

SUMMARY
Electron flux in the mitochondrial electron transport chain is determined by the superassembly of mitochondrial respiratory complexes. Different superassemblies are dedicated to receive electrons derived from NADH or FADH2, allowing cells to adapt to the particular NADH/FADH2 ratio generated from available fuel sources. When several fuels are available, cells adapt to the fuel best suited to their type or functional status (e.g., quiescent versus proliferative). We show that an appropriate proportion of superassemblies can be achieved by increasing CII activity through phosphorylation of the complex II catalytic subunit FpSDH. This phosphorylation is mediated by the tyrosine-kinase Fgr, which is activated by hydrogen peroxide. Ablation of Fgr or mutation of the FpSDH target tyrosine abolishes the capacity of mitochondria to adjust metabolism upon nutrient restriction, hypoxia/reoxygenation, and T cell activation, demonstrating the physiological relevance of this adaptive response.
INTRODUCTION
To utilize fuels efficiently, cells must exquisitely integrate the activities of membrane receptors and transporters, the intracellular compartmentalization of molecules, the enzymatic balance of each metabolic step, and the elimination of byproducts (Stanley et al., 2013). Appropriate orchestration of all these changes is critical for the cell’s ability to adapt to changing functional requirements, such as quiescence, proliferation, and differentiation, and to environmental changes, including survival in response to diverse insults. Factors known to influence this adaptation include the cellular response to oxygen availability (hypoxia-inducible factors HIF1α and HIF1β); regulators of energy availability such as mammalian target of rapamycin (mTOR), AMP-activated protein kinase, sirtuin, and forkhead box (FOX)O; and mediators of the response to reactive oxygen species (ROS), such as peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α). The involvement of these factors illustrates the interconnection between the use of alternate carbon substrates (carbohydrates, amino acids, fatty acids and ketone bodies) and the cellular response to stress, particularly oxidative stress.
At the core of this process are mitochondria. In response to changes in fuel source, mitochondria must modify their location, structure, and metabolite fluxes in order to balance their contribution to anabolism (lipogenesis and antioxidant defenses from citrate, gluconeogenesis, serine and glycine biosynthesis from pyruvate, nucleotide biosynthesis) and catabolism (TCA cycle, β-oxidation, oxidative phosphorylation). Mitochondria are central to ATP synthesis, redox balance, and ROS production, parameters directly dependent on fuel use. All catabolic processes converge on the mitochondrial electron transport chain (mETC) by supplying electrons in the form of NADH+H+ or FADH2. The relative proportion of electrons supplied via NADH and FADH2 varies with the fuel used; for example, oxidative metabolism of glucose generates a NADH/FADH2 electron ratio of 5, whereas for a typical fatty acid (FA) such as palmitate the ratio is ≈2 (Speijer, 2011).
Our recent work on the dynamic architecture of the mETC reveals that supercomplex formation defines specific pools of CIII, CIV, CoQ, and cyt c for the receipt of electrons derived from NADH or FAD (Lapuente-Brun et al., 2013). Since CIII preferentially interacts with CI, the amount of CI determines the relative availability of CIII for FADH2- or NADH-derived electrons. The regulation of CI stability is thus central to cellular adaptation to fuel availability. A substrate shift from glucose to FA requires greater flux from FAD, and this is achieved by a reorganization of the mETC superstructure in which CI is degraded, releasing CIII to receive FAD-derived electrons (Lapuente-Brun et al., 2013; Stanley et al., 2013). Failure of this adaptation results in the harmful generation of reactive oxygen species (ROS) (Speijer, 2011). The proportion of supercomplexes dedicated to receiving NADH electrons is further dependent on the structure and dynamics of mitochondrial cristae (Cogliati et al., 2013; Lapuente-Brun et al., 2013), so that reducing the number of cristae favors flux from FAD. In agreement with this, ablation of the mitochondrial protease OMA1, which prevents optic atrophy 1 (OPA1)-specific proteolysis and cristae remodeling, impairs FA degradation in mice, resulting in obesity and impaired temperature control (Quirós et al., 2012).
Cells are normally exposed to a mixed supply of fuels, but despite this, cells are often predisposed to preferentially use one source over another, according to their physiological role or status (Stanley et al., 2013). T cells, for example, switch from oxidative to glycolytic metabolism upon activation, coinciding with entry into a proliferative state, and later increase FA oxidation when they differentiate into regulatory T cells. These changes require remodeling of the mETC NADH/FADH2 flux capacity, but how cells regulate this choice of carbon source is not understood.
Here, we show that fuel choice is regulated via tyrosine phosphorylation of complex II (CII) subunit FpSDH, mediated by ROS-activation of the tyrosine kinase Fgr. This activation is required to adjust the level of complex I (CI) to optimize NADH/FADH2 electron use. Our data show this mechanism operating in three physiological situations: upon T lymphocyte activation, in the adaptation of liver and cultured cells to starvation, and in the adaptation of cells to hypoxia/reoxygenation.
RESULTS
Above-Normal CII Activity in Cells Expressing Mutant CI
Our laboratory has isolated mouse cell lines carrying different proportions of a null ND6 mutation (Acín-Pérez et al., 2003): EB2615 (30% mutant mtDNA), E23 (66%), E12 (80%), FG12-1 (95%), and FG23-1 (98%). Mitochondria from ND6 mutants showed reductions in CI proportional to the mutation load (Figure 1A). Interestingly, the lines with the highest mutation loads showed elevated activity of CII (Figure 1B). Similar observations have been reported in human cells derived from patients with CI deficiencies (Cardol et al., 2002; Fan et al., 2008; Majander et al., 1991; Pitkanen and Robinson, 1996) and in CI-deficient mice (Kruse et al., 2008). Increased CII activity was proposed to compensate for impaired activity of CI, but the underlying mechanism is unknown. This compensatory phenomenon is specific for CI-deficient cells, since cells lacking CIII or CIV showed no changes in CII activity (Figure S1A).
Figure 1. Phosphorylation of FpSDH Increases CII Activity in CI-Deficient Cells.
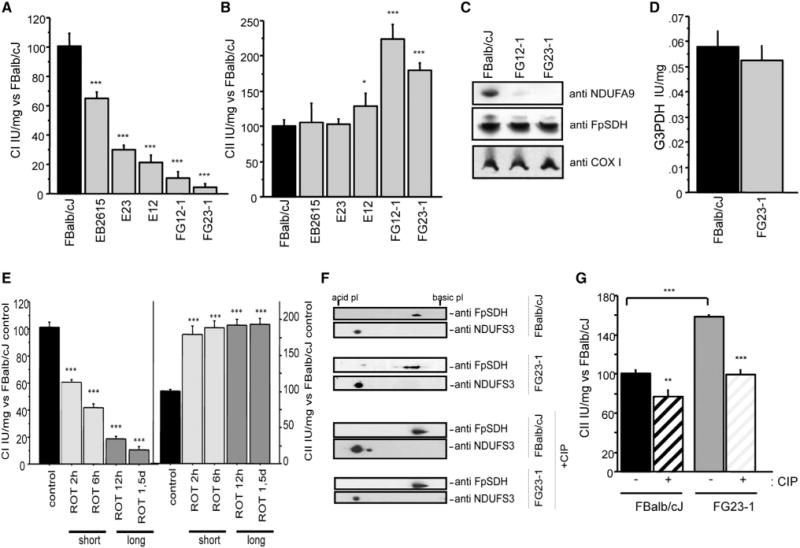
(A) Fibroblast lines with mutations in ND6 show decreased CI activity in proportion to the mutation load. Activity is expressed as the percentage of activity in FBalb/cJ cells (WT, no mutation; n = 12): EB2615 (70%; n = 6); E23 (34%; n = 6); E12 (20%; n = 9); FG12-1 (5%; n = 9); and FG23-1 (2%; n = 9).
(B) CII activity (CoQ reduction) in cell lines with high loads of the iC13887 ND6 mutation, expressed as the percentage of activity in FBalb/cJ cells (n = 18): EB2615 (n = 9); E23 (9 n = 9); E12 (n = 12); FG12-1 (n = 9); and FG23-1 (n = 20).
(C) Western blot after BNGE, detecting subunits of CI (NDUFA9), CII (FpSDH), and CIV (COX I).
(D) G3PDH activity in FG23-1 and FBalb/cJ (n = 6).
(E) Activities of CI (left) and CII (right) in a time course rotenone treatment of FBalb/cJ (200 nM) as a percentage of activity in untreated cells (n ≥ 6). Short: 2 and 6 hr treatment; long: 12 hr and 1.5 day treatment.
(F) FpSDH immunoblot after 2D IEF/SDS-PAGE (IEF strip pH 3–10) of control and calf intestine phosphatase (CIP)-treated mitochondrial protein (100 μg) from FBalb/cJ and FG23-1 cells. The acidic shift of FpSDH spots in FG23-1 cells is blocked by CIP. The CI subunit NDUFS3 was used to align and compare the blots.
(G) CII activity in CIP-treated mitochondria from FBalb/cJ and FG23-1 cells (n = 6). Data are presented as the percentage of activity in untreated FBalb/cJ. **p < 0.001; ***p < 0.0001. Enzyme activities were determined spectrophotometrically.
All data are presented as mean ± SD. See also Figure S1.
As reported (Acín-Pérez et al., 2003; Bai and Attardi, 1998), the amount of CI was reduced in cells with a high ND6 mutant load (FG12-1, FG23-1; Figure 1C, upper panel), whereas CII content was unchanged, despite above-normal activity determined spectrophotometrically (Figure 1B) and by in-gel assay (data not shown). G3PDH activity did not differ between WT and ND6 mutants (Figure 1D), indicating that increased CII activity does not reflect a generalized response of enzymes that donate electrons to CIII and is a specific response to CI deficiency.
As a further control, we treated WT FBalb/cJ cells with the specific inhibitor rotenone (200 nM), which blocks CI activity without affecting CI assembly. Rotenone-mediated inhibition of CI was accompanied by a parallel increase in CII activity, after short and long rotenone treatment, indicating that loss of CI function, not its physical absence, is responsible for the high CII activity (Figure 1E).
CII Activity Is Regulated by Phosphorylation
Several groups have proposed that CII subunit A (FpSDH) is a kinase target (Bykova et al., 2003; Salvi et al., 2007; Schulenberg et al., 2003). To investigate FpSDH phosphorylation, we separated mitochondrial proteins by 2D IEF/SDS-PAGE (isoelectric focusing followed by SDS polyacrylamide gel electrophoresis) and detected FpSDH protein by immunoblot (Figure 1F). The CI subunit NDUFS3 was used as a reference since the stability and migration of this protein are unaffected by the failure of CI assembly (Figure S1B). FpSDH in FG23-1 samples migrated as multiple spots, running at more acidic positions than samples from WT FBalb/cJ cells. This is compatible with increased phosphorylation of the protein in mutant cells. Treatment of permeabilized FG23-1 mitochondria with calf-intestine phosphatase (CIP) to remove phosphoryl residues restored the WT migration pattern, confirming that the altered pattern is due to phosphorylation (Figure 1F, lower panels). Moreover, blockade of CI activity in FBalb/cJ cells reproduced the FpSDH mobility pattern seen in FG23-1 cells (not shown). These observations indicate that FpSDH is phosphorylated when CI activity is impaired. Consistent with these findings, most FpSDH from FG23-1 mitochondrial samples eluted in the phosphorylated fraction after separation on phosphoprotein-enrichment columns, and this effect was blocked by pretreatment of permeabilized mitochondria with CIP (Figure S1C). In contrast, FpSDH from FBalb/cJ mitochondria was more concentrated in nonphosphorylated fractions (Figure S1C). The activity of CII in mitochondria from FBalb/cJ and FG23-1 cells was sensitive to CIP-mediated dephosphorylation, but this reduction was proportionally more severe in FG23-1 mitochondria, suggesting that phosphorylation increases CII activity (Figure 1G).
Phosphorylation of FpSDH Is Triggered by ROS
Cells lacking CI contain high levels of ROS (Robinson, 1998), and ND6-deficient cells produce abundant hydrogen peroxide (Moreno-Loshuertos et al., 2006) (Figure 2A), an effect mimicked by treatment with the CI inhibitor rotenone (Dlasková et al., 2008; Radad et al., 2006). Short and long rotenone treatment of WT cells showed an increase in CII activity (Figure 1E) that was accompanied by a parallel increase in ROS production (Figure S2A). ROS modulate several signaling pathways (Dröge, 2002; Hamanaka and Chandel, 2010), prompting us to evaluate the involvement of ROS in CII activation. Culturing cells for 1 week with N-acetyl cysteine (NAC, 5 mM) lowered basal H2O2 levels and decreased CII activity in control and mutant cells (Figures 2A and 2B), but the reduction was proportionally stronger in FG23-1 cells (Figure 2B). Since NAC is not a direct ROS scavenger but rather a precursor of glutathione, which changes the redox status of the cell, we investigated H2O2-dependent CII activity stimulation in isolated mitochondria. Addition of H2O2 to WT mouse liver mitochondria increased CII activity (Figure 2C) and the phosphorylation of FpSDH (Figure 2D). CII activation was blunted by the presence of catalase, which catalyzes H2O2 decomposition (Figure 2C). As a further control, we analyzed the response to moderate ROS production of fibroblasts lacking supercomplex assembly factor I (SCAFI), which is required for association between CIII and CIV (Lapuente-Brun et al., 2013). The response of WT and SCAFI-deficient fibroblasts to ROS (generated by incubation in the presence of xanthine and xanthine oxidase) was indistinguishable, indicating that ROS-induced upregulation of CII is a general response independent of the assembly of supercomplexes containing CIV (Lapuente-Brun et al., 2013) (Figure 2E). To determine whether CII is activated by superoxide or hydrogen peroxide produced in the mitochondrial matrix, we generated FBalb/cJ- and FG23-1-derived lines expressing mitochondria-targeted catalase (mt-cat) or MnSOD (Figure S2B). ROS production as well as catalase and MnSOD activities measured before and after overexpression confirmed that the cells were expressing functional enzymes (Figures S2C–S2F). Control and CI mutant cells also exhibited basal differences in catalase and MnSOD activities, as previously shown (Moreno-Loshuertos et al., 2006). The expected elevation in H2O2 production and CII activity in FG23-1 cells and rotenone-treated FBalb/cJ cells was decreased by expression of mt-cat, but not MnSOD (Figures 2F and 2G), suggesting that the key activator of the pathway is H2O2 and not superoxide.
Figure 2. FpSDH Phosphorylation and CII Activity Are ROS Dependent.
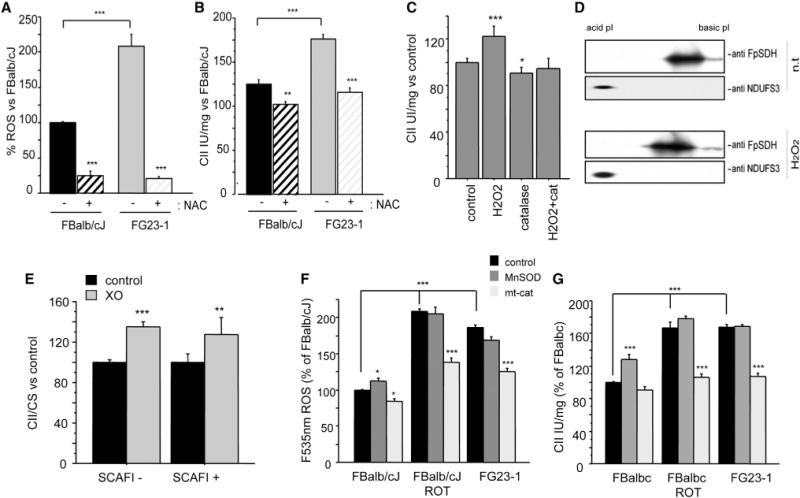
(A) ROS (H2O2) levels measured in nontreated (n.t.) FG23-1 and FBalb/cJ cells and in cells treated with NAC (5 mM, 7 days) (control, n = 35; NAC, n = 12).
(B) Effect of NAC treatment on CII activity in FBalb/cJ and in FG23-1 cells (untreated, n = 15; NAC, n = 4). Data in (A) and (B) are presented as the percentage of nontreated FBalb/cJ cells.
(C) CII activity in isolated mouse liver mitochondria treated for 10 min with H2O2 (50 μM), catalase (25 U/ml), or both (n ≥ 4).
(D) 2D IEF/SDS-PAGE (IEF strips pH 4–7) Western analysis of FpSDH from H2O2-treated mouse liver mitochondria.
(E) CII activity in mitochondria isolated from SCAFI– (left) and SCAFI+ (right) fibroblasts treated with the ROS generator xanthine oxidase (XO) (n = 4). Data are presented as the percentage of activity in nontreated fibroblasts.
(F) H2O2 production in FG23-1 and rotenone-treated FBalb/cJ fibroblasts is prevented by expression of mitochondrially targeted catalase (mt-cat, n = 5).
(G) Elevated CII activity in FG23-1 and rotenone-treated FBalb/cJ cells is prevented by expression of mt-cat but not MnSOD (n = 3). *p < 0.01; **p < 0.001; ***p < 0.0001.
All data are presented as mean ± SD. See also Figure S2.
CII Is Activated by Phosphorylation on FpSDH Mediated by a Src-Type Tyrosine Kinase
We next analyzed immunoprecipitated FpSDH by western blot with anti-phospho-Tyr, anti-phospho-Ser, and anti-phosphoThr antibodies. Only Tyr phosphorylation of FpSDH was detected in FBalb/cJ and FG23-1 cells, but the signal was stronger in FG23-1 cells (Figure 3A). Incubation of isolated mouse liver mitochondria with protein kinase inhibitors revealed that CII activity and FpSDH phosphorylation were reduced by PP2, an inhibitor of Src-family kinases (SFK), whereas no effect was observed with the cAMP/cGMP-dependent protein kinase inhibitor H89 or the PKA agonist 8Br-cAMP (Figures 3B and 3C). These results thus suggest that FpSDH is phosphorylated in vivo by a Src-type tyrosine kinase.
Figure 3. The Src-Type Kinase Fgr Phosphorylates CII.
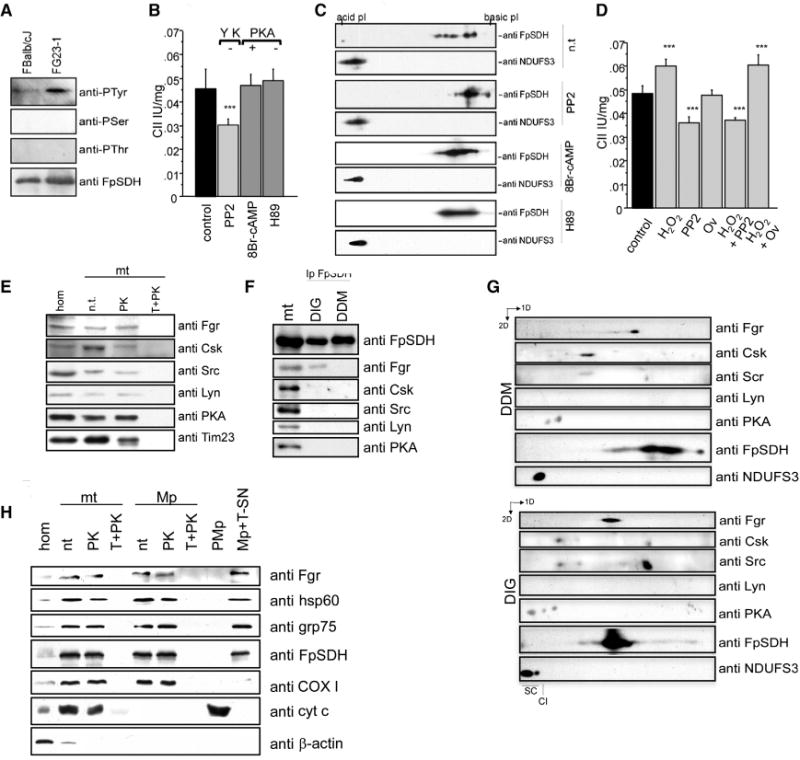
(A) Immunoblot phoshoprotein analysis in FpSDH immunoprecipitates from FBalb/cJ and FG23-1 cells.
(B) Complex II activity in mouse liver mitochondria incubated with the Tyr kinase inhibitor PP2, the PKA agonist 8Br-cAMP, or the PKA antagonist H89 (n = 5).
(C) 2D IEF/SDS-PAGE (3–10 IEF strips) Western analysis of mouse mitochondria treated with PP2, 8Br-cAMP, or H89. PP2 induces a basic shift in FpSDH compared with nontreated mitochondria (n.t.). Modulation of PKA activity had no effect on FpSDH phosphorylation. NDUFS3 (CI) was detected to align and compare blots.
(D) H2O2-induced activation of complex II is blocked by the Tyr kinase inhibitor PP2, but not by the Tyr phosphatase inhibitor orthovanadate (Ov) (n = 6).
(E) Intact mouse mitochondria (mt) protect a portion of Fgr, Csk, Src, Lyn, and PKA kinases from digestion by proteinase K: hom, cell homogenate; n.t., non-treated; PK, proteinase K; T+PK, proteinase K treatment of mitochondria solubilized with Triton X-100. Tim23 was used as a marker of intact mitochondria.
(F) Anti-FpSDH specifically coimmunoprecipitates Fgr kinase from mitochondria solubilized with digitonin (DIG), which preserves intact supercomplexes, but not dodecyl maltoside (DDM), which resolves individual respiratory complexes. mt, fully solubilized mitochondria.
(G) 2D BNGE/SDS-PAGE of DDM- and DIG-solubilized mitochondria, showing comigration of Fgr and FpSDH in DIG-treated mitochondria. Note comigration of Csk and Src in both preparations.
(H) Mitochondrial Fgr is located in the matrix. Fractionation experiment showing protection of Fgr from PK digestion in intact mitochondria (mt) and mitoplasts (Mp) but not in either fraction solubilized with Triton X-100 (T+PK). Fgr was present in the supernatant fraction of Triton X-100-solubilized mitoplasts (Mp+T-SN). P-Mp, post-mitoplast fraction. Markers used: hsp60 and Grp75, mitochondrial matrix; FpSDH, association with inner membrane; COXI, inner membrane; cyt c, intermembrane space; β-actin, cytoplasm.
All data are presented as mean ± SD.
H2O2 can promote phosphorylation on Tyr residues by activating Tyr kinases (Balamurugan et al., 2002; Chiarugi, 2008; Minetti et al., 2002) or by inhibiting Tyr phosphatases (Chiarugi, 2008). We incubated isolated mouse liver mitochondria with H2O2 in the presence of either PP2, to inhibit SFKs, or orthovanadate (Ov), a general inhibitor of Tyr-phosphatases. PP2 reduced the activity of CII and prevented the activation induced by H2O2, whereas Ov had no effect. If we assume that Ov is able to inhibit the phosphatase involved, these results are compatible with a model in which H2O2 promotes FpSDH phosphorylation by activating a PP2-sensitive kinase (Figure 3D). Full demonstration of this mechanism would require the identification of the phosphatase involved.
The Src-Type Tyrosine Kinase Fgr Interacts with CII In Vivo
The SFKs Lyn, Fyn, Fgr, and Csk have been reported to localize in mitochondria (Augereau et al., 2005; Salvi et al., 2005; Tibaldi et al., 2008). Immunoblot analysis of mitochondrial preparations pretreated with proteinase K detected several tyrosine protein kinases (Src, Lyn, and Fgr), the regulator of Src-type tyrosine kinases Csk (itself a Tyr kinase), and the Ser/Thr kinase PKA (Figure 3E). To establish whether any of these interact with CII, we performed coimmunoprecipitation experiments targeting FpSDH. Mitochondrial membranes were solubilized either with the nonionic detergent dodecyl-maltoside (DDM), to isolate individual respiratory complexes, or with the milder detergent digitonin (DIG), to isolate supercomplexes. In the DDM lysates, none of the probed protein-kinases was coimmunoprecpitated with CII (Figure 3F), whereas in DIG lysates anti-FpSDH specifically coimmunoprecipitated the Src-family kinase Fgr, suggesting physical interaction between Fgr and CII in the mitochondrial inner membrane. To confirm this, we separated DIG and DDM lysates by BNGE followed by denaturing SDS-PAGE (Figure 3G). Fgr kinase was detectable in both preparations but only comigrated with CII in the DIG-lysed samples. None of the other kinases analyzed comigrated with FpSDH, but Src/Csk and PKA both appeared to be associated with high molecular weight complexes (Figure 3G). Interestingly, Src comigrated with its regulator Csk in both preparations, suggesting strong interaction between these kinases. The mitochondrial matrix localization of Fgr kinase was confirmed by subfractionation of pure mouse liver mitochondria (Figure 3H).
Ablation of Fgr Abolishes the Activation of CII
To demonstrate the role of Fgr in the regulation of CII we examined liver mitochondria from fgr−/− mice (Lowell et al., 1994), in which Fgr is undetectable but CII content is normal (Figure 4A). Basal CII activity in these mitochondria was below normal and insensitive to H2O2 or PP2 (Figure 4B), and FpSDH phosphorylation was prevented (Figure 4C). CII activity was normal in mitochondria from Lyn and Hck Tyr kinase knockout mice (Figure 4D). The absence of Fgr affected CII activity in all analyzed tissues (Figure 4E), whereas activities of the other mitochondrial respiratory complexes remained unchanged or, in the case of CI or combined CI+III, increased (Figures 4F and 4G). We can thus conclude that Fgr is the tyrosine kinase responsible for FpSDH phosphorylation, that it interacts with its substrate, and that FpSDH phosphorylation activates CII.
Figure 4. CII Is Regulated by Fgr Phosphorylation at Tyr604.
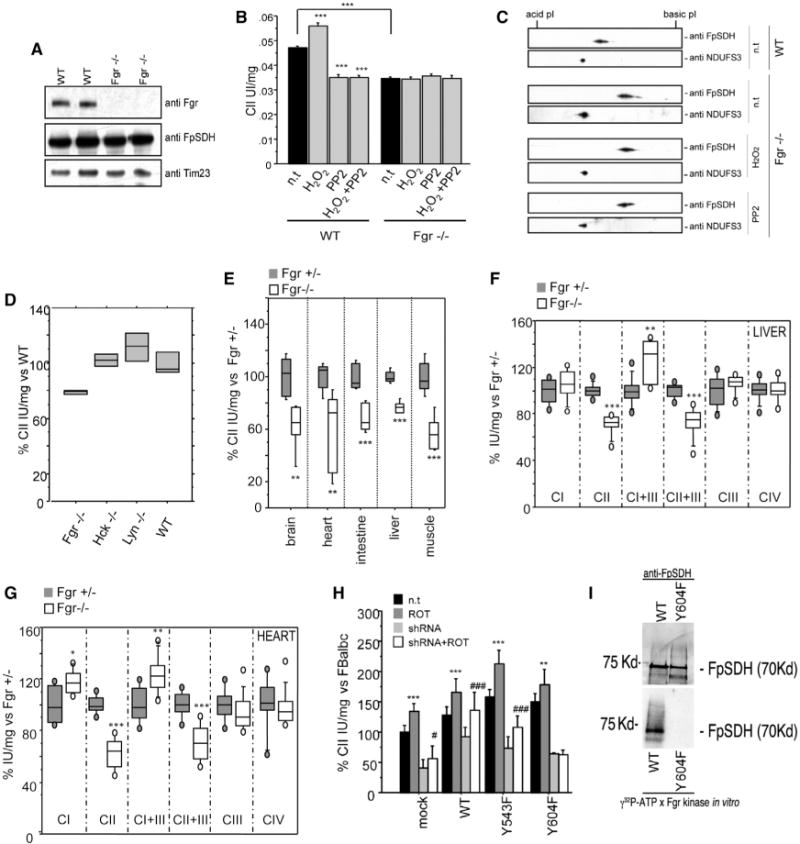
(A) SDS-PAGE western analysis of Fgr tyrosine kinase in liver mitochondrial protein preparations from control or Fgr−/− mice.
(B) CII activity in isolated liver mitochondria from WT or Fgr−/− mice in the presence of H2O2, PP2, or both. Data are presented as mean ± SD.
(C) 2D IEF/SDS-PAGE (IEF strips pH 4–7) Western analysis of FpSDH from WT liver mitochondria and from nontreated and H2O2- or PP2-treated Fgr−/− liver mitochondria. NDUFS3 (CI) is used to align and compare blots.
(D) Complex II activity in liver mitochondria from different Tyr kinase null mice.
(E) Complex II activity in tissue homogenates from Fgr+/− and Fgr−/− null mice (n = 4).
(F and G) Individual and combined mitochondrial complex activities in isolated mitochondria from liver (F) and heart (G) (n ≥ 4). In(D)–(G), lines extending from the boxes indicate the variability outside the upper and lower quartiles.
(H) CII activity in FBalb/cJ cells silenced for endogenous FpSDH and re-expressing WT and mutant FpSDH variants (n = 4). ROT, rotenone (200 nM); shRNA, silencing of endogenous FpSDH. Data are presented as the percentage of activity in nontreated, mock-infected FBalb/cJ (mean ± SD). Statistical significance versus nontreated: **p < 0.001; ***p < 0.0001. Statistical significance versus shRNA: #, p < 0.01; ###, p < 0.0001.
(I) Fgr in vitro phosphorylation (bottom) and FpSDH immunodetection (top) of immunocaptured complex II from FBalb/cJ cells expressing WT FpSDH (left line) or the Y604F mutant (right line). See also Figure S3.
Fgr Activates CII through FpSDH Phosphorylation on Y604
Analysis of mouse FpSDH identified six candidate tyrosines for phosphorylation (Figure S3A), three of them in Fgr target motifs predicted by Scansite and Prediction of PK phosphorylation site (PPSP). One residue, Y604, which is conserved among mammals (Figure S3B), was previously identified as an Fgr target in in vitro phosphorylation assays (Salvi et al., 2007). The same report showed phosphorylation of Y543, but in silico predictions indicate this residue is not an Fgr target.
To identify the residue involved in CII regulation, we measured CII activity in FBalb/cJ cells transfected with WT FpSDH or one of the mutant FpSDH variants Y543F and Y604F. To ascertain the effect of the specific residues, we also silenced endogenous FpSDH expression (Figure S3C) and analyzed the response to treatment with rotenone (Figure 4H). In cells silenced for endogenous FpSDH, re-expressed WT and Y543F FpSDH restored rotenone-activated CII activity, but this effect was not seen in cells re-expressing the single mutant Y604F (Figure 4H). We next immunocaptured CII from cells expressing WT or Y604F FpSDH and performed Fgr in vitro phosphorylation assays. As proposed for rat (Salvi et al., 2007), mouse FpSDH was phosphorylated by Fgr. In contrast, Fgr was unable to phosphorylate the Y604F FpSDH variant (Figure 4I). No other CII proteins were labeled by this approach. These results confirm the in silico prediction that FpSDH Y604 is the unique Fgr target in CII.
ROS/Fgr/CII Regulatory Pathway in T Lymphocyte Activation
Activation of resting T cells with anti-CD3/CD28 antibodies (Abs), concanavalin A/IL2 (ConA/IL2), or phorbol-12-myristate-13-acetate (PMA) plus ionomycin triggers ROS production (Kamiński et al., 2010; Nagy et al., 2003). In response to anti-CD3/CD28 Abs or ConA/IL2, T cells isolated from Fgr+/− and Fgr−/− produced similar levels of ROS (Figures 5A and 5B). However, whereas CII activity was increased in Fgr+/− T cells, it remained unaffected in Fgr−/− cells (Figure 5C). Consistent with this result, restimulation of T lymphoblasts with PMA/ionomycin increased CII activity in Fgr+/− but not Fgr−/− cells (not shown).
Figure 5. T Cell Activation and CII Activity Response.
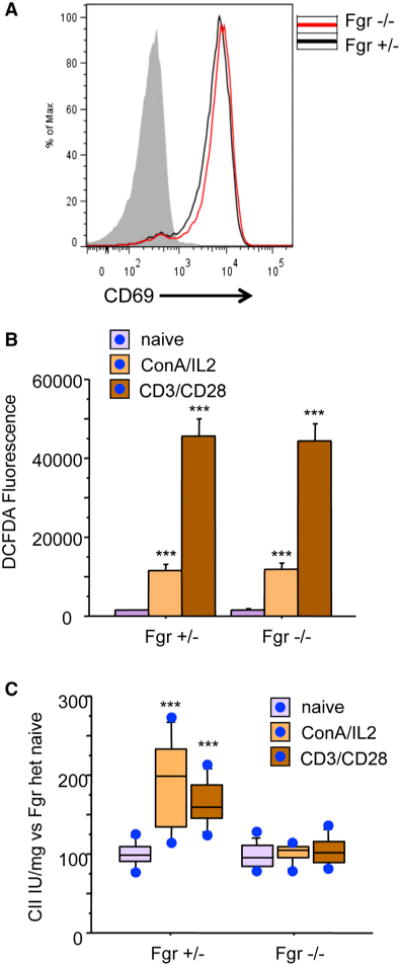
(A) Flow cytometry analysis of T cell activation. Naive T cells from Fgr+/− and Fgr−/− mice were activated with anti-CD3/CD28, and activation was assessed by detection of CD69. Gray line: negative control. The chart shows data from one representative experiment of four.
(B) Flow cytometry analysis of ROS production (2,7-DCFH2-DA) in resting T cells and upon CD3/CD28 or ConA activation (n = 3). Data are presented as mean ± SD.
(C) CII activity measured in resting and activated T cells (n = 4). Data are presented as the percentage of activity compared to resting T cells. Lines extending from the boxes indicate the variability outside the upper and lower quartiles.
ROS/Fgr/CII Pathway in Starvation and Serum Deprivation
One of the better-characterized ways in which liver cells adapt to starvation is through phosphorylation-mediated inhibition of pyruvate dehydrogenase (Huang et al., 2003), which reduces mitochondrial utilization of pyruvate and has been proposed to increase ROS production (Ten and Starkov, 2012). We recently reported that electrons derived from NADH and FADH2 follow separate routes along the mETC, and that this is important for metabolic adaptation to starvation, with an increase in the rate of FADH2 oxidization occurring at the expense of the NADH route (Lapuente-Brun et al., 2013). Since Fgr-kinase activates CII (and hence use of FADH2 electrons), we conducted a more detailed evaluation of the consequences of the lack of Fgr-kinase for liver mitochondria function. The significantly lower maximum CII activity in mitochondrial preparations from Fgr null liver was accompanied by lower-succinate-driven respiration and ATP synthesis (Figure 6A). Maximum CI activity was similar for both genotypes (Figure 6B). Surprisingly, pyruvate-plus-malate- and glutamate-plus-malate-driven respiration rates were below normal in well-fed Fgr null mice, and in the case of pyruvate-plus-malate this was accompanied by significantly lower ATP synthesis (Figure 6B). Lack of Fgr kinase thus appears to have a broader than anticipated effect on liver energy metabolism. No significant differences in citrate synthase activity were detected that could explain this decline in CI dependent respiration (Figure 6A). But although both the CI (NADH route) and the CII (FADH2 route) are altered in Fgr−/− liver mitochondria, the balance of electron supply potential to the mETC from NADH and FADH2 is shifted, increasing the CI/CII activity ratio (Figure 6C). Starving Fgr−/− mice overnight increased CI activity in liver mitochondria and reduced CII activity (Figures 6A, 6B, and S4), sharply increasing the CI/CII activity ratio, whereas the ratio in mitochondria from starved control mice decreased (Figure 6C). Feeding mitochondria with pyruvate or glutamate should generate intramitochondrial NADH, for delivery of electrons to CI. As expected, starvation-induced downregulation of pyruvate dehydrogenase activity reduced pyruvate-plus-malate-driven respiration and ATP synthesis in Fgr+/− and Fgr null mice (Figure 6B). In contrast, starvation only decreased respiration and ATP synthesis driven by glutamate-plus-malate in Fgr−/− mice, despite the higher maximal CI respiration (Figure 6B). These observations unexpectedly show that lack of Fgr kinase affects liver glutamate utilization. To avoid the complex pleiotropic effects of Fgr-kinase ablation in liver, we mimicked starvation in cultured embryonic fibroblasts (derived from Fgr+/− and Fgr−/− littermates) by overnight serum deprivation. Serum-deprived Fgr+/− fibroblasts showed the typical mitochondrial hyperfusion phenotype required to maintain ATP production upon nutrient deprivation (Gomes et al., 2011), whereas Fgr−/− mitochondria were fragmented (Figure 6D). Consistently, mitochondria from serum-deprived Fgr−/− cells showed higher processing of OPA1 (Figure 6E). Serum deprivation increased CII activity in Fgr+/− but not Fgr−/− cells (Figure 6F), recapitulating the lack of CII activation in Fgr−/− liver mitochondria from overnight-starved mice. To assess whether starvation responses were due only to Fgr-dependent CII phosphorylation, we analyzed the effects of serum deprivation in fibroblasts silenced for endogenous FpSDH and exogenously re-expressing WT or Y604F FpSDH. Serum deprivation triggered OPA-1 processing in Y604F cells (Figure 6E), mimicking the result in Fgr null fibroblasts, and only cells re-expressing WT FpSDH upregulated CII activity after serum deprivation (Figure 6G). The blunted CII activation in Y604F cells compromised cell survival after serum deprivation, revealed by a higher proportion of apoptotic annexin V positive Y604F cells (Figures 6H and S5C).
Figure 6. Mitochondria Lacking Fgr or Expressing Y604F FpSDH Respond Abnormally to Starvation and Serum Deprivation.
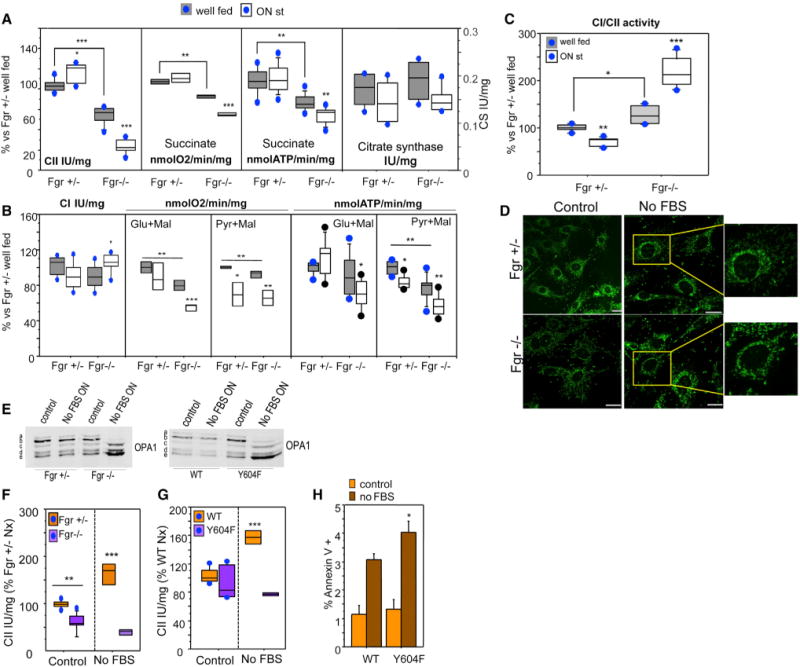
(A) Succinate-driven OXPHOS function in mouse liver mitochondria from Fgr+/− and Fgr−/− mice fed a normal diet (well fed) or starved overnight (ON st, n ≥ 4). CII activity (left), succinate-driven respiration (center left), succinate-driven ATP synthesis (center right), and citrate synthase activity (CS, right).
(B) NADH-driven OXPHOS function in mouse liver mitochondria from Fgr+/− and Fgr−/− mice fed a normal diet or starved overnight (n ≥ 4). CI activity (left), glutamate-driven respiration (center left), pyruvate-driven respiration (center), glutamate-driven ATP synthesis (center right), and pyruvate-driven ATP synthesis (right) are shown.
(C) Rate of use of NADH/FADH reducing equivalents in mouse liver mitochondria from Fgr+/− and Fgr−/− mice (n ≥ 4). Data in (A)–(C) are presented as the percentage of values obtained in well-fed Fgr+/− mice. Lines extending from the boxes indicate the variability outside the upper and lower quartiles.
(D) Immunostaining of mitochondria (Tom20, green) in Fgr+/− and Fgr−/− fibroblasts cultured with serum or without serum overnight (No FBS ON).
(E) Immunoblot showing the migration of OPA1 in lysates from control and serum-deprived Fgr+/− and Fgr−/− cells (left) or control and serum-deprived FpSDH-silenced fibroblasts re-expressing WT or Y604F FpSDH (right).
(F)CII activity in Fgr+/− and Fgr−/− fibroblasts grown in 10 mM glucose in the indicated conditions (n ≥ 5). Data are presented as the percentage activity in Fgr+/− cells grown in normoxia; lines extending from the boxes indicate the variability outside the upper and lower quartiles.
(G)CII activity in FpSDH-silenced fibroblasts re-expressing WT or Y604F FpSDH, cultured at 5 mM glucose under the indicated oxygenation conditions (n ≥ 5). Data are presented as the percentage of activity in WT-expressing cells grown in normoxia; lines extending from the boxes indicate the variability outside the upper and lower quartiles.
(H) Apoptotic events in FpSDH-silenced fibroblasts re-expressing WT or Y604F FpSDH and grown with or without serum. Apoptosis was determined by flow cytometry as the percentage of cells positive for annexin V and propidium iodide (n = 3). Data are presented as mean ± SD. *p < 0.01; **p < 0.001; ***p< 0.0001.
See also Figures S4 and S5.
ROS/Fgr/CII Pathway in Reoxygenation-Induced Metabolic Reprogramming
A drop in O2 availability triggers several adaptive mechanisms, including reduction in the activities and protein levels of OXPHOS components and in ROS production (Ali et al., 2012; Heather et al., 2012; Papandreou et al., 2006), and a notable accumulation of succinate (Cascarano et al., 1976). However, sudden reoxygenation, as occurs in reperfusion after ischemia, is accompanied by a sharp increase in ROS production as the electron transport chain readapts to oxygen availability. To evaluate the role of ROS-mediated phosphorylation of FpSDH in this adaptation, we cultured Fgr+/− and Fgr−/− fibroblasts for 48 hr at 21% O2 (normoxia), 1% O2 (hypoxia), or 1% O2 followed by reoxygenation at 21% O2 for an additional 48 hr. Immunostaining and western blot analysis indicated that ROS-mediated mitochondrial biogenesis upon reoxygenation was impaired in Fgr−/− cells, with only Fgr+/− cells recovering normoxic mitochondrial numbers and shape (Figures 7A and 7B). In both genotypes, hypoxia reduced the amount of the mitochondrial proteins Tom20 and FpSDH (Figure 7B), consistent with the reported loss of mitochondria upon hypoxia (Kim et al., 2011). In the FpSDH re-expression model, hypoxia reduced mitochondrial content (measured as the FpSDH:actin and Tom20:actin ratios) in FpSDH-silenced fibroblasts re-expressing WT or Y604F FpSDH. As predicted, reoxygenation restored or increased mitochondrial protein content in cells re-expressing WT FpSDH, and this recovery was impaired in Y604F cells; however, Y604F cells did show partial mitochondrial recovery, differing from the more severe phenotype in Fgr−/− fibroblasts (Figure 7B). The reason for this difference is likely that germline lack of Fgr affects targets other than CII required for full recovery.
Figure 7. Mitochondria Lacking Fgr or Expressing Y604F FpSDH Respond Abnormally to Hypoxia-Reoxygenation.
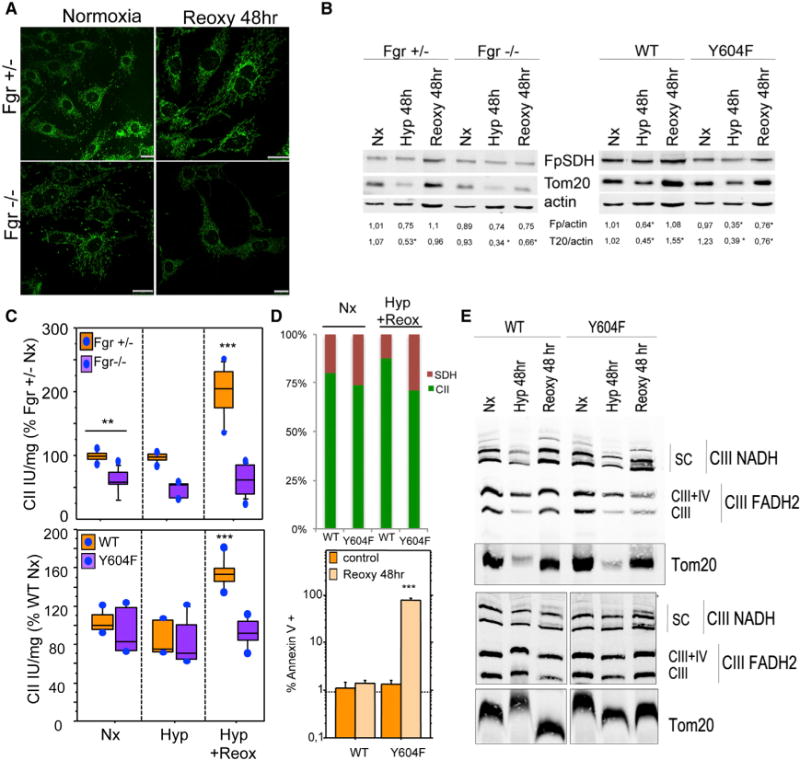
(A) Immunostaining of mitochondria (Tom20, green) in Fgr+/− and Fgr−/− fibroblasts cultured in normoxia (21% O2) or hypoxia (1 % O2)for48 hr followed by 48 hr normoxia (Reoxy 48 hr).
(B)Left: Immunoblot analysis of Fgr+/− and Fgr−/− cells cultured under normoxia (Nx), hypoxia for 48 hr(Hyp 48 hr), or hypoxia followed by normoxia (Reoxy 48 hr). Right: Immunoblot analysis of FpSDH-silenced fibroblasts re-expressing WT or Y604F FpSDH and cultured under the indicated oxygenation conditions. Numbers beneath blots show FpSDH (Fp):actin and Tom20 (T20):actin ratios (n = 4).
(C) Top: CII activity in Fgr+/− and Fgr−/− fibroblasts grown in 10 mM glucose under the indicated conditions (n ≥ 5). Bottom: FpSDH-silenced fibroblasts re-expressing WT or Y604F FpSDH, cultured with 5 mM glucose under the indicated oxygenation conditions (n ≥ 5). Data are presented as the percentage activity in Fgr+/− cells or WT-FpSDH-re-expressing cells grown in normoxia; lines extending from the boxes indicate the variability outside the upper and lower quartiles.
(D) Top: Relative activities of CII (CoQ reduction) and SDH in WT- and Y604F-re-expressing FpSDH-silenced cells grown in normoxia or through a hypoxia/reoxygenation cycle. For each cell line and condition, 100% = the sum of CII and SDH activities; absolute SDH activity did not differ between cell lines and conditions. Bottom: Apoptotic events in FpSDH-silenced fibroblasts re-expressing WT or Y604F FpSDH and grown in normoxia or through a hypoxia/reoxygenation cycle. Apoptosis was determined by flow cytometry as the percentage of annexin V- and PI-positive cells (n = 3). Data are presented as mean ± SD.
(E) BNGE of FpSDH-silenced fibroblasts re-expressing WT or Y604F FpSDH cultured at 5 mM glucose under the indicated oxygenation conditions; the blot reveals the distribution of CIII (anti-core 1 immunodetection) among the different forms of free complex and supercomplexes. The outer membrane protein Tom20 is used as a mitochondrial protein loading control. Upper and lower panels are taken from two independent experiments. Note that upon reoxygenation the amount of CIII super assembled with CI is abnormally elevated in the Y604F mutant. See also Figure S5.
To test the metabolic effect of reoxygenation, we measured CII activity in Fgr+/− and Fgr−/− fibroblasts cultured with 25 mM glucose or the more physiological 10 mM. Hypoxia did not alter CII activity under any conditions, and reoxygenation increased CII activity only in Fgr+/− cells (Figures 7C and S5A). Likewise, in re-expression assays only WT FpSDH fibroblasts upregulated CII activity upon posthypoxia reoxygenation (Figures 7C and S5B). CII has been proposed to trigger apoptosis, depending on its attachment to the inner mitochondrial membrane (reviewed in Grimm, 2013). When detached, CII is not assembled as a holocomplex, and its succinate ubiquinol reductase (SQR or CII) activity, which involves coenzyme Q reduction, is decreased. However, succinate dehydrogenase (SDH) activity is unaltered, resulting in superoxide leakage that leads to apoptosis (Albayrak et al., 2003; Lemarie et al., 2011). SDH activity was slightly higher in Y604F-expressing cells in normoxia, but the proportion of apoptotic cells (annexin V positive) was unaffected. Upon reoxygenation, the balance between CII and SDH activity in cells re-expressing WT FpSDH shifted toward CII, whereas in cells re-expressing Y604F it shifted more toward SDH (Figure 7D). This was reflected in significantly more severe apoptosis after hypoxia/reoxygenation in Y604F-re-expressing cells, in which CII activation is blunted, but not in cells re-expressing WT FpSDH (Figures 7D and S5C).
BNGE revealed a hypoxia-induced generalized decrease in the content of assembled OXPHOS complexes and Tom20 (consistent with loss of mitochondrial proteins evident in Figure 7B), with no alteration in the proportion of CIII dedicated to each coenzyme Q pool (Figure 7E). Posthypoxia reoxygenation of WT re-expressing cells restored OXPHOS complexes and supercomplex assembly while maintaining these proportions. In contrast, reoxygenated Y604F-expressing cells restored the normoxic level of CIII dedicated to NADH but not the amount dedicated to FADH2 (CIII+IV and free CIII) (Figure 7E), indicating a higher-than-normal dedication to processing electrons from the NADH-CoQ pool than from the FADH2-CoQ pool (Lapuente-Brun et al., 2013).
DISCUSSION
The data presented here demonstrate that Tyr phosphorylation of FpSDH increases CII activity in vivo, and that this mechanism triggers remodeling of the mETC to reset its capacity for processing NADH- versus FADH2-derived electrons. This Tyr phosphorylation is H2O2 mediated, is catalyzed by the Src-family kinase Fgr, and specifically targets Y604 in FpSDH. The finding that the catalytic subunit of CII can be phosphorylated is consistent with earlier observations (Bykova et al., 2003; Schulenberg et al., 2003). Moreover, our proposal that Fgr is the tyrosine kinase responsible for this phosphorylation concurs with a previous report demonstrating that Fgr, but not Lyn, is able to promote the in vitro phosphorylation of Y535 and Y596 of rat FpSDH, which correspond to Y543 and Y604 in mouse (Salvi et al., 2007).
H2O2-triggered activation of CII provides a mechanism for the association of increased CII activity and defective CI, observed in human patients and in a range of organisms from Chlamydomonas reinhardtii (Cardol et al., 2002) and Rhodobacter capsulatus (Dupuis et al., 1998) to mouse and humans (Esteitie et al., 2005; Fan et al., 2008; Majander et al., 1991). Another feature of ROS-driven activation of CII is that since hydrogen peroxide can permeate through cell membranes, any extramitochondrial source of H2O2 can potentially activate CII, suggesting a mechanism to promote metabolic adaptation in response to signals that increase H2O2. Our results thus show that activation of CII by Fgr kinase in response to a primary wave of extramitochondrial ROS can trigger a secondary wave of ROS production as a consequence of CII activation. This pathway provides a mechanism for amplifying ROS signals within the cell. The increase in CII activity triggered by H2O2 is a quick response mechanism, independent of gene expression and therefore not involving any increase in mitochondrial biogenesis or regulation through PGC-1α, a common feature of mitochondrial disease (Moreno-Loshuertos et al., 2006; Acín-Perez et al., 2009; Srivastava et al., 2009; Wenz et al., 2008).
FpSDH activity is regulated by acetylation on several lysine residues, and deacetylation mediated by sirtuin 3 (Cimen et al., 2010; Finley et al., 2011) increases CII activity independently of ROS. The regulation of CII acetylation is incompletely understood, but fuel availability is likely to play a part, probably in a complex tissue-specific pattern (Boyle et al., 2013; Finley et al., 2011). The convergence of multiple posttranslational modifications on the catalytic subunit of CII highlights the importance of fine-tuning CII activity to ensure correct cell metabolism. This role has remained unappreciated despite the considerable knowledge accumulated on the function of the tricarboxylic acid (TCA) cycle. Two other TCA cycle enzymes, aconitase and KGDH, are known to be reversibly downregulated by physiological increases in ROS levels (Bulteau et al., 2003; Moreno-Loshuertos et al., 2006). Our current results show that CII should be included among the TCA cycle enzymes regulated by ROS.
Our results also show that Src-family kinase signaling operates within mitochondria, regulating the fundamental metabolic processes of the TCA cycle and oxidative phosphorylation. SFKs are implicated in a wide variety of signaling pathways, regulating cell growth, differentiation, cell shape, migration, and survival (Ingley, 2008; Parsons and Parsons, 2004). Moreover, the physiological role of SFK activation by H2O2 has also been shown in another model system: the role of Lyn kinase as a redox sensor activated by H2O2 in leukocyte wound attraction (Yoo et al., 2011). The role of reversible protein phosphorylation in mitochondria is an emerging field (Pagliarini and Dixon, 2006; Pagliarini et al., 2005), and several mitochondrial proteins have been proposed as Tyr phosphorylation targets (Augereau et al., 2005; Salvi et al., 2005, 2007; Tibaldi et al., 2008). Moreover, a number of SFKs have been proposed to localize in mitochondria (Pagliarini and Dixon, 2006; Tibaldi et al., 2008). One previous report has described the function of a mitochondrial SFK, demonstrating that c-Src enhances the activity of subunit II of cytochrome c oxidase, the terminal mETC enzyme (Miyazaki et al., 2003). Thus the TCA cycle and the mETC are both positively regulated by the action of SFKs: Fgr for the TCA cycle and c-Src for the mETC. By bringing the regulation of mitochondrial energetic metabolism within the remit of cellular signaling pathways regulated by SFKs, these findings have broad implications for the integration of cell adaptation and energy regulation. The results presented here highlight the importance of this regulation in the adaptation to loss of CI, regulation of CII activity during the metabolic switch upon activation of naive T cells, and metabolic adaptation of mitochondria to starvation and hypoxia/reoxygenation. CII has also been proposed to act as universal oxygen sensor (Baysal, 2006), suggesting that this pathway is also critically important in situations where cells need to respond rapidly to changes in fuel availability or O2 concentration.
EXPERIMENTAL PROCEDURES
Further details of methods are provided in Supplemental Experimental Procedures.
Cell Culture
Cell lines were grown in Dulbecco’s modified Eagle’s medium (DMEM; GIBCOBRL) supplemented with 5% fetal bovine serum (FBS; GIBCOBRL). Fibroblasts from Fgr+/− and Fgr null mice were isolated from mouse ear and immortalized by transfection with pLOX-Ttag-iresTK (Addgene) and grown in DMEM with 10% FBS. FBalb/cJ and FG23-1 cells expressing MnSOD or HA-tagged mt-catalase and FBalb/cJ cells overexpressing different forms of FpSDH and silenced for the endogenous FpSDH were generated by viral infection using polybrene (8 μg/ml).
Isolation of Mitochondria and Mitochondrial Fractions from Mouse Liver and Cell Lines
Mitochondria were isolated from cell lines as described (Schägger and von Jagow, 1991) with some modifications, and from mouse liver as described (Fernández-Vizarra et al., 2002).
OXPHOS Function and Enzyme Activities
O2 consumption was measured in mouse liver mitochondria (100 μg) as described (Hofhaus et al., 1996). ATP synthesis in isolated mitochondria (15–25 μg mitochondrial protein) was measured using a kinetic luminescence assay (Vives-Bauza et al., 2007). Mitochondrial fractions were prepared and the activities of individual complexes measured spectrophotometrically (Birch-Machin and Turnbull, 2001). Catalase activity was measured in total cell lysates (300 μg) (Moreno-Loshuertos et al., 2006). Total SOD and MnSOD (KCN insensitive) activities were assessed in total cell lysates (50 mg) using the SOD Assay Kit (Sigma-Aldrich).
Blue-Native Gel Electrophoresis
Cell-culture-derived mitochondria (50–75 μg) were separated on 5%–13% gradient blue native gels (Schägger and von Jagow, 1991).
Isoelectric Focusing and 2D SDS-PAGE
Mitochondrial preparations (100 μg) were processed with by Ready Prep 2D Cleanup (BioRad) and applied to pH 3–10 or pH 4–7 IPG strips (BioRad) and incubated overnight at room temperature. Isoelectric focusing and second dimension SDS-PAGE were run under standard conditions.
Phoshoprotein Enrichment
Phosphoprotein-enriched mitochondrial fractions were isolated on phosphoprotein enrichment columns (QIAGEN).
Immunocapture
Complex II and FpSDH were immunocaptured from mitochondria isolated from either cells or mouse liver. For details see Supplemental Experimental Procedures.
In Vitro Fgr Phosphorylation
Recombinant Fgr kinase (Abnova) was used to phosphorylate complex II immunocaptured from cells expressing WT and Y604F FpSDH (Salvi et al., 2007).
Immunological Analysis
Antibodies used in this study are listed in Supplemental Experimental Procedures.
H2O2 Production
H2O2 production was measured in cultured cells grown in the absence or presence of 5 mM NAC for 7 days (Moreno-Loshuertos et al., 2006).
Isolation and Stimulation of Naive CD4+ and CD8+ T Cells
CD4+ and CD8+ T cells were purified from splenic cell suspensions obtained from 8- to 10-week-old Fgr+/− and Fgr−/− male mice. To obtain differentiated T lymphoblasts, naive T cells were cultured with concanavalin A (Sigma) and human recombinant IL-2 (50 U/ml, Glaxo).
Flow Cytometry ROS Production Determination
ROS production in naive or activated T lymphocytes and in cells before and after overexpression of detoxifying enzymes was assessed by flow cytometry of 2,7-DCFH2-DA or MitoSOX staining (Kamiński et al., 2012).
Determination of Apoptosis in Cultured Cells
Apoptosis was monitored by flow cytometry detection of annexin V and propidium iodide (PI) staining in 106 cells resuspended in 10 mM HEPES/NaOH (pH 7.4), 140 mM NaCl, 2.5 mM CaCl2.
Mouse Strains
Animal studies were approved by the local ethics committee. Analyses were performed in 8- to 11-week-old male mice.
In Silico Analysis
See Supplemental Experimental Procedures.
Statistical Analysis
Comparisons between groups were made by one-way ANOVA. Pairwise comparisons were made by Fisher’s PLSD post hoc test. Differences were considered statistically significant at p < 0.05. Data were analyzed with StatView (Adept Scientific, UK).
Acknowledgments
We thank Dr. Concepción Jimenez and Andrés Gonzalez-Guerra for technical assistance, Anatoly A. Starkov for a valuable discussion, and Dr. Simon Bartlett (CNIC) for English editing. This study was supported by grants from the Ministerio de Economía y Competitividad (SAF2012-1207 and CSD2007-00020), the Comunidad de Madrid (CAM/API1009), and the EU (UE0/MCA1108 and UE0/MCA1201). R.A.-P. is recipient of a Ramon y Cajal contract. The CNIC is supported by the Ministerio de Economía y Competitividad and the Pro-CNIC Foundation.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes five figures and Supplemental Experimental Procedures and can be found with this article online at http://dx.doi.org/10.1016/j.cmet.2014.04.015.
References
- Acín-Pérez R, Bayona-Bafaluy MP, Bueno M, Machicado C, Fernández-Silva P, Pérez-Martos A, Montoya J, López-Pérez MJ, Sancho J, Enríquez JA. An intragenic suppressor in the cytochrome c oxidase I gene of mouse mitochondrial DNA. Hum Mol Genet. 2003;12:329–339. doi: 10.1093/hmg/ddg021. [DOI] [PubMed] [Google Scholar]
- Acín-Perez R, Salazar E, Brosel S, Yang H, Schon EA, Manfredi G. Modulation of mitochondrial protein phosphorylation by soluble adenylyl cyclase ameliorates cytochrome oxidase defects. EMBO Mol Med. 2009;1:392–406. doi: 10.1002/emmm.200900046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albayrak T, Scherhammer V, Schoenfeld N, Braziulis E, Mund T, Bauer MKA, Scheffler IE, Grimm S. The tumor suppressor cybL, a component of the respiratory chain, mediates apoptosis induction. Mol Biol Cell. 2003;14:3082–3096. doi: 10.1091/mbc.E02-10-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali SS, Hsiao M, Zhao HW, Dugan LL, Haddad GG, Zhou D. Hypoxia-adaptation involves mitochondrial metabolic depression and decreased ROS leakage. PLoS ONE. 2012;7:e36801. doi: 10.1371/journal.pone.0036801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augereau O, Claverol S, Boudes N, Basurko MJ, Bonneu M, Rossignol R, Mazat JP, Letellier T, Dachary-Prigent J. Identification of tyrosine-phosphorylated proteins of the mitochondrial oxidative phosphorylation machinery. Cell Mol Life Sci. 2005;62:1478–1488. doi: 10.1007/s00018-005-5005-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Attardi G. The mtDNA-encoded ND6 subunit of mitochondrial NADH dehydrogenase is essential for the assembly of the membrane arm and the respiratory function of the enzyme. EMBO J. 1998;17:4848–4858. doi: 10.1093/emboj/17.16.4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balamurugan K, Rajaram R, Ramasami T, Narayanan S. Chromium(III)-induced apoptosis of lymphocytes: death decision by ROS and Src-family tyrosine kinases. Free Radic Biol Med. 2002;33:1622–1640. doi: 10.1016/s0891-5849(02)01115-2. [DOI] [PubMed] [Google Scholar]
- Baysal BE. A phenotypic perspective on Mammalian oxygen sensor candidates. Ann N Y Acad Sci. 2006;1073:221–233. doi: 10.1196/annals.1353.024. [DOI] [PubMed] [Google Scholar]
- Birch-Machin MA, Turnbull DM. Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Methods Cell Biol. 2001;65:97–117. doi: 10.1016/s0091-679x(01)65006-4. [DOI] [PubMed] [Google Scholar]
- Boyle KE, Newsom SA, Janssen RC, Lappas M, Friedman JE. Skeletal muscle MnSOD, mitochondrial complex II, and SIRT3 enzyme activities are decreased in maternal obesity during human pregnancy and gestational diabetes mellitus. J Clin Endocrinol Metab. 2013;98:E1601–E1609. doi: 10.1210/jc.2013-1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulteau AL, Ikeda-Saito M, Szweda LI. Redox-dependent modulation of aconitase activity in intact mitochondria. Biochemistry. 2003;42:14846–14855. doi: 10.1021/bi0353979. [DOI] [PubMed] [Google Scholar]
- Bykova NV, Egsgaard H, Møller IM. Identification of 14 new phosphoproteins involved in important plant mitochondrial processes. FEBS Lett. 2003;540:141–146. doi: 10.1016/s0014-5793(03)00250-3. [DOI] [PubMed] [Google Scholar]
- Cardol P, Matagne RF, Remacle C. Impact of mutations affecting ND mitochondria-encoded subunits on the activity and assembly of complex I in Chlamydomonas. Implication for the structural organization of the enzyme. J Mol Biol. 2002;319:1211–1221. doi: 10.1016/S0022-2836(02)00407-2. [DOI] [PubMed] [Google Scholar]
- Cascarano J, Ades IZ, O’Conner JD. Hypoxia: a succinate-fumerate electron shuttle between peripheral cells and lung. J Exp Zool. 1976;198:149–153. doi: 10.1002/jez.1401980204. [DOI] [PubMed] [Google Scholar]
- Chiarugi P. Src redox regulation: there is more than meets the eye. Mol Cells. 2008;26:329–337. [PubMed] [Google Scholar]
- Cimen H, Han MJ, Yang Y, Tong Q, Koc H, Koc EC. Regulation of succinate dehydrogenase activity by SIRT3 in mammalian mitochondria. Biochemistry. 2010;49:304–311. doi: 10.1021/bi901627u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes LC, et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 2013;155:160–171. doi: 10.1016/j.cell.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dlasková A, Hlavatá L, Ježek P. Oxidative stress caused by blocking of mitochondrial complex I H(+) pumping as a link in aging/disease vicious cycle. Int J Biochem Cell Biol. 2008;40:1792–1805. doi: 10.1016/j.biocel.2008.01.012. [DOI] [PubMed] [Google Scholar]
- Dröge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- Dupuis A, Chevallet M, Darrouzet E, Duborjal H, Lunardi J, Issartel JP. The complex I from Rhodobacter capsulatus. Biochim Biophys Acta. 1998;1364:147–165. doi: 10.1016/s0005-2728(98)00025-5. [DOI] [PubMed] [Google Scholar]
- Esteitie N, Hinttala R, Wibom R, Nilsson H, Hance N, Naess K, Teär-Fahnehjelm K, von Döbeln U, Majamaa K, Larsson NG. Secondary metabolic effects in complex I deficiency. Ann Neurol. 2005;58:544–552. doi: 10.1002/ana.20570. [DOI] [PubMed] [Google Scholar]
- Fan W, Waymire KG, Narula N, Li P, Rocher C, Coskun PE, Vannan MA, Narula J, Macgregor GR, Wallace DC. A mouse model of mitochondrial disease reveals germline selection against severe mtDNA mutations. Science. 2008;319:958–962. doi: 10.1126/science.1147786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Vizarra E, López-Pérez MJ, Enriquez JA. Isolation of biogenetically competent mitochondria from mammalian tissues and cultured cells. Methods. 2002;26:292–297. doi: 10.1016/S1046-2023(02)00034-8. [DOI] [PubMed] [Google Scholar]
- Finley LWS, Haas W, Desquiret-Dumas V, Wallace DC, Procaccio V, Gygi SP, Haigis MC. Succinate dehydrogenase is a direct target of sirtuin 3 deacetylase activity. PLoS ONE. 2011;6:e23295. doi: 10.1371/journal.pone.0023295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13:589–598. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm S. Respiratory chain complex II as general sensor for apoptosis. Biochim Biophys Acta. 2013;1827:565–572. doi: 10.1016/j.bbabio.2012.09.009. [DOI] [PubMed] [Google Scholar]
- Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci. 2010;35:505–513. doi: 10.1016/j.tibs.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heather LC, Cole MA, Tan JJ, Ambrose LJA, Pope S, Abd-Jamil AH, Carter EE, Dodd MS, Yeoh KK, Schofield CJ, Clarke K. Metabolic adaptation to chronic hypoxia in cardiac mitochondria. Basic Res Cardiol. 2012;107:268. doi: 10.1007/s00395-012-0268-2. [DOI] [PubMed] [Google Scholar]
- Hofhaus G, Shakeley RM, Attardi G. Use of polarography to detect respiration defects in cell cultures. Methods Enzymol. 1996;264:476–483. doi: 10.1016/s0076-6879(96)64043-9. [DOI] [PubMed] [Google Scholar]
- Huang B, Wu P, Popov KM, Harris RA. Starvation and diabetes reduce the amount of pyruvate dehydrogenase phosphatase in rat heart and kidney. Diabetes. 2003;52:1371–1376. doi: 10.2337/diabetes.52.6.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingley E. Src family kinases: regulation of their activities, levels and identification of new pathways. Biochim Biophys Acta. 2008;1784:56–65. doi: 10.1016/j.bbapap.2007.08.012. [DOI] [PubMed] [Google Scholar]
- Kamiński MM, Sauer SW, Klemke CD, Süss D, Okun JG, Krammer PH, Gülow K. Mitochondrial reactive oxygen species control T cell activation by regulating IL-2 and IL-4 expression: mechanism of ciprofloxacin-mediated immunosuppression. J Immunol. 2010;184:4827–4841. doi: 10.4049/jimmunol.0901662. [DOI] [PubMed] [Google Scholar]
- Kamiński MM, Sauer SW, Kamiński M, Opp S, Ruppert T, Grigaravičius P, Grudnik P, Gröne HJ, Krammer PH, Gülow K. T cell activation is driven by an ADP-dependent glucokinase linking enhanced glycolysis with mitochondrial reactive oxygen species generation. Cell Rep. 2012;2:1300–1315. doi: 10.1016/j.celrep.2012.10.009. [DOI] [PubMed] [Google Scholar]
- Kim H, Scimia MC, Wilkinson D, Trelles RD, Wood MR, Bowtell D, Dillin A, Mercola M, Ronai ZA. Fine-tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol Cell. 2011;44:532–544. doi: 10.1016/j.molcel.2011.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse SE, Watt WC, Marcinek DJ, Kapur RP, Schenkman KA, Palmiter RD. Mice with mitochondrial complex I deficiency develop a fatal encephalomyopathy. Cell Metab. 2008;7:312–320. doi: 10.1016/j.cmet.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapuente-Brun E, Moreno-Loshuertos R, Acín-Pérez R, Latorre-Pellicer A, Colás C, Balsa E, Perales-Clemente E, Quirós PM, Calvo E, Rodríguez-Hernández MA, et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science. 2013;340:1567–1570. doi: 10.1126/science.1230381. [DOI] [PubMed] [Google Scholar]
- Lemarie A, Huc L, Pazarentzos E, Mahul-Mellier AL, Grimm S. Specific disintegration of complex II succinate:ubiquinone oxidoreductase links pH changes to oxidative stress for apoptosis induction. Cell Death Differ. 2011;18:338–349. doi: 10.1038/cdd.2010.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowell CA, Soriano P, Varmus HE. Functional overlap in the src gene family: inactivation of hck and fgr impairs natural immunity. Genes Dev. 1994;8:387–398. doi: 10.1101/gad.8.4.387. [DOI] [PubMed] [Google Scholar]
- Majander A, Huoponen K, Savontaus ML, Nikoskelainen E, Wikström M. Electron transfer properties of NADH:ubiquinone reductase in the ND1/3460 and the ND4/11778 mutations of the Leber hereditary optic neuroretinopathy (LHON) FEBS Lett. 1991;292:289–292. doi: 10.1016/0014-5793(91)80886-8. [DOI] [PubMed] [Google Scholar]
- Minetti M, Mallozzi C, Di Stasi AMM. Peroxynitrite activates kinases of the src family and upregulates tyrosine phosphorylation signaling. Free Radic Biol Med. 2002;33:744–754. doi: 10.1016/s0891-5849(02)00891-2. [DOI] [PubMed] [Google Scholar]
- Miyazaki T, Neff L, Tanaka S, Horne WC, Baron R. Regulation of cytochrome c oxidase activity by c-Src in osteoclasts. J Cell Biol. 2003;160:709–718. doi: 10.1083/jcb.200209098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Loshuertos R, Acín-Pérez R, Fernández-Silva P, Movilla N, Pérez-Martos A, Rodriguez de Cordoba SR, Gallardo ME, Enríquez JA. Differences in reactive oxygen species production explain the phenotypes associated with common mouse mitochondrial DNA variants. Nat Genet. 2006;38:1261–1268. doi: 10.1038/ng1897. [DOI] [PubMed] [Google Scholar]
- Nagy G, Koncz A, Perl A. T cell activation-induced mitochondrial hyperpolarization is mediated by Ca2+- and redox-dependent production of nitric oxide. J Immunol. 2003;171:5188–5197. doi: 10.4049/jimmunol.171.10.5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliarini DJ, Dixon JE. Mitochondrial modulation: reversible phosphorylation takes center stage? Trends Biochem Sci. 2006;31:26–34. doi: 10.1016/j.tibs.2005.11.005. [DOI] [PubMed] [Google Scholar]
- Pagliarini DJ, Wiley SE, Kimple ME, Dixon JR, Kelly P, Worby CA, Casey PJ, Dixon JE. Involvement of a mitochondrial phosphatase in the regulation of ATP production and insulin secretion in pancreatic beta cells. Mol Cell. 2005;19:197–207. doi: 10.1016/j.molcel.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3:187–197. doi: 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Parsons SJ, Parsons JT. Src family kinases, key regulators of signal transduction. Oncogene. 2004;23:7906–7909. doi: 10.1038/sj.onc.1208160. [DOI] [PubMed] [Google Scholar]
- Pitkanen S, Robinson BH. Mitochondrial complex I deficiency leads to increased production of superoxide radicals and induction of superoxide dismutase. J Clin Invest. 1996;98:345–351. doi: 10.1172/JCI118798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirós PM, Ramsay AJ, Sala D, Fernández-Vizarra E, Rodríguez F, Peinado JR, Fernández-García MS, Vega JA, Enríquez JA, Zorzano A, López-Otín C. Loss of mitochondrial protease OMA1 alters processing of the GTPase OPA1 and causes obesity and defective thermogenesis in mice. EMBO J. 2012;31:2117–2133. doi: 10.1038/emboj.2012.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radad K, Rausch WD, Gille G. Rotenone induces cell death in primary dopaminergic culture by increasing ROS production and inhibiting mitochondrial respiration. Neurochem Int. 2006;49:379–386. doi: 10.1016/j.neuint.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Robinson BH. Human complex I deficiency: clinical spectrum and involvement of oxygen free radicals in the pathogenicity of the defect. Biochim Biophys Acta. 1998;1364:271–286. doi: 10.1016/s0005-2728(98)00033-4. [DOI] [PubMed] [Google Scholar]
- Salvi M, Brunati AM, Toninello A. Tyrosine phosphorylation in mitochondria: a new frontier in mitochondrial signaling. Free Radic Biol Med. 2005;38:1267–1277. doi: 10.1016/j.freeradbiomed.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Salvi M, Morrice NA, Brunati AM, Toninello A. Identification of the flavoprotein of succinate dehydrogenase and aconitase as in vitro mitochondrial substrates of Fgr tyrosine kinase. FEBS Lett. 2007;581:5579–5585. doi: 10.1016/j.febslet.2007.11.005. [DOI] [PubMed] [Google Scholar]
- Schägger H, von Jagow G. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal Biochem. 1991;199:223–231. doi: 10.1016/0003-2697(91)90094-a. [DOI] [PubMed] [Google Scholar]
- Schulenberg B, Aggeler R, Beechem JM, Capaldi RA, Patton WF. Analysis of steady-state protein phosphorylation in mitochondria using a novel fluorescent phosphosensor dye. J Biol Chem. 2003;278:27251–27255. doi: 10.1074/jbc.C300189200. [DOI] [PubMed] [Google Scholar]
- Speijer D. Oxygen radicals shaping evolution: why fatty acid catabolism leads to peroxisomes while neurons do without it: FADH2/NADH flux ratios determining mitochondrial radical formation were crucial for the eukaryotic invention of peroxisomes and catabolic tissue differentiation. Bioessays. 2011;33:88–94. doi: 10.1002/bies.201000097. [DOI] [PubMed] [Google Scholar]
- Srivastava S, Diaz F, Iommarini L, Aure K, Lombes A, Moraes CT. PGC-1alpha/beta induced expression partially compensates for respiratory chain defects in cells from patients with mitochondrial disorders. Hum Mol Genet. 2009;18:1805–1812. doi: 10.1093/hmg/ddp093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley IA, Ribeiro SM, Giménez-Cassina A, Norberg E, Danial NN. Changing appetites: the adaptive advantages of fuel choice. Trends Cell Biol. 2013;24:118–127. doi: 10.1016/j.tcb.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ten VS, Starkov A. Hypoxic-ischemic injury in the developing brain: the role of reactive oxygen species originating in mitochondria. Neurol Res Int. 2012;2012:542976. doi: 10.1155/2012/542976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibaldi E, Brunati AM, Massimino ML, Stringaro A, Colone M, Agostinelli E, Arancia G, Toninello A. Src-Tyrosine kinases are major agents in mitochondrial tyrosine phosphorylation. J Cell Biochem. 2008;104:840–849. doi: 10.1002/jcb.21670. [DOI] [PubMed] [Google Scholar]
- Vives-Bauza C, Yang L, Manfredi G. Assay of mitochondrial ATP synthesis in animal cells and tissues. Methods Cell Biol. 2007;80:155–171. doi: 10.1016/S0091-679X(06)80007-5. [DOI] [PubMed] [Google Scholar]
- Wenz T, Diaz F, Spiegelman BM, Moraes CT. Activation of the PPAR/PGC-1alpha pathway prevents a bioenergetic deficit and effectively improves a mitochondrial myopathy phenotype. Cell Metab. 2008;8:249–256. doi: 10.1016/j.cmet.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Yoo SK, Starnes TW, Deng Q, Huttenlocher A. Lyn is a redox sensor that mediates leukocyte wound attraction in vivo. Nature. 2011;480:109–112. doi: 10.1038/nature10632. [DOI] [PMC free article] [PubMed] [Google Scholar]