

Abstract
We analyzed the in situ molecular correlates of infection from cancer patients treated with reovirus. Melanoma, colorectal, and ovarian cancer samples from such patients showed variable infection of the cancer cells but not the intermingled benign cells. RT in situ PCR showed most cancer cells contained the viral genome with threefold less having productive viral infection as documented by either tubulin or reoviral protein co-expression. Productive infection in the cancer cells was strongly correlated with co-expression of p38 and caspase-3 as well as apoptosis-related death (P<0.001). The cancer cell apoptotic death was due to a marked viral-induced inhibition of microRNA-let-7d that, in turn, upregulated caspase-3 activity. In summary, reovirus shows a striking tropism to cancer cells in clinical samples. A rate-limiting factor of reovirus-induced cancer cell death is productive viral infection that operates via the marked reduction of microRNA-let-7d and concomitant elevated caspase-3 expression.
Keywords: apoptosis, caspase-3, microRNA-let-7d, PKR, Ras, reovirus
Reovirus is a naked double-stranded RNA virus that may cause infections in humans that are typically self-limited. To evade the PKR (protein kinase RNA-activated (also known as interferon-inducible RNA-dependent protein kinase) antiviral defense system, the virus has evolved to have enhanced infectivity for cells with elevated proliferation-related protein kinases, such as Ras, EGFR, pERK, and p38, as these inactivate PKR via dephosphorylation.1 As these protein kinases are typical of cancer cells, there has been strong interest in the potential use of reovirus as an anti-tumor agent.2–4
Reolysin (Oncolytics), which contains serotype 3 of the Dearing strain, has been used in a wide variety of tumors, including ovarian, colon, prostate, and head and neck cancers.5,6 The resultant studies have shown that the virus has limited toxicity and demonstrates single agent and combinatorial efficacy by both radiological and/or biomarker evaluation.2–4 Similarly, reoviral treatment of azoxymethane-induced colon cancer in rats much reduced the precancers and invasive cancers associated with this carcinogenic agent.7 Combination treatment with various agents such as paclitaxel, T-regulatory cells inhibitors, interleukin-2, VEGF inhibitors, and cell cycle synchronization agents such as hydroxyurea have been shown to potentiate the anti-tumor effect of reovirus.8–12 Recent evidence suggests that chemotherapy and VEGF inhibitors are able to potentiate reovirus-related killing of tumor cells by increasing the VEGFR2 receptor in endothelial cells, which makes them more permissive to viral entry.11 This study also highlighted the possible role of the immune system in general and NK activation in particular in the reovirus-mediated antitumor response, which involves activation of VEGFR1.8–11
Reovirus can upregulate the expression of certain proteins such as Ras that, in turn, can makes the cancer cells more permissive for viral-induced apoptotic death.13 Park et al14 showed that NIH 3T3 cells were highly resistant to reovirus but, after stably transfected with the oncogene CUG2, the p38 and Ras were upregulated, and the cancer cells then became highly susceptible to reovirus infection with an associated marked increase in the apoptotic death. It has been well established that p38 expression is a key molecular event in fostering reovirus replication in cancer cells.15 The molecular events that follow from viral infection to increased cancer cell death are poorly understood and are important to understand given the growing interest in reovirus as an adjunct treatment for cancer.1–4 The purpose of this paper was to dissect the molecular correlates of reovirus infection in cancer cells with a focus on the virus-induced changes that led to the death of the malignant cells.
Materials and methods
Cell Lines and Validation Studies
Over 50 different cell samples used for in vitro studies with reovirus were available for study and came from eleven different cell lines (kindly provided by Victoria Roulstone, Alan Melcher, and Matt Coffey). The cell lines were grown as previously described16 and were exposed to various regimens of reovirus, no virus controls, and different chemotherapeutic agents including cisplatin and paclitaxel. The immunohistochemical analyses of reoviral protein in these cell lines were done blinded to the pretreatment regimes so as to validate the specificity of our assay.
Tissue Samples of Malignant Tumors
All tissue samples were obtained via Internal Review Board (IRB) approved internal reviews (OSU protocol 2009 E0409). The tissues were fixed in 10% buffered formalin and embedded in paraffin for routine processing. A hematoxylin- and eosin-stained slide was reviewed by a board certified anatomic pathologist (GJN) to verify the presence of the tumor and to quantify the degree of inflammation and necrosis in the tumor sample.
Immunohistochemistry
Our immunohistochemistry protocol has been previously published.17 The reovirus antibody to a caspid protein was a kind gift of Dr Matt Coffey (Oncolytics, Alberta, Canada). The following antibodies were used in this study (with optimal conditions and source in parentheses): tubulin (1:100, antigen retrieval, Abcam); caspase-3 (1:33, antigen retrieval, Abcam); PKR–phosphorylated form (1:50, antigen retrieval, Abcam); pERK (1:100, antigen retrieval, Abcam), Ras (1:10, antigen retrieval, Abcam), and p38 (1:250, antigen retrieval, Abcam).
In Situ Hybridization
Our microRNA in situ hybridization protocol has been previously published.17 In brief, after digestion in protease, the tissue and probe microRNA (0.1–1 pmole/µl, 5′ digoxigenin tagged, Exiqon) were co-incubated at 60 °C for 5 min, then hybridized for 2–15 h at 37C. After a wash in 0.1 × SSC and 2% bovine serum albumin at 50C for 10 min, the microRNA–probe complex is visualized via NBT/BCIP (Roche) due to the action of the alkaline phosphatase that is conjugated to the antidigoxigenin antibody. The TUNEL-based in situ hybridization assay was done according to the manufacturer’s specifications (Roche Diagnostics).
RT In Situ PCR
Our RT in situ PCR protocol has been previously published.18 We used this method to ascertain how efficiently the viral genome entered malignant and benign cells in a given neoplastic tissue as the assay is theoretically able to detect one viral genome per cell.18,19 Immunohistochemistry analysis of reoviral protein is far less sensitive and most likely requires productive infection of the cell by the virus, with many genomic copies per cell, to be scored as positive.18,19
In brief, optimal protease digestion was determined by using the RT in situ PCR-positive (no DNase) and -negative (DNase, no primers) controls with direct incorporation of digoxigenin—dUTP. The presence of a strong nuclear-based signal (no DNase, due to nonspecific DNA repair) and its loss with DNase digestion and no primers confirmed that the RT in situ PCR test for reovirus was target specific.18,19 An additional control used in each run was a tissue known to be reovirus negative, such as tumor samples from patients treated with the placebo and not reovirus.
Co-Expression Analyses
Co-expression analyses were done using the Nuance system (CRI) as previously published.17 The Nuance system isolates individual colorimetric signals, converts them to a fluorescent-based signal, then mixes them to determine co-expression of two or more targets in a given cell.
Global MicroRNA Analyses
The NanoString nCounter Human miRNA Expression Assay Kit (http://www.nanostring.com/) was used to profile >700 human and human-associated viral miRNAs. In total, 100 ng of total RNAwas used as input for nCounter miRNA sample preparation reactions. All sample preparation was performed according to manufacturer’s instructions (Nano-String Technologies). Preparation of small RNA samples involves the ligation of a specific DNA tag onto the 3′ end of each mature miRNA. These tags are designed to normalize the Tm’s of the miRNAs as well as to provide a unique identification for each miRNA species in the sample. Hybridized probes were purified using the nCounter Prep Station (NanoString Technologies) following the manufacturer’s instructions to remove excess capture and reporter probes and to immobilize transcript-specific ternary complexes on a streptavidin-coated cartridge. Data collection was carried out on the nCounter Digital Analyzer (NanoString Technologies) following the manufacturer’s instructions to count individual fluorescent barcodes and quantify target RNA molecules present in each sample. For each assay, a high density scan (600 fields of view) was performed.
Luciferase Assay
The luciferase assay was done using a standard previously published protocol.20 In brief, Hela cells (5 × 103) were seeded in quadruplicate in a 96-multi-well plate and transfected after 24 h with GoClone Reporter Constructs 3′UTR Capase 3 vector (SwitchGear Genomics Menlo Park, CA, USA) plus pre-miRNA control or pre-miR let7-d to a final concentration of 20 nM. Luciferase activity was measured 48 h after transfection using the Light-Switch Luciferase Assay System.
Results
Cell Line Validation Study
In the first part of the study, we analyzed 51 samples from 11 different cell lines (HN3, HN5, SIHN 5B, PJ41, Detroit, CAL27, CHL, A375, DO4, SK-Mel28, and WM 1791C) that were exposed to either no pretreatment or reovirus with variable combinations of different chemotherapeutic agents. The analyses were done blinded to the experimental conditions. Decoding the experimental conditions showed that the immunohistochemistry assay correctly predicted whether the cells were exposed to reovirus in all 51 samples. The reovirus-positive cells showed a strong cytoplasmic signal with a range of positive cancer cells of 2.3–97.1%. Representative photos of the cell lines SK-Mel28 and CAL27 are presented in Figure 1.
Figure 1.
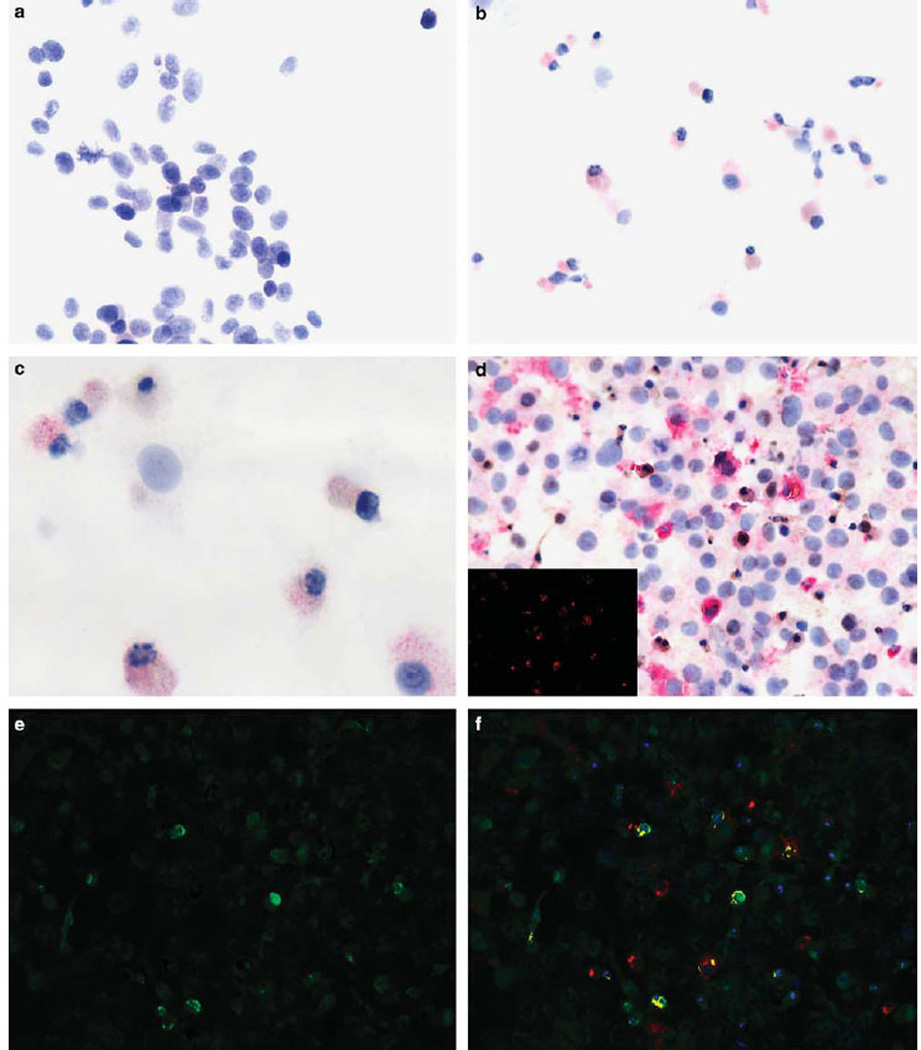
Productive reovirus infection in cell preparations. The cell line SK-Mel28 was exposed to reovirus in suspension. The uninfected cells (a) show no viral-specific signal; note the uniformity of the large neoplastic cells. After reoviral treatment, many cells show a strong red cytoplasmic signal for the viral capsid protein (b). Note that many of the cells show shrunken, darkened nuclei typical of apoptosis (c, higher magnification). The cell line CAL27 was tested for co-expression of reovirus (red signal) and tubulin (brown signal) (d); the insert shows the Nuance converted signal for reovirus (fluorescent red). (e) Shows the Nuance converted tubulin signal (fluorescent green). The merged images for reovirus and tubulin (f) show reovirus protein-positive cells that co-express tubulin as fluorescent yellow, consistent with productive infection.
The viral protein detected in the cell lines may represent passive incorporation of the viral protein, and not productive viral infection. To address this point, we exploited the observation that productive reoviral infection requires attachment of the viral protein to the microtubulin scaffold.21 Co-expression analyses of reoviral and microtubulin proteins in the cell lines CAL27 and Detroit indeed documented that there was strong co-expression (Figure 1, CAL27 cells).
As reovirus infection is correlated with RAS, pERK, and apoptosis (analyzed here by caspase-3 and the TUNEL assay), but not with PKR (phosphorylated form) expression,1,14,15 we next analyzed the percentage of cancer cells positive for these markers in the SK-Mel28 cancer cells unexposed vs exposed to the virus. As evident from Table 1, in each case, there was a significant increase (P <0.0001) in the percentage of cancer cells that expressed each marker after reovirus treatment. Further, when compared with the reovirus-infected cells alone, the combination of reovirus plus taxol in the SK-Mel28 cell line led to a >100%increase in the percentage of reoviral-infected cells that showed productive infection as documented by tubulin co-expression (11.8% (s.d. 3.9) vs 24.2% (s.d. 4.7), P = 0.0007).
Table 1.
Correlation of molecular markers and reovirus infection in the melanoma cell line SK-Mel28
| Marker | No reovirus (mean (s.d.))a |
Reovirus (mean (s.d.)) |
P-value (reovirus vs no treatment) |
|---|---|---|---|
| PKR | 0.3 (0.5)b | 7.0 (1.0) | <0.0001 |
| pERK | 1.0 (1.0) | 9.6 (1.5) | <0.0001 |
| Ras | 5.3 (2.5) | 49.3 (1.1) | <0.0001 |
| Caspase-3 | 1.6 (1.0) | 6.8 (0.7) | <0.0001 |
| TUNEL | 0.4 (0.5) | 5.0 (0.8) | <0.0001 |
Abbreviations: PKR, protein kinase RNA-activated (also known as interferon-inducible RNA-dependent protein kinase; the phosphorylated form was detected; pERK, extracellular-signal-regulated kinases; the phosphorylated form was detected.
s.d.
The values are the percentage of positive cancer cells and represent a minimum of n = 3 and a tabulation of at least 350 cells.
We next addressed the question: were the cancer cells that showed increased expression of pERK, Ras, PKR, and caspase-3, as well as showing increased apoptosis (TUNEL assay) the same cells with detectable reoviral protein? Co-expression analyses did show a striking co-expression of reoviral protein in RAS, pERK, p38, caspase-3-, and TUNEL-positive cells. Representative examples are shown in Figure 2 where it is evident that cells expressing caspase-3 invariably co-expressed abundant viral protein; strong p38 and reoviral protein co-expression is also evident. However, also note that there was virtually no co-expression between the virus protein and PKR, providing further evidence that the reoviral protein marked productive infection as phosphorylated PKR will block viral proliferation.1
Figure 2.
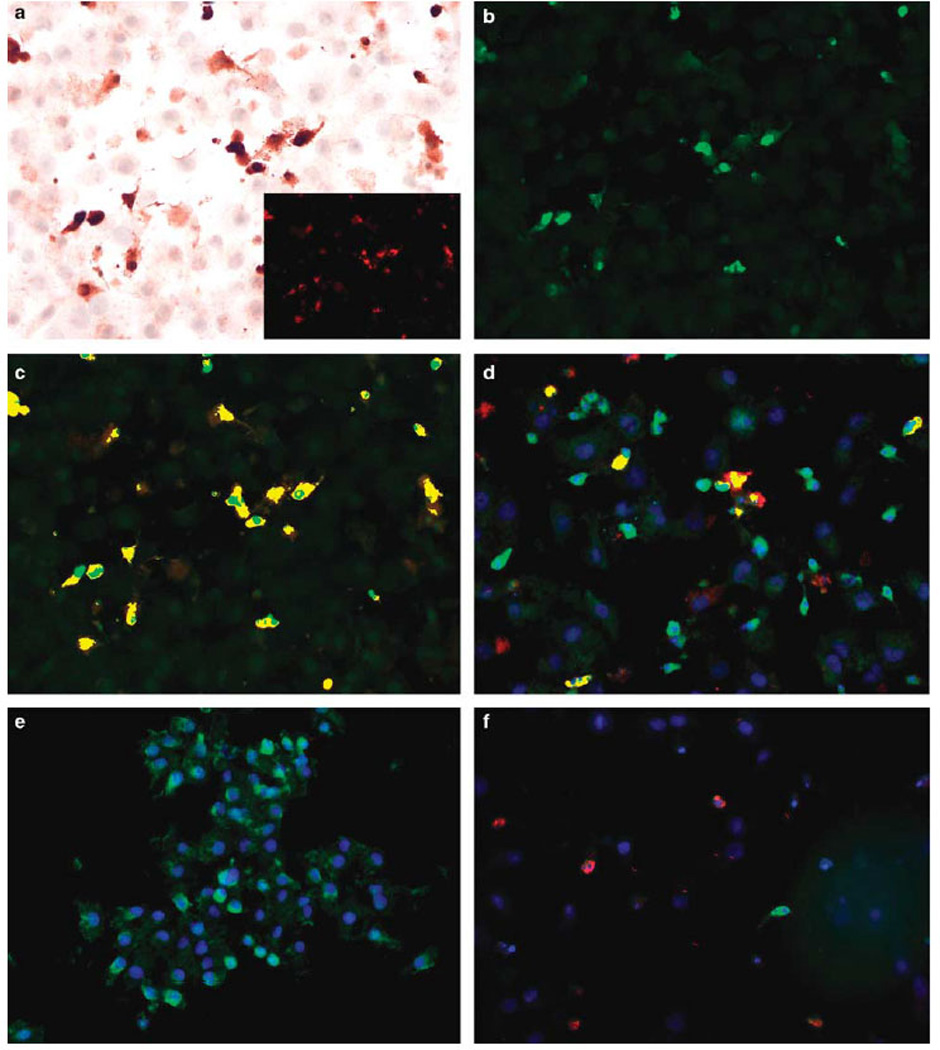
Molecular correlates of reoviral infection in cell preparations. Co-expression analysis of reovirus (red signal) and caspase-3 (brown signal) (a) in the cell line CAL27 shows that many of the cells with signal show apoptotic changes. The insert shows the Nuance isolated reovirus signal (fluorescent red) while (b) depicts the caspase-3 isolated signal (fluorescent green). When the two signals are merged, basically all cancer cells expressing detectable reoviral protein also express caspase-3 that is evident as fluorescent yellow (c). (d) Shows the merged co-expression analysis of reovirus (fluorescent red) and p38 (fluorescent green); note that most reoviral-positive cells are p38 positive as they show fluorescent yellow. A negative control (no reovirus treatment) shows the p38 signal without any reoviral signal (e). Whereas most reoviral-positive cells expressed p38 (d), Ras, and pERK (data not shown), none of the occasional cells that expressed PKR (fluorescent green, f) co-expressed reovirus (fluorescent red, f).
Tumor Tissue Analysis for Reovirus
We next examined a series of tumor samples from patients treated with reovirus. There were a total of 22 tumor samples with the following distribution: colon cancer (10 plus 3 control colon cancers from people not treated with Reovirus), ovarian cancer (2), and melanoma (7). We had an additional 10 liver samples from people who had metastatic colon cancer treated with reovirus and 3 more liver samples from people with metastatic colon cancer not exposed to the virus for a total number of 35 tissue samples from cancer patients. The patients had each received one IV or intraperitoneal cycle of Reolysin as part of a phase 1/2 trial as previously reported;16 all studies were approved by the respective IRBs.
An examination of the hematoxylin and eosin characteristics of the cancer specimens showed a variable degree of degeneration, necrosis, chronic inflammation, and histological changes consistent with apoptosis. Representative examples are given in Figure 3.
Figure 3.
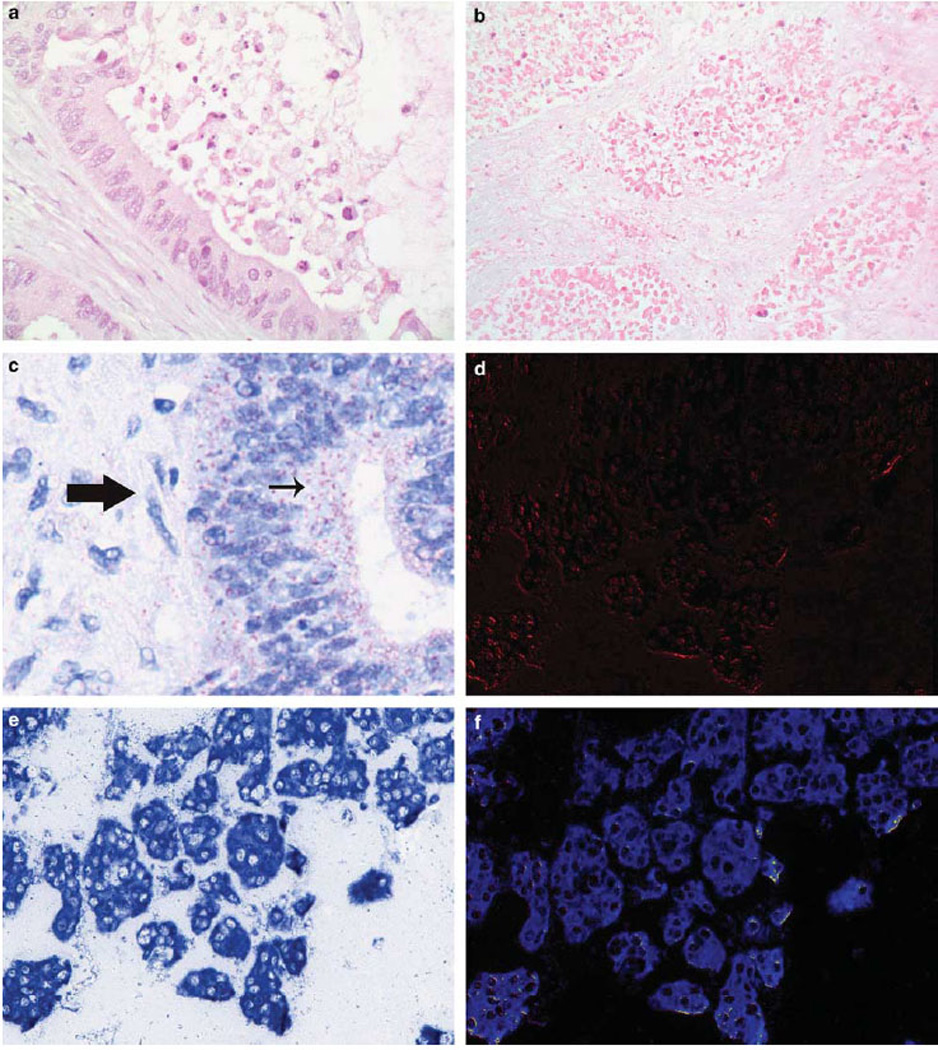
Reoviral protein and RNA in cancer tissues. Routine histological analyses of tissues from patients with colorectal cancer treated with Reovirus show foci of apoptosis in the center of the malignant gland (a) and more wide spread tumor necrosis (b). (c) Shows that the signal for reoviral protein after immunohistochemistry localized to the cancer cells (small arrow) and not the stromal cells (large arrow). (d–f) Show the results of co-expression analyses for reoviral RNA, as detected by RT in situ PCR, and reoviral protein visualized with immunohistochemistry in an ovarian cancer. (e) Shows the RGB image where it is evident that most of the tumor cells contain viral RNA. Note the striking localization of the viral RNA to the tumor cell nests. (d) Shows the Nuance converted image of the reoviral protein (fluorescent red) and (f) is the Nuance image after merging the reoviral protein and reoviral RNA (fluorescent blue). Note that, although some cancer cells contain both reoviral RNA and protein (fluorescent yellow), many only contain detectable viral genome, suggesting a relatively poor rate of productive infection in this tumor.
We next analyzed the 35 tissues for reovirus by immunohistochemistry. Of the cancer tissues from the patients treated with the virus, reoviral protein was detected in 9/10 of the colon cancers, 2/2 ovarian cancers, and 2/7 melanomas for a total of 13/19 (68%). Each of the three tumors from patients not treated with reovirus was viral protein negative. In all, 4 of the 13 liver tissues were positive for reovirus. However, the positive cases showed signal in very few hepatocytes and only in people treated with the virus. Representative examples of reovirus detection in the tumor biopsies of the patients are provided in Figure 3. No virus-specific inclusions were evident. Note the strong propensity for the viral protein to localize to the cancer cells and not the surrounding stromal cells. Surprisingly, it was also very rare to detect reovirus in mononuclear cells, including cells with the cytological features of macrophages and lymphocytes. Reoviral protein signal was seen in endothelial cells, but this too was very rare (<1%) when compared with the number of reoviral-positive cancer cells. The viral load, as defined by the percentage of positive cancer cells, varied a lot among the reoviral-positive cancers, ranging from 5 to 59%. By using 20% as the threshold of low vs high viral load, it was noted that 6/13 (46%) reoviral-positive cases were high viral load and the other 54% were low viral load. Strong apoptosis was defined as multiple islands of at least 30 apoptotic cells, and was confirmed by the TUNEL assay. Using this criteria, a strong apoptotic response was seen in 1/7 of the low viral load and 4/6 of the high viral load cases.
It is documented that for many viruses, especially RNA viruses and retroviruses, viral protein detection by immunohistochemical may not accurately reflect the actual distribution of the viral genome in a given tissue.18,19 This is because the viral RNA may be transcriptionally silent or minimally active and, thus, many more cells may contain the viral genome than the viral protein. Thus, the next question we addressed was whether the reovirus immunohistochemistry data accurately reflected the distribution of the viral genome in the malignant tumors and the admixed benign cells. To this end, we analyzed a series of the tissues for reoviral RNA using the highly sensitive RT in situ PCR method. There were sufficient tissue samples to analyze the viral genome distribution in 10 of the cancer samples. Representative data are provided in Figure 3. Note that in the reovirus-positive cases the reoviral protein immunohistochemistry analyses underestimated the percentage of cancer cells that contained the reoviral RNA. When analyzed by immunohistochemistry, the mean % of cancer cells in the positive tissues was 31.0 with a s.d. of 17.8. When analyzed by RTin situ PCR, the mean%of cancer cells in the positive tissues was 88.9 with a s.d. of 4.9. Thus, overall, there was about a threefold increase in the cancer cells that contained the viral genome as compared with the viral protein. However, the RT in situ PCR, such as the immunohistochemistry, again showed that the reovirus was markedly targeting the cancer cells and not the surrounding benign cells. We then examined a series of mouse tissues that were exposed to a cell line (B16.OVA) that is relatively resistant to reovirus infection in vivo. As recently published,8 relatively few cancer cells were reovirus protein positive (commonly around 1–3%) and they showed a strong perivascular distribution. When we did RT in situ PCR for the reoviral genome on these samples, the pattern was likewise very different when compared with the clinical samples. Specifically, only 20–40% of the tumor cells in the mouse models showed the viral genome, as compared with the majority of tumor cells in the clinical samples. However, the same striking perivascular pattern was evident for the reoviral RNA-positive cells in the mouse model as was evident with the reovirus protein (data not shown). Also, in the mouse model the ratio of reoviral RNA positive/reoviral protein positive was similar to the human clinical samples at about 3:1.
Evidence of Productive Reoviral Infection in the Clinical Cancer Tissues
Next, we did reovirus protein/tubulin protein co-expression analyses in the reovirus exposed clinical tumor samples. These analyses indeed documented that the cancer cells that expressed reovirus protein in the clinical samples also routinely co-expressed tubulin (Figure 4). To obtain further evidence of productive reoviral infection, we combined RT in situ PCR analysis of reoviral RNA with immunohistochemistry for reoviral protein. This data confirmed the presence of reoviral protein in the reovirus RNA-positive cells (Figure 3).
Figure 4.
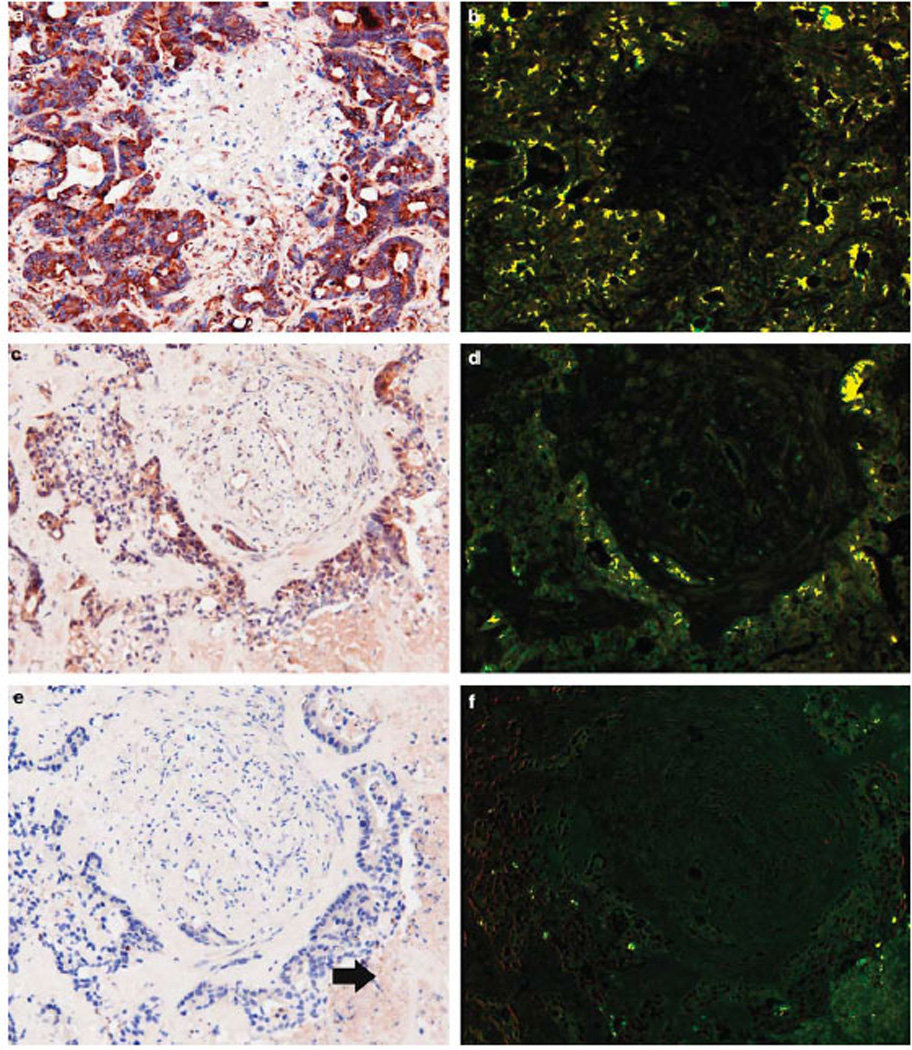
Molecular correlates of reoviral infection in serial tissue sections. Serial sections from a metastatic colon cancer from a patient treated with reovirus were analyzed for co-expression of reovirus and tubulin (a), reovirus and p38 (c), plus reovirus and caspase-3 (e); note the small area of apoptosis (arrow). The images were then analyzed with the Nuance system to assess the degree of co-expression; reoviral protein is fluorescent red and the cellular protein fluorescent green. There is extensive co-expression of reoviral and tubulin protein (b, seen as fluorescent yellow), indicative of productive infection. Similarly, reoviral and p38 proteins are often detected in the same cells (d); note that the co-expression is evident only in cancer cells and not in the surrounding stromal cells. Co-expression analyses between reovirus and caspase-3 showed rare cells positive for both proteins in the area of apoptosis (f).
Molecular Correlates of Cells Productively Infected by Reovirus
After having established that the colorectal and ovarian cancer tissues and the melanoma samples from patients exposed to reovirus showed a striking predilection of other viral genome for cancer cells, but a much lower rate of productively infected cancer cells, we next sought to determine the molecular correlates of the cells productively infected by the virus.
Several investigators have shown that reoviral infection in vitro is enhanced by the presence of a variety of proteins that make up the Ras-RalGEF-p38 pathway.1,14,15 Thus, we examined a series of the reoviral-positive cancer tissues for the expression of the pERK, PKR, Ras, and p38. When analyzed separately in serial sections it was evident that there was a strong similarity in the distribution of p38, pERK, or Ras when compared with histological distribution of the reoviral protein-positive cells. Co-expression analysis showed that, as with the reovirus-infected cell lines, there was strong co-expression between reovirus protein and tubulin as well as with pERK and p38 in the cancer tissue samples (Figure 4). Similarly, there was basically no co-expression in these same samples between reovirus protein and phosphorylated PKR protein that, thus, served as an internal negative control (data not shown).
To further address the association between apoptosis and productive reoviral infection that was noted in the cell samples, we analyzed the tissues for caspase-3 by immunohistochemistry and for apoptosis using the TUNEL assay. Caspase-3 expression was readily evident in the tissues where reovirus protein was noted by immunohistochemistry and showed a similar distribution pattern. Co-expression analyses of reoviral and caspase-3 protein showed that the caspase-3-positive cells in the reovirus-infected cancers invariably co-expressed reoviral protein (Figure 4). Reoviral protein also co-expressed with cells positive for apoptosis by the TUNEL assay (data not shown).
MicroRNA Analyses of the Reoviral-Infected Cell Lines
Given the marked association of reoviral and caspase-3 expression/apoptosis in the cell lines and clinical samples, we decided to see if the reovirus was modulating certain microRNAs that, in turn, could upregulate caspase-3 expression. Hence, we performed global microRNA analyses on the melanoma cancer cell line SK –Mel28 as they showed either 0% reovirus-positive cells or over 85% reovirus-positive cells. Representative data are presented in Table 2. Note that several microRNAs were markedly downregulated in the reovirus-positive samples. The microRNA most downregulated by reovirus was miR-let-7d. Target scan analyses confirmed that the 3′UTR of caspase-3 contains sequences that would interact with microRNA-let-7d. Further, RNA scan analyses demonstrated that part of the reovirus genome was strongly homologous with microRNA-let-7d, (Figure 5) which could inactivate it and, concomitantly, explain in part the associated upregulation of caspase-3.
Table 2.
MicroRNA dysregulation associated with reovirus infection
| MicroRNA | Mean control |
Mean reovirus infected |
Fold change control vs reovirus |
|---|---|---|---|
| hsa-miR-520g | 1 | 160.7085065 | −160.7085065 |
| hsa-miR-516a-3p | 1 | 107.3253392 | −107.3253392 |
| hsa-miR-218 | 1 | 97.48225048 | −97.48225048 |
| hsa-miR-548h | 1 | 94.49078931 | −94.49078931 |
| hsa-miR-200b | 1 | 86.57758264 | −86.57758264 |
| hsa-miR-487a | 1 | 84.05077481 | −84.05077481 |
| hsa-miR-1266 | 1 | 63.48320414 | −63.48320414 |
| hsa-miR-571 | 1 | 51.94547276 | −51.94547276 |
| hsa-miR-33b | 1 | 51.94547276 | −51.94547276 |
| hsa-miR-620 | 1 | 50.66350261 | −50.66350261 |
| hsa-miR-138 | 1 | 50.17635193 | −50.17635193 |
| hsa-miR-410 | 1 | 48.9975499 | −48.9975499 |
| hsa-miR-656 | 1 | 47.81874787 | −47.81874787 |
| hsa-miR-339-5p | 1 | 47.81874787 | −47.81874787 |
| hsa-miR-519e | 1 | 46.63994584 | −46.63994584 |
| hsa-miR-1308 | 51.56146402 | 1 | 51.56146402 |
| hsa-miR-296-5p | 53.86708094 | 1 | 53.86708094 |
| hsa-miR-187 | 55.51959193 | 1 | 55.51959193 |
| hsa-miR-30e | 57.69091082 | 1 | 57.69091082 |
| hsa-miR-582-5p | 65.02048101 | 1 | 65.02048101 |
| hsa-miR-9 | 74.1055701 | 1 | 74.1055701 |
| hsa-miR-374b | 78.79269762 | 1 | 78.79269762 |
| hsa-miR-23a | 83.23959837 | 1 | 83.23959837 |
| hsa-miR-34a | 91.46539779 | 1 | 91.46539779 |
| hsa-miR-93 | 93.29197913 | 1 | 93.29197913 |
| hsa-miR-363 | 106.2343414 | 1 | 106.2343414 |
| hsa-miR-15b | 116.5409176 | 1 | 116.5409176 |
| hsa-miR-186 | 117.6316242 | 1 | 117.6316242 |
| hsa-miR-374a | 199.5630123 | 1 | 199.5630123 |
| hsa-let-7d | 240.5104802 | 1 | 240.5104802 |
The cell line SK-Mel28 was exposed to reovirus in suspension. The chart depicts the 15 microRNAs that were most increased in response to reovirus infection (top) and the 15 microRNAs that were most decreased (bottom); note that microRNA-let-7d was the most downregulated of all the over 600 microRNAs that were tested.
Figure 5.
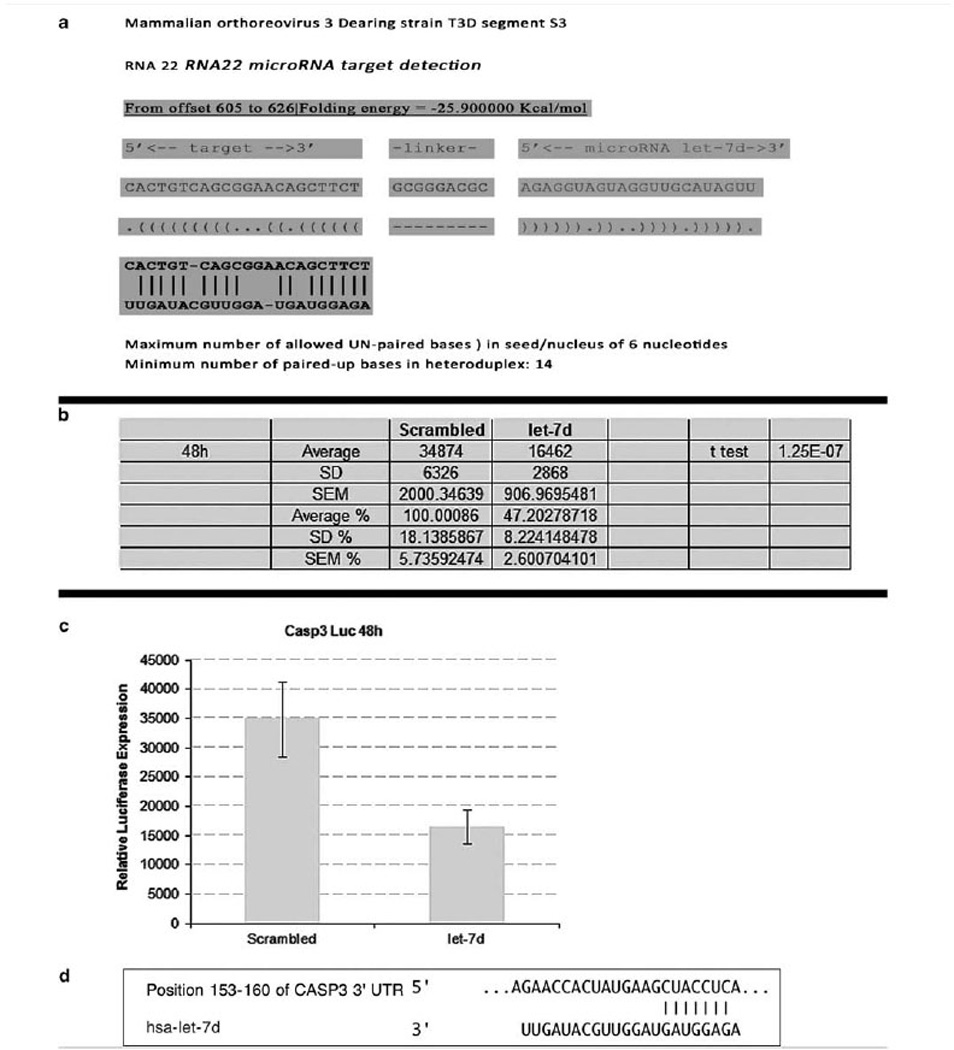
Sequence homology between microRNA-let-7d and reovirus and luciferase assay for caspase-3 regulation by the microRNA. (a) Shows a representative analysis between the reovirus strain used in this study (serotype-3, Dearing strain) and microRNA-let-7d as analyzed by RNA 22 microRNA target detection; note the strong sequence homology. (b, c) Show the raw data and graphical representation, respectively, for caspase-3 downregulation by microRNA-let-7d as determined by the luciferase assay at 48 h. (d) shows the seed sequence interactions with regards to miRNA-let-7d and caspase-3.
To further address the issue of whether miR-let-7d could indeed downregulate the activated form of caspase-3 expression, we performed the luciferase assay with the GoClone Reporter Constructs 3′UTR Capase 3 vector (SwitchGear Genomics Menlo Park, CA, USA) plus pre-miR control or pre-miR let7-d to final concentration of 20 nM. Luciferase activity was measured 48 h after transfection using the Light-Switch Luciferase Assay System. These data showed that microRNA-let-7d produced a 50% reduction in caspase-3 expression in this time period (Figure 5).
Next, to see if the in vitro data translated to the in vivo systems, we did co-expression analyses in the tissues between miR-let-7d and reovirus as well as caspase-3. These analyses did indeed confirm a strong inverse correlation between both microRNAlet-7d and reovirus in both the cell lines and tissue (data not shown). Similarly, the cancer cells positive for microRNA-let-7d in the cell lines and tissue samples did not express caspase-3, but did co-express PKR (data not shown). Thus, the in vivo data corroborated the in vitro data in showing that expression of miR-let-7d was associated with the loss of the active form of caspase-3 or productive reovirus infection, but was strongly correlated with the expression of the phosphorylated form of PKR. We did not perform any additional in situ co-expression analysis with reovirus and the most increased microRNA associated with viral infection (microRNA-520 g, Table 2) because nearly 100% of the reoviral-infected cells contained the microRNA (data not shown).
Discussion
The focus of this study was to dissect the molecular correlates of reovirus infection in cancerous tissues. Both the immunohistochemical and RT in situ PCR analyses of reovirus in the tumors showed a striking predilection of the virus for the malignant cells. A possible explanation relates to the strong co-expression observed in this study between reoviral protein and several proteins that would be far more common in cancer cells, such as p38 and Ras. As documented by Norman et al,15 an activated Ras-RaIGEF-p38 pathway is an important event for reoviral infection due to the concomitant inactivation of PKR via its dephosphorylation. Consistent with this hypothesis was the observation in this study that cancer cells with reoviral protein did not co-express with phosphorylated PKR. Interestingly, when RT in situ PCR was used, it was clear that overall threefold more cancer cells contained viral RNA when compared with the viral protein. The smaller percentage of tumor cells with reoviral protein as compared with the viral genome suggested that, although the viral genome avidly enters cancer cells, the process of viral activation, with the associated abundant viral protein production, is the rate limiting event with regard to the clinical efficacy of reovirus treatment. This suggests that efforts to increase the productive infectivity of reovirus in malignant tissues may increase the efficacy of the virus to destroy tumor cells. The data, including reoviral protein/tubulin co-expression, strongly support the hypothesis that reoviral protein detection by immunohistochemistry can be used as a measure of the effectiveness of reovirus to actively proliferate in cancer cells as it is a good indicator of productive infection.
This study did not address the molecular events that are required in the viral RNA-positive cells to induce productive infection in those cells. The co-expression analyses, however, clearly indicated that there was a very strong correlation between productive reoviral infection and the expression of p38, Ras, as well as caspase-3 in the cancer cells. This study did generate data that support a hypothesis to explain the key molecular events of reovirus that can lead to the cancer cell’s death. First, it is known that the reovirus virion is not infectious; rather, modifications to the capsid viral coat are required to activate the lambda 3 RNA-dependent RNA polymerase required for productive viral infection.21,22 Thus, one can speculate that this capsid modification may be protease digestion, such as can be achieved with caspase-3. When the viral RNA does get synthesized in abundance, the viral genome is capable of inactivating microRNA-let-7d. This, in turn, can activate caspase-3 activity that, in turn, may continue to transform the intact virion to a modified virion that is capable of generating many more viral particles. Of course, this would be associated with detectable viral protein production with tubulin co-expression. Further, microRNA-let-7d deactivation would also upregulate Ras expression23 that, in turn, would also facilitate productive viral infection and apoptosis-related death of the cancer cells, as demonstrated by other groups.1,14,15 This theory is also consistent with the findings of Torrisani et al24 who showed that reovirus-induced death of melanoma cells was blocked by caspase-3 inhibition. Interestingly, reduced microRNA let 7 expression in general is typically associated with worse prognoses in various cancers, as well as increased cancer stem cell production.23,25 However, in reovirus-treated cancers there is most likely a much more rapid microRNA-let-7d downregulation than in nonviral-treated cancer cells. This, in turn, will rapidly increase Ras activation, allowing the cell to be more permissive to productive viral infection, as well as more rapidly increase caspase-3 activation and, ultimately, destroy the cancer cell via apoptosis.
Multiple studies have emphasized that reoviral-induced killing of cancer cells is enhanced by pretreatment with various agents, especially chemotherapeutic agents.6,8–11 One potential mechanism may be the stabilization of themicrotubulin scaffold by certain chemotherapeutic agent, such as the taxanes.6 We indeed noted that pretreatment with paclitaxel and reovirus in a cancer cell line did indeed increase the percentage of tumor cells with productive viral infection compared with reovirus alone treated cells. Another theory as to the potentiating effect of chemotherapy on reovirus-induced apoptosis of cancer cells is that the former much reduces the T-cell-based cytokine response against reovirus that, in turn, may increase the effective concentration of the reovirus in the cancer tissue. Our work does not preclude additional factors, such as an increased NK anti-tumor response,23 or reduced reovirus inactivation by the immune system, as explanations for the ability of reovirus to slow down the growth of cancers.8
In sum, large amount of work is on-going using anti-microRNA therapy that has been shown effective, at times, in markedly reducing tumor size.26 This paper suggests that a key reason reovirus may be effective in destroying cancer cells is that the productive viral infection basically functions as an acute ‘anti-microRNA’ therapy. That is, the proliferating viral genome can acutely and markedly reduce microRNA-let-7d that, in turn, bypasses the normal ‘caspase-apoptosis cascade’ by markedly increasing the expression of caspase-3, that is the principle caspase responsible for the apoptotic death of these cancer cells.
Acknowledgements
This study is supported by a grant from the Lewis Foundation (GJN). Oncolytics has provided technical and financial support to GJN, AM, and KR. We thank Dr Margaret Nuovo for expert photomicroscopy.
Footnotes
Disclosure/conflict of interest
The authors declare no conflict of interest.
References
- 1.Strong JE, Coffey MC, Tang D, et al. The molecular basis of viral oncolysis: usurpation of the Ras signaling pathway by reovirus. EMBO J. 1998;17:3351–3362. doi: 10.1093/emboj/17.12.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Comins C, Heinemann L, Harrington K, et al. Reovirus: viral therapy for cancer ‘as nature intended’. Clin Oncol (R Coll Radiol) 2008;20:548–554. doi: 10.1016/j.clon.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 3.Gollamudi R, Ghalib MH, Desai KK, et al. Intravenous administration of Reolysin, a live replication competent RNA virus is safe in patients with advanced solid tumors. Invest New Drugs. 2010;28:641–649. doi: 10.1007/s10637-009-9279-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yap TA, Brunetto A, Pandha H, et al. Reovirus therapy in cancer: has the orphan virus found a home? Expert Opin Investig Drugs. 2008;17:1925–1935. doi: 10.1517/13543780802533401. [DOI] [PubMed] [Google Scholar]
- 5.Hirasawa K, Nishikawa SG, Norman KL, et al. Oncolytic reovirus against ovarian and colon cancer. Cancer Res. 2002;62:1696–1701. [PubMed] [Google Scholar]
- 6.Heinemann L, Simpson GR, Boxall A, et al. Synergistic effects of oncolytic reovirus and docetaxel chemotherapy in prostate cancer. BMC Cancer. 2011;11:221. doi: 10.1186/1471-2407-11-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alain T, Wong JF, Endersby R, et al. Reovirus decreases azoxymethane-induced aberrant crypt foci and colon cancer in a rodent model. Cancer Gene Ther. 2007;14:867–872. doi: 10.1038/sj.cgt.7701068. [DOI] [PubMed] [Google Scholar]
- 8.Sei S, Mussio JK, Yang QE, et al. Synergistic antitumor activity of oncolytic reovirus and chemotherapeutic agents in non-small cell lung cancer cells. Mol Cancer. 2009;8:47–61. doi: 10.1186/1476-4598-8-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kottke T, Thompson J, Diaz RM, et al. Improved systemic delivery of oncolytic reovirus to established tumors using preconditioning with cyclophosphamide-mediated Treg modulation and interleukin-2. Clin Cancer Res. 2009;15:561–569. doi: 10.1158/1078-0432.CCR-08-1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heinemann L, Simpson GR, Annels NE, et al. The effect of cell cycle synchronization on tumor sensitivity to reovirus oncolysis. Mol Ther. 2010;18:2085–2093. doi: 10.1038/mt.2010.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kottke T, Hall G, Pulido J, et al. Antiangiogenic cancer therapy combined with oncolytic virotherapy leads to regression of established tumors in mice. J Clin Invest. 2010;120:1551–1560. doi: 10.1172/JCI41431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pandha HS, Heinemann L, Simpson GR, et al. Synergistic effects of oncolytic reovirus and cisplatin chemotherapy in murine malignant melanoma. Clin Cancer Res. 2009;15:6158–6166. doi: 10.1158/1078-0432.CCR-09-0796. [DOI] [PubMed] [Google Scholar]
- 13.Kawaguchi K, Etoh T, Suzuki K, et al. Efficacy of oncolytic reovirus against human gastric cancer with peritoneal metastasis in experimental animal model. Int J Oncol. 2010;37:1433–1438. doi: 10.3892/ijo_00000795. [DOI] [PubMed] [Google Scholar]
- 14.Park EH, Park EH, Cho IR, et al. CUG2, a novel oncogene confers reoviral replication through Ras and p38 signaling pathway. Cancer Gene Ther. 2010;17:307–314. doi: 10.1038/cgt.2009.83. [DOI] [PubMed] [Google Scholar]
- 15.Norman KL, Hirasawa K, Yang AD, et al. Reovirus oncolysis: the Ras/RalGEF/p38 pathway dictates host cell permissiveness to reovirus infection. Proc Natl Acad Sci USA. 2004;101:11099–11104. doi: 10.1073/pnas.0404310101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harrington KJ, Karapanagiotou EM, Roulstone V, et al. Two-stage phase I dose-escalation study of intratumoral reovirus type 3 dearing and palliative radiotherapy in patients with advanced cancers. Clin Cancer Res. 2010;16:3067–3077. doi: 10.1158/1078-0432.CCR-10-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nuovo GJ. In situ detection of microRNAs in paraffin embedded, formalin fixed tissues and co-localization of their putative targets. Methods. 2010;52:307–315. doi: 10.1016/j.ymeth.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 18.Nuovo GJ. The utility of in situ-based methodologies including in situ polymerase chain reaction for the diagnosis and study of viral infections. Hum Pathol. 2007;38:1123–1136. doi: 10.1016/j.humpath.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 19.Bagasra O, Pomerantz RJ. In situ polymerase chain reaction and HIV-1. Clin Lab Med. 1994;14:351–365. [PubMed] [Google Scholar]
- 20.Iliopoulos D, Rotem A, Struhl K. Inhibition of miR-193a expression by Max and RXRa activates K-Ras and PLAU to mediate distinct aspects of cellular transformation. Cancer Res. 2011;71:5144–5153. doi: 10.1158/0008-5472.CAN-11-0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Farsetta DL, Chandran K, Nibert ML. Transcriptional activities of reovirus RNA polymerase in recoated cores. Initiation and elongation are regulated by separate mechanisms. J Biol Chem. 2000;275:39693–39701. doi: 10.1074/jbc.M004562200. [DOI] [PubMed] [Google Scholar]
- 22.Parker JS, Broering TJ, Kim J, et al. Reovirus core protein μ2 determines the filamentous morphology of viral inclusion bodies by interacting with and stabilizing microtubules. J Virol. 2002;76:4483–4496. doi: 10.1128/JVI.76.9.4483-4496.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prestwich RJ, Errington F, Steele LP, et al. Reciprocal human dendritic cell-natural killer cell interactions induce antitumor activity following tumor cell infection by oncolytic reovirus. J Immunol. 2009;183:4312–4321. doi: 10.4049/jimmunol.0901074. [DOI] [PubMed] [Google Scholar]
- 24.Torrisani J, Bournet B, du Rieu MC, et al. let-7 MicroRNA transfer in pancreatic cancer-derived cells inhibits in vitro cell proliferation but fails to alter tumor progression. Hum Gene Ther. 2009;20:831–844. doi: 10.1089/hum.2008.134. [DOI] [PubMed] [Google Scholar]
- 25.Yang X, Lin X, Zhong X, et al. Double-negative feedback loop between reprogramming factor LIN28 and microRNA let-7 regulates aldehyde dehydrogenase 1-positive cancer stem cells. Cancer Res. 2010;70:9463–9472. doi: 10.1158/0008-5472.CAN-10-2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garofalo M, Croce CM. microRNAs: master regulators as potential therapeutics in cancer. Annu Rev Pharmacol Toxicol. 2011;51:25–43. doi: 10.1146/annurev-pharmtox-010510-100517. [DOI] [PubMed] [Google Scholar]