

Abstract
Costimulation by B7-1 and B7-2 molecules results in divergent biological effects. This is particularly striking in the NOD mouse, since the lack of B7-2 leads to complete protection from autoimmunity, whereas the B7-1 deficiency causes exacerbation of disease. We tested the hypothesis that B7-1 costimulation suppresses pancreatic autoimmunity. We describe that the lack of B7-1 not only causes aberrant thymocyte maturation but also significantly enhances expansion, survival, and effector function of islet specific T cells in periphery. We also observed a significant reduction in the proportion of T-regulatory (T-regs) cells. Immunophenotypic analysis of T and APCs revealed a significantly lower frequency of T cells expressing the negative costimulatory receptor PD-1 in B7-1KO mice whereas the proportion of B7-H1 positive APCs was found to be significantly higher. Blocking studies in B7-1KO mice suggest that B7-H1 provides negative signals for anti islet CD4 and CD8 T cell expansion but is differentially required for their priming. Our data demonstrate that deficiency of B7-1 mediated costimulation causes multitude of immunological defects, which involve reduction in T-regs and a concomitant enhancement of expansion, survival and effector potential of auto reactive T cells.
Keywords: Costimulation, B7-1, type 1 diabetes, NOD, autoimmunity
1 Introduction
Activation of naïve T cells requires the supplementation of primary TCR signal with costimulatory input. B7 ligands (B7-1/CD80 and B7-2/CD86) bind to both activating (CD28) and deactivating (CTLA-4) receptors in T cells (Sharpe and Freeman, 2002). In addition to the positive costimulation provided by B7-1 and B7-2, two new members of B7 family, B7-H1 (PD-L1) and B7-DC (PD-L2) have been identified as negative costimulators, which can interact with their receptor PD-1 on T cells leading to the transduction of negative signals, altered positive selection and peripheral tolerance (Chen, 2004; Keir et al., 2006; Khoury and Sayegh, 2004). However, a second, as yet uncharacterized non-PD-1 receptor has been suspected to provide positive signals to T cells (Kanai et al., 2003). The dynamic fine-tuning of immune response also involves B7-1 and B7-2 mediated preferential localization of CTLA-4 and CD28, respectively, to the synapse (Pentcheva-Hoang et al., 2004). Although B7-1 and B7-2 can provide similar signals (Lanier et al., 1995; Santra et al., 2000; Schweitzer et al., 1997), several reports also suggest that these molecules have the capacity to induce functionally distinct effects (Guerder et al., 1998; Slavik et al., 1999). The final outcome of immune response is directly linked to the quality of costimulatory signals provided by APCs to T cells. B7-1 displays a distinct temporal expression pattern and its expression is inducible, unlike B7-2, which is constitutively expressed on antigen presenting cells (APCs) (Larsen et al., 1994) suggesting that B7-2 plays a role in the initial priming of T cells, whereas B7-1 may help sustain the active phase of the disease (Salomon and Bluestone, 2001). Furthermore, B7-1 costimulation alone has been shown to be insufficient to induce T1D in nonautoimmune disease-prone mice, but its expression on APCs is important in both the T cell priming and effector phases of T1D (Camacho et al., 2001; Guerder et al., 1994).
The development of type 1 diabetes in the NOD mouse is distinctly regulated by B7-1 and B7-2. For example, the absence of B7-2 leads to complete protection, whereas, B7-1 neutralization or deficiency causes exacerbation of disease (Salomon and Bluestone, 2001; Salomon et al., 2001; Yadav et al., 2004). These results suggest that B7-1 may play a role in suppressing the development of autoimmunity in the NOD mouse. However, the underlying mechanisms explaining B7-1 mediated regulation of autoimmunity have not been fully described. The central goal of this study is to define the immunological parameters underlying the exacerbation of autoimmunity in the absence of B7-1. Our study demonstrates that enhanced disease severity in mice lacking B7-1 is associated with reduced levels of the immunnoregulatory factor PD-1, altered expression of PD-1 ligands, increased effector function, expansion, and survival of autoreactive T cells. We propose that B7-1 exerts a counter-regulatory function, diminishing autoreactivity in the periphery.
2 Materials and Methods
2.1 Mice
Female NODshi, BDC2.5 NOD, 8.3NODscids were obtained from The Scripps Research Institute animal facility. In our NOD colony, diabetes starts at 15 weeks of age and ~84% of mice get diabetic by 26 weeks of age. The B7-1KORag−/− mice on the NOD genetic background were a kind gift from Dr. J. Bluestone (Univ. California, San Francisco, CA). The B7-1KORag−/− NOD mice were crossed with NOD. The progeny was screened for the B7-1 allele using the following primers for triplex PCR: B7-1 (sense) CTGTCCAAGTCAGTGAAAGAT, B7-1 (anti sense) GGACAACTTTACTAAAGCCA, Stat Neo GCTACCCGTGATATT GCTGAAGAG. The B7-1 wild type allele band was 300bp and B7-1 mutant allele was 750bp. The progeny was also typed for RAG-2 wild type and mutant allele separately using the following primers. RAG-2 (sense) TTAATTCAACCAGGCTTCTCACTT, RAG2 (anti-sense) GCCTGCTTATTGTCTCCTGGTAT, gave a 974bp band. For the mutant allele, a primer was chosen for the neo gene: CACCATGATATTCGGCAAGC and used with the RAG anti sense primer to obtain a product of 1034bp. Finally B7-1KORag+/+ NOD mice were chosen for the study and are described as B7-1KO in the text. We confirmed the incidence of diabetes in these mice and found that 100% of B7-1KO mice become diabetic by 20 weeks of age compared to NOD control group in which ~35% became diabetic (p=0.015, Kaplan Meier test) at this age in our colony (Fig. 1). All experiments were conducted in accordance with institutional guidelines for animal care and use.
Fig. 1.
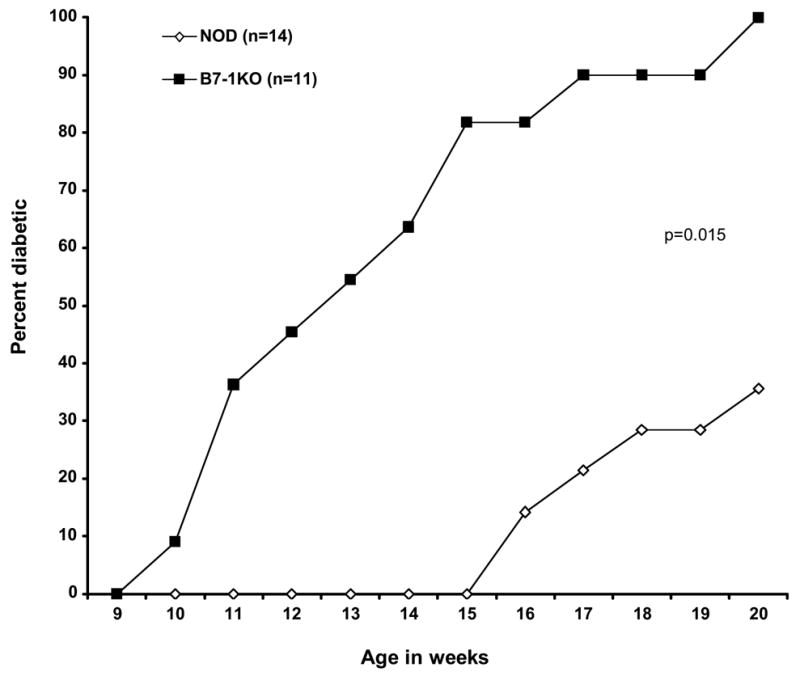
Exacerbation of type 1 diabetes in mice lacking B7-1. NOD and B7-1KO were monitored for blood glucose on weekly basis and were considered diabetic if mice exhibited two consecutive blood glucose observations of 250mg/dl.
2.2 Antibodies and flow cytometric analyses
RBC lysed single cell suspensions from spleen, pancreatic lymph nodes (PLN) were incubated with appropriate amount of monoclonal antibodies (Pharmingen, La Jolla, CA) and stained for indicated cell markers and analyzed by flowcytometry. Appropriate isotype controls were used to determine the background staining.
2.3 Determination of IFN-γ production by T cells using TCR/CD28 mediated stimulation of PLN cells in vitro
The intracellular IFN-γ staining was performed according to Mendel and Shevach (Mendel and Shevach, 2006) with some modifications. In brief, PLN cells (1–2×106 cells/well) from B7-1KO and NOD mice were in vitro activated with plate (48-well) bound anti-CD3 antibody (1 μg/ml, 145-2C11 clone) in complete RPMI 1640 (containing 10% FCS, 5U/ml and 50□g/ml of penicillin-streptomycin respectively, 10mM HEPES buffer, 2mM L-glutamine) in the presence of GolgiPlug™ (pharmingen, 1 μl/ml). Soluble anti-CD28 antibody (1 μg/ml, 7.51 clone) was also added for costimulation. Five hours post stimulation; cells were harvested, washed and processed for intracellular IFN-γ staining according to the manufacturer’s protocol (pharmingen).
2.4 CFSE labeling and cell survival
1.2–2.5 × 107 CFSE labeled (as described previously (Yadav et al., 2004)) total splenocytes from either BDC2.5 or 8.3NODscids (4–7 week old) in 200μl of sterile PBS were injected intravenously per recipient (5–8 week old NOD, and B7-1KO mice). For monitoring cell death in adoptively transferred cells, PLN cells (1–2×106) from the recipients were also stained for annexin and Vβ4+CFSElow / Vβ8.1/8.2+ CFSElow cells were analyzed for the cells positive for annexin.
2.5 In vivo blockade of B7-H1 and flow cytometry
For in vivo blocking of B7-H1, 5–8 weeks old B7-1KO mice were treated with anti-B7-H1antibody (clone MIH5, a kind gift from Dr. Miyuki Azuma) i.p. in two doses of 250–300μg each, two days apart. With the first dose, CFSE labeled splenocytes/animal were also administered i.v either from BDC2.5 (1.3–1.8 × 107/animal) or 8.3NODscids (0.8–1.3 × 107/animal) donors (as described above). The control group of B7-1KO mice was injected with the same amount of CFSE labeled BDC2.5/8.3NODscids cells but was treated with rat-IgG instead. Four days post cell transfer the animals were sacrificed and PLN cells were analyzed by flow cytometry for expansion (CFSE dilution) and activation (CD69 expression).
2.6 Diabetes incidence
Female mice were screened weekly for hyperglycemia using Glucometer Elite strips. Mice with two successive weekly blood glucose levels greater than 250 mg/dl were considered diabetic.
2.7 Statistical analyses
The cumulative diabetes incidence was compared using the Kaplan-Meier method. Determination of level of significance in other set of data was carried out by employing non-parametric Mann-Whitney U test (unpaired), taking into consideration the sample size and the range of variation. A value of p<0.05 was considered as significant and are depicted as mean± SEM in the text.
3 Results
3.1 B7-1 deficiency causes exacerbation of diabetes and aberrant thymocyte maturation
NOD mice, when devoid of B7-1, have been reported to exhibit exacerbation of type 1 diabetes (Salomon and Bluestone, 2001). Deficiency in B7-1/2 mediated costimulation can affect thymic selection (Gao et al., 2002) and subtle abnormalities in T cell selection and function may initiate or perpetuate autoreactivity (Firestein, 2004). To ask whether this occurred in the NOD genetic background, we checked the percentages and absolute cell numbers of single positive (SP) double positive (DP) and double negative (DN) CD4 and CD8 thymocytes in NOD and B7-1KO mice. Although the percentages of CD4SP, CD8SP, DN and DP thymocytes were not significantly affected in B7-1KO mice (not shown), significantly higher numbers of CD4SP (p=0.023) and DP thymocytes (p=0.023) were observed in mice lacking B7-1. However, the numbers of CD8SP and DN thymocytes did not exhibit any significant change between the two groups (Table 1). Besides B7, B7-HI has also been suggested to play an important role in positive selection by binding its ligand PD-1 (Keir et al., 2005). We therefore asked if thymic B7-HI expression is affected in B7-1KO mice. Interestingly, a significantly reduced frequency of B7-HI positive T cells were observed in B7-1KO mice in all thymic T cell subsets (Table 1). No significant difference in the levels of PD-1+ thymocytes was observed (not shown). This suggests that B7-1KO mice exhibit a deficiency of B7-H1 mediated signals in the thymus, which in turn can lead to perturbed thymocyte maturation.
Table 1.
Comparison of thymic cellularity and B7-H1 expression in NOD and B7-1KO mice.
| NOD | B7-1KO | n | p value | |
|---|---|---|---|---|
| Cellularity of thymic subsets (numbers) | ||||
|
| ||||
| CD4+CD8− (CD4 single positive) | 6.6±0.4 × 106 | 11.8±1.6 × 106 | 10 | p= 0.023 |
| CD4−CD8+ (CD8 single positive) | 2.3±0.2 × 106 | 2.9±0.4 × 106 | 10 | NS |
| CD4−CD8− (double negative) | 0.4±0.03 × 106 | 0.5±0.06 × 106 | 10 | NS |
| CD4+CD8+ (double positive) | 1.9±0.1 × 106 | 3.5±0.5 × 106 | 10 | p= 0.023 |
|
| ||||
| B7-H1 expression in thymic subsets (%) | ||||
|
| ||||
| CD4+CD8−B7-H1+ (CD4 single positive) | 68.5±2.6 | 56.8±0.8 | 6 | p=0.003 |
| CD4−CD8+B7-H1+ (CD8 single positive) | 57.0±4.3 | 46.4±1.4 | 6 | p=0.010 |
| CD4−CD8−B7-H1+ (double negative) | 20.3±1.4 | 15.7±0.4 | 6 | p=0.045 |
| CD4+CD8+B7-H1+ (double positive) | 26.3±2 | 21.2±1.0 | 6 | p=0.025 |
n indicates number of pooled animals from individual experiments for statistical analysis of data. The data are pooled from 2–3 independent experiments with 4–6 weeks old mice. For the B7-H1 data, the thymic subsets are gated on total (CD3+) T cells. The values corresponding to NOD and B7-1KO are mean± SEM. NS means not significant.
3.2 Increased peripheral cellularity and anti-islet CD4 and CD8 T cell expansion in B7-1KO mice
We hypothesized that the increased numbers of DP and CD4 SP T cells in the thymus of B7-1KO mice may result in enhanced cellularity in the periphery. Indeed, the size of the CD4 compartment was found to be significantly increased both in spleen (p=0.021) and PLNs (p=0.034) in B7-1KO mice. The total number of CD8 T cells was increased in the spleens of B7-1KO mice (p=0.034) (Table 2).
Table 2.
Comparison of peripheral total CD4 and CD8 T cell numbers in NOD and B7-1KO mice.
| NOD | B7-1KO | n | p value | |
|---|---|---|---|---|
| Total cellular yield | ||||
| PLN | 2.9±0.3 × 106 | 4.2±0.3 × 106 | 11 | p= 0.038 |
| Spleen | 6.7±0.8 × 107 | 11.0±1.3 × 107 | 14–15 | p= 0.038 |
| Total CD4 T cell numbers | ||||
| PLN | 1.5±0.1 × 106 | 2.1±0.1 × 106 | 10–11 | p= 0.034 |
| Spleen | 1.8±0.2 × 107 | 3.0±0.3 × 107 | 14–15 | p= 0.021 |
| Total CD8 T cell numbers | ||||
| PLN | 0.6±0.06 × 106 | 0.9±0.09 × 106 | 10–11 | p= 0.067 |
| Spleen | 0.9±0.1 × 107 | 1.4±0.1 × 107 | 15–16 | p= 0.034 |
n indicates number of pooled animals from individual experiments for statistical analysis of data. The data are pooled from 3–4 independent experiments with 6–9 weeks old mice. The values corresponding to NOD and B7-1KO are mean± SEM. NS means not significant.
To test responses to islet antigens, NOD and B7-1KO mice were injected with CFSE labeled splenocytes from either BDC2.5 or from 8.3NODscid transgenic NOD mice, which harbor an anti-islet CD4 or CD8 T cell repertoire respectively (Katz et al., 1993; Lieberman et al., 2003). On day 4 post transfer, the PLN cells were harvested and analyzed for proliferation based on CFSE dilution. Interestingly, B7-1KO mice exhibited a significantly increased percentage of proliferating 8.3NODscid (p=0.037) and BDC2.5 (p=0.011) TCR transgenic T cells (Fig 2A).
Fig. 2.
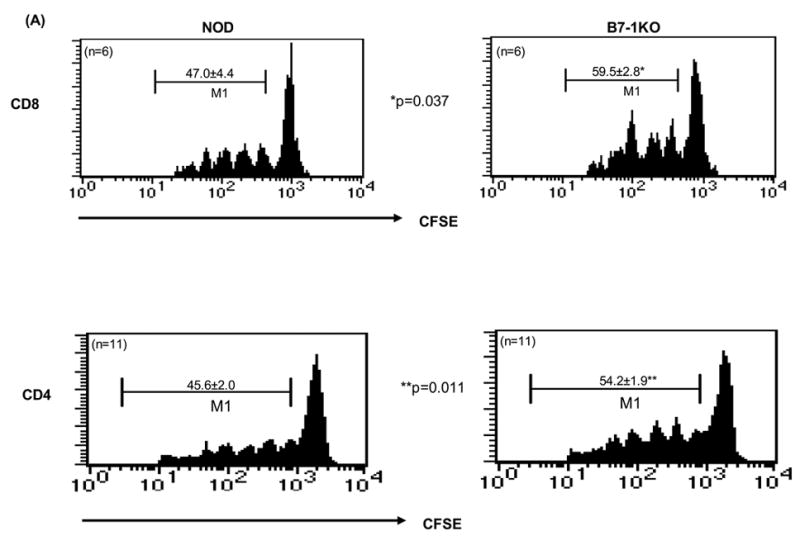
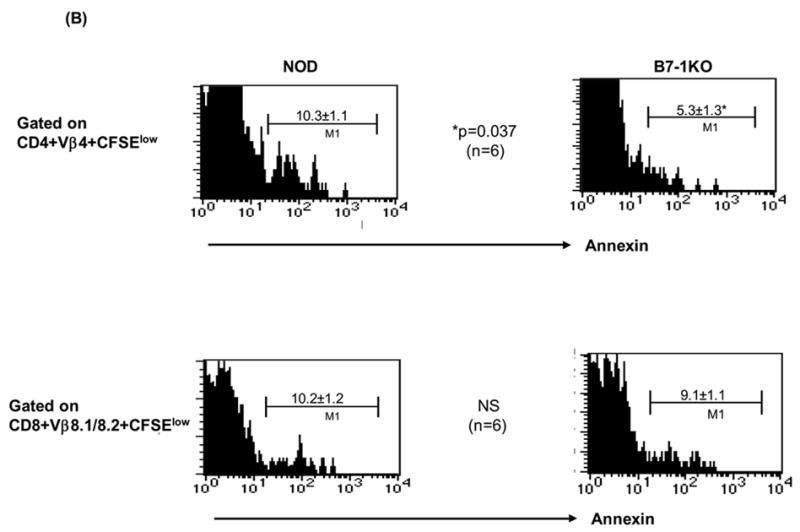
Enhanced expansion of islet-specific CD4 and CD8 T cells and increased survival of anti islet CD4 T cells in B7-1KO NOD mice. (A): CFSE-labeled splenocytes either from BDC2.5 or from 8.3NODscids were administered into NOD and B7-1KO hosts. After 4 days, the PLN cells were analyzed by flow cytometry for CFSE dilution to assess proliferation. The CFSE profiles of Vβ4+CD4+CFSE+ and Vβ8.1/8.2+CD8+CFSE+ T cells in NOD and B7-1KO recipients are represented. M1 depicts dividing cells. Analysis of cells before transfer indicated >80% of them were naive (i.e. CD44low). The values represent mean percentage± SEM. B: For in vivo screening of apoptosis of anti-islet CD4 and CD8 T cells, adoptively transferred CFSE labeled BDC2.5 and 8.3 T cells were tracked in vivo (as described for Fig. 1). Histograms depict annexin staining in the PLNs of cells gated on M1 (CFSElow, proliferating). The data are pooled from 2–3 independent experiments and are represented as mean percentage± SEM. NS, not significant.
3.3 B7-1 differentially regulates anti-islet CD4 and CD8 T cell survival
The increase in T lymphocyte cellularity in B7-1KO mice may reflect their prolonged survival. To test this, the adoptively transferred CFSE labeled BDC2.5 and 8.3NODscids cells were stained with annexin. A significantly reduced (p=0.037) frequency of CD4+CFSElowannexin+ T cells was observed in B7-1KO recipients compared to NOD controls. However, analysis of adoptively transferred 8.3NODscid cells revealed no significant change in the frequency of CD8+ CFSElowannexin+ T cells between B7-1KO and NOD mice (Fig 2B). This suggests that survival of islet specific CD4 T cells is negatively regulated by B7-1.
3.4 B7-1KO mice display reduced proportions of T-regulatory cells and increased proportions of effector cells
CD4+CD25+CD62L+ T cells have been shown to possess regulatory potential (Szanya et al., 2002). We found a significantly reduced proportion of these cells in B7-1KO mice both in the thymus (p=0.0005) as well as in the PLN compartment (p=0.009) (Table 3 and Fig. 4A). Pro-inflammatory Th1 cytokines like IFN-γ has been implicated in the development of type 1 diabetes (Wenner et al., 1996). We tested in vitro activated PLN cells from NOD and B7-1KO mice for their production of IFN-γ and found a significantly higher frequency of IFN-γ + CD4 (p=0.009) and CD8 (p=0.010) T cells (Fig 3). These results suggest that B7-1 mediated signals inhibit the generation of effector T cells in the periphery.
Table 3.
Immunophenotypic characterization of PLN T cells in NOD and B7-1KO mice.
| Cellular subsets | NOD | B7-1KO | n | p-value |
|---|---|---|---|---|
| CD4+CD28hi | 5.2±0.3 | 8.8±0.7 | 8 | p=0.001 |
| CD8+CD28hi | 5.1±0.2 | 6.9±0.4 | 8 | p=0.006 |
| CD4+PD-1+ | 9.8±0.8 | 7.2±0.3 | 8 | p=0.015 |
| CD8+PD-1+ | 5.9±0.7 | 6.6±0.4 | 5 | NS |
| CD4+CD25+CD62L+ | 5.5±0.1 | 3.9±0.1 | 5 | p=0.009 |
| CD4+CD25+CD62L+ (Thymus) | 2.1±0.03 | 1.4±0.07 | 8 | p=0.0005 |
n indicates number of pooled animals from individual experiments for statistical analysis of data. 6–8 weeks old mice were used. The values corresponding to NOD and B7-1KO are mean percentage ± SEM. NS means not significant.
Fig. 4.
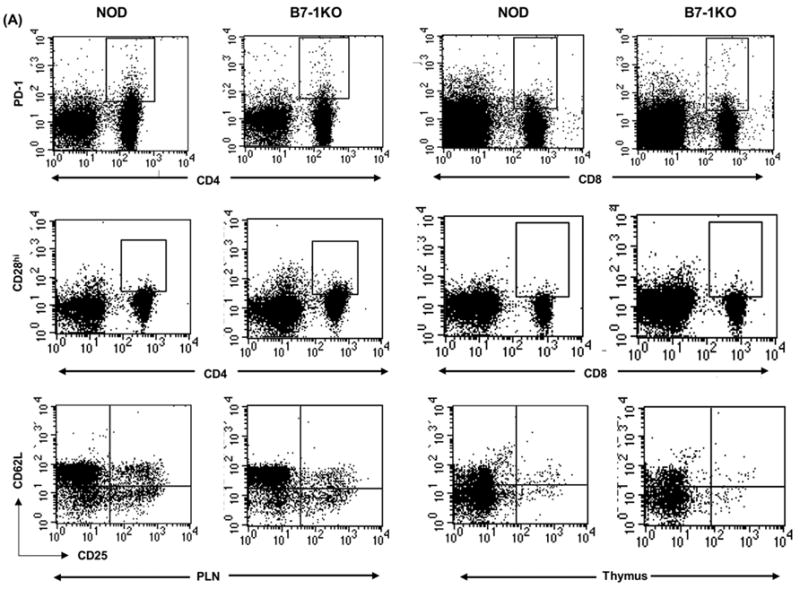
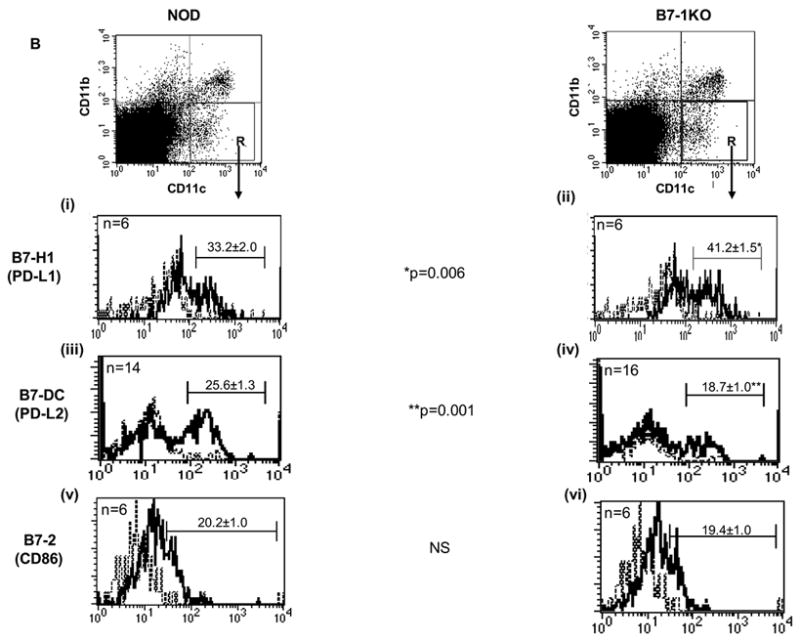
Immunophenotypic characterization of T cell and APCs in B7-1KO versus NOD mice. A: This is the dot plot representation of data described in table 2. The thymic and PLN T-reg dot plots are gated on CD3+CD4+ and CD4+ T cells respectively. B: PLN cells from 5 to 8 weeks old NOD and B7-1KO mice were stained to determine the costimulatory expression profiles of B7-H1 (i and ii), B7-DC (iii and iv), and B7-2 (v and vi). The dot plots are a typical representation of CD11b and CD11c distribution profile in NOD and B7-1KO mice. The histograms are gated on the cells depicted by region R in the dot plot and are described as CD11c+ cells in the text. The dotted lines in the histograms represent the isotype control staining and solid lines represent the actual staining for the corresponding marker. The data are pooled from 2-4 independent experiments. Values represent mean percentage± SEM. NS, not significant.
Fig. 3.
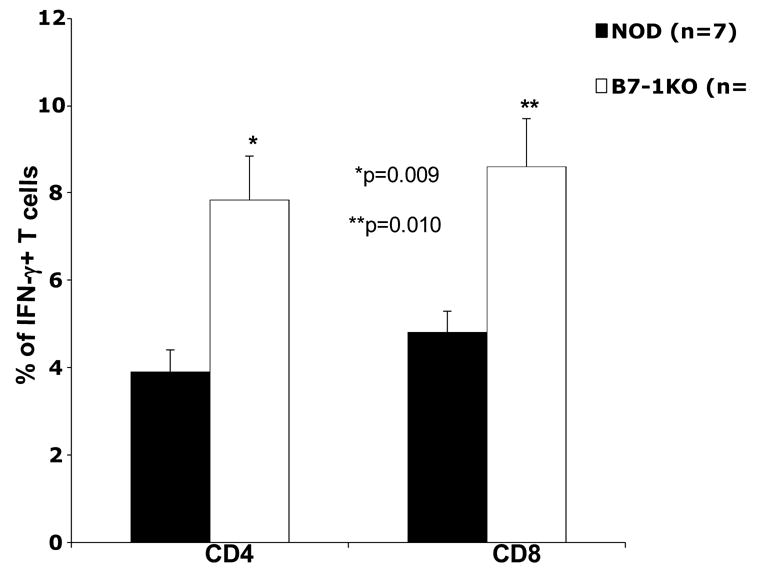
CD4 and CD8 T cells show enhanced effector cytokine production in B7-1KO mice. PLN cells from NOD and B7-1KO mice were stimulated in vitro and analyzed for IFN-γ staining (as described in material and methods) by flow cytometry. The bars depict the percentage of CD4+IFN-γ+ and CD8+IFN-γ+ T cells. Staining with corresponding isotype controls exhibited negligible staining. Data are pooled from 2 independent experiments and the values represent mean percentage± SEM.
3.5 Distinct ratios of CD28 and PD-1 Ligands in the periphery of B7-1KO mice
Variation in the levels of CD28 impact immune responses by controlling the threshold of T cell activation (Umlauf et al., 1993; Zhang et al., 2002). T cell costimulation is a fine tuned process and even a subtle phenotypic differences imparted by APCs may enhance the aggressiveness of auto reactive T cell response. We asked whether the absence of B7-1 affected the proportion of T cells expressing CD28 and found a significantly increased frequency of CD28hi cells among CD4 (p=0.001) and CD8 T cells (p=0.006) in B7-1KO mice (Table 3 and Fig. 4A). However, no significant change in CD28hi cells was observed in spleens (not shown). This data demonstrates that the B7-1 negatively regulates cell surface levels of its binding partner CD28. However, no significant difference in the B7-2 expression was observed in the APCs of B7-1 KO mice (Fig 4Bv,vi).
We also asked whether the enhanced pathogenic potential of T cells in B7-1KO mice is associated with alterations in the levels of PD-l/B7-H1/B7-DC molecules. Interestingly, we observed a significantly lower (p=0.015) percentage of CD4+PD-1+ T cells in B7-1KO mice (Table 3 and Fig. 4A). Analysis of PD ligands on APCs in the PLNs revealed a significantly enhanced (p=0.006) proportion of CD11c+B7-H1+ cells in B7-1KO mice compared to NOD mice (Fig 4Bi,ii). However, a significantly lower (p=0.001) frequency of CD11c+B7-DC+ cells was observed in B7-1KO mice (Fig 4Biii,iv) suggesting that B7-1 restricts expression of B7-H1, but may promote expression of B7-DC.
3.6 B7-H1 regulates priming and expansion of autoreactive T cells in periphery
We ascertained the role played by B7-H1 in modulating autoreactive T cell responses. CFSE labeled BDC2.5 transgenic CD4 T cells or 8.3NODscid transgenic CD8 T cells were injected into B7-1KO mice treated with blocking anti-B7-H1 antibody (Tsushima et al., 2003). We analyzed the CFSE dilution profiles on the basis of and resting/undivided cells (CFSEhigh, M1) and dividing cells (CFSElow, M2). Significantly higher percentages of dividing cells were observed for both anti-islet CD4 (p=0.041) (Fig 5A) and CD8 T cells (p=0.015) (Fig 5C) in anti B7-H1 treated mice.
Fig. 5.
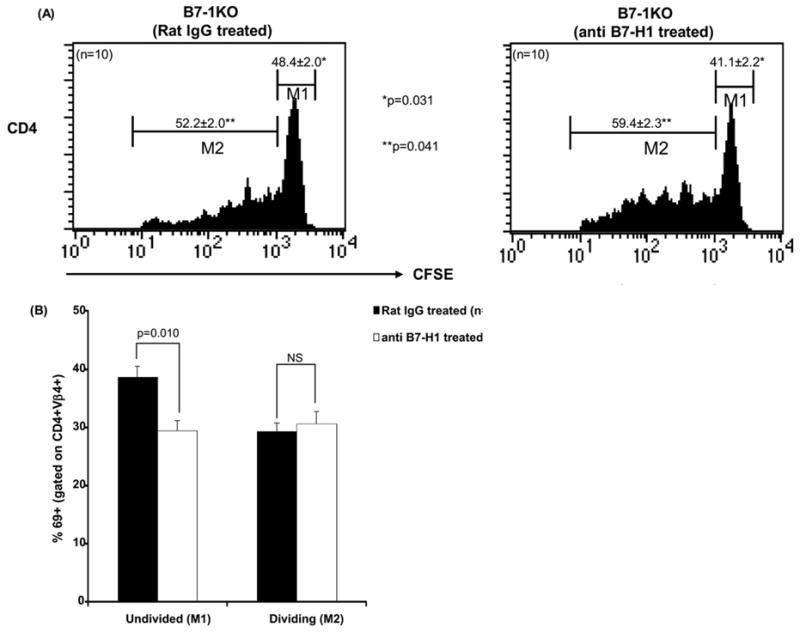
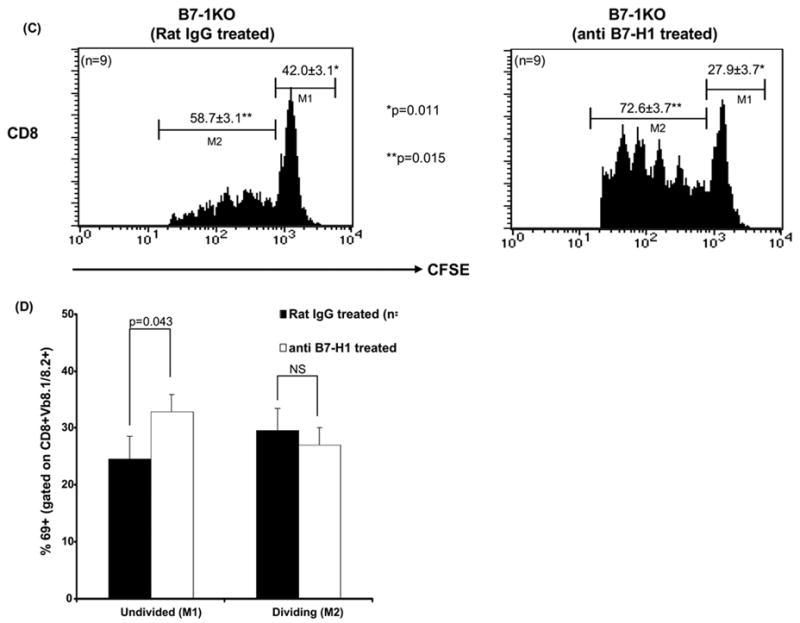
B7-H1 controls the priming and expansion of anti islet T cells. CFSE labeled transgenic 8.3NODscid/BDC2.5 anti-islet cells were adoptively transferred in B7-1KO mice which were also treated with either anti B7-H1 or rat-IgG (controls) and the PLN cells from the treated mice were analyzed for expansion (CFSE dilution) and priming (CD69 expression). Histograms depict the profiles of CD4+Vβ4+CFSE+ (A) and CD8+Vβ8.1/8.2+CFSE+ (C) T cells and the values represent mean percentage± SEM. Bars represents the CD69 expression in CD4+Vβ4+cells (B) and CD8+Vβ8.1/8.2+ T cells (D) gated on M1 (undivided) and M2 (divided) and the values represent mean percentage± SEM. The data are pooled from 2–3 independent experiments.
CD69 expression is elevated upon TCR ligation (Isogawa et al., 2005). We asked whether the priming of T cells was affected by B7-H1. The adoptively transferred CFSE labeled T cells were also monitored for CD69 expression in B7-1KO mice treated with B7-H1 Interestingly, a significantly lower (p=0.010) percentage of CD69+CFSEhigh+CD4+ undivided T cells was observed in anti B7-H1 treated mice, suggesting that in this context B7-H1 costimulation promotes TCR signaling and activation (Fig. 5B). Conversely, anti-islet CD8 T cells showed a significantly higher (p=0.043) percentage of CD69+CFSEhigh+CD8+ undivided T cells in anti B7-H1 treated mice (Fig. 5D), indicating that B7-H1 costimulation inhibits TCR legation in CD8 T cells. Therefore, when B7-1 is absent B7-H1 promotes the activation of CD4 T cells, while inhibiting CD8 T cell autoimmunity.
4 Discussion
Our work demonstrates that B7-1 expression ultimately acts to suppress the emergence of pathogenic autoimmune repertoire by regulating availability of pathogenic signals.
Costimulation by B7-1/2 is required for the intrathymic deletion of autoreactive T cells (Gao et al., 2002). Our thymic data showed significantly higher numbers of CD4SP and DP thymocytes in B7-1KO mice, indicating altered thymocyte maturation. In addition, we also observed a higher proportion of CD8+CD5hi single positive thymocytes in B7-1KO mice (Yadav and Sarvetnick, unpublished observations) indicating increased numbers of high-avidity T cells in the thymus. The reduction in B7-HI signals in thymic T cell subsets in B7-1KO mice may play a role in the increased cell numbers we observed in the thymus. Indeed, a similar increase in the DP and CD4SP thymocytes has been described in B7-H1 deficient mice (Keir et al., 2005).
We observed significantly enhanced expansion and survival of autoreactive T cells in B7-1KO mice. B7-1 preferentially interacts with CTLA-4, (Walunas et al., 1994) inducing CD4 T cell death (Scheipers and Reiser, 1998). It is possible that enhanced survival and expansion of CD4 T cells creates a more stable pathogenic CD4 T cell repertoire, which in turn can provide additional help to CD8 T cells thereby augmenting their effector potential. Higher proportions of T cells from the PLN of B7-1KO mice produced the pro-inflammatory cytokine IFN-γ (Wenner et al., 1996). Thus, the loss of B7-1 mediated costimulation helps create an inflammatory microenvironment, further augmenting the pathogenic potential of T cells. One reason for this may be the loss of CD4+CD25+CD62L+ T-regulatory cells both in PLN and thymus compartments in B7-1KO mice since this cell type controls pathogenic cells in the periphery (Szanya et al., 2002). B7/CD28 costimulation has been shown to play a critical role in the maintaining homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes (Salomon et al., 2000). Morever, in order to transmit suppressive signals, CD4CD25+ T-regs requires engagement of the B7 molecules expressed on target T cells (Paust et al., 2004), and this mechanism of immunoregulation may be defective in B7-1KO mice. Therefore, not only is the emergence of pathogenic repertoire enhanced but T-reg mediated control of anti islet T cells is lost in B7-1KO mice (Bour-Jordan et al., 2004).
The engagement of PD-1, an immunoreceptor belonging to the CD28/CTLA-4 family can lead to cell cycle arrest (Latchman et al., 2001). Loss of PD-1 has been shown to cause exacerbation of diabetes in NOD mice (Wang et al., 2005). CD28 is a major positive costimulatory receptor on T cells and CD28hi cells have been shown to preferentially migrate to inflammatory sites (Salazar-Fontana et al., 2001). We observed an increase in CD28hi T cells and a concomitant reduction in the frequency of PD-1+ CD4 T cells in mice lacking B7-1. B7-1 and B7-2 engagement has been shown to modulate CD28 expression on T cells (Eck et al., 1997) and ligation of CD28 with B7-1 has been reported to down regulate CD28 transcription. Thus, mice lacking B7-1 harbor an increased proportion T cells expressing CD28 and a decreased proportion of cells expressing PD-1.
The quality of costimulation provided by APCs to T cells determines the final outcome of any T cell response. We observed a significantly higher expression of B7-H1, whereas, the B7-DC expression was reduced in mice lacking B7-1. B7-H1 and B7-DC can engage with PD-1 providing negative costimulatory signals (Chen, 2004; Khoury and Sayegh, 2004). The role of B7-H1 in regulating T cell responses in vivo is not fully understood as its blockade precipitates diabetes (Ansari et al., 2003) but has also been shown to provide positive signals for naïve T cells to promote allograft rejection and autoimmunity (Subudhi et al., 2004). Our B7-H1 blocking studies confirmed the role of B7-H1 in providing negative signals for T cell expansion in the periphery of B7-1KO mice. However, in terms of T cell priming, the role of B7-H1 is more diverse. B7-H1 exerts a stimulatory role on CD4 T cells and an inhibitory role CD8 T cells. The differential modulation of PD-1/B7-H1 pathway on CD4 and CD8 T cells has been suggested previously (Carter et al., 2002). It is tempting to speculate that increase in the B7-H1 availability in B7-1KO mice may represent a mechanism to compensate for the reduction in the expression of its receptor PD-1 on T cells albeit unsuccessfully as these mice exhibit enhanced autoreactivity. Whether a concomitant reduction of B7-DC on B7-1KO APCs plays any significant role in the exacerbation of diabetes still needs further investigation.
The enhancement of autoreactivity due to loss of B7-1 in already autoimmune NOD mice signifies two important points. First, B7-1 plays an important role in immunoregulation of anti islet T cell reactivity but is not fully successful in its regulating. Second, even subtle changes in the availability of APC mediated costimulatory signals and expansion of autorective T cells is enough to tilt the nature of autoreactivity from chronic in NOD mice to more acute nature in B7-1KO mice. In sum, our results demonstrate that exacerbation of diabetes in the absence of B7-1 mediated costimulation involves the failure of immunological checks and balances. Not only is the availability of T-regs reduced in B7-1KO mice, but also due to consolidation of pathogenic signals, T cells exhibit a concomitant enhancement in their expansion, survival and effector capabilities. This creates a microenvironment conducive for the emergence of the anti islet T cell repertoire.
Acknowledgments
The authors wish to thank Hideo Yagita, Malin F. Tullberg, Natasha Hill, Sandrine Dabernat and members of the lab for their helpful comments and Amy Maday, Patrick Secrest for technical help. Caroline Lanigan, Ph.D. is thanked for her expert advice regarding statistical analysis. Deepak Yadav is the recipient of postdoctoral fellowship awards from Myasthenia Gravis Foundation of America (MGFA, 2003) and Juvenile Diabetes Research Foundation International (JDRF, 2005). This work was supported by funding from NIH grants DK054063 and AI51973 to Nora Sarvetnick. This is manuscript # 18206 from Dept. of Immunology, The Scripps Research Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ansari MJ, Salama AD, Chitnis T, Smith RN, Yagita H, Akiba H, Yamazaki T, Azuma M, Iwai H, Khoury SJ, Auchincloss H, Jr, Sayegh MH. The programmed death-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J Exp Med. 2003;198:63–9. doi: 10.1084/jem.20022125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bour-Jordan H, Salomon BL, Thompson HL, Szot GL, Bernhard MR, Bluestone JA. Costimulation controls diabetes by altering the balance of pathogenic and regulatory T cells. J Clin Invest. 2004;114:979–87. doi: 10.1172/JCI20483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho SA, Heath WR, Carbone FR, Sarvetnick N, LeBon A, Karlsson L, Peterson PA, Webb SR. A key role for ICAM-1 in generating effector cells mediating inflammatory responses. Nat Immunol. 2001;2:523–9. doi: 10.1038/88720. [DOI] [PubMed] [Google Scholar]
- Carter L, Fouser LA, Jussif J, Fitz L, Deng B, Wood CR, Collins M, Honjo T, Freeman GJ, Carreno BM. PD-1:PD-L inhibitory pathway affects both CD4(+) and CD8(+) T cells and is overcome by IL-2. Eur J Immunol. 2002;32:634–43. doi: 10.1002/1521-4141(200203)32:3<634::AID-IMMU634>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Chen L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat Rev Immunol. 2004;4:336–47. doi: 10.1038/nri1349. [DOI] [PubMed] [Google Scholar]
- Eck SC, Chang D, Wells AD, Turka LA. Differential down-regulation of CD28 by B7-1 and B7-2 engagement. Transplantation. 1997;64:1497–9. doi: 10.1097/00007890-199711270-00025. [DOI] [PubMed] [Google Scholar]
- Firestein GS. The T cell cometh: interplay between adaptive immunity and cytokine networks in rheumatoid arthritis. J Clin Invest. 2004;114:471–4. doi: 10.1172/JCI22651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao JX, Zhang H, Bai XF, Wen J, Zheng X, Liu J, Zheng P, Liu Y. Perinatal blockade of b7-1 and b7-2 inhibits clonal deletion of highly pathogenic autoreactive T cells. J Exp Med. 2002;195:959–71. doi: 10.1084/jem.20011948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerder S, Eynon EE, Flavell RA. Autoimmunity without diabetes in transgenic mice expressing beta cell-specific CD86, but not CD80: parameters that trigger progression to diabetes. J Immunol. 1998;161:2128–40. [PubMed] [Google Scholar]
- Guerder S, Picarella DE, Linsley PS, Flavell RA. Costimulator B7-1 confers antigen-presenting-cell function to parenchymal tissue and in conjunction with tumor necrosis factor alpha leads to autoimmunity in transgenic mice. Proc Natl Acad Sci U S A. 1994;91:5138–42. doi: 10.1073/pnas.91.11.5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isogawa M, Furuichi Y, Chisari FV. Oscillating CD8(+) T cell effector functions after antigen recognition in the liver. Immunity. 2005;23:53–63. doi: 10.1016/j.immuni.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Kanai T, Totsuka T, Uraushihara K, Makita S, Nakamura T, Koganei K, Fukushima T, Akiba H, Yagita H, Okumura K, Machida U, Iwai H, Azuma M, Chen L, Watanabe M. Blockade of B7-H1 suppresses the development of chronic intestinal inflammation. J Immunol. 2003;171:4156–63. doi: 10.4049/jimmunol.171.8.4156. [DOI] [PubMed] [Google Scholar]
- Katz JD, Wang B, Haskins K, Benoist C, Mathis D. Following a diabetogenic T cell from genesis through pathogenesis. Cell. 1993;74:1089–100. doi: 10.1016/0092-8674(93)90730-e. [DOI] [PubMed] [Google Scholar]
- Keir ME, Latchman YE, Freeman GJ, Sharpe AH. Programmed death-1 (PD-1):PD-ligand 1 interactions inhibit TCR-mediated positive selection of thymocytes. J Immunol. 2005;175:7372–9. doi: 10.4049/jimmunol.175.11.7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, Koulmanda M, Freeman GJ, Sayegh MH, Sharpe AH. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med. 2006;203:883–95. doi: 10.1084/jem.20051776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury SJ, Sayegh MH. The roles of the new negative T cell costimulatory pathways in regulating autoimmunity. Immunity. 2004;20:529–38. doi: 10.1016/s1074-7613(04)00116-5. [DOI] [PubMed] [Google Scholar]
- Lanier LL, O’Fallon S, Somoza C, Phillips JH, Linsley PS, Okumura K, Ito D, Azuma M. CD80 (B7) and CD86 (B70) provide similar costimulatory signals for T cell proliferation, cytokine production, and generation of CTL. J Immunol. 1995;154:97–105. [PubMed] [Google Scholar]
- Larsen CP, Ritchie SC, Hendrix R, Linsley PS, Hathcock KS, Hodes RJ, Lowry RP, Pearson TC. Regulation of immunostimulatory function and costimulatory molecule (B7-1 and B7-2) expression on murine dendritic cells. J Immunol. 1994;152:5208–19. [PubMed] [Google Scholar]
- Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, Iwai Y, Long AJ, Brown JA, Nunes R, Greenfield EA, Bourque K, Boussiotis VA, Carter LL, Carreno BM, Malenkovich N, Nishimura H, Okazaki T, Honjo T, Sharpe AH, Freeman GJ. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–8. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- Lieberman SM, Evans AM, Han B, Takaki T, Vinnitskaya Y, Caldwell JA, Serreze DV, Shabanowitz J, Hunt DF, Nathenson SG, Santamaria P, DiLorenzo TP. Identification of the beta cell antigen targeted by a prevalent population of pathogenic CD8+ T cells in autoimmune diabetes. Proc Natl Acad Sci U S A. 2003;100:8384–8. doi: 10.1073/pnas.0932778100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendel I, Shevach EM. Activated T cells express the OX40 ligand: requirements for induction and costimulatory function. Immunology. 2006;117:196–204. doi: 10.1111/j.1365-2567.2005.02279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paust S, Lu L, McCarty N, Cantor H. Engagement of B7 on effector T cells by regulatory T cells prevents autoimmune disease. Proc Natl Acad Sci U S A. 2004;101:10398–403. doi: 10.1073/pnas.0403342101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pentcheva-Hoang T, Egen JG, Wojnoonski K, Allison JP. B7-1 and B7-2 selectively recruit CTLA-4 and CD28 to the immunological synapse. Immunity. 2004;21:401–13. doi: 10.1016/j.immuni.2004.06.017. [DOI] [PubMed] [Google Scholar]
- Salazar-Fontana LI, Sanz E, Merida I, Zea A, Sanchez-Atrio A, Villa L, Martinez AC, de la Hera A, Alvarez-Mon M. Cell surface CD28 levels define four CD4+ T cell subsets: abnormal expression in rheumatoid arthritis. Clin Immunol. 2001;99:253–65. doi: 10.1006/clim.2001.5003. [DOI] [PubMed] [Google Scholar]
- Salomon B, Bluestone JA. Complexities of CD28/B7: CTLA-4 costimulatory pathways in autoimmunity and transplantation. Annu Rev Immunol. 2001;19:225–52. doi: 10.1146/annurev.immunol.19.1.225. [DOI] [PubMed] [Google Scholar]
- Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–40. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- Salomon B, Rhee L, Bour-Jordan H, Hsin H, Montag A, Soliven B, Arcella J, Girvin AM, Padilla J, Miller SD, Bluestone JA. Development of spontaneous autoimmune peripheral polyneuropathy in B7-2-deficient NOD mice. J Exp Med. 2001;194:677–84. doi: 10.1084/jem.194.5.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santra S, Barouch DH, Jackson SS, Kuroda MJ, Schmitz JE, Lifton MA, Sharpe AH, Letvin NL. Functional equivalency of B7-1 and B7-2 for costimulating plasmid DNA vaccine-elicited CTL responses. J Immunol. 2000;165:6791–5. doi: 10.4049/jimmunol.165.12.6791. [DOI] [PubMed] [Google Scholar]
- Scheipers P, Reiser H. Fas-independent death of activated CD4(+) T lymphocytes induced by CTLA-4 crosslinking. Proc Natl Acad Sci U S A. 1998;95:10083–8. doi: 10.1073/pnas.95.17.10083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer AN, Borriello F, Wong RC, Abbas AK, Sharpe AH. Role of costimulators in T cell differentiation: studies using antigen-presenting cells lacking expression of CD80 or CD86. J Immunol. 1997;158:2713–22. [PubMed] [Google Scholar]
- Sharpe AH, Freeman GJ. The B7-CD28 superfamily. Nat Rev Immunol. 2002;2:116–26. doi: 10.1038/nri727. [DOI] [PubMed] [Google Scholar]
- Slavik JM, Hutchcroft JE, Bierer BE. CD80 and CD86 are not equivalent in their ability to induce the tyrosine phosphorylation of CD28. J Biol Chem. 1999;274:3116–24. doi: 10.1074/jbc.274.5.3116. [DOI] [PubMed] [Google Scholar]
- Subudhi SK, Zhou P, Yerian LM, Chin RK, Lo JC, Anders RA, Sun Y, Chen L, Wang Y, Alegre ML, Fu YX. Local expression of B7-H1 promotes organ-specific autoimmunity and transplant rejection. J Clin Invest. 2004;113:694–700. doi: 10.1172/JCI19210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szanya V, Ermann J, Taylor C, Holness C, Fathman CG. The subpopulation of CD4+CD25+ splenocytes that delays adoptive transfer of diabetes expresses L-selectin and high levels of CCR7. J Immunol. 2002;169:2461–5. doi: 10.4049/jimmunol.169.5.2461. [DOI] [PubMed] [Google Scholar]
- Tsushima F, Iwai H, Otsuki N, Abe M, Hirose S, Yamazaki T, Akiba H, Yagita H, Takahashi Y, Omura K, Okumura K, Azuma M. Preferential contribution of B7-H1 to programmed death-1-mediated regulation of hapten-specific allergic inflammatory responses. Eur J Immunol. 2003;33:2773–82. doi: 10.1002/eji.200324084. [DOI] [PubMed] [Google Scholar]
- Umlauf SW, Beverly B, Kang SM, Brorson K, Tran AC, Schwartz RH. Molecular regulation of the IL-2 gene: rheostatic control of the immune system. Immunol Rev. 1993;133:177–97. doi: 10.1111/j.1600-065x.1993.tb01516.x. [DOI] [PubMed] [Google Scholar]
- Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, Thompson CB, Bluestone JA. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1:405–13. doi: 10.1016/1074-7613(94)90071-x. [DOI] [PubMed] [Google Scholar]
- Wang J, Yoshida T, Nakaki F, Hiai H, Okazaki T, Honjo T. Establishment of NOD-Pdcd1−/− mice as an efficient animal model of type I diabetes. Proc Natl Acad Sci U S A. 2005;102:11823–8. doi: 10.1073/pnas.0505497102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenner CA, Guler ML, Macatonia SE, O’Garra A, Murphy KM. Roles of IFN-gamma and IFN-alpha in IL-12-induced T helper cell-1 development. J Immunol. 1996;156:1442–7. [PubMed] [Google Scholar]
- Yadav D, Judkowski V, Flodstrom-Tullberg M, Sterling L, Redmond WL, Sherman L, Sarvetnick N. B7-2 (CD86) controls the priming of autoreactive CD4 T cell response against pancreatic islets. J Immunol. 2004;173:3631–9. doi: 10.4049/jimmunol.173.6.3631. [DOI] [PubMed] [Google Scholar]
- Zhang J, Bardos T, Li D, Gal I, Vermes C, Xu J, Mikecz K, Finnegan A, Lipkowitz S, Glant TT. Cutting edge: regulation of T cell activation threshold by CD28 costimulation through targeting Cbl-b for ubiquitination. J Immunol. 2002;169:2236–40. doi: 10.4049/jimmunol.169.5.2236. [DOI] [PubMed] [Google Scholar]