. Software programs such as PLINK (Purcell et al. 2007) and various packages written for the R software (http://cran.r-project.org/web/views/Genetics.html) make genome-wide data analysis computationally feasible, even on standard desktop computers (Aulchenko et al. 2007). Quality control procedures have been standardized—many publicly available datasets include “cleaned” genetic data and/or detailed instructions on how to perform data quality checks. Rules of thumb have been established (Laurie et al. 2010). a And techniques have been developed to address the unique structure of genomic data (Hamer and Sirota 2000). b
Third, data are meaningful—and not just in the abstract sense that the genome must be important because virtually all of human behavior, health, and disease are at least partly “heritable, ” i.e. influenced by genetic factors (Plomin et al. 2008). The data are meaningful because large-scale genetic association studies, in particular genome-wide association studies (GWAS), have produced a library of genetic associations that link thousands of common DNA sequence variants with a range health and disease processes and outcomes (Hindorff et al.; Yu et al. 2008). This is critically important. Most social scientists don’t want to be gene-hunters (although there are some notable exceptions (Boardman et al. 2013; Rietveld et al. 2013; Fowler, Settle, and Christakis 2011; Benjamin et al. 2012)). But once geneticists establish connections between specific variants and phenomena that social scientists study, those variants can become powerful tools (Belsky, Moffitt, and Caspi 2013).
Selecting genetic variants to study: Hypothesis-driven and hypothesis-free approaches
The first challenge social scientists face in integrating genetics into their research is figuring out what genes or genetic variants they should study. Broadly speaking, there are two approaches to addressing this challenge. The first is based on extant biological evidence (hypothesis-driven). The second is based on statistical evidence from whole-genome data mining (hypothesis-free). The sections below discuss these two approaches and suggest criteria social scientists can use to evaluate the genetic evidence base. Our aim is to help guide social scientists in identifying promising genetic variants to incorporate into their research.
The hypothesis-driven approach reverse engineers the biology of some phenomenon of interest (hereafter, the “phenotype”), identifies proteins critical to that biology, and then finds genes that contribute to the production of those proteins. If possible, the next step is to identify “functional polymorphisms” in the selected genes—sequence variations within or around the genes that affect quantity or structure of the protein produced (Collier et al. 1996; Lesch et al. 1996; Tol et al. 1992; Asghari et al. 1995). If functional polymorphisms are not evident, variation within and around the gene can be surveyed and tested for association to identify variants for use in subsequent research (Israel et al. 2009).
The hypothesis-driven approach has a natural affinity with other biomarker research insofar as genetic associations can be interpreted as evidence for the role of a specific biological process in the etiology of the phenotype. A strength of the hypothesis-driven approach is that it provides, at least in theory, indirect measures of hard to observe biological processes, such as processes in brain (Hariri 2009). For example, addiction is understood partly as a dysregulation of reward-based learning (Koob and Volkow 2010). Dopaminergic neurotransmission has an established role in the reinforcement of reward in learning (Hyman, Malenka, and Nestler 2006). A number of genes encoding proteins that mediate dopaminergic neurotransmission have been identified, including several with well-characterized functional variants (Hariri 2009). Studies have used these genes to implicate dysregulation of dopaminergic neurotransmission in the pathogenesis of addiction (Nikolova et al. 2011; Bogdan, Hyde, and Hariri 2013).
The limitation of a hypothesis-driven approach is that it depends on existing knowledge—knowledge of the biology giving rise to the phenotype and knowledge of how genes influence that biology. So, when should social scientists consider a hypothesis-driven approach to selecting genetic variants for their research? Three useful criteria are
When there is extensive knowledge of the biological pathway in which the gene is implicated.
When there is evidence for modulation of the function of that pathway by the gene, e.g. from model organism studies where the gene is selectively ablated/ inactivated or from human monogenic disorders arising from mutations in the gene.
When there is evidence that a specific variant alters the function of the gene within the targeted pathway.
These criteria are not intended as hard rules. Instead, they are meant as a starting point for evaluating the quality of the evidence base.
The hypothesis-free approach substitutes comprehensive measurement of the genome and statistical rigor in place of biological plausibility. Hypothesis-free approaches survey variants across the entire genome and test associations between each variant and a phenotype. This approach is referred to as a genome-wide association study (GWAS). Although GWAS can refer to a range of study designs (Hirschhorn and Daly 2005), the usual approach is to assay one or two million single-nucleotide polymorphisms (SNPs) selected to measure common variation throughout the genomec and to correlate each variant with a trait or disease state (Pearson and Manolio 2008).
The strength of the hypothesis-free approach is that it leapfrogs current knowledge of biology to make discoveries that push the frontiers of science (Hindorff et al. 2009). For example, in the cases of obesity and asthma, the first and best-replicated GWAS discoveries, FTO for obesity and the ORMDL3/GSDMB locus on chromosome 17 for asthma, were unanticipated in existing biological models of disease pathogenesis (Herbert et al. 2006; Moffatt et al. 2007). With this strength comes substantial ambiguity about the function of discovered SNPs. The FTO case is instructive. Follow-up research linked the obesity-risk variant in FTO to behavioral measures of appetite (Wardle et al. 2008). But the biological link between FTO and appetite eluded researchers until recently. Now, evidence suggests that the GWAS discovered SNP rs9930506 in the FTO gene gives rise to obesity not through changes in the structure or function of FTO, but through a change in function of a nearby gene, IRX3 (Smemo et al. 2014). The implication for social science researchers is that biological inference about the function of GWAS discoveries is fluid even as the risk information furnished in the discovered SNPs remains constant. In other words, the hypothesis-free approach furnishes measurements of genetic risk, broadly defined, not necessarily specific information about the pathways through which that risk operates.
The limitation of the hypothesis-free approach is that it is vulnerable to statistical artifacts. To begin with, stringent correction for multiple testing is needed; the p-value corresponding to an alpha-value of 0.05 in standard GWAS analysis is p<5×10-8 (Pearson and Manolio 2008). As a result, GWAS design is almost entirely focused on maximizing statistical power. Extreme group comparisons (e.g. severe cases versus super-controls with no pathology (Schwartz and Susser 2010)) are one approach to increasing statistical power in GWAS, but the dominant strategy is to increase sample size (Sullivan, Daly, and O’Donovan 2012). As a result, effective hypothesis-free discovery research depends on the organization of large-scale consortia that can assemble samples of tens or even hundreds of thousands of individuals and mount multiple replication attempts (Sullivan 2010; Benjamin et al. 2012).
So, when should social scientists consider a hypothesis-free approach to selecting genetic variants for their research? Three useful criteria are
When discovery sample sizes are very large. (Just how large sample sizes need to be will vary depending on the phenotype. In general, sample sizes of thousands are a minimum requirement.)
When independent replications are positive. Top journals require independent replication for publication of GWAS. In addition to such “internal” replications, studies conducted by other groups, studies or sub-analyses using population-representative samples (Manolio 2009) or samples with more refined measures of a phenotype add confidence that a GWAS discovery is legitimate.
When the technical quality of the GWAS is high. This can be difficult for a non-geneticist to determine. Guidance on evaluating GWAS quality control steps can be found here (Laurie et al. 2010). Criteria 1 and 2 are highly correlated with this third criterion.
In our view, both hypothesis-driven and hypothesis-free approaches to select inggenetic variants to study are valid. The critical issue is the alignment of the approach used to select variants to study with the research question. A hypothesis-driven approach is aligned with questions about mechanism, e.g. “Do differences in a specific biological process explain differences in an outcome?” The hypothesis-driven approach can furnish genetic variants that measure individual differences in difficult to observe biological processes. For example, a recent study of smoking cessation used a hypothesis-driven approach to construct a genetic measure of dopamine signaling in the brain (David et al. 2013). The authors then used that genetic measure to test the hypothesis that dopamine signaling capacity would predict smoking cessation therapy outcome. A hypothesis-free approach is aligned with questions about risk, e.g. “Do individuals at higher genetic risk fare differently from their lower genetic risk peers?” The hypothesis-free approach furnishes genetic variants that measure individual differences in genetic risk. For example, another recent study of smoking cessation derived a measure of genetic predisposition to quit success from hypothesis-free GWAS of clinical trial data (Uhl et al. 2012). The authors then used that genetic measure test the hypothesis that genetic predisposition to quit success would predict smoking cessation therapy outcome.
Both hypothesis-driven and hypothesis-free approaches have unique advantages for social science research. Hypothesis-driven approaches can leverage DNA, which is accessible from any tissue in the body, to ask research questions about aspects of the biology of social processes that would otherwise be inaccessible, e.g. because they are located in brain. Meanwhile, a hypothesis-free approach can provide a point of entry to asking genetic questions about social problems for which the biology is as yet poorly understood.
Measuring a continuum of genetic risk: Genetic risk scores
Once a set of genetic variants are identified, the next challenge is how to use them. In genetics, the usual approach is to examine one genetic variant at time, i.e. tests of each genetic variant are conducted independently. We argue that for social scientists, this approach is generally sub-optimal for two reasons:
The complex traits, behaviors, and health outcomes of interest to social scientists are highlypolygenic (Visscher, Hill, and Wray 2008); they reflect the influence of many different genes.
Individual genetic loci influencing the etiology of complex phenotypes have low penetrance (Gibson 2011); no single genotype determines phenotype, or is even highly predictive.
These two reasons highlight technical and conceptual problems with studying genetic variants one at a time in social science research.
The technical problem is that studying one genetic variant at a time requires a great deal of statistical power (effect-sizes are small, many variants must be tested). Such power can be obtained by brute force, as in the use of massive samples (Kilpelainen et al. 2011), or by surgical precision, as in carefully designed experiments (Shalev et al. 2009). But in either case, achieving sufficient power often precludes the incorporation of design features that social scientists need to address the substantive questions motivating genetic inquiry in the first place (e.g. detailed measurements of phenotype and environmental context, population-representative sampling, and longitudinal follow-up) (Belsky, Moffitt, and Caspi 2013).
The conceptual problem is that studying one genetic variant at a time rarely captures the genetic influence social scientists wish to study. Genetic predisposition to any behavior/trait/outcome of interest to social scientists is quantitative; there is a continuum of genetic risk that reflects small contributions from many genetic loci. Some individuals carry very few genetic risk factors, others carry many, and most of the population is somewhere in the middle (Figure 1) (Plomin, Haworth, and Davis 2009). The genotype of any one variant may not be informative about where an individual is located on the continuum of genetic risk.
Figure 1. Polygenic risk for complex health conditions is continuously and normally distributed.
A solution that addresses the technical and conceptual limitations of studying genetic variants one at a time is to combine information across variants into a quantitative measurement of the continuum of genetic risk: a genetic risk score. A genetic risk score is a summary measure of a set of risk-associated genetic variants. Depending on the discipline, phenotype, and use of the score, a variety of names are used, e.g. “multi-locus genetic profile” (Nikolova et al. 2011), “polygenic risk score” (Llewellyn et al. 2014), “allelic score” (Spycher et al. 2012), “SNP score” (Vrieze, McGue, and Iacono 2012), “genotype score” (Meigs et al. 2008), “genetic prediction score” (Zhao et al. 2014) among others. Some genetic risk scores include thousands of variants only weakly linked with disease (Wray, Goddard, and Visscher 2007; Purcell et al. 2009; Evans, Visscher, and Wray 2009). Others are composed of a handful of markers with well established evidence of association with a phenotype of interest (Morrison et al. 2007; Kathiresan et al. 2008; Ripatti et al. 2010). In our view, the defining characteristic of a genetic risk score is that it provides a quantitative measure of genetic predisposition that is calculated using information from multiple genetic variants.
Genetic risk scores are most often additive summaries of risk information from a set of genetic variants (e.g. a count of risk-associated alleles across a panel of SNPs (Morrison et al. 2007)). This approach assumes that the individual genetic variants in the score make additive contributions to the phenotype. We know this assumption of additive contribution is probably wrong. Epistasis—interaction between genetic loci—is important and pervasive (Zuk et al. 2012). Unfortunately, we know very little about epistatic interactions. Given the specifics of the interactions are unknown, additivity provides a workable default. As epistatic interactions are characterized, they can be incorporated into genetic risk scores.
Genetic risk scores can be computed from genetic variants selected using either hypothesis-driven or hypothesis-free approaches. In a hypothesis-driven approach, the goal should be biological coherence of the variants included in the score. For example, scores developed to measure the continuum of genetic influence on dopaminergic neurotransmission have been used to study neural reactivity to reward (Nikolova et al. 2011; Stice et al. 2012). In a hypothesis-free approach, the goal should be statistical coherence—the same standard of evidence should be applied to all variants in the genome to select SNPs into the score. We describe a formal, three-stage approach to this process in an earlier article (Belsky, Moffitt, Sugden, et al. 2013). A simple example is to select all genome-wide significant SNPs reported in a GWAS (Belsky, Sears, et al. 2013). Although the most common method of hypothesis-free variant selection for a genetic risk score is GWAS of SNPs (which are common DNA sequence variants), whole-genome sequencing studies that analyzer are-variants can also be used (Purcell et al. 2014). Hybrids of hypothesis-free and hypothesis-driven approaches to constructing genetic risk scores also exist. Results from GWAS can be screened using databases that link variants with genes and genes with biological pathways to construct pathway-specific genetic risk scores. For example, Li and colleagues conducted GWAS of lung function within a cohort of asthma cases, applied pathway analysesd to the subset of SNPs with p-values<10-4, and then constructed a score based on SNPs that met their p-value criterion and were linked with a biologically plausible pathway (X. Li et al. 2013).
In the remainder of this section, we provide an overview of approaches to constructing genetic risk scores from GWAS results. The optimal strategy for constructing a specific genetic risk score will depend on the nature of the GWAS evidence and the data available to construct the score. Here, we highlight a handful of key issues and provide some guidance based on our own experience.
There are two broad classes of approaches to constructing genetic risk scores from GWAS results. One approach is to construct the score from SNPs with strong evidence for association with the phenotype of interest—usually SNPs with p-values<5×10-8. We call this the “top-hits” approach. Top-hits scores are conservative. They include only a subset of the genetic variants that contribute to a phenotype. But they are designed to have a high signal to noise ratio. Some top hits scores are simple counts of risk-associated alleles across a set of SNPs. Some top hits scores incorporate weights that customize the contribution of each SNP to the score. In our view, such weights are most useful when the GWAS phenotype is closely aligned with the phenotype that will be analyzed using the genetic risk score. For example we used published GWAS estimates of SNP effects on BMI to weight SNPs in a genetic risk score for obesity (Belsky et al. 2012; Belsky, Moffitt, Sugden, et al. 2013).
A second approach is to construct the score from all or at least a very large number of measured genetic variants. We call this the “whole-genome” approach. (Whole-genome genetic risk scores are sometimes called “Purcell scores” after the geneticist and statistician Shaun Purcell, who pioneered their use in psychiatric genetics (Purcell et al. 2009).) Whole-genome scores are liberal. They are based on a polygenic model that assumes very large numbers of genomic loci make “infinitesimal” contributions to phenotypic variation (Visscher, Hill, and Wray 2008; Wray, Goddard, and Visscher 2007; Gibson 2011). Whole-genome genetic risk scores seek to capture these infinitesimal contributions by incorporating information from all SNPs in a GWAS that meet some nominal threshold of statistical significance (e.g. p<0. 1) or even the complete set of SNPs analyzed. To minimize the statistical noise generated by including so many SNPs (many of which will not be truly related to the phenotype), whole-genome genetic risk scores weight each SNP by the effect-size estimated in a GWAS (Wray, Goddard, and Visscher 2007). The highly inclusive approach of whole-genome genetic risk scores has been met with skepticism. But there is now evidence that the whole-genome approach to constructing a genetic risk score captures more signal than noise and in many cases outperforms genetic risk scores computed from top hits (Dudbridge 2013; Evans, Visscher, and Wray 2009).
The first task in constructing a genetic risk score from GWAS results is to select the GWAS(s) that will form the basis of the score. Now that genome-wide data and computational tools for analyzing them are widely available, GWAS studies are proliferating and some are of higher quality than others. A brief primer on how to evaluate a GWAS can be found here (Pearson and Manolio 2008). A more in depth overview along with detailed discussion of theoretical issues is here (Bush and Moore 2012). A further consideration beyond GWAS quality is to match the racial/ethnic population in the GWAS to the target population for the genetic risk score analysis. We discuss this issue in detail elsewhere (Belsky, Moffitt, Sugden, et al. 2013; Belsky, Moffitt, and Caspi 2013). Briefly, subtle differences in the structures of genomes between racial/ethnic populations can cause a given GWAS SNP to tag different regions of DNA sequence in different populations (Frazer et al. 2007; Cardon and Palmer 2003). This means that a particular SNP may measure a disease-causing variant in one population, but not in another. Ultimately, the extent to which SNPs discovered in GWAS of one population provide valid measures of risk in another is an empirical question (Domingue, Belsky, et al. 2014). Absent empirical evidence, we encourage matching the race/ethnicity of the GWAS population to the population that will be analyzed using the genetic risk score.
After GWAS have been selected, the primary decision point in constructing a genetic risk score is what p-value threshold should be used to select SNPs for inclusion in the score. For top-hits scores, genome-wide significance is an appropriate starting point. For whole-genome genetic risk scores, studies often test a series of p-value thresholds. Scores are generated from sets of SNPs selected using increasingly liberal p-value cut-offs until maximum predictive power in the training dataset is achieved. (Ideally, the GWAS sample that provides the p-values and weights for a whole-genome genetic risk score should be different from the sample used to test p-value thresholds.) The optimal p-value threshold is a function of the genetic architecture (Gibson 2011; Flint and Kendler 2014) of the trait being analyzed and the statistical power of the GWAS. In cases of highly polygenic traits where most genetic effects are very small, more inclusive p-value thresholds will yield better score performance. In studies of psychiatric phenotypes, where most whole-genome genetic risk score research has been conducted, increasingly liberal p-value thresholds improve genetic risk score performance up to a certain level, after which score performance plateaus or sometimes modestly declines, e.g. (Ripke et al. 2013; Demirkan et al. 2011; Rietveld et al. 2013). The p-value level at which score performance reaches a plateau is negatively correlated with GWAS power. This can be seen in the successive iterations of the Psychiatric Genomics Consortium’s GWAS of schizophrenia. In the first iteration, including about 7,000 individuals, the performance of a genetic risk score (measured as Nagelkereke’s Pseudo R2) increased as the p-value threshold for inclusion was relaxed from p<0. 1 to p<0.5, achieving a peak performance of between R2=0.02 and 0.03 across replication datasets (Purcell et al. 2009). In the second iteration, including 15,000 individuals, score performance plateaued at a p-value threshold of 0.2, with maximum R2 ranging between 0.03 and 0.06 across replication datasets (Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium 2011). In the most recent iteration, including 21,000 individuals, score performance plateaued at a p-value threshold of 0.1, with maximum R2 exceeding 0.06 (Ripke et al. 2013).
A secondary decision point in constructing genetic risk scores is the linkage threshold used to define whether selected SNPs provide independent information. GWAS chips include many SNPs with highly correlated allele frequencies. SNPs that covary in excess of chance are referred to as being in “linkage disequilibrium” (LD). SNPs that are spatially proximate on the genome are often in high LD, but LD can extend over large expanses of sequence. In the context of GWAS, LD is typically quantified using the metric R2, which is computed as the squared correlation of genotypes between a pair of SNPs. Just as a p-value must be set to determine which SNPs are associated with the phenotype, an R2 value must be set to establish which SNPs are independent and which are redundant. The R2 threshold must balance two competing threats to score quality. One threat is over-representation of regions characterized by high LD and dense GWAS coverage. In such regions, a single causal variant could give rise to a large number of strong association signals all providing essentially the same information. Too liberal an R2 threshold will result in over-counting risk alleles in the region. The other threat is the presence of multiple causal variants within a given genomic region (sometimes referred to as “allelic heterogeneity”) (Lango Allen et al. 2010; McClellan and King 2010). For example in the case of smoking, there is evidence for multiple causal signals within the region on chromosome 15 harboring the CHRNA5/A3/B4 gene cluster (Saccone et al. 2010; Liu et al. 2010). Too conservative an R2 threshold would under-count risk alleles in this region. In general, R2 thresholds for top-hits genetic risk scores tend to be relatively liberal. We used a threshold of R2<0.60 in constructing genetic risk scores for smoking and for asthma (Belsky, Sears, et al. 2013; Belsky, Moffitt, Baker, et al. 2013). Others have used thresholds as high as 0.80 (Milton et al. 2014). We discuss the issue in more detail in a previous article (Belsky, Moffitt, Sugden, et al. 2013). R2 thresholds for whole-genome genetic risk scores tend to be more conservative. R2<0.20 is standard (Ripke et al. 2013), although we are not aware of empirical examinations of this threshold.
The guidance we offer here is intended as a starting point. Constructing genetic risk scores involves multiple decision points and gold standard practices have not been established. Top hits genetic risk scores, especially to the extent that they are composed of SNPs reaching genome-wide significance in multiple samples and that are in relatively weak LD, are relatively easy to implement. Whole genome-genetic risk scores require more technical sophistication and are more sensitive to the quality of the source GWAS. Several international consortia make their GWAS results available for download.e These data are an excellent resource for constructing genome-wide genetic risk scores.
Genetic risk scores in social science research: 3 suggestions
Genetic risk scores have been studied primarily as tools to predict whether apparently healthy individuals will develop disease (Dudbridge 2013; Wray et al. 2013). Despite recent advances in genomic medicine (Manolio 2013), genetic risk assessment for common health conditions remains an area under development. Age-related macular degeneration is one case where genetic risk assessment may already have clinical value (Seddon et al. 2009). And evidence is growing for the application of genetic risk scores in clinical assessments of cardiovascular disease risk (Ganna et al. 2013). But, in general, the potential to use common genetic variants to predict whether an individual will develop a common health condition remains uncertain (Manolio 2010). (For discussion of why genetic variants with well replicated evidence for association with a disease may not aid in clinical prediction, see (Kraft et al. 2009; Jakobsdottir et al. 2009).) Studies that examine how genetic risk scores combine with other risk information to predict disease outcomes will be needed to determine their ultimate clinical utility (Belsky, Moffitt, and Caspi 2013). Parallel to this integration genetic risk information into medicine, social scientists can test how genetic risk scores combine with the elements of their research paradigms to improve understanding of social and behavioral processes and their outcomes. Below we outline three applications of genetic risk scores in social science research.
First, genetic risk scores can be used to study developmental processes. The DNA sequence is fixed a birth. Most genetic research is conducted on large samples of adults. Genetic risk score studies can shed light on what happens in between. For example, we studied how a genetic risk score derived from GWAS of adult BMI related to processes of growth in-utero (measured as birth weight), during infancy, and in childhood, and then tested whether early-life growth mediated genetic influences on the pathogenesis of obesity in adolescence, young adulthood, and middle life (Belsky et al. 2012). We found that children at high genetic risk were born at similar weights to their low genetic risk peers, but subsequently grew more rapidly. In turn, this rapid growth mediated genetic influence on obesity during adolescence through midlife. Building on this finding, other researchers then showed that increased appetite beginning early in childhood was one path through which genetic influences contributed to accelerated weight gain, suggesting a potential intervention target (Llewellyn et al. 2014; Belsky 2014). This same model can be applied to a range of phenotypes. For example, we also studied genetic influences on smoking and asthma in this way (Belsky, Moffitt, Baker, et al. 2013; Belsky, Sears, et al. 2013). And the phenotypes need not be health-related. In the case of genetic discoveries for educational attainment (Rietveld et al. 2013), genetic risk score studies could investigate genetic influences on intermediate phenotypes such as academic performance, in-grade retention, disciplinary actions, etc. and the extent to which these early developmental manifestations mediate genetic influences on degree attainment.
Second, genetic risk scores can be used to study how genetic factors contribute to relationships between behavioral and social processes and health outcomes. For example, cognitive ability and certain personality traits are associated with risk of developing a range of chronic physical and mental health problems (Israel et al. 2014; Meier et al. 2014; Jokela et al. 2011). GWAS have identified many genetic variants predisposing to these chronic physical and mental health problems (Hindorff et al.). Genetic risk score studies can test whether psychological traits function to mediate or modify genetic influences in chronic disease etiology. To our knowledge such studies have not yet been conducted. But proof of concept can be seen in two recent studies showing that genetic risks discovered in GWAS of schizophrenia are related to cognitive ability and cognitive decline (McIntosh et al. 2013; Lencz et al. 2014), although this finding remains contentious (van Scheltinga et al. 2013).
Third, genetic risk scores can be used to study how individuals become exposed to certain environments and what the outcomes of those exposures are, i.e. to study polygenic risk-environment correlations (rGE) and interactions (GxE). rGE occurs where genetic risks are patterned over specific social or physical environments. To our knowledge the only published examples of polygenic rGE deal with the assortment of genetically similar individuals into friendship and spousal pairs (Christakis and Fowler 2014; Domingue, Fletcher, et al. 2014), but there is growing molecular genetic evidence that rGE is a real and an empirically tractable phenomenon (Fowler, Settle, and Christakis 2011; Boardman, Domingue, and Fletcher 2012; Conley et al. 2014). Now that data measuring individuals’ genomes and their the social and physical environments are available, analysis of rGE should be a priority.
GxE occurs where genetic differences between individuals cause variation in their responses to certain social or physical environments (or vice versa). The best documented polygenic GxE is of lifestyle risk factors modifying polygenic risk for obesity; physically active individuals appear to be protected against polygenic risk for obesity, whereas those living a sedentary lifestyle with unhealthy diet appear to be especially susceptible (S. Li et al. 2010; Ahmad et al. 2013 Qi, Li, et al. 2012; Qi, Chu, et al. 2012). A second example is of trauma exposure modifying polygenic risk for smoking (Meyers et al. 2013; Belsky, Moffitt, and Caspi 2013). In these cases and for other emerging GxE findings, further research is needed to refine causal inference about the specific environmental exposures that function to mitigate or amplify genetic risks (Fletcher and Conley 2013; Moffitt, Caspi, and Rutter 2005). In addition, there are opportunities to investigate how genetic risks affect response to social policy interventions (Fletcher 2012) and how genetic influences are shaped by cultural and temporal contexts (Kim et al. 2010; Chiao and Blizinsky 2010; Demerath et al. 2013).
In our view, among the most important contributions to be made from the integration of genetics into social science is improved understanding of the causes and means to treat social gradients in health. We recognize that this is a controversial assertion. The specter of eugenics movement looms large in our scientific history—rightly so we feel. But a healthy regard for past sins need not impede progress, nor should concern for what that progress might yield. If certain genetic risks are concentrated in population strata with high disease burden, this is motivation for research to understand how those genetic risks operate and how they can be mitigated. If genetic risks are not patterned in this way, it is a powerful argument for modifying environments.
In a recent article in the American Journal of Public Health (Belsky, Moffitt, and Caspi 2013), we articulated 3 possible models of how genetic factors contribute to social gradients in health (Figure 2): One possibility is that they do not. We call this model G+E: Genetic and environmental risks make independent contributions to morbidity; social gradients arise from a concentration of environmental risks in socially disadvantaged individuals. A second possibility is that genetic risks may cause social gradients in health directly. We call this model rGE: Genetic risks are unequally distributed in the population; social gradients arise from a concentration of genetic risks in socially disadvantaged individuals. A third possibility is that genetic risks may cause social gradients in health in interaction with environmental risks. We call this model GxE: Genetic risks are amplified by/ act to amplify environmental risks; social gradients arise from synergies between genetic and environmental risks.
Figure 2. Three models of genetic contributions to social gradients in health.
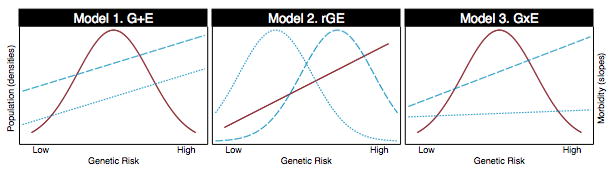
In each panel, the densities graph hypothesized distributions of polygenic risk (left-side y-axis) and the slopes graph hypothesized associations between polygenic risk and morbidity (right-side y-axis). Red lines show cases in which the distribution of polygenic risk (density) or the genetic gradient in disease risk (slope) are the same in socially advantaged and socially disadvantaged population strata. For example, in Models 1 and 3, the distribution of polygenic risk is shared across social strata. Blue lines show where the distribution of polygenic risk or the genetic gradient in disease risk are different in socially advantaged and socially disadvantaged population strata. For example, in Model 2, the distribution of polygenic risk is shifted to the right in the socially disadvantaged population stratum and to the left in the socially advantaged population stratum (the socially disadvantaged population stratum carries a higher burden of genetic risk as compared to the socially advantaged stratum).

These models are testable. GWAS have discovered hundreds of variants associated with the chronic diseases that mediate social gradients in health. Genetic risk scores can translate these discoveries into measurements that map directly to the distributions of genetic risk hypothesized in each of the 3 models in Figure 2. Survey data together with a range of data tracking air pollution (Logue et al. 2014), the built environment (Gordon-Larsen et al. 2006), crime (Gómez et al. 2004), and even more sociologically evocative measures, such as of neighborhood disorder and decay (Odgers et al. 2012), provide an unprecedented view of social disadvantage. Studies that bring these data together are needed to chart a new course for social science and genetics.
Conclusion
The genome represents a unique and valuable source of information for social scientists. Genomic data are becoming available on an unprecedented scale. Analyzing such data has been compared to drinking from a fire hose (Hunter and Kraft 2007). We view genetic risk scores as a means of regulating the flow of genetic information into social science research in a way that is both conceptually appealing and analytically powerful.
Acknowledgments
DWB is supported by grants from the National Institute on Aging (T32 AG000029, P30 AG028716-08). SI is supported by grants from the National Institute of Child Health & Human Development (HD061298 and HD077482) and is grateful to the Yad Hanadiv Rothschild Foundation for the award of a Rothschild Fellowship.
Footnotes
For example, the p-value threshold recommended for detecting deviations from Hardy Weinberg equilibrium, the expected distribution of genotypes given the allele frequency, is 10-4
Population stratification arises when ancestry is associated with a phenotype of interest. There are two approaches to addressing population stratification. One approach is use family-based designs. (Boardman et al. 2013) As second approach is to model population structure, for example using principal components analysis. (Price et al. 2006; Novembre et al. 2008)
Variation in DNA sequence arises through the combination of chromosome segments inherited from mother and father. As a result, spatially proximate DNA sequence variants tend to be highly correlated. GWAS chips assay SNPs selected to “tag” clusters of correlated variants. As a result, a single SNP may be used to measure several or even a few hundred variants.
Pathway analyses mine databases that describe existing knowledge about molecular processes to attach genes and genetic variants to coherent networks reflecting higher-level biological phenomena.
The Social Science Genetic Association Consortium results for educational attainment can be found at http://ssgac.org/Data.php; The Psychiatric Genomics Consortium results for a range of mental disorders can be found at https://pgc.unc.edu/Sharing.php#SharingOpp; Global Lipids Genetics Consortium results for cholesterol traits can be found at http://www.sph.umich.edu/csg/abecasis/public/lipids2013/; GABRIEL Consortium results for asthma can be found at http://www.cng.fr/gabriel/results.html; GIANT Consortium results anthropometric traits can be found at http://www.broadinstitute.org/collaboration/giant/index.php/GIANT_consortium_data_files.
References
- Ahmad S, Rukh G, Varga TV, Ali A, Kurbasic A, Shungin D, Ericson U, et al. Gene × Physical Activity Interactions in Obesity: Combined Analysis of 111,421 Individuals of European Ancestry. PLoS Genet. 2013 Jul 25;9(7) doi: 10.1371/journal.pgen.1003607. e1003607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asghari V, Sanyal S, Buchwaldt S, Paterson A, Jovanovic V, Van Tol HHM. Modulation of Intracellular Cyclic AMP Levels by Different Human Dopamine D4 Receptor Variants. Journal of Neurochemistry. 1995;65(3):1157–1165. doi: 10.1046/j.1471-4159.1995.65031157.x. [DOI] [PubMed] [Google Scholar]
- Aulchenko YS, Ripke S, Isaacs A, van Duijn CM. GenABEL: An R Library for Genome-Wide Association Analysis. Bioinformatics. 2007 May 15;23(10):1294–1296. doi: 10.1093/bioinformatics/btm108. [DOI] [PubMed] [Google Scholar]
- Belsky DW. Appetite for Prevention: Genetics and Developmental Epidemiology Join Forces in Obesity Research. JAMA Pediatrics. 2014 Apr;168(4):309–311. doi: 10.1001/jamapediatrics.2013.5291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsky DW, Moffitt TE, Baker TB, Biddle AK, Evans JP, Harrington H, Houts R, et al. Polygenic Risk and the Developmental Progression to Heavy, Persistent Smoking and Nicotine Dependence: Evidence from a 4-Decade Longitudinal Study. JAMA Psychiatry. 2013 Mar;70(5):534–542. doi: 10.1001/jamapsychiatry.2013.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsky DW, Moffitt TE, Caspi A. Genetics in Population Health Science: Strategies and Opportunities. American Journal of Public Health. 2013 Oct;103(Suppl 1):S73–83. doi: 10.2105/AJPH.2012.301139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsky DW, Moffitt TE, Houts R, Bennett GG, Biddle AK, Blumenthal JA, Evans JP, et al. Polygenic Risk, Rapid Childhood Growth, and the Development of Obesity: Evidence from a 4-Decade Longitudinal Study. Archives of Pediatrics and Adolescent Medicine. 2012;166(6):515–521. doi: 10.1001/archpediatrics.2012.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsky DW, Moffitt TE, Sugden K, Williams B, Houts R, McCarthy J, Caspi A. Development and Evaluation of a Genetic Risk Score for Obesity. Biodemography & Social Biology. 2013 May 23;59(1):85–100. doi: 10.1080/19485565.2013.774628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsky DW, Sears MR, Hancox RJ, Harrington H, Houts R, Moffitt TE, Sugden K, Williams B, Poulton R, Caspi A. Polygenic Risk and the Development and Course of Asthma: An Analysis of Data from a Four-Decade Longitudinal Study. The Lancet Respiratory Medicine. 2013;1(6):453–361. doi: 10.1016/S2213-2600(13)70101-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin DJ, Cesarini D, van der Loos MJHM, Dawes CT, Koellinger PD, Magnusson PKE, Chabris CF, et al. The Genetic Architecture of Economic and Political Preferences. Proceedings of the National Academy of Sciences of the United States of America. 2012 May;109(21):8026–31. doi: 10.1073/pnas.1120666109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boardman JD, Domingue BW, Blalock CL, Haberstick BC, Harris KM, McQueen MB. Is the Gene-Environment Interaction Paradigm Relevant to Genome-Wide Studies? The Case of Education and Body Mass Index. Demography. 2013 Nov 27; doi: 10.1007/s13524-013-0259-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boardman JD, Domingue BW, Fletcher JM. How Social and Genetic Factors Predict Friendship Networks. Proceedings of the National Academy of Sciences of the United States of America. 2012 Oct;109(43):17377–81. doi: 10.1073/pnas.1208975109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdan R, Hyde LW, Hariri AR. A Neurogenetics Approach to Understanding Individual Differences in Brain, Behavior, and Risk for Psychopathology. Molecular Psychiatry. 2013 Mar;18(3):288–99. doi: 10.1038/mp.2012.35. [DOI] [PubMed] [Google Scholar]
- Bush WS, Moore JH. Chapter 11: Genome-Wide Association Studies. PLoS Computational Biology. 2012;8(12) doi: 10.1371/journal.pcbi.1002822. e1002822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardon LR, Palmer LJ. Population Stratification and Spurious Allelic Association. The Lancet. 2003 Feb 15;361(9357):598–604. doi: 10.1016/S0140-6736(03)12520-2. [DOI] [PubMed] [Google Scholar]
- Carey JR, Vaupel JW. Biodemography. In: Poston DL, Micklin M, editors. Handbook of Population, Handbooks of Sociology and Social Research. Springer; US: 2005. pp. 625–658. http://dx.doi.org/10.1007/0-387-23106-4_22. [Google Scholar]
- Chiao JY, Blizinsky KD. Culture–gene Coevolution of Individualism–collectivism and the Serotonin Transporter Gene. Proceedings of the Royal Society B: Biological Sciences. 2010 Feb 22;277(1681):529–537. doi: 10.1098/rspb.2009.1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christakis NA, Fowler JH. Friendship and Natural Selection. Proceedings of the National Academy of Sciences. 2014 Jul 14; doi: 10.1073/pnas.1400825111. 201400825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier DA, Stöber G, Li T, Heils A, Catalano M, Di Bella D, Arranz MJ, et al. A Novel Functional Polymorphism within the Promoter of the Serotonin Transporter Gene: Possible Role in Susceptibility to Affective Disorders. Molecular Psychiatry. 1996 Dec;1(6):453–460. [PubMed] [Google Scholar]
- Conley D, Siegal ML, Domingue B, Mulan Harris K, McQueen M, Boardman J. Testing the Key Assumption of Heritability Estimates Based on Genome-Wide Genetic Relatedness. Journal of Human Genetics. 2014 Mar 6; doi: 10.1038/jhg.2014.14. http://www.nature.com/jhg/journal/vaop/ncurrent/full/jhg201414a.html. [DOI] [PMC free article] [PubMed]
- Crimmins E, Kim JK, Vasunilashorn S. Biodemography: New Approaches to Understanding Trendsand Differences in Population Health and Mortality. Demography. 2010 Mar 1;47(1):S41–S64. doi: 10.1353/dem.2010.0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David SP, Strong DR, Leventhal AM, Lancaster MA, McGeary JE, Munafò MR, Bergen AW, et al. Influence of a Dopamine Pathway Additive Genetic Efficacy Score on Smoking Cessation: Results from Two Randomized Clinical Trials of Bupropion. Addiction. 2013;108(12):2202–2211. doi: 10.1111/add.12325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demerath EW, Choh AC, Johnson W, Curran JE, Lee M, Bellis C, Dyer TD, Czerwinski SA, Blangero J, Towne B. The Positive Association of Obesity Variants with Adulthood Adiposity Strengthens over an 80-Year Period: A Gene-by-Birth Year Interaction. Human Heredity. 2013;75(2-4):175–185. doi: 10.1159/000351742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demirkan A, Penninx BWJH, Hek K, Wray NR, Amin N, Aulchenko YS, van Dyck R, et al. Genetic Risk Profiles for Depression and Anxiety in Adult and Elderly Cohorts. Molecular Psychiatry. 2011 Jul;16(7):773–83. doi: 10.1038/mp.2010.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingue BW, Belsky DW, Harris KM, Smolen A, McQueen MB, Boardman JD. Polygenic Risk Predicts Obesity in Both White and Black Young Adults. PLoS ONE. 2014 Jul 3;9(7) doi: 10.1371/journal.pone.0101596. e101596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingue BW, Fletcher J, Conley D, Boardman JD. Genetic and Educational Assortative Mating among US Adults. Proceedings of the National Academy of Sciences. 2014 May 19; doi: 10.1073/pnas.1321426111. 201321426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudbridge F. Power and Predictive Accuracy of Polygenic Risk Scores. PLoS Genetics. 2013 Mar;9(3):e1003348. doi: 10.1371/journal.pgen.1003348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans DM, Visscher PM, Wray NR. Harnessing the Information Contained within Genome-Wide Association Studies to Improve Individual Prediction of Complex Disease Risk. Human Molecular Genetics. 2009 Sep 15;18(18):3525–31. doi: 10.1093/hmg/ddp295. [DOI] [PubMed] [Google Scholar]
- Fletcher JM. Why Have Tobacco Control Policies Stalled? Using Genetic Moderation to Examine Policy Impacts. PloS One. 2012;7(12):e50576. doi: 10.1371/journal.pone.0050576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher JM, Conley D. The Challenge of Causal Inference in Gene–Environment Interaction Research: Leveraging Research Designs From the Social Sciences. American Journal of Public Health. 2013 Oct;103(S1):S42–S45. doi: 10.2105/AJPH.2013.301290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint J, Kendler KS. The Genetics of Major Depression. Neuron. 2014 Feb 5;81(3):484–503. doi: 10.1016/j.neuron.2014.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg LA, Absher D, Dumanski JP. Non-Heritable Genetics of Human Disease: Spotlight on Post-Zygotic Genetic Variation Acquired during Lifetime. Journal of Medical Genetics. 2013 Jan;50(1):1–10. doi: 10.1136/jmedgenet-2012-101322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler JH, Settle JE, Christakis NA. Correlated Genotypes in Friendship Networks. Proceedings of the National Academy of Sciences. 2011 Feb 1;108(5):1993–1997. doi: 10.1073/pnas.1011687108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, et al. A Second Generation Human Haplotype Map of over 3.1 Million SNPs. Nature. 2007 Oct 18;449(7164):851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganna A, Magnusson PKE, Pedersen NL, de Faire U, Reilly M, Arnlöv J, Sundström J, Hamsten A, Ingelsson E. Multilocus Genetic Risk Scores for Coronary Heart Disease Prediction. Arteriosclerosis, Thrombosis, and Vascular Biology. 2013 May 16; doi: 10.1161/ATVBAHA.113.301218. [DOI] [PubMed] [Google Scholar]
- Gibson G. Rare and Common Variants: Twenty Arguments. Nature Reviews Genetics. 2011 Feb;13(2):135–45. doi: 10.1038/nrg3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez JE, Johnson BA, Selva M, Sallis JF. Violent Crime and Outdoor Physical Activity among Inner-City Youth. Preventive Medicine. 2004 Nov;39(5):876–881. doi: 10.1016/j.ypmed.2004.03.019. [DOI] [PubMed] [Google Scholar]
- Gordon-Larsen P, Nelson MC, Page P, Popkin BM. Inequality in the Built Environment Underlies Key Health Disparities in Physical Activity and Obesity. Pediatrics. 2006 Feb 1;117(2):417–424. doi: 10.1542/peds.2005-0058. [DOI] [PubMed] [Google Scholar]
- Hamer D, Sirota L. Beware the Chopsticks Gene. Molecular Psychiatry. 2000 Jan;5(1):11–13. doi: 10.1038/sj.mp.4000662. [DOI] [PubMed] [Google Scholar]
- Hariri AR. The Neurobiology of Individual Differences in Complex Behavioral Traits. Annual Review of Neuroscience. 2009;32:225–47. doi: 10.1146/annurev.neuro.051508.135335. 19400720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert A, Gerry NP, McQueen MB, Heid IM, Pfeufer A, Illig T, Wichmann HE, et al. A Common Genetic Variant Is Associated with Adult and Childhood Obesity. Science. 2006 Apr 14;312:279–83. doi: 10.1126/science.1124779. 16614226. [DOI] [PubMed] [Google Scholar]
- Hindorff LA, Junkins HA, Mehta JP, Manolio TA. A Catalog of Published Genome-Wide Association Studies. http://www.genome.gov/gwastudies/
- Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential Etiologic and Functional Implications of Genome-Wide Association Loci for Human Diseases and Traits. Proc Natl Acad Sci U S A. 2009 Jun 9;106:9362–7. doi: 10.1073/pnas.0903103106. 19474294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschhorn JN, Daly MJ. Genome-Wide Association Studies for Common Diseases and Complex Traits. Nature Reviews Genetics. 2005 Feb;6(2):95–108. doi: 10.1038/nrg1521. [DOI] [PubMed] [Google Scholar]
- Hunter DJ, Kraft P. Drinking from the Fire Hose--Statistical Issues in Genomewide Association Studies. New England Journal of Medicine. 2007 Aug 2;357:436–9. doi: 10.1056/NEJMp078120. 17634446. [DOI] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural Mechanisms of Addiction: The Role of Reward-Related Learning and Memory. Annual Review of Neuroscience. 2006;29(1):565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Israel S, Lerer E, Shalev I, Uzefovsky F, Riebold M, Laiba E, Bachner-Melman R, et al. The Oxytocin Receptor (OXTR) Contributes to Prosocial Fund Allocations in the Dictator Game and the Social Value Orientations Task. PloS One. 2009;4(5):e5535. doi: 10.1371/journal.pone.0005535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel S, Moffitt TE, Belsky DW, Hancox RJ, Poulton R, Roberts B, Murray W, Caspi A. Translating Personality Psychology to Help Personalize Preventive Medicine for Young Adult Patients. Journal of Personality and Social Psychology. 2014;106(3):484–498. doi: 10.1037/a0035687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsdottir J, Gorin MB, Conley YP, Ferrell RE, Weeks DE. Interpretation of Genetic Association Studies: Markers with Replicated Highly Significant Odds Ratios May Be Poor Classifiers. PLoS Genetics. 2009 Feb;5(2):e1000337. doi: 10.1371/journal.pgen.1000337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jokela M, Batty GD, Deary IJ, Silventoinen K, Kivimäki M. Sibling Analysis of Adolescent Intelligence and Chronic Diseases in Older Adulthood. Annals of Epidemiology. 2011 Jul;21(7):489–496. doi: 10.1016/j.annepidem.2011.01.008. [DOI] [PubMed] [Google Scholar]
- Kathiresan S, Melander O, Anevski D, Guiducci C, Burtt NP, Roos C, Hirschhorn JN, et al. Polymorphisms Associated with Cholesterol and Risk of Cardiovascular Events. The New England Journal of Medicine. 2008 Mar 20;358(12):1240–9. doi: 10.1056/NEJMoa0706728. [DOI] [PubMed] [Google Scholar]
- Kilpelainen TO, Qi L, Brage S, Sharp SJ, Sonestedt E, Demerath E, Ahmad T, et al. Physical Activity Attenuates the Influence of FTO Variants on Obesity Risk: A Meta-Analysis of 218,166 Adults and 19,268 Children. PLoS Medicine. 2011 Nov;8:e1001116. doi: 10.1371/journal.pmed.1001116. 22069379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Sherman DK, Sasaki JY, Xu J, Chu TQ, Ryu C, Suh EM, Graham K, Taylor SE. Culture, Distress, and Oxytocin Receptor Polymorphism (OXTR) Interact to Influence Emotional Support Seeking. Proceedings of the National Academy of Sciences. 2010 Sep 7;107(36):15717–15721. doi: 10.1073/pnas.1010830107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Volkow ND. Neurocircuitry of Addiction. Neuropsychopharmacology : Official Publication of the American College of Neuropsychopharmacology. 2010 Jan;35:217–38. doi: 10.1038/npp.2009.110. 19710631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft P, Wacholder S, Cornelis M, Hu FB, Hayes RB, Thomas G, Hoover R, Hunter DJ, Chanock S. Beyond Odds Ratios - Communicating Disease Risk Based on Genetic Profiles. Nature Reviews Genetics. 2009;10:264–269. doi: 10.1038/nrg2516. [DOI] [PubMed] [Google Scholar]
- Lango Allen H, Estrada K, Lettre G, Berndt SI, Weedon MN, Rivadeneira F, Willer CJ, et al. Hundreds of Variants Clustered in Genomic Loci and Biological Pathways Affect Human Height. Nature. 2010 Oct 14;467:832–8. doi: 10.1038/nature09410. 20881960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurie CC, Doheny KF, Mirel DB, Pugh EW, Bierut LJ, Bhangale T, Boehm F, et al. Quality Control and Quality Assurance in Genotypic Data for Genome-Wide Association Studies. Genetic Epidemiology. 2010 Sep;34(6):591–602. doi: 10.1002/gepi.20516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Wray NR, Goddard ME, Visscher PM. Estimating Missing Heritability for Disease from Genome-Wide Association Studies. American Journal of Human Genetics. 2011 Mar 11;88:294–305. doi: 10.1016/j.ajhg.2011.02.002. 21376301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lencz T, Knowles E, Davies G, Guha S, Liewald DC, Starr JM, Djurovic S, et al. Molecular Genetic Evidence for Overlap between General Cognitive Ability and Risk for Schizophrenia: A Report from the Cognitive Genomics consorTium (COGENT) Molecular Psychiatry. 2014 Feb;19(2):168–174. doi: 10.1038/mp.2013.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesch KP, Bengel D, Heils A, Sabol SZ, Greenberg BD, Petri S, Benjamin J, Muller CR, Hamer DH, Murphy DL. Association of Anxiety-Related Traits with a Polymorphism in the Serotonin Transporter Gene Regulatory Region. Science. 1996 Nov;274:1527–1531. doi: 10.1126/science.274.5292.1527. ISI:A1996VV77500048. [DOI] [PubMed] [Google Scholar]
- Li S, Zhao JH, Luan J, Ekelund U, Luben RN, Khaw K-T, Wareham NJ, Loos RJF. Physical Activity Attenuates the Genetic Predisposition to Obesity in 20,000 Men and Women from EPIC-Norfolk Prospective Population Study. PLoS Medicine. 2010 Jan;7(8):1–9. doi: 10.1371/journal.pmed.1000332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Hawkins GA, Ampleford EJ, Moore WC, Li H, Hastie AT, Howard TD, et al. Genome-Wide Association Study Identifies TH1 Pathway Genes Associated with Lung Function in Asthmatic Patients. The Journal of Allergy and Clinical Immunology. 2013 Aug;132(2):313–320.e15. doi: 10.1016/j.jaci.2013.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JZ, Tozzi F, Waterworth DM, Pillai SG, Muglia P, Middleton L, Berrettini W, et al. Meta-Analysis and Imputation Refines the Association of 15q25 with Smoking Quantity. Nature Genetics. 2010 May;42:436–40. doi: 10.1038/ng.572. 20418889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llewellyn CH, Trzaskowski M, van Jaarsveld CHM, Plomin R, Wardle J. Satiety Mechanisms in Genetic Risk of Obesity. JAMA Pediatrics. 2014 Feb 17; doi: 10.1001/jamapediatrics.2013.4944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue JM, Klepeis NE, Lobscheid AB, Singer BC. Pollutant Exposures from Natural Gas Cooking Burners: A Simulation-Based Assessment for Southern California. Environmental Health Perspectives. 2014 Jan 1;122(1):43–50. doi: 10.1289/ehp.1306673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA. Cohort Studies and the Genetics of Complex Disease. Nature Genetics. 2009 Jan;41(1):5–6. doi: 10.1038/ng0109-5. 19112455. [DOI] [PubMed] [Google Scholar]
- Manolio TA. Genomewide Association Studies and Assessment of the Risk of Disease. New England Journal of Medicine. 2010 Jul 8;363:166–76. doi: 10.1056/NEJMra0905980. 20647212. [DOI] [PubMed] [Google Scholar]
- Manolio TA. Bringing Genome-Wide Association Findings into Clinical Use. Nature Reviews Genetics. 2013 Aug;14(8):549–558. doi: 10.1038/nrg3523. [DOI] [PubMed] [Google Scholar]
- McClellan J, King M-C. Genetic Heterogeneity in Human Disease. Cell. 2010 Apr;141(2):210–7. doi: 10.1016/j.cell.2010.03.032. [DOI] [PubMed] [Google Scholar]
- McIntosh AM, Gow A, Luciano M, Davies G, Liewald DC, Harris SE, Corley J, et al. Polygenic Risk for Schizophrenia Is Associated with Cognitive Change Between Childhood and Old Age. Biological Psychiatry. 2013 Feb;:1–6. doi: 10.1016/j.biopsych.2013.01.011. [DOI] [PubMed] [Google Scholar]
- Meier MH, Caspi A, Reichenberg A, Keefe RSE, Fisher HL, Harrington H, Houts R, Poulton R, Moffitt TE. Neuropsychological Decline in Schizophrenia From the Premorbid to the Postonset Period: Evidence From a Population-Representative Longitudinal Study. American Journal of Psychiatry. 2014 Jan 1;171(1):91–101. doi: 10.1176/appi.ajp.2013.12111438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meigs JB, Shrader P, Sullivan LM, McAteer JB, Fox CS, Dupuis J, Manning AK, et al. Genotype Score in Addition to Common Risk Factors for Prediction of Type 2 Diabetes. New England Journal of Medicine. 2008 Nov;359:2208–2219. doi: 10.1056/NEJMoa0804742. ISI:000260994000004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers JL, Cerdá M, Galea S, Keyes KM, Aiello AE, Uddin M, Wildman DE, Koenen KC. Interaction between Polygenic Risk for Cigarette Use and Environmental Exposures in the Detroit Neighborhood Health Study. Translational Psychiatry. 2013 Aug 13;3(8):e290. doi: 10.1038/tp.2013.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton JN, Gordeuk VR, Taylor JG, Gladwin MT, Steinberg MH, Sebastiani P. Prediction of Fetal Hemoglobin in Sickle Cell Anemia Using an Ensemble of Genetic Risk Prediction Models. Circulation: Cardiovascular Genetics. 2014 Mar 1; doi: 10.1161/CIRCGENETICS.113.000387. CIRCGENETICS.113.000387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffatt MF, Kabesch M, Liang L, Dixon AL, Strachan D, Heath S, Depner M, et al. Genetic Variants Regulating ORMDL3 Expression Contribute to the Risk of Childhood Asthma. Nature. 2007 Jul;448(7152):470–3. doi: 10.1038/nature06014. [DOI] [PubMed] [Google Scholar]
- Moffitt TE, Caspi A, Rutter M. Strategy for Investigating Interactions between Measured Genes and Measured Environments. Archives of General Psychiatry. 2005 May;62(5):473–81. doi: 10.1001/archpsyc.62.5.473. [DOI] [PubMed] [Google Scholar]
- Morrison AC, Bare La, Chambless LE, Ellis SG, Malloy M, Kane JP, Pankow JS, Devlin JJ, Willerson JT, Boerwinkle E. Prediction of Coronary Heart Disease Risk Using a Genetic Risk Score: The Atherosclerosis Risk in Communities Study. American Journal of Epidemiology. 2007 Jul 1;166(1):28–35. doi: 10.1093/aje/kwm060. [DOI] [PubMed] [Google Scholar]
- Nikolova YS, Ferrell RE, Manuck SB, Hariri AR. Multilocus Genetic Profile for Dopamine Signaling Predicts Ventral Striatum Reactivity. Neuropsychopharmacology : Official Publication of the American College of Neuropsychopharmacology. 2011 Aug;36(9):1940–7. doi: 10.1038/npp.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novembre J, Johnson T, Bryc K, Kutalik Z, Boyko AR, Auton A, Indap A, et al. Genes Mirror Geography within Europe. Nature. 2008 Nov 6;456(7218):98–101. doi: 10.1038/nature07331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odgers CL, Caspi A, Bates CJ, Sampson RJ, Moffitt TE. Systematic Social Observation of Children’s Neighborhoods Using Google Street View: A Reliable and Cost-Effective Method. Journal of Child Psychology and Psychiatry, and Allied Disciplines. 2012 Oct;53(10):1009–17. doi: 10.1111/j.1469-7610.2012.02565.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson TA, Manolio TA. How to Interpret a Genome-Wide Association Study. JAMA. 2008 Mar 19;299:1335–44. doi: 10.1001/jama.299.11.1335. 18349094. [DOI] [PubMed] [Google Scholar]
- Plomin R, DeFries JC, McClearn GE, McGuffin P. Behavioral Genetics. 5. New York: Worth Publishers; 2008. [Google Scholar]
- Plomin R, Haworth CMa, Davis OSP. Common Disorders Are Quantitative Traits. Nature Reviews Genetics. 2009 Dec;10(12):872–8. doi: 10.1038/nrg2670. [DOI] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal Components Analysis Corrects for Stratification in Genome-Wide Association Studies. Nature Genetics. 2006 Aug;38:904–909. doi: 10.1038/ng1847. ISI:000239325700019. [DOI] [PubMed] [Google Scholar]
- Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, O’Dushlaine C, et al. A Polygenic Burden of Rare Disruptive Mutations in Schizophrenia. Nature. 2014 Jan 22; doi: 10.1038/nature12975. advance online publication, http://www.nature.com/nature/journal/vaop/ncurrent/full/nature12975.html?utm_content=buffer77ff4&utm_medium=social&utm_source=twitter.com&utm_campaign=buffer. [DOI] [PMC free article] [PubMed]
- Purcell SM, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. American Journal of Human Genetics. 2007 Sep;81:559–575. doi: 10.1086/519795. ISI:000249128200012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, Sullivan PF, Sklar P. Common Polygenic Variation Contributes to Risk of Schizophrenia and Bipolar Disorder. Nature. 2009 Aug 6;460(7256):748–52. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Q, Chu AY, Kang JH, Jensen MK, Curhan GC, Pasquale LR, Ridker PM, et al. Sugar-Sweetened Beverages and Genetic Risk of Obesity. The New England Journal of Medicine. 2012 Oct;367(15):1387–96. doi: 10.1056/NEJMoa1203039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Q, Li Y, Chomistek AK, Kang JH, Curhan GC, Pasquale LR, Willett WC, Rimm EB, Hu FB, Qi L. Television Watching, Leisure Time Physical Activity, and the Genetic Predisposition in Relation to Body Mass Index in Women and Men. Circulation. 2012 Oct 9;126(15):1821–1827. doi: 10.1161/CIRCULATIONAHA.112.098061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rietveld Ca, Medland SE, Derringer J, Yang J, Esko T, Martin NW, Westra H-J, et al. GWAS of 126,559 Individuals Identifies Genetic Variants Associated with Educational Attainment. Science (New York, N Y) 2013 Jul;340(6139):1467–71. doi: 10.1126/science.1235488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripatti S, Tikkanen E, Orho-Melander M, Havulinna AS, Silander K, Sharma A, Guiducci C, et al. A Multilocus Genetic Risk Score for Coronary Heart Disease: Case-Control and Prospective Cohort Analyses. Lancet. 2010 Oct 23;376(9750):1393–400. doi: 10.1016/S0140-6736(10)61267-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke S, O’Dushlaine C, Chambert K, Moran JL, Kähler AK, Akterin S, Bergen SE, et al. Genome-Wide Association Analysis Identifies 13 New Risk Loci for Schizophrenia. Nature Genetics. 2013 Oct;45(10):1150–1159. doi: 10.1038/ng.2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccone NL, Culverhouse RC, Schwantes-An T-H, Cannon DS, Chen X, Cichon S, Giegling I, et al. Multiple Independent Loci at Chromosome 15q25.1 Affect Smoking Quantity: A Meta-Analysis and Comparison with Lung Cancer and COPD. PLoS Genetics. 2010 Aug;6(8) doi: 10.1371/journal.pgen.1001053. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2916847&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanScheltinga aFT, Bakker SC, vanHaren NEM, Derks EM, Buizer-Voskamp JE, Cahn W, Ripke S, Ophoff Ra, Kahn RS. Schizophrenia Genetic Variants Are Not Associated with Intelligence. Psychological Medicine. 2013 Feb;:1–8. doi: 10.1017/S0033291713000196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium. Genome-Wide Association Study Identifies Five New Schizophrenia Loci. Nature Genetics. 2011 Oct;43(10):969–976. doi: 10.1038/ng.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Susser E. Genome-Wide Association Studies: Does Only Size Matter? American Journal of Psychiatry. 2010 Jul 1;167(7):741–744. doi: 10.1176/appi.ajp.2010.10030465. [DOI] [PubMed] [Google Scholar]
- Seddon JM, Reynolds R, Maller J, Fagerness Ja, Daly MJ, Rosner B. Prediction Model for Prevalence and Incidence of Advanced Age-Related Macular Degeneration Based on Genetic, Demographic, and Environmental Variables. Investigative Ophthalmology & Visual Science. 2009 May;50(5):2044–53. doi: 10.1167/iovs.08-3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalev I, Lerer E, Israel S, Uzefovsky F, Gritsenko I, Mankuta D, Ebstein RP, Kaitz M. BDNF Val66Met Polymorphism Is Associated with HPA Axis Reactivity to Psychological Stress Characterized by Genotype and Gender Interactions. Psychoneuroendocrinology. 2009 Apr;34(3):382–388. doi: 10.1016/j.psyneuen.2008.09.017. [DOI] [PubMed] [Google Scholar]
- Smemo S, Tena JJ, Kim K-H, Gamazon ER, Sakabe NJ, Gómez-Marín C, Aneas I, et al. Obesity-Associated Variants within FTO Form Long-Range Functional Connections with IRX3. Nature. 2014 Mar 12; doi: 10.1038/nature13138. advance online publication, http://www.nature.com/nature/journal/vaop/ncurrent/full/nature13138.html. [DOI] [PMC free article] [PubMed]
- Spycher BD, Henderson J, Granell R, Evans DM, Smith GD, Timpson NJ, Sterne JaC. Genome-Wide Prediction of Childhood Asthma and Related Phenotypes in a Longitudinal Birth Cohort. The Journal of Allergy and Clinical Immunology. 2012 Aug;130(2):503–9.e7. doi: 10.1016/j.jaci.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stice E, Yokum S, Burger K, Epstein L, Smolen A. Multilocus Genetic Composite Reflecting Dopamine Signaling Capacity Predicts Reward Circuitry Responsivity. The Journal of Neuroscience. 2012 Jul 18;32(29):10093–10100. doi: 10.1523/JNEUROSCI.1506-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PF. The Psychiatric GWAS Consortium: Big Science Comes to Psychiatry. Neuron. 2010 Oct 21;68(2):182–186. doi: 10.1016/j.neuron.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PF, Daly MJ, O’Donovan M. Genetic Architectures of Psychiatric Disorders: The Emerging Picture and Its Implications. Nature Reviews Genetics. 2012 Aug;13(8):537–51. doi: 10.1038/nrg3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tol HHMV, Wu CM, Guan H-C, Ohara K, Bunzow JR, Civelli O, Kennedy J, Seeman P, Niznik HB, Jovanovic V. Multiple Dopamine D4 Receptor Variants in the Human Population. Nature. 1992 Jul 9;358(6382):149–152. doi: 10.1038/358149a0. [DOI] [PubMed] [Google Scholar]
- Uhl GR, Walther D, Musci R, Fisher C, Anthony JC, Storr CL, Behm FM, Eaton WW, Ialongo N, Rose JE. Smoking Quit Success Genotype Score Predicts Quit Success and Distinct Patterns of Developmental Involvement with Common Addictive Substances. Molecular Psychiatry. 2012 Nov;(May):1–5. doi: 10.1038/mp.2012.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visscher PM, Hill WG, Wray NR. Heritability in the Genomics Era--Concepts and Misconceptions. Nature Reviews Genetics. 2008 Apr;9:255–66. doi: 10.1038/nrg2322. 18319743. [DOI] [PubMed] [Google Scholar]
- Vrieze SI, McGue M, Iacono WG. The Interplay of Genes and Adolescent Development in Substance Use Disorders: Leveraging Findings from GWAS Meta-Analyses to Test Developmental Hypotheses about Nicotine Consumption. Human Genetics. 2012 Jun;131(6):791–801. doi: 10.1007/s00439-012-1167-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardle J, Carnell S, Haworth CMA, Farooqi IS, O’Rahilly S, Plomin R. Obesity Associated Genetic Variation in FTO Is Associated with Diminished Satiety. Journal of Clinical Endocrinology & Metabolism. 2008 Sep 1;93(9):3640–3643. doi: 10.1210/jc.2008-0472. [DOI] [PubMed] [Google Scholar]
- Wray NR, Goddard ME, Visscher PM. Prediction of Individual Genetic Risk to Disease from Genome-Wide Association Studies. Genome Research. 2007 Oct;17(10):1520–8. doi: 10.1101/gr.6665407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray NR, Yang J, Hayes BJ, Price AL, Goddard ME, Visscher PM. Pitfalls of Predicting Complex Traits from SNPs. Nature Reviews Genetics. 2013 Jun;14(7):507–515. doi: 10.1038/nrg3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Gwinn M, Clyne M, Yesupriya A, Khoury MJ. A Navigator for Human Genome Epidemiology. Nat Genet. 2008 Feb;40:124–5. doi: 10.1038/ng0208-124. 18227866. [DOI] [PubMed] [Google Scholar]
- Zhao J, Jiang C, Lam TH, Liu B, Cheng KK, Xu L, Yeung SLA, Zhang W, Leung GM, Schooling CM. Genetically Predicted Testosterone and Cardiovascular Risk Factors in Men: A Mendelian Randomization Analysis in the Guangzhou Biobank Cohort Study. International Journal of Epidemiology. 2014 Feb 1;43(1):140–148. doi: 10.1093/ije/dyt239. [DOI] [PubMed] [Google Scholar]
- Zuk O, Hechter E, Sunyaev SR, Lander ES. The Mystery of Missing Heritability: Genetic Interactions Create Phantom Heritability. Proceedings of the National Academy of Sciences of the United States of America. 2012 Jan;109(4):1193–8. doi: 10.1073/pnas.1119675109. [DOI] [PMC free article] [PubMed] [Google Scholar]