

Abstract
Traumatic spinal cord injury causes an inflammatory reaction involving blood-derived macrophages and central nervous system (CNS)-resident microglia. Intra-vital two-photon microscopy enables the study of macrophages and microglia in the spinal cord lesion in the living animal. This can be performed in adult animals with a traumatic injury to the dorsal column. Here, we describe methods for distinguishing macrophages from microglia in the CNS using an irradiation bone marrow chimera to obtain animals in which only macrophages or microglia are labeled with a genetically encoded green fluorescent protein. We also describe a injury model that crushes the dorsal column of the spinal cord, thereby producing a simple, easily accessible, rectangular lesion that is easily visualized in an animal through a laminectomy. Furthermore, we will outline procedures to sequentially image the animals at the anatomical site of injury for the study of cellular interactions during the first few days to weeks after injury.
Keywords: Cellular Biology, Issue 93, Intravital, spinal cord crush injury, chimera, microglia, macrophages, dorsal column crush, axonal dieback
Introduction
The inflammatory reaction to disease or injury in the central nervous system (CNS) is poorly understood, especially with regards to the interactions among immune and resident cells within the tissue. Investigations of these cellular interactions in the spinal cord are of particular interest in the living animal. The only easily accessible CNS white matter tract is in the dorsal columns of the spinal cord, making this an important area on which to focus efforts on improving feasible experimental approach. These investigations have been limited due to the technical difficulty in accessing and stabilizing the CNS for imaging. Live imaging in the spinal cord has been described previously1-13, however, few studies have addressed cellular movement in a spinal cord injury beyond a few hours after the initial insult. The traumatic spinal cord lesion is a complex environment, with neurons, astrocytes, fibroblasts, NG2 progenitor cells, and immune cells including microglia, neutrophils, macrophages, T cells, B cells and dendritic cells14,15. Macrophages are the subset of immune cells responsible for phagocytosis in the lesion, infiltrating from the circulation. The role of these phagocytic cells has been debated, with reports indicating that these cells can take on both damaging and protective roles in the injured tissue. These roles range from increasing secondary axonal dieback after an injury and acting in a phagocytic manner16-22, to taking on a wound healing phenotype and decreasing functional deficits in the injured animal21,23,24.
Previously, attempts to distinguish macrophages from microglia have relied primarily on morphology in healthy tissue. However, activated microglia and macrophages express many of the same markers and display indistinguishable morphology after injury25-29, making the separation of different activities of these cells difficult to study based on these factors alone30. These cells can be separated by differential expression of CD45 using flow cytometry33,34, although this approach is less useful in discriminating these cell types in vivo. Utilizing differential expression of CCR2 and CX3CR1 in microglia and macrophages has also been explored, although the dynamic changes in the expression of these markers as monocytes differentiate into macrophages can complicate accurate analysis35,36. Microglia are the resident immune cells in the CNS and arise from yolk sac progenitors during fetal development, while macrophages are derived from bone marrow progenitors and enter the injured CNS after an insult31,32. An alternative approach to using morphology to distinguish these two cell types is to replace the bone marrow with progenitors from a donor animal expressing a traceable marker in the macrophages derived from the donor progenitors, while preserving the CNS resident, recipient-derived microglia. These bone marrow chimeric models are commonly utilized in many other applications37-40. This method has unique caveats associated with damage to the integrity of the blood-brain barrier caused by irradiation or cytotoxic drugs used to eradicate marrow progenitors in the host, thereby limiting its use in certain applications. Recently, functional distinctions between microglia and macrophages in the CNS are beginning to be discerned by using flow cytometry, gene chip array analysis and chimerism methods24,26,41-44, which will prove very useful in the future. Although separating these two cell types remains difficult, understanding their unique functions is important in leading to more targeted treatments of disorders that involve both microglia and macrophages within the CNS.
Description of cellular interactions within the spinal cord lesion has primarily come from studies involving cell culture models. This is due to the challenges involved with imaging both the spinal cord lesion and immune cells together in a living animal. Current rodent models of spinal cord injury of varying type and severity include contusion models45,46, pin-prick injuries2, laceration47, and the dorsal column crush injury described here16,18,48. Inflammation in the meninges increases with the severity of injury and poses unique challenges for the implantation of windows or surgeries for serial imaging. Some of these challenges include the infiltration of phagocytic cells as well as the generation of fibrous tissue over the surgical site. Some of these issues can be overcome by creating smaller lesions and covering the exposed spinal cord with non-immunogenic surgical dressing between the dura and the paraspinous muscles to allow for subsequent re-opening surgery, as is done here to study the dorsal column crush injury. The ability to image only the dorsal portion of the spinal cord also limits the choice of injury, since contusion models primarily cause damage to the central cord, often sparing the most dorsal part of the dorsal columns, especially at early time points after injury45,49. Therefore, we describe a simple injury model that yields a clean, repeatable, rectangular shaped lesion that is useful for both the observation of cell movement within the lesion and for easy quantification of axonal position relative to the lesion.
Protocol
NOTE: All animals should be housed and utilized in accordance with the animal care and use committee (IACUC) at the institution. All procedures described here are approved by the Case Western Reserve University IACUC.
1. Transgenic Animals
Use commercially available fluorescent reporter mice to label axons (Thy-1 YFP H)50 and microglia / monocytes (CX3CR1GFP/GFP 51; see list of materials). These mice should be intercrossed and maintained as individual breeding colonies according to institutional animal care policies.
2. Bone Marrow Chimeras
Identify animals that are at least 8 weeks of age for use as recipient animals.
Place host animals in a circular irradiation holding cage designed to hold the mouse in a position to ensure even, whole body irradiation.
Set timer on a cesium (Cs-137) irradiator to administer a whole-body radiation dosage of 800-1,000 Rads to the animals according to manufacturer’s instructions (examples shown here are 980 Rads).
Return animals to their cages to rest for a minimum of 4 hr.
Choose donor animals of the appropriate genotype (see Figure 2A) as the source of marrow progenitor cells. NOTE: The appropriate donors are animals expressing different lineage-specific fluorescent reporters from the host to identify different cell populations, or one of the same genotype as controls.
Fill one Petri dish with 70% alcohol, and a second dish with 10 ml Roswell Park Memorial Institute (RPMI) media and a 40 µm cell strainer soaked in the media. Fill a 15 ml conical tube with RPMI media. Keep the dishes and tube on ice.
Sacrifice the donor animals by CO2 asphyxiation according to institutional guidelines. Clean the skin and fur by soaking the entire animal with 70% ethanol until all fur is wet.
Remove the skin from lower half of animal by cutting around the mid-section and pulling skin down over the hind legs to fully expose the leg muscles.
Remove the tibia by anterior overextension at the knee joint to dislocate the bone, then using sterile forceps and scissors, peel and cut muscles away from both ends of the tibia. Place the bone in the Petri dish with alcohol for up to 30 sec then transfer to the cell strainer inside the Petri dish containing the media.
Dislocate the hip joint and peel away the muscles attached to the femur, placed the femur briefly in alcohol and then place it in the same cell strainer inside the dish.
In a tissue culture hood, use sterile scissors to cut off both ends of the bones and insert a 21 gauge needle on a 1 ml syringe into the end of the bone. Flush the marrow into the 15 ml conical with media. Repeat the procedures for the remaining bones.
Using the back of the plunger from a 3 ml syringe, crush bone ends in the cell strainer on the petri dish to extract more cells.
Place the filter on top of a 50 ml conical tube and transfer media with aspirated cells from the 15 ml conical tube. Use the back of the plunger to crush the marrow in order to obtain a single cell suspension. Wash the 15 ml conical tube, bone fragments and the cell strainer with 30 ml of media into a 50 ml conical tube.
Centrifuge the cells for 5 min at 500 x g to pellet cells.
Lyse red blood cells with 2 ml of Ammonium-Chloride-Potassium (ACK) lysis buffer for 2 min. Quench the lysis buffer with 10 ml of media containing 10% fetal bovine serum. Centrifuge at 500 x g for 5-10 min to pellet.
Wash cells once in 10 ml of RPMI media and twice in saline, centrifuging at 500 x g for 5-10 min to pellet for each wash.
Resuspend cells to a final concentration of 3 x 106 cells in 200 µl of saline.
Transfer 3 x 106 marrow cells via tail vein injection into irradiated recipient mice.
Place acidified water (pH 3.0) in recipient cage and allow mice to recover. NOTE: Mice that fail the transplantation will die within 10-14 days after the procedure.
After 8 weeks, verify marrow reconstitution by examining peripheral blood immune cells using flow cytometry (Figure 2B-C).
3. Dorsal Column Crush Injury
NOTE: Choose appropriate chimera animals or double transgenic animals for surgery based on the experiment desired. Animals with either labeled resident or bone marrow derived cells were created in previous steps.
Prepare and autoclave surgical tools, including rongeurs, Dumont #4 forceps, iris scissors, forceps for holding tissues, needle drivers and wound clips.
Prepare medications to administer to the mice for pain control. Bupernex and Marcaine are commonly used for pain control. Use appropriate medications as required and approved by the Institutional Animal Care and Use Committee (IACUC).
Induce anesthesia with 4% isoflurane and maintain with 1-2% isoflurane to achieve a breathing rate of 60-100 breaths per min. Apply eye lubricant to the animal’s eyes to prevent dryness under anesthesia and confirm lack of response by foot pinch. Maintain the temperature using a heating pad and monitor the temperature using a rectal probe.
Shave the back of the mouse with an electric shaver over the targeted spinal cord level, and then swab the area three times with povidone-iodine followed by twice with 70% alcohol. Place the animal on a sterile drape, and cover the animal with another sterile drape with a hole cut in an appropriate location to visualize the surgical site. Maintain all tools on sterile drapes throughout the surgery.
Cut the skin along the midline at the desired spinal level with iris scissors to expose the muscle on the back of the animal.
Under a dissecting microscope, cut the back muscles, including the trapezius and the ligaments of the latissimus dorsi along the midline where they attach to the dorsal spinal processes, and expose the vertebral column.
Remove the rotator muscles between the dorsal and transverse processes of the vertebra to reveal the lamina of the specific vertebra to be removed (T11 in this case). Identify T11 by the insertion of the last non-floating rib onto this process.
Insert tips of rongeurs into the space above and below the process to be removed and lift slightly to make sure they are securely in place around the vertebral lamina but not too deep to injure the spine. Close rongeurs in one movement to remove the lamina.
Enlarge the laminectomy as needed by using the rongeurs to remove small parts of the remaining vertebra.
Measure 1 mm from the tips of a #4 forceps by holding them against a ruler and marking with a permanent marker. Measure a 1 mm in separation between the two tines by holding the forceps against the ruler and fix the maximum width of the tips to 1 mm by sliding a rubber forceps ring into place over the shaft of the forceps.
Create a small hole by inserting a 30 gauge needle through the dura at the two insertion points for the tines of the forceps to access the spinal cord.
Insert the forceps tines at a 90° angle to the dura through the holes to the depth of 1 mm marked on the sides of the forceps, making sure the mark is above the dura. Pinch close the forceps and hold for 10 sec. Release, and repeat forceps squeeze two more times for a total of three holds. Remove forceps from the spinal cord.
Soak a piece of absorbable gelatin compressed sponge in saline and place over the injury site.
Using a running stitch, suture the muscle in place over the laminectomy and soaked gelatin sponge with #4 synthetic non-absorbable nylon sutures.
Clip the skin in place using 5 mm wound clips.
Inject 10 µl Marcaine (1.0 mg/kg) in 490 µl of saline subcutaneously around the incision site and 5 µl Bupernex (0.1 mg/kg) intramuscularly into the gluteus muscles.
Allow animal to recover on a warm pad and monitor for any difficulties in movement or signs of paralysis in the hind limbs. Do not leave animal unattended or in the presence of other animals until fully recovered from anesthesia. Do not use any animals that display observable motor deficits. NOTE: Although this crush lesion can be performed at any level along the spinal cord, practically, T10–L4 pose fewer technical challenges for imaging with respect to breathing and stabilizing body movement using clamps. Clamp placement must be modified around the rib cage to image at thoracic levels.
4. Intravital Imaging
Prepare and autoclave surgical tools, including Dumont #4 forceps, iris scissors, forceps for holding tissue, needle drivers and wound clips.
Anesthetize the mouse as before with isoflurane. Induce anesthesia at 4-5% and maintain at 1-2% as needed to maintain a breath rate of 60-100 breaths per minute and a lack of response to foot pinch. Keep the mouse heated, monitor breath rate when not imaging and continuously monitor animal temperature with a rectal probe.
Clean the skin around the wound clips with Betadine and then alcohol and remove the wound clips according to manufacturer instructions. Once wound clips are removed, remove hair with Nair if needed and clean the site again with povidone-iodine and 70% alcohol.
Re-open the skin with scissors. Remove any remaining sutures from muscle and pull muscle apart with forceps, cutting with scissors if needed.
Under a dissecting microscope, create pockets for the spinal cord clamps by cutting the muscle with scissors at the side of the transverse processes on the vertebra one vertebral level above and below the laminectomy.
Place clamps around each of these vertebra and tighten. Adjust as needed to allow room for microscope objective to reach the spinal cord. Make sure that the spinal cord is parallel to the imaging platform, and maintain tissue stability for imaging. If breathing artifact is seen, readjust clamps accordingly to minimize motion in the imaging field.
Cover the clamps with parafilm.
Remove the absorbable gelatin compressed sponge from the spinal cord with forceps, delicately picking off as many pieces as possible, also removing as much scar tissue from over the meninges as possible. Cover exposed spinal cord with saline and new sponge if needed. NOTE: If any bleeding occurs from the surrounding muscle, either staunch with sponge or cauterize as needed. However, take care not to touch the spinal cord with the cautery. Cover the spinal cord with either a piece of dampened tissue or gelatin compressed sponge when cauterizing.
Create a well to hold the immersion fluid by first sealing the skin and tissue with Vetbond, and then using dental acrylic to build up a well between the two clamps of the spinal cord holder and the sides of the animal. Wait until the acrylic cures.
Fill the well with artificial cerebral spinal fluid (aCSF) and check again for any bleeding around the surgery site.
Optionally at this point, inject fluorescent vessel dyes intravenously by tail vein injection to highlight the blood vessels during imaging. NOTE: Fluorescent dextran above 70 kDa is often used, although quantum dots and fluorescently labeled lectins also serve as useful vessel dyes. 50 µl of 150,000 WM TRITC-dextran is typically injected intravenously.
To obtain physiologically relevant immune cell motility, the temperature of the mouse must be maintained at 37 °C. Move the mouse to a temperature controlled environment under the microscope for imaging and monitor the animal for correct anesthesia level by breath rate and foot pinch. Monitor the animal frequently during imaging to determine depth of anesthesia, aiming to achieve a respiratory rate of 60 - 100 breaths per minute.
Locate the imaging site through the microscope eyepiece.
Adjust the 2-photon laser scanning microscope to take a z-stack image every 30-60 sec to obtain dynamic imaging data.
Once imaging session is completed remove the animal from the heated box.
Loosen the spinal clamps and remove the mouse from the clamps. Pull the acrylic well away from the skin while removing the clamps.
If the mouse is to be imaged again, place saline soaked gelatin compressed sponge over the spinal cord. Close the muscle and skin over the surgery site. Do not leave the animal unattended or in the presence of other animals while recovering from anesthesia. If the animal shows any signs of neurologic deficit after recovery, the animal should be euthanized. Otherwise, euthanize the mouse in a CO2 chamber before it emerges from anesthesia according to IACUC-approved euthanasia protocol.
Analyze fluorescent images using image analysis software by following the manufacturer’s instructions.
Representative Results
A schematic diagram depicting the dorsal column crush lesion is shown in Figure 1A. After the lesion, the animal is prepared and stabilized for intravital microscopy using spinal clamps (Figure 1B). An image taken immediately after the dorsal column crush injury shows a clearly visible rectangular lesion with forceps tine insertion points that are clearly identifiable. Axons at the injury site are severed across the span of the entire dorsal column (Figure 1C). The integrity of blood vessels remains intact, and no evidence of vessel dye leakage was detected by fluorescence. To perform serial imaging of the same lesion over weeks, the muscles and skin can be sutured over the laminectomy for subsequent re-exposure. Similar lesion morphology is seen at later time points (Figures 1D, E). 5 days after injury, the forceps insertion sites are still visible but the lesion has begun to increase in size due to secondary injury caused by inflammation. The axons at the caudal end of the lesion have retracted from the initial site of the injury via a process called “axonal dieback”. The axons at the rostral end of the lesion exhibited Wallerian-like degeneration (Figure 1D, E). Between days 5 and 22 after injury, the size of the lesion has stabilized. Concurrently, a large influx of CX3CR1+ cells can be seen infiltrating in and around the crush lesion during these time points, although the identity of these cells (microglia versus macrophages) can not be distinguished in the preparation as shown in Figure 1.
In order to distinguish the behavior of microglia and macrophages within the crush lesion microenvironment, a radiation chimera model was used (outlined in Figure 2A). First, the bone marrow progenitor cells of a CX3CR1+/GFP mouse are replaced with non-fluorescent donor cells, thus only GFP+ CNS-resident microglia are visible by fluorescent imaging. The efficiency of marrow reconstitution can be confirmed by flow cytometry examination of the peripheral blood 8 weeks after marrow transplant (Figure 2B, C). In Figure 2B, F4/80 and GFP positive macrophages, highlighted in blue, are detected in a CX3CR1+/GFP → C57BL/6 chimeric mouse, in which some GFP negative but F4/80 positive monocytes are also present. In Figure 2C, no double positive F4/80 and GFP positive cells are left after replacement of CX3CR1+/GFP recipient bone marrow with wild type bone marrow. Before injury, microglia are evenly distributed within the spinal cord, displaying a resting, ramified morphology (Figure 2D). 8 days after injury, only a few microglia can be detected in and around the lesion. These microglia display an amoeboid morphology (Figure 2E). Second, bone marrow progenitor cells in a non-fluorescent mouse are replaced by marrow cells from a CX3CR1+/GFP mouse, therefore rendering bone marrow derived CX3CR1+ monocytes / macrophages visible by GFP fluorescence (Figure 2A). Before injury, the only GFP+ cells detected are largely perivascular, which have been reported to be bone marrow derived and continually turn over on a regular basis28 (Figure 2F). After a crush injury, CX3CR1+/GFP cells can be seen along the inside of blood vessels surrounding the lesion (Figure 2H-J), and the lesion center is filled with infiltrating CX3CR1+/GFP macrophages (Figure 2G).
Time lapse imaging of the dorsal columns can be performed for up to 6 hr. In Figure 3, we show a non-chimeric CX3CR1+/GFP mouse immediately after injury in which cells from the circulation can be seen moving out of the blood vessel towards the lesion core and moving within the injury site (Figure 3A, B; Supplemental Movie 1). Image analysis utilizing fluorescence intensity to identify cells can track the paths taken by macrophages (Figure 3A, B; Supplemental Movie 1). Statistics derived from the imaging analysis show the average motility of macrophages in the lesion is 3.6 µm/min (Figure 3D). Finally, at 22 days after injury, axons, microglia and macrophages can still be detected within the lesion demonstrating the area of imaging is stable over long periods of time (Supplementary Movie 2).
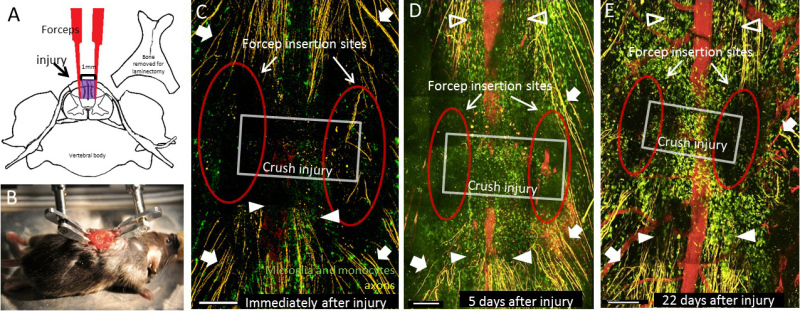 Figure 1: Creation and sequential imaging of the dorsal column crush injury. (A) The dorsal column crush injury is performed by removing the dorsal process of the vertebra at the level of interest. Forceps are inserted into the dorsal column of the spinal cord 1 mm apart and are then closed 3 times to produce the lesion shown in purple. (B) The animal is then positioned with spinal cord clamps to obtain stability for intravital imaging. (C) Immediately after injury, the forceps insertion sites (red oval) and the rectangular crush injury volume (grey box) can be clearly identified. Axons in the dorsal roots can be seen (white arrows), as well as the injured ends of the axons on the caudal side of the lesion (filled arrow heads). (D) 5 days after injury, the forceps insertion sites (red oval), and the extent of the lesion (grey box) can still be easily visualized. The lesion has increased in size since the initial insult. Axons undergoing Wallerian-like degeneration can be seen rostral to the lesion (open arrow heads) in addition to axons in the dorsal roots (white arrows) and the ends of injured axons (white arrowheads). (E) 22 days after injury, the lesion site is still identifiable with similar dimensions to the injury after 5 days. Note the large number of CX3CR1+ cells in and around the injured site. Features labeled are the same as in (D). All scale bars = 200 μm. Please click here to view a larger version of this figure.
Figure 1: Creation and sequential imaging of the dorsal column crush injury. (A) The dorsal column crush injury is performed by removing the dorsal process of the vertebra at the level of interest. Forceps are inserted into the dorsal column of the spinal cord 1 mm apart and are then closed 3 times to produce the lesion shown in purple. (B) The animal is then positioned with spinal cord clamps to obtain stability for intravital imaging. (C) Immediately after injury, the forceps insertion sites (red oval) and the rectangular crush injury volume (grey box) can be clearly identified. Axons in the dorsal roots can be seen (white arrows), as well as the injured ends of the axons on the caudal side of the lesion (filled arrow heads). (D) 5 days after injury, the forceps insertion sites (red oval), and the extent of the lesion (grey box) can still be easily visualized. The lesion has increased in size since the initial insult. Axons undergoing Wallerian-like degeneration can be seen rostral to the lesion (open arrow heads) in addition to axons in the dorsal roots (white arrows) and the ends of injured axons (white arrowheads). (E) 22 days after injury, the lesion site is still identifiable with similar dimensions to the injury after 5 days. Note the large number of CX3CR1+ cells in and around the injured site. Features labeled are the same as in (D). All scale bars = 200 μm. Please click here to view a larger version of this figure.
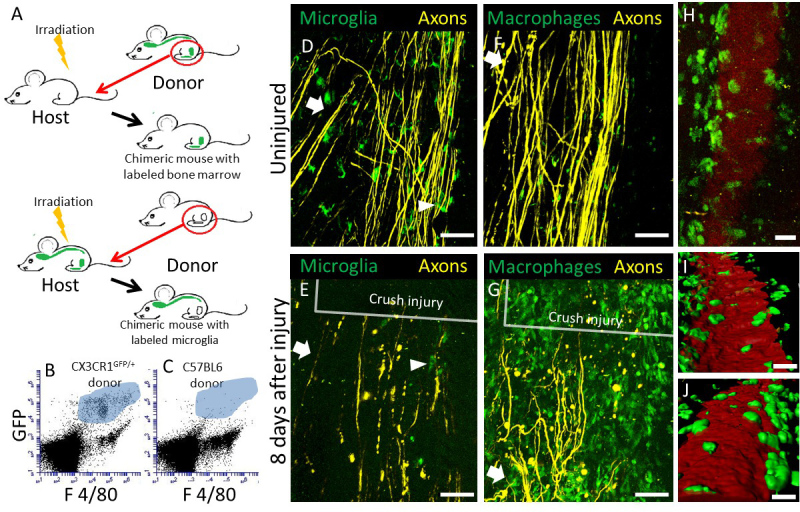 Figure 2: Construction of chimeric mice to track CX3CR1+/GFP expressing CNS-resident microglia or bone marrow-derived macrophages in the dorsal column crush lesion. (A) A schematic diagram of chimeric mice generation with either CX3CR1+/GFP expressing microglia or macrophages. First, Thy-1 YFP H mice are irradiated and the bone marrow is reconstituted with marrow cells isolated from a CX3CR1+/GFP mouse, resulting in a chimeric mouse whose macrophages are GFP+. Second, Thy-1 YFP H / CX3CR1+/GFP mice are irradiated and the bone marrow is reconstituted with marrow cells isolated from a non-fluorescent mouse, producing a chimeric mouse whose microglia are GFP+. (B) Example of flow cytometry data with F4/80+ and CX3CR1 GFP+ cells in the blood from a chimeric animal with a CX3CR1+/GFP marrow donor transplanted into an irradiated C57BL/6 recipient. Cells derived from the CX3CR1 positive donors that are positive for both GFP and F4/80 are shown in the blue highlighted area. (C) Example of flow cytometry data with F4/80+ and CX3CR1 GFP+ cells in a chimeric animal with a C57BL6 donor marrow transplanted into an irradiated CX3CR1+/GFP recipient. Note the lack of double positive CX3CR1+/GFP and F4/80 cells within the blue highlighted area. (D) An intravital two-photon microscopic snapshot of a mouse from the first chimeric scheme, showing GFP+ microglia with ramified morphology within the dorsal column (arrow head). These cells are interspersed among axons (yellow) in the dorsal root and the dorsal column. Scale bar = 100 µm. (E) Intravital imaging of the same mouse in (B) 8 days after dorsal column injury reveals a few CX3CR1+ microglia with large and amoeboid shaped cell bodies that are lacking processes (arrow head). Scale bar = 100 µm. (F) Snapshot of a mouse from the second chimeric scheme in (A), showing scant GFP+ macrophages, within the CNS parenchyma. Scale bar = 100 µm. (G) Intravital imaging of the same mouse in (F) 8 days after dorsal column injury reveals a large influx of macrophages in and around the lesion. Scale bar = 100 µm. (H) A higher magnification picture of CX3CR1+ blood derived cells in contact with the vessels in the uninjured animal. Scale bar = 30 µm. (I) A reconstruction of CX3CR1 cells in contact with the vessel, viewed from inside the vessel. Scale bar = 30 µm. (J) A reconstruction of CX3CR1 cells in contact with the vessel, viewed from outside of the vessel. Scale bar = 20 µm. Please click here to view a larger version of this figure.
Figure 2: Construction of chimeric mice to track CX3CR1+/GFP expressing CNS-resident microglia or bone marrow-derived macrophages in the dorsal column crush lesion. (A) A schematic diagram of chimeric mice generation with either CX3CR1+/GFP expressing microglia or macrophages. First, Thy-1 YFP H mice are irradiated and the bone marrow is reconstituted with marrow cells isolated from a CX3CR1+/GFP mouse, resulting in a chimeric mouse whose macrophages are GFP+. Second, Thy-1 YFP H / CX3CR1+/GFP mice are irradiated and the bone marrow is reconstituted with marrow cells isolated from a non-fluorescent mouse, producing a chimeric mouse whose microglia are GFP+. (B) Example of flow cytometry data with F4/80+ and CX3CR1 GFP+ cells in the blood from a chimeric animal with a CX3CR1+/GFP marrow donor transplanted into an irradiated C57BL/6 recipient. Cells derived from the CX3CR1 positive donors that are positive for both GFP and F4/80 are shown in the blue highlighted area. (C) Example of flow cytometry data with F4/80+ and CX3CR1 GFP+ cells in a chimeric animal with a C57BL6 donor marrow transplanted into an irradiated CX3CR1+/GFP recipient. Note the lack of double positive CX3CR1+/GFP and F4/80 cells within the blue highlighted area. (D) An intravital two-photon microscopic snapshot of a mouse from the first chimeric scheme, showing GFP+ microglia with ramified morphology within the dorsal column (arrow head). These cells are interspersed among axons (yellow) in the dorsal root and the dorsal column. Scale bar = 100 µm. (E) Intravital imaging of the same mouse in (B) 8 days after dorsal column injury reveals a few CX3CR1+ microglia with large and amoeboid shaped cell bodies that are lacking processes (arrow head). Scale bar = 100 µm. (F) Snapshot of a mouse from the second chimeric scheme in (A), showing scant GFP+ macrophages, within the CNS parenchyma. Scale bar = 100 µm. (G) Intravital imaging of the same mouse in (F) 8 days after dorsal column injury reveals a large influx of macrophages in and around the lesion. Scale bar = 100 µm. (H) A higher magnification picture of CX3CR1+ blood derived cells in contact with the vessels in the uninjured animal. Scale bar = 30 µm. (I) A reconstruction of CX3CR1 cells in contact with the vessel, viewed from inside the vessel. Scale bar = 30 µm. (J) A reconstruction of CX3CR1 cells in contact with the vessel, viewed from outside of the vessel. Scale bar = 20 µm. Please click here to view a larger version of this figure.
Figure 3:. Representative cell tracking data in a non-chimeric CX3CR1+/GFP mouse immediately after dorsal column crush injury. (A) A snapshot of CX3CR1+ microglia and macrophages around damaged axons (yellow) from Supplementary Movie 1 immediately after a dorsal column crush injury. (B) The paths (grey) taken by the CX3CR1+ cells in (A) during a 110 min intravital imaging session. (C) A time coded representation of the tracks in (B). Tracks are colored based on their time within the whole 110 minute movie, as shown in the time bar. (D) Histogram of overall motility of the CX3CR1+ cells. All scale bars are 30 µm. Please click here to view a larger version of this figure.
Supplementary Movie 1: Representative intravital movie immediately after dorsal column crush injury in the double heterozygote Thy-1YFP/+ / CX3CR1GFP/+ mouse. Intravital spinal imaging was performed immediately after a dorsal column crush injury at T11 level. Right panel shows the movement of CX3CR1+ microglia and macrophages. The paths of the cell movement are shown as white tracks on the left panel. The injury is located to the upper left corner. CX3CR1+ cells can be seen moving into the site of the injury along these tracks. Axons are shown in yellow. Scale bar = 40 µm. Total time: 110 min. Playback speed: 300x.
Supplementary Movie 2: Representative intravital movie at 22 days after dorsal column crush lesion in the double heterozygote Thy-1YFP/+ / CX3CR1GFP/+ mouse. Shown here is a representative movie taken in a mouse 22 days after the initial dorsal column crush injury at spinal level T11. The injury is located at the upper right corner of the frame. Axons (yellow) ascending rostrally are shown at the bottom right. The dorsal vein and blood vessels are labeled with TRITC-dextran (red). CX3CR1+ cells within the lesion move at a slower speed compared to those immediately after injury. Scale bar = 50 µm. Total time: 90 min. Playback speed: 600x.
Discussion
Imaging interactions of different cell types in their native tissue compartments during ensuing pathology in real-time has generated great interest. Within the dense network of the CNS, cell-cell contact and signaling with adjacent cells are essential for normal function and for understanding CNS pathology. Here, we described the use of 2-photon laser scanning microscopy for the observation of cellular movement within a mechanical lesion in the spinal cord. In addition to the quality of the surgery and tissue preparation, mechanical stability of the tissue is paramount for successful time-lapse microscopy, particularly the isolation of the spinal cord from breathing motion artifact. Stability can be assessed by looking for movement of the spinal cord under a dissecting microscope that corresponds to the heart rate or breathing of the animal. Stability should be assessed both while performing the surgery and also immediately before beginning imaging. If the animal is not stable, adjustments should be made to the placement and tightness of the spinal cord clamps. Cell-type specific fluorescent reporter mouse models coupled with bone marrow chimera approaches have allowed the identification of monocytic cells versus microglia and axons. Previous studies have revealed that blood derived monocytes and not microglia are responsible for secondary axonal damage after trauma using the methodology described here43. Dextran conjugated vessel dye allows for vessel identification both to provide landmarks and to identify breaches in vessel integrity. The selection of the appropriate fluorescent reporter mouse model is critical to allow the proper identification of the desired cell types to be imaged.
Development of fluorescent mouse models that are appropriate for the studies being undertaken is also critical to the success of experiments. Distinguishing between the two phagocytic populations of CX3CR1+/GFP cells in the CNS has traditionally been difficult. As shown here, a common immunological technique, irradiation chimeras, can be utilized to distinguish radio-resistant, CNS-resident microglia from bone marrow-derived macrophages. The irradiation procedure has the potential to damage the blood brain barrier and alter cell phenotypes, so use of this model should be considered carefully. The extent of these changes differs with irradiation dose as well as duration of recovery period, and their impact on different inflammatory models has not been fully studied. The effects of irradiation on CNS blood brain barrier can be minimized by shielding the head during irradiation, and this has been shown to decrease cell infiltration into the spinal cord after irradiation, even if the spinal cord is not directly shielded52. Here we have observed that the number of cells infiltrating into the CNS at steady state as a result of chimera generation is insignificant in contrast to the number of cells entering the lesion. Other alternative models to consider include drug-induced chimeras52 or parabiotic mouse models53.
Here, we have presented a specific, simple and reproducible small injury model that results in a lesion to the dorsal white matter of the spinal cord that is easy to image using the 2-photon microscopy. This method also provides a quantifiable lesion to assay the degree of axonal dieback in the dorsal columns that in the literature has been coupled with complementary fixed tissue analysis16,18,43,48,54. In this model, animals display only minimal deficits and require no special care after injury. Future studies utilizing the dorsal column crush injury and imaging techniques described here may be powerful screening tools to assess the efficacy of treatment for spinal cord injury. These treatments may include small molecule inhibitors, drugs, cell products, tissue grafts and combinatorial treatments. The interplay between macrophages, microglia and neurons are also likely to play a role in other disease models in the spinal cord, including multiple sclerosis, tumors, meningitis and amyotrophic lateral sclerosis, and this technique may be useful in the study of these diseases as well.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The following agencies provided critical funding support for this study: MSTP T32 GM007250 (T.A.E.), 5T32EB7509 (D.S.B.), NCI R01 CA154656 (A.Y.H.), Dana Foundation (A.Y.H.), St. Baldrick’s Foundation (A.Y.H.), Alex’s Lemonade Stand Foundation (A.Y.H.), Gabrielle’s Angel Foundation (A.Y.H.) and Hyundai Hope-on-Wheels Program (A.Y.H.). The authors are thankful for the indispensable help of Jerry Silver and Sarah Busch in learning the Dorsal Column Crush (DCC) injury. The authors are also grateful to Jingquang You, Elisabeth Hare and Hongmei Hu for technical help with genotyping and Ross Anderson for mounting the spinal stabilizing unit.
References
- Farrar MJ, et al. Chronic in vivo imaging in the mouse spinal cord using an implanted chamber. Nat Methods. 2012;9(3):297–302. doi: 10.1038/nmeth.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerschensteiner M, Schwab M, Lichtman E, Misgeld T. In vivo imaging of axonal degeneration and regeneration in the injured spinal cord. Nat Med. 2005;11(5):572–577. doi: 10.1038/nm1229. [DOI] [PubMed] [Google Scholar]
- Davalos D, Akassoglou K. In vivo imaging of the mouse spinal cord using two-photon microscopy. J Vis Exp. 2012. p. 2760. [DOI] [PMC free article] [PubMed]
- Davalos D, et al. Stable in vivo imaging of densely populated glia, axons and blood vessels in the mouse spinal cord using two-photon microscopy. J Neurosci Methods. 2008;169(1):1–7. doi: 10.1016/j.jneumeth.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8(6):752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- Figley SA, et al. A spinal cord window chamber model for in vivo longitudinal multimodal optical and acoustic imaging in a murine model. PLoS One. 2013;8(3):58081. doi: 10.1371/journal.pone.0058081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffens H, Nadrigny F, Kirchhoff F. In vivo two-photon imaging of neurons and glia in the mouse spinal cord. Cold Spring Harb Protoc. 2012;2012(12) doi: 10.1101/pdb.prot072264. [DOI] [PubMed] [Google Scholar]
- Ertürk A, et al. Three-dimensional imaging of the unsectioned adult spinal cord to assess axon regeneration and glial responses after injury. Nat Med. 2012;18(1):166–171. doi: 10.1038/nm.2600. [DOI] [PubMed] [Google Scholar]
- Dibaj P, et al. In Vivo imaging reveals distinct inflammatory activity of CNS microglia versus PNS macrophages in a mouse model for ALS. PLoS One. 2011;6(3) doi: 10.1371/journal.pone.0017910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibaj P, et al. NO mediates microglial response to acute spinal cord injury under ATP control in vivo. Glia. 2010;58(9):1133–1144. doi: 10.1002/glia.20993. [DOI] [PubMed] [Google Scholar]
- Fenrich KK, Weber P, Rougon G, Debarbieux F. Long- and short-term intravital imaging reveals differential spatiotemporal recruitment and function of myelomonocytic cells after spinal cord injury. J Physiol. 2013;591(19):4895–4902. doi: 10.1113/jphysiol.2013.256388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenrich KK, et al. Long-term in vivo imaging of normal and pathological mouse spinal cord with subcellular resolution using implanted glass windows) J Physiol. 2012;590(16):3665–3675. doi: 10.1113/jphysiol.2012.230532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dray C, Rougon G, Debarbieux F. Quantitative analysis by in vivo imaging of the dynamics of vascular and axonal networks in injured mouse spinal cord) Proc Natl Acad Sci U S A. 2009;106(23):9459–9464. doi: 10.1073/pnas.0900222106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovich PG, Jones TB. Manipulating neuroinflammatory reactions in the injured spinal cord: back to basics. Trends Pharmacol Sci. 2003;24(1):13–17. doi: 10.1016/s0165-6147(02)00006-8. [DOI] [PubMed] [Google Scholar]
- Donnelly DJ, Popovich PG. Inflammation and its role in neuroprotection, axonal regeneration and functional recovery after spinal cord injury. Exp Neurol. 2008;209(2):378–388. doi: 10.1016/j.expneurol.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn KP, Busch SA, Hawthorne AL, van Rooijen N, Silver J. Another barrier to regeneration in the CNS: activated macrophages induce extensive retraction of dystrophic axons through direct physical interactions. J Neurosci. 2008;28(38):9330–9341. doi: 10.1523/JNEUROSCI.2488-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch MT, Silver J. CNS injury, glial scars, and inflammation: Inhibitory extracellular matrices and regeneration failure. Exp Neurol. 2008;209:294–301. doi: 10.1016/j.expneurol.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch SA, Horn KP, Silver DJ, Silver J. Overcoming macrophage-mediated axonal dieback following CNS injury. J Neurosci. 2009;29(2):9967–9976. doi: 10.1523/JNEUROSCI.1151-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovich PG, van Rooijen N, Hickey WF, Preidis G, McGaughy V. Hematogenous macrophages express CD8 and distribute to regions of lesion cavitation after spinal cord injury. Exp Neurol. 2003;182(2):275–287. doi: 10.1016/s0014-4886(03)00120-1. [DOI] [PubMed] [Google Scholar]
- Popovich PG, et al. Depletion of hematogenous macrophages promotes partial hindlimb recovery and neuroanatomical repair after experimental spinal cord injury. Exp Neurol. 1999;158(2):351–365. doi: 10.1006/exnr.1999.7118. [DOI] [PubMed] [Google Scholar]
- Kigerl KA, et al. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29(43):13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gensel JC, et al. Macrophages promote axon regeneration with concurrent neurotoxicity. J Neurosci. 2009;29(12):3956–3968. doi: 10.1523/JNEUROSCI.3992-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter R, et al. Recruitment of beneficial M2 macrophages to injured spinal cord is orchestrated by remote brain choroid plexus. Immunity. 2013;38(3):555–569. doi: 10.1016/j.immuni.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter R, et al. Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med. 2009;6(7):1000113. doi: 10.1371/journal.pmed.1000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann F, et al. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327(5966):656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovich PG, Hickey WF. Bone marrow chimeric rats reveal the unique distribution of resident and recruited macrophages in the contused rat spinal cord. J Neuropathol Exp Neurol. 2001;60(7):676–685. doi: 10.1093/jnen/60.7.676. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM. Microglia and monocytes: 'tis plain the twain meet in the brain. Nat Neurosci. 2011;14(9):1098–1100. doi: 10.1038/nn.2917. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Cardona AE. The myeloid cells of the central nervous system parenchyma. Nature. 2010;468(7321):253–262. doi: 10.1038/nature09615. [DOI] [PubMed] [Google Scholar]
- Prinz M, Priller J, Sisodia SS, Ransohoff RM. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat Neurosci. 2011;14(10):1227–1235. doi: 10.1038/nn.2923. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 1152;91(2):461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- Kierdorf K, et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci. 2013;16(3):273–280. doi: 10.1038/nn.3318. [DOI] [PubMed] [Google Scholar]
- Ginhoux F, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford AL, Goodsall AL, Hickey WF, Sedgwick JD. Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J Immunol. 1995;154(9):4309–4321. [PubMed] [Google Scholar]
- Herber DL, et al. Diverse microglial responses after intrahippocampal administration of lipopolysaccharide. Glia. 2006;53(4):382–391. doi: 10.1002/glia.20272. [DOI] [PubMed] [Google Scholar]
- Saederup N, et al. Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS One. 2010;5(10):13693. doi: 10.1371/journal.pone.0013693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani M, et al. The fractalkine receptor but not CCR2 is present on microglia from embryonic development throughout adulthood. J Immunol. 2012;188(1):29–36. doi: 10.4049/jimmunol.1100421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A. The use of bone marrow-chimeric mice in elucidating immune mechanisms. Methods Mol Med. 2006;127:281–292. doi: 10.1385/1-59745-168-1:281. [DOI] [PubMed] [Google Scholar]
- Spangrude GJ. Assessment of lymphocyte development in radiation bone marrow chimeras. Curr Protoc Immunol. 2008;4(4.6) doi: 10.1002/0471142735.im0406s81. [DOI] [PubMed] [Google Scholar]
- Duran-Struuck R, Dysko RC. Principles of bone marrow transplantation (BMT): providing optimal veterinary and husbandry care to irradiated mice in BMT studies. J Am Assoc Lab Anim Sci. 2009;48(1):11–22. [PMC free article] [PubMed] [Google Scholar]
- Koon HW, Ho S, Cheng M, Ichikawa R, Pothoulakis C. Differentiating functional roles of gene expression from immune and non-immune cells in mouse colitis by bone marrow transplantation. J Vis Exp. e4208. 2012. p. 4208. [DOI] [PMC free article] [PubMed]
- London A, Cohen M, Schwartz M. Microglia and monocyte-derived macrophages: functionally distinct populations that act in concert in CNS plasticity and repair. Front Cell Neurosci. 2013;7 doi: 10.3389/fncel.2013.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Schwartz M. Non-identical twins - microglia and monocyte-derived macrophages in acute injury and autoimmune inflammation. Front Immunol. 2012;3:89. doi: 10.3389/fimmu.2012.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans TA, et al. High-resolution intravital imaging reveals that blood-derived macrophages but not resident microglia facilitate secondary axonal dieback in traumatic spinal cord injury. Exp Neurol. 2014;254:109–120. doi: 10.1016/j.expneurol.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, et al. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat Neurosci. 2014;17(1):131–143. doi: 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, et al. A contusive model of unilateral cervical spinal cord injury using the infinite horizon impactor. J Vis Exp. 2012;65:3313–3310. doi: 10.3791/3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna V, et al. A contusion model of severe spinal cord injury in rats. J Vis Exp. 2013. [DOI] [PMC free article] [PubMed]
- Zhang YP, et al. Controlled cervical laceration injury in mice. J Vis Exp. 2013;75:50030. doi: 10.3791/50030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, et al. PTPsigma is a receptor for chondroitin sulfate proteoglycan, an inhibitor of neural regeneration. Science. 1126;326(5952):592–596. doi: 10.1126/science.1178310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ek CJ, et al. Spatio-temporal progression of grey and white matter damage following contusion injury in rat spinal cord. PLoS One. 2010;5(8):e12021. doi: 10.1371/journal.pone.0012021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G, et al. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28(1):41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Jung S, et al. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20(11):4106–4114. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mildner A, et al. Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of Alzheimer's disease. J Neurosci. 2011;31(31):11159–11171. doi: 10.1523/JNEUROSCI.6209-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kierdorf K, Katzmarski N, Haas CA, Bone Prinz M. marrow cell recruitment to the brain in the absence of irradiation or parabiosis bias. PLoS One. 2013;8(3):e58544. doi: 10.1371/journal.pone.0058544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filous AR, Evans TA, Lang BT, Silver J. Neuroscience 2013, Society for Neuroscience. San Diego: Abstracts; 2013. Synaptic-like connections between dystrophic axons and NG2+ cells: Another reason for regeneration failure after spinal cord injury. [DOI] [PMC free article] [PubMed] [Google Scholar]