

Abstract
The objective of this video protocol is to discuss how to perform and analyze a three-dimensional fluorescent orbital particle tracking experiment using a modified two-photon microscope1. As opposed to conventional approaches (raster scan or wide field based on a stack of frames), the 3D orbital tracking allows to localize and follow with a high spatial (10 nm accuracy) and temporal resolution (50 Hz frequency response) the 3D displacement of a moving fluorescent particle on length-scales of hundreds of microns2. The method is based on a feedback algorithm that controls the hardware of a two-photon laser scanning microscope in order to perform a circular orbit around the object to be tracked: the feedback mechanism will maintain the fluorescent object in the center by controlling the displacement of the scanning beam3-5. To demonstrate the advantages of this technique, we followed a fast moving organelle, the lysosome, within a living cell6,7. Cells were plated according to standard protocols, and stained using a commercially lysosome dye. We discuss briefly the hardware configuration and in more detail the control software, to perform a 3D orbital tracking experiment inside living cells. We discuss in detail the parameters required in order to control the scanning microscope and enable the motion of the beam in a closed orbit around the particle. We conclude by demonstrating how this method can be effectively used to track the fast motion of a labeled lysosome along microtubules in 3D within a live cell. Lysosomes can move with speeds in the range of 0.4-0.5 µm/sec, typically displaying a directed motion along the microtubule network8.
Keywords: Bioengineering, Issue 92, fluorescence, single particle tracking, laser scanning microscope, two-photon, vesicle transport, live-cell imaging, optics
Introduction
A large number of approaches have been developed to date to track fluorescent particles in three dimensions using a microscope. Most approaches rely on the use of fast cameras, ideally suited to track in two dimensions, typically combined with customized modifications of the emission optics of the microscope to achieve tracking in the axial direction. Laser scanning microscopes (either confocal or two photons) conventionally can track a fluorescent particle by performing a time sequence of z-stacks, although this process is typically time consuming, and yields reasonable time resolution (10 Hz) only if the particle being tracked is kept in the center of a small raster imaging region by an active feedback mechanism2.
The idea of locking-in to the particle is the base of the orbital tracking method. Instead of a raster scan a circular orbit is performed around the fluorescent particle. The intensity of the fluorescence along the orbit precisely localizes the particle position4.
The localized position of the particle can then be used to actuate the microscope scanners and re-center the orbit on the particle position. The galvanometer scanners of the microscope are driven by an analog voltage. The AC component of this voltage allows performing an orbit with the focused laser beam, i.e. a sine and a cosine wave applied to the X and Y scanning mirrors will allow performing a circular orbit. The DC offset of the signal facilitates changing the position of the orbit center. Once an orbit period is determined and the waveform for the AC signal is configured, the feedback system needs to update only the DC component of the signal.
A software able to read the fluorescent signal collected from the detectors is required to control the scanners and the detectors, to calculate the position of the particle and update the DC offset. For a successful feedback imaging a careful choice of the orbit parameters (size and timing) is required and these parameters have to be adjusted based upon the characteristics of the fluorescent particles that it is necessary to track.
The physical and mathematical foundations of the technique were described in the past4,5 for both 2D and 3D applications. In this protocol we briefly describe the main hardware components of the setup, and in more detail the choice of the parameters and the sample preparation required for a typical experiment allowing us following the displacement of a lysosome at a high temporal resolution within a living cell.
Protocol
1. Sample Preparation
Maintain CHOK1 cells in tissue culture flasks using DMEM supplemented with 10% fetal bovine serum and 100 I.U/ml of penicillin 50 µg/ml of streptomycin. Incubate the cells in a 5% CO2 humidified incubator at 37 °C.
Harvest and then plate CHOK1 cells on a 14 mm diameter micro-well with a surface thickness of 0.16 mm. Seed cells for optimal density for imaging, around 60-70% confluency.
Incubate cells overnight at 37 °C, 5% CO2. Wash the cells three times in HBSS (Hank Buffered Saline Solution), and incubate cells in a solution containing 50 nM of Lysotracker DND26 green and 150 nM of tubulin tracker green. Incubate cells for 1 hr at 37 °C. Optional step: Prior to incubation, perform an additional stain with a mitochondrial matrix staining dye (25 nM).
Wash the cells three times in HBSS to remove any unbound dye. Add fresh DMEM growth medium prior to imaging the cells.
2. Microscope Configurations
The microscope for the particle tracking described in the video protocol is assembled on the frame of a commercially available inverted microscope (Figure 4). However commercial modules for 3D orbital particle tracking are now available.
Use a Coherent Chameleon-Ultra II Ti:Sa femtosecond laser excitation light source, with a tunable output wavelength range between 690 nm-1,040 nm.
Ensure that the laser beam is aligned in the rear port of the microscope using IR-coated mirrors. The laser beam is attenuated using a rotating half-wave plate followed by a calcite linear polarizer. Attenuate the beam so that the average power at the sample is between 0.5 and 2 mW. NOTE: The laser beam is reflected by a pair of galvanometer-motor actuated mirrors that allow control of the position and trajectory of the focused beam in the sample plane. In a typical configuration the collimated laser beam exiting of the galvanometer mirrors is expanded (10X) passing through a beam expander, before entering the rear of the microscope objective after reflection on short-pass dichroic mirror.
Collect the fluorescence light from the sample by placing a high numerical aperture water objective (60X, NA 1.2) into the light path. Choose a fluorescence filter cube according to the desired emission wavelengths in either one or two channel configuration. Employ emission bandpass filters to further select the spectral range of the emitted fluorescence.
Place the sample onto a motorized stage, and adjust the fine motion of the microscope objective using an objective piezo-controller.
Direct the light from the filter cube into photomultiplier tubes where the signal is discriminated and sent to a digital I/O data acquisition card. NOTE: Computer generated waveforms are the analog output of the I/O card and are provided to the scanners control electronics. The photon counting input from the photomultipliers and the output signal to the scanners are measured and controlled via the I/O card by the Laboratory for Fluorescence Dynamics SimFCS software.
3. Imaging
Position the cells on the stage and focus using transmitted light illumination.
Switch to raster scan imaging to identify the cells that have incorporated the dye and to visualize the underlying microtubules. Identify the initial area where vesicle movement is present.
- Once an isolated vesicle is identified, set the following parameters for orbital tracking:
- Select the radius of the orbit to define the size of the circular scan according to the size of the particle being tracked. For a point emitter, set the radius of the orbit equal to the waist of the Point Spread Function (PSF) of the excitation beam to maximize sensitivity and response.
- Set the axial distance to define the distance between the two orbits that are performed to localize the particle position along the axial direction. Set the axial distance to 1.5-3 times the PSF waist.
- Define the dwell time according to the brightness of the particle to set the time spent on each point of the orbit, which will also determine the photo-bleaching rate. Use a dwell time between 10-100 µsec.
- Set the number of points in each orbit to 64 or 128 to yield orbit periods in the order of 4-32 msec and provide a high temporal resolution for determining the particle position.
Change the DC offset signal sent to the mirrors in order to center the beam on the particle through the graphical user interface via a cursor selection in the raster-scanned image.
Begin tracking by switching the microscope mode from raster-imaging to orbital scanning.
Activate the feedback and data collection.
4. Trajectory Analysis
Use the software to display the fluorescence collected at each point along the orbit and at each time point in the form of an ‘intensity carpet’. The intensity collected along the carpet provides information on the interaction of the particle with other bright objects. Use the software to display both the trajectory information (i.e. the DC displacement over time of the x,y scanners and of the z-piezo) as well as the fluorescence intensity collected from the photomultiplier tube over time in the form of time series.
Use the time series representation and the ‘intensity carpet’ information to select only a region of interest in the trajectory.
Use the software to display a 2D projection of the selected portion of the particle trajectory. Select the appropriate controls to color-code the trajectory according to the particle fluorescence intensity.
Select the option to display the particle trajectory in 3D, and color-code it according to the fluorescence intensity. Rotate the trajectory in 3D using the controls to visualize the features of the lysosome motion along the microtubule.
Representative Results
According to this protocol fast 3D single particle tracking can be performed inside living cells using a modified two-photon microscope to track the displacement of fluorescently labeled lysosomes. The experiment performed consists of tracking an isolated lysosome moving inside the cell after the endosome maturation process9. The lysosomes were stained using a fluorescent green dye and excited at 930 nm exploiting 2-photon excitation. Our data show that it is possible to obtain x,y,z displacement trajectories with a temporal resolution of 32 msec (Figure 1a). The carpet analysis displays the intensity recorded along each orbit over time (Figure 1b). During the tracking the integrated intensity of the emitted fluorescence can be recorded (Figure 2a). The reconstructed 3D trajectory displays a directed motion, as can be expected from a vesicle moving on the microtubule network. Additionally, it is possible to correlate the 3D trajectory to a raster scan image of the surrounding region (Figure 2b). This confirms the movement of the particle along the direction of a microtubule bundle.
The cells were additionally stained with a red marker for mitochondria to record eventual interaction of the vesicle with these organelles during their movement. It is possible to correlate the 3D trajectory of the particle inside the cell (Figure 3a) to the intensity carpet recorded from the mitotracker fluorescence emission (Figure 3b). This allows us to record the proximity interactions of the vesicle with the mitochondrial network as a function of their position within the cell. The emission peak recorded in the “mitochondria channel” does not give functional information about the way the organelles interact. It can provide only information on the time of their interaction and the relative spatial position of the fluorescent object with respect to the particle being tracked. This relative position is extracted from the angle within the orbit where the emission peak is recorded. A schematic representation of the microscope we used to perform this experiment can be observed in Figure 4.
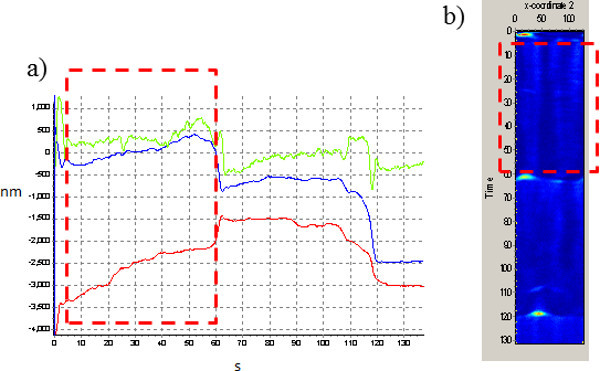 Figure 1. Displacement vs time trajectories and intensity carpet. a) x, y and z lysosome displacement vs. time traces. b) The intensity trajectory (i.e. the intensity collected along each circular orbit vertical axis is time) provides information on the fluorescence surroundings of the particle. Bright spots outside of the boxed correspond to interactions of the particle with other bright objects, and are associated to jumps in the trajectory as the orbits re-centers on the brightest fluorescence source. The boxed region corresponds to the portion of the trajectory in between two jumps.
Figure 1. Displacement vs time trajectories and intensity carpet. a) x, y and z lysosome displacement vs. time traces. b) The intensity trajectory (i.e. the intensity collected along each circular orbit vertical axis is time) provides information on the fluorescence surroundings of the particle. Bright spots outside of the boxed correspond to interactions of the particle with other bright objects, and are associated to jumps in the trajectory as the orbits re-centers on the brightest fluorescence source. The boxed region corresponds to the portion of the trajectory in between two jumps.
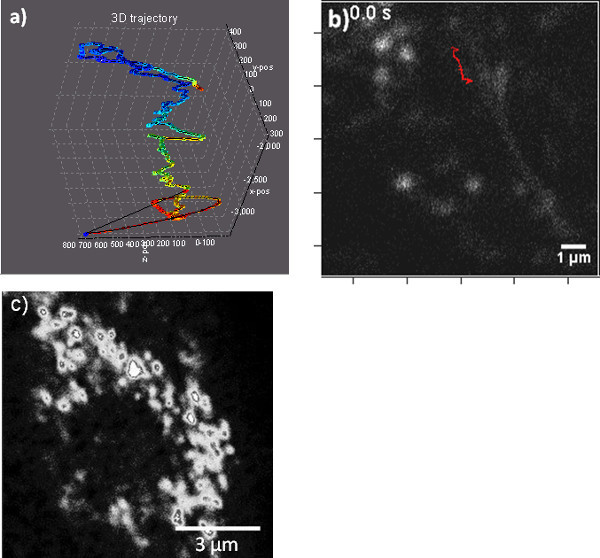 Figure 2. Following the 3D trajectory of a lysosome. a) 3D reconstruction of a portion of the trajectory of a lysosome collected using particle tracking. The trajectory is color coded for the fluorescence intensity collected from the particle (red, high; blue, low photon counts). Axes scales are in nm. Note that x,y,z axes have different ranges. b) 2D projection of the 3D trajectory, displaying an overlay of the trajectory on top of the microtubule imaged in a raster scan immediately after the trajectory was collected. c) Mitochondria stained with mitotracker.
Figure 2. Following the 3D trajectory of a lysosome. a) 3D reconstruction of a portion of the trajectory of a lysosome collected using particle tracking. The trajectory is color coded for the fluorescence intensity collected from the particle (red, high; blue, low photon counts). Axes scales are in nm. Note that x,y,z axes have different ranges. b) 2D projection of the 3D trajectory, displaying an overlay of the trajectory on top of the microtubule imaged in a raster scan immediately after the trajectory was collected. c) Mitochondria stained with mitotracker.
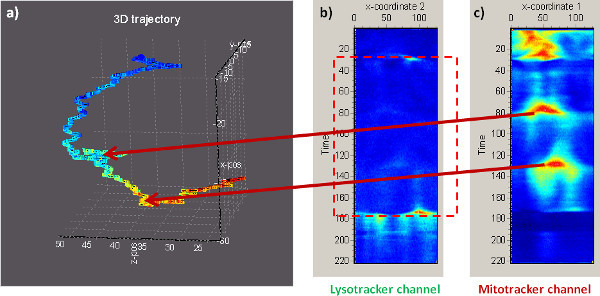 Figure 3. Extending the orbital tracking to more than one channel (excitation at 930 nm). a) 3D trajectory of a lysosome within the cell. b) Intensity carpet in the green channel (the fluorescence collected in this channel is used for tracking). c) Intensity carpet in the red channel, where mitochondria were stained with a red fluorescent matrix targeted dye. The regions where the trajectory gets in contact with the mitochondria (arrowheads) appear as intensity bumps in the carpet, as the beam orbiting around the lysosome crosses into the mitochondrial matrix.
Figure 3. Extending the orbital tracking to more than one channel (excitation at 930 nm). a) 3D trajectory of a lysosome within the cell. b) Intensity carpet in the green channel (the fluorescence collected in this channel is used for tracking). c) Intensity carpet in the red channel, where mitochondria were stained with a red fluorescent matrix targeted dye. The regions where the trajectory gets in contact with the mitochondria (arrowheads) appear as intensity bumps in the carpet, as the beam orbiting around the lysosome crosses into the mitochondrial matrix.
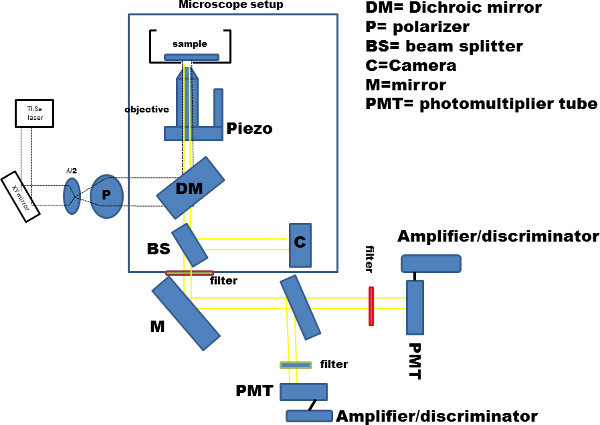 Figure 4. Schematics of the experimental setup: modified two-photon microscope. Schematic representation of the laser set-up: a 2-Photon Ti:Sa laser with a range 690-1,040 nm was used, a polarizer and an half waveplate are used to control the excitation power. All the other items are listed in the figure using the indicated abbreviations. We used a 60X objective with a NA of 1.2.
Figure 4. Schematics of the experimental setup: modified two-photon microscope. Schematic representation of the laser set-up: a 2-Photon Ti:Sa laser with a range 690-1,040 nm was used, a polarizer and an half waveplate are used to control the excitation power. All the other items are listed in the figure using the indicated abbreviations. We used a 60X objective with a NA of 1.2.
Discussion
Despite the tremendous progresses of fluorescence microscopy techniques and instrumentations over the last years, achieving trajectories of fluorescent particles in three dimensions with a high temporal resolution has remained a challenge in the field. If high temporal resolution has been achieved tracking particles in two dimensions, extension to the axial direction typically brings a drastic reduction in the frequency response of the system10.
In this video-protocol we focused on the demonstrative application of tracking an individual particle in three dimensions within a living cell with a high temporal resolution (32 msec in this experiment) using a two-photon microscope. The setup required for performing such an experiment is typically available to laboratories performing laser scanning microscopy. Both instrument add-ons for 3D orbital tracking as well as the control software are available on the market, for commercial laser scanning microscopes such as those manufactured by Olympus. The use of a pulsed high power infrared laser source, where available, is advantageous in the design of the experimental setup since it does not require de-scanning of the emitted fluorescence. Furthermore it was demonstrated in the past that infrared irradiation is more benign towards the cell, under specific conditions11, given the reduction of out of focus fluorescence and photo-bleaching.
Although fast tracking of a fluorescent particle in two dimensions can be performed using a sensitive camera, the absence of any three-dimensional information can be often limiting in interpreting the biophysical meaning of the trajectory information. For instance, vesicles moving along microtubule frequently run up against microtubules intersection regions8,12. In this situation, the use of a 3D tracking technique overcomes the limit of using 2D projections of a 3D structure.
Due to the time response of our objective nanopositioner piezo device, the period of the movement in the z-axis is limited to about 30 msec, while in the x,y directions it can go down to a few msec. The localization precision of the method scales as the signal to noise ratio 4,5. The only apparent limitation of the method, i.e. the loss of a global view of the environment of the particle, is compensated by the possibility of reconstructing a temporal intensity carpet13, tracing the history of the interactions of the fluorescent particle with neighboring fluorescent sources, displaying either the same or a different spectral emission.
We show that the motion of lysosomes, vesicles that undergo directed motion along microtubules, powered by either kinesins or dyneins, can be followed within a living cell after staining using a commercially available dye. Recently, 2D tracking of lysosomes was correlated to the super-resolution imaging of the microtubule network, in the effort of detecting the routes that the vesicles take at microtubule-microtubule intersections8. 3D trajectories of the lysosomes display a more complex behavior than unidirectional motion. Although an average directed motion can be observed, bending, twisting and possible rotations along the tubules can be observed from these trajectories.
We have identified two critical steps in performing these experiments: first, it is necessary to achieve a labeling density of the lysosomes that is at the same time high enough to obtain brightly stained particles, and that at the same time provides a density of particles low enough to allow tracking of individual particles. In the case of two crossing particles, the microscope may switch from one particle to another one while tracking. The second critical step concerns the choice of the pixel dwell time, allowing a reasonable balance between high signal to noise ratio and tracking speed. We have observed that tracking speed in the order of 32 msec per tracking cycle are typically sufficient to follow lysosomes undergoing fast directed motion at speeds below 1 µm/sec.
In summary, orbital tracking allows to measure in three dimensions and with a temporal resolution of <40 msec the intracellular motion of fluorescently labeled vesicles. The 3D orbital tracking has the potential to be employed in the future for the study of different highly dynamic cellular processes such us the study of the chromatin dynamics real-time transcription and diffusion of plasmonic nanoparticles in the intracellular medium.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This project was supported by grants NIH NIGMS 8P41 GM103540-28 and P50-GM076516
References
- So PT, Dong CY, Masters BR, Berland KM. Two photon excitation fluorescence microscopy. Annu Rev Biomed Eng. 2000;2:399–429. doi: 10.1146/annurev.bioeng.2.1.399. [DOI] [PubMed] [Google Scholar]
- Dupont A, Lamb DC. Nanoscale three dimensional single particle tracking. Nanoscale. 2011;3:4532–4541. doi: 10.1039/c1nr10989h. [DOI] [PubMed] [Google Scholar]
- Estrada LC, Gratton E. 3D nanometer images of biological fibers by directed motion of gold nanoparticles. Nano Lett. 2011;11:4656–4660. doi: 10.1021/nl2022042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kis-Petikova K, Gratton E. Distance measurement by circular scanning of the excitation beam in the two photon microscope. Microscopy Research and Technique. 2004;63:34–49. doi: 10.1002/jemt.10417. [DOI] [PubMed] [Google Scholar]
- Levi V, Ruan Q, Gratton E. 3 D particle tracking in a two photon microscope application to the study of molecular dynamics in cells. Biophys J. 2005;88:2919–2928. doi: 10.1529/biophysj.104.044230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund FW, Wustner D. A comparison of single particle tracking and temporal image correlation spectroscopy for quantitative analysis of endosome motility. Journal of Microscopy. 2013;252:169–188. doi: 10.1111/jmi.12080. [DOI] [PubMed] [Google Scholar]
- Nath S, et al. Spreading of Neurodegenerative Pathology via Neuron to Neuron Transmission of beta Amyloid. Journal of Neuroscience. 2012;32:8767–8777. doi: 10.1523/JNEUROSCI.0615-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balint S, Vilanova IV, Alvarez AS, Lakadamyali M. Correlative live cell and superresolution microscopy reveals cargo transport dynamics at microtubule intersections. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:3375–3380. doi: 10.1073/pnas.1219206110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huotari J, Helenius A. Endosome maturation. Embo Journal. 2011;30:3481–3500. doi: 10.1038/emboj.2011.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragan T, So PT, Kwon HS, Gratton E. 3D particle tracking on the two photon microscope. Multiphoton Microscopy in the Biomedical Sciences. 2001;2:247–258. [Google Scholar]
- Konig K, So PTC, Mantulin WW, Gratton E. Cellular response to near-infrared femtosecond laser pulses in two photon microscopes. Optics Letters. 1997;22:135–136. doi: 10.1364/ol.22.000135. [DOI] [PubMed] [Google Scholar]
- Caviston JP, Holzbaur EL. Microtubule motors at the intersection of trafficking and transport. Trends Cell Biol. 2006;16:530–537. doi: 10.1016/j.tcb.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Cardarelli F, Lanzano L, Gratton E. Capturing directed molecular motion in the nuclear pore complex of live cells. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:9863–9868. doi: 10.1073/pnas.1200486109. [DOI] [PMC free article] [PubMed] [Google Scholar]