

Abstract
Background
Epigenetic alterations of specific genes have been reported to be related to colorectal cancer (CRC) transformation and would also appear to be involved in the early stages of colorectal carcinogenesis. Little data are available on the role of these alterations in determining a different risk of colorectal lesion recurrence. The aim of the present study was to verify whether epigenetic alterations present in pre-neoplastic colorectal lesions detected by colonoscopy can predict disease recurrence.
Methods
A retrospective series of 78 adenomas were collected and classified as low (35) or high-risk (43) for recurrence according to National Comprehensive Cancer Network guidelines. Methylation alterations were analyzed by the methylation-specific multiplex ligation probe assay (MS-MLPA) which is capable of quantifying methylation levels simultaneously in 24 different gene promoters. MS-MLPA results were confirmed by pyrosequencing and immunohistochemistry.
Results
Higher levels of methylation were associated with disease recurrence. In particular, MLH1, ATM and FHIT gene promoters were found to be significantly hypermethylated in recurring adenomas. Unconditional logistic regression analysis used to evaluate the relative risk (RR) of recurrence showed that FHIT and MLH1 were independent variables with an RR of 35.30 (95% CI 4.15-300.06, P = 0.001) and 17.68 (95% CI 1.91-163.54, P = 0.011), respectively.
Conclusions
Histopathological classification does not permit an accurate evaluation of the risk of recurrence of colorectal lesions. Conversely, results from our methylation analysis suggest that a classification based on molecular parameters could help to define the mechanisms involved in carcinogenesis and prove an effective method for identifying patients at high risk of recurrence.
Keywords: Colorectal adenoma, Gene promoter methylation profile, Risk of recurrence, Prognostic marker, Pre-neoplastic lesion classification
Background
Colorectal cancer (CRC), a disease arising from complex and heterogeneous etiological factors and pathogenetic mechanisms, develops in a multi-step manner from normal epithelium, through a pre-malignant lesion (adenoma), into a malignant lesion (carcinoma) [1]. Histopathological evaluation of early stage CRC in many cases reveals areas of adenomatous mucosa, but the presence of tissue with histological features ranging from pure tubular to pure villous adenomas accompanied by dysplasia is also frequently detected in invasive colorectal cancer [1],[2]. Although individuals with syndromes that strongly predispose to adenomas, e.g. familial adenomatous polyposis (FAP), invariably develop CRC by the third to fifth decade of life if these lesions are not removed [3], most adenomas (not FAP) have a low risk of progressing into cancer (about 5%) if not resected. An adenomatous polyp is a much more frequent finding than CRC and polypectomy has a distinctly protective effect on the subsequent development of CRC. It has been estimated that in the first 10 years after polypectomy, the risk of CRC is reduced to a level similar to that of individuals whose colonoscopy does not reveal the presence of polyps [4],[5].
Different molecular mechanisms seem to be related to CRC development. The vast majority of tumors (about 50-80%), present chromosomal instability (CIN) [3],[6],[7], while a smaller fraction (10-15%) is characterized by microsatellite instability (MSI) [3],[6],[7]. In recent years, epigenetic alterations have gained recognition as a key mechanism in carcinogenesis. In particular, hypermethylation of CpG islands present in gene promoter sequences leads to the inactivation of tumor suppressor genes, working in a different way with respect to genetic mutations [8],[9].
This aberrant methylation status occurs at the same time as genetic alterations which drive the initiation and progression of colorectal cancer, suggesting that methylation plays an important role in many stages of tumor transformation [10]-[14]. The existence of a methylator phenotype could be related to distinctive biological and/or clinical characteristics [15].
CRCs that show hypermethylation changes in numerous different CpG-rich DNA regions are defined as showing the CpG island methylator phenotype (CIMP) [16]. CIMP-positive cancers have distinct clinical pathological characteristics such as proximal colon location, mucinous and poorly differentiated histology, female preponderance and older age [17]. This phenotype also seems to be associated with MSI and BRAF mutations [18],[19]. Conversely, hypomethylation of specific sequences may decrease the fidelity of chromosomal segregation [20], suggesting that it may be involved in the chromosomal instability phenotype [21]. DNA methylation changes probably lead adenomatous precursor lesions to progress into malignant tumors. In fact, sessile serrated adenomas, considered important precursors of cancer, are often CIMP-positive.
Taking the above considerations into account, a better understanding of the epigenetic mechanisms associated with adenoma-carcinoma transition could represent an important tool for CRC prevention. In accordance with international guidelines, pre-neoplastic lesions of the colon and rectum are classified according to pathological parameters (size, histology, number of polyps and dysplasia) as having high or low risk of recurrence. In high risk patients a new colonoscopy is performed after 3 years, while in low risk subjects the time interval is extended to 5 years. However, this type of subdivision is unable to predict the real risk of developing a new lesion. In fact, it has been seen that patients who are classified as high risk may not experience any further problems, while those who are classed as low risk may relapse after a short time.
Little data is available on the relationship between risk of recurrence of pre-neoplastic lesions and molecular alterations of colorectal lesions, whereas a great deal is known about the mechanisms of CRC transformation. Although a number of gene promoter methylation profiles have been shown to characterize specific stages of tumor progression, no data are available on epigenetic alterations or risk of disease evolution/recurrence. The identification of these specific epigenetic profiles could help us to better understand the mechanisms of adenoma recurrence and, possibly, adenoma-carcinoma transition, resulting in a more accurate classification of the risk of recurrence of pre-neoplastic and permitting a personalized program of cancer prevention.
The aim of this study was to evaluate whether altered methylation profiles in pre-neoplastic lesions sampled by colonoscopy is capable of identifying patients at high risk of recurrence with greater accuracy than conventional clinical pathological parameters.
Methods
Case series
We evaluated formalin fixed paraffin-embedded (FFPE) tissue samples of pre-neoplastic colorectal lesions endoscopically identified and surgically removed from a series of 78 patients who underwent follow up for at least 5 years. Lesions were classified as adenomas at low risk (3 tubular polyps with a diameter < 1 cm) or high risk (high-risk dysplasia, > 3 adenomatous villous or tubulovillous polyps, at least one of which with a diameter of ≥ 1 cm, or an in situ carcinoma) of recurrence according to National Comprehensive Cancer Network guidelines. All tissue samples were obtained from the Pathology Unit of Morgagni-Pierantoni Hospital (Forlì, Italy). Informed consent for the use of biological samples was obtained from all individuals who agreed to take part in the study for research purposes. The study protocol was reviewed and approved by the IRST Ethics Committee.
DNA extraction
DNA was extracted using a digestion buffer (50 mM KCl, 10 mM Tris–HCl pH8, 2.5 mM MgCl2, 0.45% v/v TWEEN-20 and proteinase K 25 mg/ml). Approximately three 5-μm slices of paraffin-embedded tissue was added to 150 ml of home-made buffer and 10 ml of proteinase K (25 mg/ml). After overnight incubation at 58°C with gentle shaking, the sample was heated to 98°C for 10 min, cooled to room temperature and then centrifuged at 6000 rpm for 10 min. The supernatant containing DNA was transferred to a new vial and centrifuged again as per the previous step until all traces of paraffin were removed. The quality and quantity of DNA were assessed using NanoDrop ND-1000 (Thermo Fisher Scientific, Waltham, USA) and the DNA was stored at −20°C until molecular analysis was performed.
Quantitative DNA methylation analyses
Methylation-specific multiplex ligation probe analysis
Methylation-specific (MS) multiplex ligation probe analysis (SALSA MLPA ME001 Tumour Suppressor-1 kit, MLPA®; MRC-Holland, Amsterdam, The Netherlands), a high-throughput, semi-quantitative, methylation-specific enzyme-based polymerase chain reaction (PCR) assay, was performed according to the manufacturer’s instructions. Each probe corresponded to a single gene, apart from MLH1 and RASSF1, both of which contained two probes. Two ligation probe reactions were needed to calculate the percentage of methylation, one of which contained the methylation-sensitive enzyme HhaI. Briefly, 200 ng of each sample was diluted to 5 μl with TE buffer and heated at 98°C for 10 min followed by incubation at 25°C for 5 min in a thermocycler. Following the addition of ligation probes, samples were first incubated at 98°C for 1 min and then at 60°C for 16–18 h to permit hybridization. Samples were split equally into two vials, each containing the same amount of DNA (volume 10 ul). Ligase-65 mix (Ligase-65 buffer, Ligase-65 enzyme and water) was added to the first vial, and Ligase-Digestion mix (Ligase-65 buffer, Ligase- 65 enzyme, HhaI enzyme [Promega, Southampton, UK] and water) to the second.
Both samples were incubated at 49°C for 30 min, after which the ligase enzyme was inactivated by heating at 98°C for 5 min. PCR buffer, deoxynucleoside 5-triphosphates (dNTPs) and Taq polymerase were added to the samples during preheating at 72°C. The PCR reaction was performed in a thermocycler preheated to 72°C, under the following conditions: 35 cycles at 95°C for 30 s, 60°C for 30 s and 72°C for 60 s. The final incubation was at 72°C for 20 min. Amplification products were analyzed on an ABI-3130 DNA Analyzer (Applied Biosystems, Warrington, UK). Negative water controls were included to ensure no contamination. Internal validation was performed using unmethylated and methylated genomic DNA (Millipore, Watford, UK).
Intrasample normalization was performed to address peak variations due to fluctuations in the assay run, such as amount of DNA, ploidy variations and PCR conditions, The relative peak height of each probe was determined by dividing the absolute peak height by the mean height of all 15 control probes. A methylation percentage for each probe was obtained using the following calculation, as described previously [22]:
| (1) |
Validation of MS-MLPA results
Validation of MS-MLPA results was only performed for the three most significant genes: ATM, FHIT and MLH1. ATM and MLH1 were confirmed by pyrosequencing CpG analysis, while FHIT was validated by immunohistochemistry (IHC) staining.
Twenty microliters of extracted DNA were converted using Epitect Bisulphite kit (Qiagen, Hilden, Germany) in accordance with the “Sodium Bisulphite Conversion of Unmethylated Cytosines in DNA” protocol. Converted DNA was eluted in 20 μl of elution buffer. Five microliters of bisulphite-treated DNA were used to amplify the specific promoter regions of ATM and MLH1 genes with primer sets designed to amplify the same CpG sites as those of the MS-MLPA approach. Primer sets for amplification and sequencing were designed by Diatech Pharmacogenetics (Jesi, Italy) (Table 1).
Table 1.
Validation of MS-MLPA results for ATM, MLH1 and FHIT
| Gene | Method | Primer sequence/polyclonal antibody | No. samples examined | Overall concordance (%) |
|---|---|---|---|---|
|
ATM |
Pyrosequencing CpG analysis |
Fw: 5′-AGAAGTGGGAGTTGGGTAGTT-3′ |
77/78 |
73% |
| Rv: 5′-biotinCTCCCCCCCCCTACCACTACACTC-3′ | ||||
| Seq: 5′-AGGAGGAGAGAGGAGT-3′ | ||||
|
MLH1 |
Pyrosequencing CpG analysis |
Fw: 5′-biotinGGGAGGTAAGTTTAAGTGGAATAT-3′ |
72/78 |
79% |
| Rv: 5′-CCAATCCCCACCCTAAAACCCTC-3′ | ||||
| Seq: 5′-CTAAACTCCCAAATAATAACCT-3′ | ||||
| FHIT | Immunohistochemistry | Rabbit polyclonal anti-FHIT; clone PA1-37690; Thermo Scientific Pierce; working dilution: 1/200 | 57/78 | 84% |
Abbreviations: Fw Forward Primer, Rv Reverse primer, Seq sequence analyzed.
Each PCR reaction was performed in a final volume of 50 μl containing 2 μl of each primer (5 μM), 1 μl of Takara dNTP mixture (10 mM of each dNTP) (Takara Bio Inc., Otsu, Japan), 1 μl of Takara 50 mM Mg++ solution (Takara Bio Inc.), 2.5 μl of EvaGreen™ Dye (20X), 10 μl of Takara 5X R-PCR Buffer (Mg++ free) (Takara Bio Inc.), 0.5 μl of Takara Ex Taq™ HS (5 U/μl) (Takara Bio Inc.), 26 μl of water and 5 μl of bisulphite-treated DNA. Amplification was done by quantitative Real Time PCR on Rotor Gene™ 6000 (Corbett Life Science, Cambridge, UK) equipped with Rotor Gene 6000 Series Software 1.7 Build 87. The cycling programme for ATM and MLH1 consisted of one hold cycle at 95°C for 5 min, the second hold cycle at 72°C for 5 min, one pre-melting cycle at 65°C for 90 s and then one melting cycle from 65°C to 95°C with an increase of 1°C every 5 s, with fluorescence acquisition. Between the first two holding cycles there were 45 cycles. For ATM gene, these cycles consisted of: denaturation at 95°C for 30 s, annealing 56°C for 30 s and elongation 72°C for 20 s. For MLH1, the 45 cycles comprised denaturation at 95°C for 30 s, annealing at 56°C for 60 s and an elongation cycle at 72°C for 30 s.
Promoter CpG sites were analyzed by PyroQ-CpG™ 1.0.9 software (Biotage, Uppsala, Sweden) on Pyromark Q96 ID (Qiagen). 40 μl of PCR products were added to 37 μl of binding buffer and 3 μl of Sepharose beads and mixed at 1400 rpm for 10 min at room temperature. The Sepharose beads with single-stranded templates attached were released into a plate containing an annealing mixture composed of 38.4 μl of annealing buffer and 1.6 μl of the corresponding sequencing primers. All the experimental procedures were carried out according to the manufacturer’s instructions. We added water as negative control and universal methylated and unmethylated samples as positive control.
Four-μm-thick FFPE adenoma sections were used for immunodetection. Sections were deparaffinized in xylene and dehydrated in a graded alcohol series, according to the local protocol. Antigen retrieval was achieved by microwaving in 10 mM of sodium citrate buffer at pH 6 for 30 min. Sections were incubated with rabbit polyclonal anti-FHIT (clone PA1-37690; Thermo Fisher Scientific, Waltham, USA) at a 1/200 working dilution. From this point onwards, all the steps were performed automatically by Autostainer Plus Staining System (Dako Cytomatic, Glostrop, Denmark). LSAB protein block (Dako; Carpinteria, USA) was performed for 15 min. The staining of the primary antibody was performed for 130 min. Sections were immunostained with anti-rabbit biotinylated secondary antibody LSAB (Dako) for 10 min. Visualization was performed using DAB chromogen (Dako). Sections were counterstained with hematoxylin, dehydrated in the same graded alcoholic scale and mounted. On the basis of antibody datasheet instructions, negative and positive control sections were incubated with the secondary antibody in the presence or not of the primary antibody, respectively.
Statistical analysis
In order to evaluate the correlation between methylation status and prognosis for adenoma/disease recurrence, patients were subdivided into relapsed (R) or not relapsed (NR) at 60 months of follow-up. The relationship between clinical pathological characteristics and patient status was analyzed using the chi-square test.
Methylation was evaluated as both a continuous variable and binary variable. In particular, a cut off of 20% of methylated DNA was used to classify a promoter as hypermethylated. Hypermethylation frequencies in NR and R samples were compared using Fisher’s exact test. The student’s T test was used to compare the mean methylation levels of NR and R samples. Methylation status of multiple genes was evaluated to determine the presence of hypermethylation. Its accuracy (the proportion of R and NR patients correctly identified by the hypermethylated profile) in detecting recurrent lesions using the defined hypermethylation cut off was expressed in terms of sensitivity (proportion of R patients correctly identified by the hypermethylated profile) and specificity (proportion of NR patients correctly identified by the hypermethylated profile) in relation to the total series. For both indicators, 95% confidence intervals (95% CI) were calculated. Logistic regression was used to analyze the Relative Risks (RR) and their 95% CI for patient status and methylation status as dichotomous variables. All analyses were performed using SAS Statistical software (version 9.3, SAS Institute, Cary, North Carolina, USA) or Graphpad Prism software version 5.0d. Statistical significance for all tests was taken as P < 0.05. The validation of the MS-MLPA results was done considering the results obtained by pyrosequencing CpG analysis and IHC considered as dichotomous variables.
Results
MS-MLPA analysis
Tissue specimens of adenomas from 78 patients were collected for methylation status analysis (Table 2). Patients were divided into relapsed (R) or not relapsed (NR) on the basis of disease recurrence at 5 years of follow up. In particular, 47 patients (27 with high risk and 20 with low risk adenomas) did not show disease recurrence (NR), while 31 patients (16 with high risk and 15 low risk adenomas) developed new colorectal lesions (R) during this period. No differences in terms of recurrence were noted on the basis of pathological classification (high or low risk adenoma) and no correlation was found between the grade of dysplasia and development of new lesions during follow up. Conversely, the site of the first lesion was significantly related to risk of disease relapse (P = 0.015).
Table 2.
Clinical pathological characteristics of the case series
| Total n (%) | Disease recurrence n (%) | No. of disease recurrence n (%) | P | |
|---|---|---|---|---|
|
Gender |
|
|
|
|
| Male |
56 (71.8) |
24 (77.4) |
32 (68.1) |
|
| Female |
22 (28.2) |
7 (22.6) |
15 (31.9) |
0.523 |
|
Median age, years (range) |
|
|
|
|
| Male |
61 (42–85) |
64 (48–85) |
61 (42–79) |
0.263 |
| Female |
66 (40–81) |
63 (51–72) |
66 (40–81) |
0.972 |
|
Risk of recurrence |
|
|
|
|
| High risk |
43 (55.1) |
16 (51.6) |
27 (57.4) |
|
| Low risk |
35 (44.9) |
15 (48.4) |
20 (42.6) |
0.784 |
|
Dysplasia |
|
|
|
|
| Low (low and medium) grade |
61 (78.2) |
26 (83.9) |
35 (74.5) |
|
| High grade |
17 (21.8) |
5 (16.1) |
12 (25.5) |
0.481 |
|
Lesion dimension |
|
|
|
|
| 0–0.9 cm |
9 (11.5) |
3 (9.7) |
6 (12.8) |
|
| ≥ 1 cm |
29 (37.2) |
11 (35.5) |
18 (38.3) |
|
| Not specified |
40 (51.3) |
17 (54.8) |
23 (48.9) |
1.000 |
|
Lesion localization |
|
|
|
|
| Ascending colon |
19 (24.4) |
10 (32.3) |
9 (19.1) |
|
| Descending colon |
37 (47.4) |
9 (29.0) |
28 (59.6) |
|
| Mixed |
22 (28.2) |
12 (38.7) |
10 (21.3) |
0.015 |
|
Adenoma morphology |
|
|
|
|
| Tubular |
46 (59.0) |
19 (61.3) |
27 (57.4) |
|
| Villous |
3 (3.8) |
0 |
3 (6.4) |
|
| Tubulovillous (mixed) | 29 (37.2) | 12 (38.7) | 17 (36.2) | 0.441 |
MS-MLPA analysis was performed for all samples, obtaining a quantification of methylation status for the entire case series. Two probes (GSTP1 and MLH1 CpG 02) were discarded from the analysis because they were negative for methylation (0% methylation level) in 92% and 83% of cases, respectively. We first evaluated the number of hypermethylated promoters in R and NR patients using a methylation level of 20% to define a gene promoter as hypermethylated. Primary lesions that relapsed showed a higher number of hypermethylated markers (median 6, range 2–24) than non recurring lesions (median 4, range 0–12) (Figure 1A).
Figure 1.
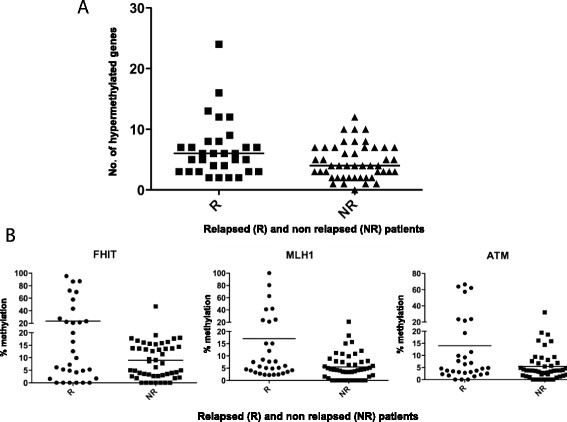
Gene methylation level distribution. A) Hypermethylated genes in the case series subdivided according to the presence or not of disease recurrence. B) Comparison of methylation levels of the three most significant genes in R and NR samples.
The promoters of three genes (FHIT, MLH1 and ATM) were found to be hypermethylated in a significantly higher fraction of adenomas that recurred compared to non recurring lesions (Figure 1B). Furthermore, TP73 and BRCA1 promoter hypermethylation were related to recurrence, albeit with low statistical significance or borderline significance, respectively (Table 3).
Table 3.
Frequency of promoter hypermethylation in patients with recurrent or non recurrent disease
| Gene ID |
% R |
% NR |
Overall series |
P |
|---|---|---|---|---|
| (Total = 31) | (Total = 47) | (Total = 78) | ||
|
FHIT
|
38.71 (12/31) |
2.13 (1/47) |
16.67 (13/78) |
3.1E-05 |
|
MLH1
|
25.81 (8/31) |
2.13 (1/47) |
11.54 (9/78) |
0.002 |
|
ATM
|
22.58 (7/31) |
2.13 (1/47) |
10.26 (8/78) |
0.006 |
|
TP73
|
35.48 (11/31) |
12.77 (6/47) |
21.79 (17/78) |
0.025 |
|
BRCA1
|
9.68 (3/31) |
0.00 (0/47) |
3.85 (3/78) |
0.059 |
|
CHFR
|
29.03 (9/31) |
10.64 (5/47) |
17.95 (14/78) |
0.068 |
|
IGSF4
|
12.90 (4/31) |
2.13 (1/47) |
6.41 (5/78) |
0.078 |
|
ESR1
|
70.97 (22/31) |
85.11 (40/47) |
79.49 (62/78) |
0.158 |
|
DAPK1
|
22.58 (7/31) |
10.64 (5/47) |
15.38 (12/78) |
0.203 |
|
CDKN2B
|
45.16 (14/31) |
29.79 (14/47) |
35.90 (28/78) |
0.228 |
|
RASSF1
CpG1 |
41.94 (13/31) |
29.79 (14/47) |
34.62 (27/78) |
0.333 |
|
RASSF1
CpG2 |
12.90 (4/31) |
6.38 (3/47) |
8.97 (7/78) |
0.427 |
|
HIC1
|
16.13 (5/31) |
8.51 (4/47) |
11.54 (9/78) |
0.471 |
|
CDKN2A
|
22.58 (7/31) |
14.89 (7/47) |
17.95 (14/78) |
0.548 |
|
CASP8
|
6.45 (2/31) |
2.13 (1/47) |
3.85 (3/78) |
0.560 |
|
CDH13
|
80.65 (25/31) |
74.47 (35/47) |
76.92 (60/78) |
0.592 |
|
CD44
|
3.23 (1/31) |
8.51 (4/47) |
6.41 (5/78) |
0.643 |
|
BRCA2
|
12.90 (4/31) |
8.51 (4/47) |
10.26 (8/78) |
0.706 |
|
RARB
|
48.39 (15/31) |
44.68 (21/47) |
46.15 (36/78) |
0.818 |
|
APC
|
45.16 (14/31) |
48.94 (23/47) |
47.44 (37/78) |
0.819 |
|
TIMP3
|
38.71 (12/31) |
36.17 (17/47) |
37.18 (29/78) |
1.000 |
|
CDKN1B
|
9.68 (3/31) |
8.51 (4/47) |
8.97 (7/78) |
1.000 |
|
VHL
|
6.45 (2/31) |
6.38 (3/47) |
6.41 (5/78) |
1.000 |
| PTEN | 3.23 (1/31) | 4.26 (2/47) | 3.85 (3/78) | 1.000 |
Abbreviations: R recurrent disease, NR non recurrent disease. P-value < 0.05.
We then compared the mean methylation levels of gene promoters in R and NR patients, confirming that MLH1, ATM and FHIT were significantly differentially methylated in adenomas on the basis of the presence or not of lesion recurrence (Figure 2).
Figure 2.
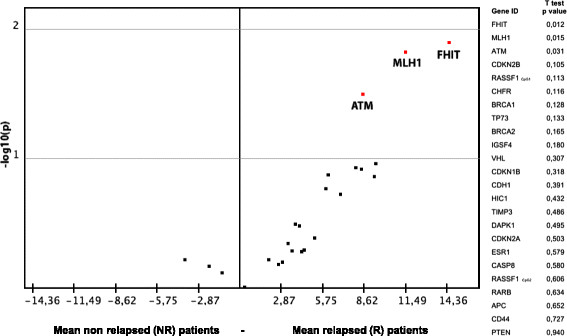
Volcano Plot representing the differences in methylation levels between relapsed and non relapsed samples plotted against their statistical significance for all gene promoters analyzed. The three promoters displaying significantly increased methylation levels in R samples (two-tailed T test, P < 0.05) are highlighted in the upper right corner. T-test P values of the comparison between methylation levels in R vs NR samples are shown to the right of the plot.
In particular, lower levels of methylation were associated with no recurrence of disease, while substantially higher values were correlated with relapse. Moreover, other genes showed differences in terms of methylation alterations. In particular, higher methylation levels of CDKN2B, RASSF1, CHFR, BRCA2 and IGSF4 were observed in adenomas that recurred.
Methylation status phenotype and clinical pathological parameters
Taking these data into account, we evaluated the methylation status, determined on the basis of the presence or not of hypermethylation in the most significantly altered gene promoters (Table 4a,b). Analysis of MLH1, ATM and FHIT genes showed that the presence of hypermethylation in at least one of these genes indicated disease recurrence with 61% sensitivity (95% CI 44–76), and 94% specificity (95% CI 83–98), with an overall accuracy of 81% (95% CI 72–90) (Table 4a). This contrasts with the conventionally used histopathological classification which highlighted a similar distribution of recurrence in high- and low-risk subgroups (Table 2). The integration of BRCA1 and TP73 markers into the panel of genes did not increase accuracy when either or both were considered in methylation status analysis (Table 4b).
Table 4.
Number of hypermethylated markers in recurrent lesions
|
Sensitivity (%) |
Specificity (%) |
Accuracy (%) |
|
|---|---|---|---|
| (95%CI) | (95%CI) | (95%CI) | |
|
a) FHIT, MLH1, ATM |
|
|
|
|
≥1 |
61.29 (43.82-76.27) |
93.61 (82.84-97.81) |
80.76 (72.02-89.52) |
|
≥2 |
22.58 (11.40-39.81) |
100 (92.44-100) |
69.23 (58.99-79.47) |
|
≥3 |
6.45 (1.79-20.72) |
100 (92.44-100) |
62.82 (52.09-73.55) |
|
b) FHIT, MLH1, ATM, TP73, BRCA1 |
|
|
|
|
≥1 |
70.96 (53.41-83.90) |
85.11 (72.31-92.59) |
79.49 (70.53-88.45) |
|
≥2 |
38.71 (23.73-56.18) |
95.74 (85.75-98.83) |
73.08 (63.24-82.92) |
|
≥3 |
16.13 (7.09-32.63) |
100 (92.44-100) |
66.66 (56.21-77.13) |
|
≥4 |
6.45 (1.79-20.72) |
100 (92.44-100) |
62.82 (52.09-73.55) |
|
≥5 |
3.22 (0.57-16.19) |
100 (92.44-100) |
61.53 (50.74-72.34) |
|
c) FHIT, MLH1 |
|
|
|
|
≥1 |
58.06 (40.77-73.58) |
95.74 (85.75-98.83) |
80.77 (72.02-89.52) |
| ≥2 | 9.68 (3.35-24.90) | 100 (92.44-100) | 64.10 (53.45-74.75) |
Sensitivity, R patients who were correctly identified by the hypermethylated profile; Specificity, NR patients who were correctly identified by the hypermethylated profile; Accuracy, R patients, correctly identified by the hypermethylated profile, and NR patients, correctly identified by the hypermethylated profile, divided by the total series; 95% CI, 95% confidence intervals.
Unconditional logistic regression analysis was carried out to evaluate the capacity of MLH1, ATM and FHIT gene methylation to predict recurrence. FHIT and MLH1 proved to be independent variables with an RR of recurrence of 35.30 (95% CI 4.15-300.06, P = 0.001) and 17.68 (95% CI 1.91-163.54, P = 0.011), respectively. CIMP analysis showed that hypermethylation of at least 1 of these gene promoters identified recurring adenomas with 58% sensitivity and 96% specificity (Table 4c).
Methylation status was not related to age or grade of dysplasia. Conversely, a higher frequency of MLH1 hypermethylation was associated with site of lesion. In particular, a higher frequency of methylated MLH1 was observed in ascending with respect to descending lesions (71% and 29%, respectively, P = 0.07).
Validation of MS-MLPA results
Pyrosequencing measures the methylation level of single promoter CpG sites and is used to confirm the results from other analytical methods [23].
The average methylation percentage of the same CpG sites as those used for the MS-MLPA approach was considered for data analysis (data not shown). This approach was only utilized for MLH1 and ATM as reliable results were not obtained for FHIT. For this reason, FHIT was evaluated by immunohistochemistry. A methylated cut-off of ≥20% was used to discriminate between the methylated and unmethylated phenotype.
Good concordance was observed between MS-MLPA and the other two methods used (Table 1). In particular, a comparison between the MS-MLPA and pyrosequencing methods showed a 79% (57/72 cases) agreement in samples for MLH1 and a 73% (56/77cases) agreement for ATM, respectively. The concordance between MS-MLPA results and IHC was 84% for FHIT (48/57 cases) (Figure 3). This validation was not performed on samples for which there was insufficient biological material.
Figure 3.
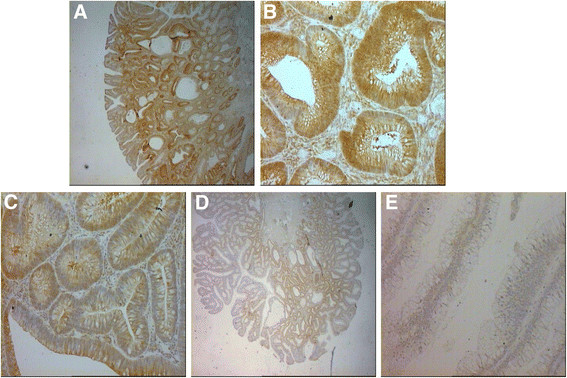
IHC staining of FHIT protein in adenoma samples. A) High cytoplasmic staining in 85% of colonic glands (grade 3+), a small fraction of glands (15%-20%) showing low intensity staining (grade 2+). Magnification 2.5 ×. B) High cytoplasmic staining in 85% of colonic glands (grade 3+). Magnification 20×. C) Medium cytoplasm staining in 80% of colonic glands (grade 2+). Magnification 20×. D) Low cytoplasmic staining in 60% of colonic glands; 40%, grade 1+ and 20%, grade 2+. Magnification 2.5×. E) Negative cytoplasmic staining of colonic glands. Magnification 2.5 x.
Conclusions
The adenoma-carcinoma sequence is accepted as the main pathway for the development of colorectal cancer. Although some genetic studies have provided evidence that CRC can develop in other ways, early stage CRCs frequently show adenomatous mucosa at the tumor periphery. Foci of different grades of dysplasia, intra-mucosal carcinoma and invasive cancer have also been observed in pre-neoplastic lesions, indicating a potential relationship between these different stages of colorectal lesions [7],[17]. A high number of adenomas are now detected in apparently healthy individuals undergoing routine colorectal cancer screening, but little information is available on the effective risk of recurrence in these patients.
For this purpose we selected a series of pre-neoplastic lesions classified histologically as high or low risk lesions from patients with a different clinical history. No statistically significant differences were found between adenomas classified as low risk and those classed as high risk with respect to recurrence during the 5-year follow up. Such data indicate that histopathological classification alone is insufficient to plan an adequate follow up of these patients. Moreover, grade of dysplasia, polyp size and other morphological parameters do not appear to be useful for predicting clinical evolution and therefore for organizing adequate patient surveillance. Although defined molecular subtypes of CRC exist, the molecular subgroups of CRC cannot be accurately distinguished histologically or clinically at this time [24].
Conversely, the results from the methylation profile analyzed in this study indicate that a molecular approach is capable of accurately predicting recurrence. In particular, we identified three genes (MLH1, ATM and FHIT) differentially methylated in adenomas that recurred during the five-year follow up. The association between the methylation of these three genes and the higher aggressiveness of pre-neoplastic lesions may be attributable to their biological functions. In fact, MLH1 and ATM genes play a key role in DNA detection and repair systems and their inactivation may cause genomic DNA to become more unstable and error-prone, increasing the risk of transformation.
The MLH1 protein is involved in the DNA mismatch repair system (MMR) and methylation of this gene has been observed in CRC, especially in tumors characterized by MSI, a molecular marker of the presence of defective MMR [25],[26]. The ATM protein, a serine/threonine kinase involved in DNA double-strand break repair, is also involved in DNA repair and its inactivation is a highly destabilizing event for the cell, promoting the progression of neoplastic disease [27],[28]. It is interesting to note that MLH1 is an independent variable, despite the molecular interaction between MLH1 and ATM in regulating DNA repair. This suggests that concurrent inactivation of both genes may also be important in cancer development.
FHIT, a tumor suppressor gene involved in numerous important mechanisms associated with cell cycle response to stress signals and DNA replication control, is another independent variable [29]. Wali reported that the FHIT gene loses its ability to produce its specific protein in the early stages of lung, head and neck, esophageal, colorectal, breast, and cervical cancer [30]. The diminution or loss of FHIT protein expression appears to be influenced by the extensive promoter methylation program manifested in CIMP-high CRC cases [31]. TP73 and BRCA1 genes, both related to a higher risk of recurrence, are also involved in cell cycle control and DNA repair. In particular, TP73 is a homolog of TP53 tumor suppressor gene, known to be involved in the regulation of cell proliferation and apoptosis [32]-[34], while BRCA1 represents a key regulator in the repair of double-stranded DNA breaks [26],[35]. In Huang et al.’s 2010 study on 110 stage I to IV CRC patients, TP73 and BRCA1 were identified from a panel of 15 radiation-related genes as prognosis-related markers on the basis of their significant correlation with clinical prognostic variables [36].
In our study, methylation status analysis of a combination of the three most significant genes (MLH1, ATM, FHIT) confirmed that they could be used to accurately identify patients at a higher risk of recurrence. Moreover, it is worthy of note that these genes (MLH1, ATM, FHIT, TP73 and BRCA1) were not among those most frequently methylated in our case series, suggesting that the risk of recurrence is related to specific molecular characteristics.
In fact, higher aberrant methylation (more than 70% of cases with methylation levels higher than 20%) was noted for ESR1 and CDH13, which are not associated with a risk of recurrence. ESR1 has been shown to occur in histologically normal colon epithelium in an age dependent fashion [16],[37], CDH13 is also frequently methylated in CRC and seems to be correlated with the adenoma-carcinoma transition, like other genes with methylation frequencies > 30% (CDKN2B, RASSF1, RARB, APC and TIMP3). Although these genes are probably related to the first step of colorectal transformation, they do not determine a molecular condition of “general colorectal instability” capable of increasing the risk of normal epithelial cell transformation. The high frequency of promoter hypermethylation of these genes confirms previously published literature data [37],[38].
The strength of our study lies in the fact that the MS-MLPA technique has the advantage of requiring a small quantity of DNA and has been shown to work well in FFPE samples [39]. However, it is also somewhat limited due to the small case series (5-year follow up records are not easily obtained in this patient setting) and to the heterogeneity of the cell population. Laser micro-dissection rather them manual macro-dissection would provide more material that is pure enough for analysis. Furthermore, when using an MS-MLPA validation approach, it must be remembered that, unlike pyrosequencing, MS-MLPA does not require bisulphate conversion and that it does not quantify the presence of protein, as does IHC.
In conclusion, a more extensive analysis is needed to confirm these preliminary data, our results would nonetheless seem to indicate that a classification based on molecular parameters could more accurately select patients at high risk of recurrence. These methylation profiles could also provide important information on the aggressiveness of the lesion and on disease evolution, useful elements when planning tailored follow up.
Abbreviations
CRC: Colorectal cancer
FAP: Familial adenomatous polyposis
CIN: Chromosomal instability
MSI: Microsatellite instability
CIMP: CpG island methylator phenotype
MS-MLPA: Methylation-specific multiplex ligation probe analysis
dNTPs: Deoxynucleoside 5-triphosphates
FFPE: Formalin fixed paraffin-embedded
R: Relapsed
NR: Not relapsed
95% CI: 95% confidence interval
RR: Relative risks
MMR: Mismatch repair
IHC: Immunohistochemistry
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CR and DC conceived and designed the study. MZ, GDM, MMT and GF carried out the immunohistochemistry assay and performed the pyrosequencing and MS-MLPA analyses. ACG and LS were responsible for patient recruitment. LS and MP interpreted the immunohistochemistry results. ES, CZ and CM performed the statistical analyses. CR, DC, GDM, MZ, GF and ES drafted the manuscript. DA and WZ reviewed the manuscript for important intellectual content. All authors read and approved the final manuscript.
Contributor Information
Claudia Rengucci, Email: claudia.rengucci@irst.emr.it.
Giulia De Maio, Email: giulia.demaio@irst.emr.it.
Andrea Casadei Gardini, Email: andrea.casadei@irst.emr.it.
Mattia Zucca, Email: mttzcc@gmail.com.
Emanuela Scarpi, Email: emanuela.scarpi@irst.emr.it.
Chiara Zingaretti, Email: chiara.zingaretti@googlemail.com.
Giovanni Foschi, Email: giovanni.foschi@irst.emr.it.
Maria Maddalena Tumedei, Email: maria.tumedei@irst.emr.it.
Chiara Molinari, Email: chiara.molinari@irst.emr.it.
Luca Saragoni, Email: lsaragoni@ausl.fo.it.
Maurizio Puccetti, Email: m.puccetti@ausl.ra.it.
Dino Amadori, Email: direzione.scientifica@irst.emr.it.
Wainer Zoli, Email: wainer.zoli@irst.emr.it.
Daniele Calistri, Email: daniele.calistri@irst.emr.it.
Acknowledgements
The authors thank Ursula Elbling for editing the manuscript and Sara Bravaccini for technical support. They also thank Gianmarco Musciano of Diatech Pharmacogenetics, Jesi (AN), Italy, for his advice and contribution to the development of the pyrosequencing assay.
References
- Jass JR. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology. 2007;50:113–130. doi: 10.1111/j.1365-2559.2006.02549.x. [DOI] [PubMed] [Google Scholar]
- Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348:919–932. doi: 10.1056/NEJMra012242. [DOI] [PubMed] [Google Scholar]
- Rustgi AK. The genetics of hereditary colon cancer. Genes Dev. 2007;21:2525–2538. doi: 10.1101/gad.1593107. [DOI] [PubMed] [Google Scholar]
- Zauber AG, Winawer SJ, O’Brien MJ, Lansdorp-Vogelaar I, Van Ballegooijen M, Hankey BF, Shi W, Bond JH, Schapiro M, Panish JF, Stewart ET, Waye JD. Colonoscopic polypectomy and long-term prevention of colorectal-cancer deaths. N Engl J Med. 2012;366:687–696. doi: 10.1056/NEJMoa1100370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simunic M, Perkovic N, Rosic-Despalatovic B, Tonkic A, Ardalic Z, Titlic M, Maras-Simunic M. Colonoscopic polypectomies and recommendations on the colonoscopy follow-up intervals depending on endoscopic and histopathological findings. Acta Inform Med. 2013;21:166–169. doi: 10.5455/aim.2013.21.166-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011;6:479–507. doi: 10.1146/annurev-pathol-011110-130235. [DOI] [PubMed] [Google Scholar]
- Ahnen DJ. The American College of Gastroenterology Emily Couric Lecture–the adenoma-carcinoma sequence revisited: has the era of genetic tailoring finally arrived? Am J Gastroenterol. 2011;106:190–198. doi: 10.1038/ajg.2010.423. [DOI] [PubMed] [Google Scholar]
- Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143–153. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- Zitt M, Zitt M, Müller HM. DNA methylation in colorectal cancer–impact on screening and therapy monitoring modalities? Dis Markers. 2007;23:51–71. doi: 10.1155/2007/891967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo Y, Issa JP. Epigenetic changes in colorectal cancer. Cancer Metastasis Rev. 2004;23:29–39. doi: 10.1023/A:1025806911782. [DOI] [PubMed] [Google Scholar]
- De Maat MF, van de Velde CJ, van der Werff MP, Putter H, Umetani N, Klein-Kranenbarg EM, Turner RR, Van Krieken JHJM, Bilchik A, Tollenaar RAEM, Hoon DSB. Quantitative analysis of methylation of genomic loci in early-stage rectal cancer predicts distant recurrence. J Clin Oncol. 2008;26:2327–2335. doi: 10.1200/JCO.2007.14.0723. [DOI] [PubMed] [Google Scholar]
- Hartmann O, Spyratos F, Harbeck N, Dietrich D, Fassbender A, Schmitt M, Eppenberger-Castori S, Vuaroqueaux V, Lerebours F, Welzel K, Maier S, Plum A, Niemann S, Foekens JA, Lesche R, Martens JW. DNA methylation markers predict outcome in node-positive, estrogen receptor-positive breast cancer with adjuvant anthracycline-based chemotherapy. Clin Cancer Res. 2009;15:315–323. doi: 10.1158/1078-0432.CCR-08-0166. [DOI] [PubMed] [Google Scholar]
- Richiardi L, Fiano V, Vizzini L, De Marco L, Delsedime L, Akre O, Tos AG, Merletti F. Promoter methylation in APC, RUNX3, and GSTP1 and mortality in prostate cancer patients. J Clin Oncol. 2009;27:3161–3168. doi: 10.1200/JCO.2008.18.2485. [DOI] [PubMed] [Google Scholar]
- Di Domenico M, Santoro A, Ricciardi C, Iaccarino M, Iaccarino S, Freda M, Feola A, Sanguedolce F, Losito S, Pasquali D, Di Spiezio Sardo A, Bifulco G, Nappi C, Bufo P, Guida M, De Rosa G, Abbruzzese A, Caraglia M, Pannone G. Epigenetic fingerprint in endometrial carcinogenesis: the hypothesis of a uterine field cancerization. Cancer Biol Ther. 2011;12:447–457. doi: 10.4161/cbt.12.5.15963. [DOI] [PubMed] [Google Scholar]
- Issa JP. CpG island methylator phenotype in cancer. Nat Rev Cancer. 2004;4:988–993. doi: 10.1038/nrc1507. [DOI] [PubMed] [Google Scholar]
- Rashid A, Issa JPJ. CpG island methylation in gastroenterologic neoplasia: a maturing field. Gastroenterology. 2004;127:1578–1588. doi: 10.1053/j.gastro.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D, Koh H, Simms L, Barker M, Leggett B, Levine J, Kim M, French AJ, Thibodeau SN, Jass J, Haile R, Laird PW. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38:787–793. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- Hinoue T, Weisenberger DJ, Pan F, Campan M, Kim M, Young J, Kim M, Young J, Whitehall VL, Leggett BA, Laird PW. Analysis of the association between CIMP and BRAF in colorectal cancer by DNA methylation profiling. PLoS One. 2009;4:e 8357. doi: 10.1371/journal.pone.0008357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji W, Hernandez R, Zhang XY, Qu GZ, Frady A, Varela M, Ehrlich M. DNA demethylation and pericentromeric rearrangements of chromosome 1. Mutat Res. 1997;379:33–41. doi: 10.1016/S0027-5107(97)00088-2. [DOI] [PubMed] [Google Scholar]
- Goel A, Nagasaka T, Arnold CN, Inoue T, Hamilton C, Niedzwiecki D, Compton C, Mayer RJ, Goldberg R, Bertagnolli MM, Boland CR. The CpG Island methylator phenotype and chromosomal instability are inversely correlated in sporadic colorectal cancer. Gastroenterology. 2007;132:127–138. doi: 10.1053/j.gastro.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Leong KJ, Wei W, Tannahill LA, Caldwell GM, Jones CE, Morton DG, Matthews GM, Bach SP. Methylation profiling of rectal cancer identifies novel markers of early-stage disease. Br J Surg. 2011;98:724–734. doi: 10.1002/bjs.7422. [DOI] [PubMed] [Google Scholar]
- Moon JW, Lee SK, Lee JO, Kim N, Lee YW, Kim SJ, Kang HJ, Kim J, Kim HS, Park SH. Identification of novel hypermethylated genes and demethylating effect of vincristine in colorectal cancer. J Exp Clin Cancer Res. 2014;33:4. doi: 10.1186/1756-9966-33-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardhan K, Liu K. Epigenetics and colorectal cancer pathogenesis. Cancers (Basel) 2013;5:676–713. doi: 10.3390/cancers5020676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, Jessup JM, Kolodner R. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997;57:808–811. [PubMed] [Google Scholar]
- Fu D, Calvo JA, Samson LD. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat Rev Cancer. 2012;12:104–120. doi: 10.1038/nrc3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol. 2008;9:759–769. doi: 10.1038/nrm2514. [DOI] [PubMed] [Google Scholar]
- Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer. 2003;3:155–168. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- Huebner K, Saldivar JC, Sun J, Shibata H, Druck T. Hits, Fhits and Nits: beyond enzymatic function. Adv Enzyme Regul. 2011;51:208–217. doi: 10.1016/j.advenzreg.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wali A. FHIT: doubts are clear now. Scientific World Journal. 2010;10:1142–1151. doi: 10.1100/tsw.2010.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Temaimi RA, Jacob S, Al-Ali W, Thomas DA, Al-Mulla F. Reduced FHIT expression is associated with mismatch repair deficient and high CpG island methylator phenotype colorectal cancer. J Histochem Cytochem. 2013;61:627–638. doi: 10.1369/0022155413497367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28:1057–1068. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- Herreros-Villanueva M, Muñiz P, García-Girón C, Cavia-Saiz M, Del Corral MJ. TAp73 is one of the genes responsible for the lack of response to chemotherapy depending on B-Raf mutational status. J Transl Med. 2010;8:15. doi: 10.1186/1479-5876-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allocati N, Di Ilio C, De Laurenzi V. p63/p73 in the control of cell cycle and cell death. Exp Cell Res. 2012;318:1285–1290. doi: 10.1016/j.yexcr.2012.01.023. [DOI] [PubMed] [Google Scholar]
- Murphy CG, Moynahan ME. BRCA gene structure and function in tumor suppression: a repair-centric perspective. Cancer J. 2010;16:39–47. doi: 10.1097/PPO.0b013e3181cf0204. [DOI] [PubMed] [Google Scholar]
- Huang MY, Wang JY, Chang HJ, Kuo CW, Tok TS, Lin SR. CDC25A, VAV1, TP73, BRCA1 and ZAP70 gene overexpression correlates with radiation response in colorectal cancer. Oncol Rep. 2011;25:1297–1306. doi: 10.3892/or.2011.1171. [DOI] [PubMed] [Google Scholar]
- Lao VV, Grady WM. Epigenetics and colorectal cancer. Nat Rev Gastroenterol Hepatol. 2011;8:686–700. doi: 10.1038/nrgastro.2011.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda H, Kato Y, Yoshikawa H, Arai M, Togashi K, Nagai H, Konishi F, Miki Y. Frequent involvement of ras-signalling pathways in both polypoid-type and flat-type early-stage colorectal cancers. J Exp Clin Cancer Res. 2006;25(2):235–242. [PubMed] [Google Scholar]
- Casadio V, Molinari C, Calistri D, Tebaldi M, Gunelli R, Serra L, Falcini F, Zingaretti C, Silvestrini R, Amadori D, Zoli W. DNA Methylation profiles as predictors of recurrence in non muscle invasive bladder cancer: an MS-MLPA approach. J Exp Clin Cancer Res. 2013;32:94. doi: 10.1186/1756-9966-32-94. [DOI] [PMC free article] [PubMed] [Google Scholar]