

Abstract
The voltage-gated Kv1.3 K+ channel is a novel target for immunomodulation of autoreactive effector memory T (TEM) cells that play a major role in the pathogenesis of autoimmune diseases. We describe the characterization of the novel peptide ShK(L5) that contains l-phosphotyrosine linked via a nine-atom hydrophilic linker to the N terminus of the ShK peptide from the sea anemone Stichodactyla helianthus. ShK(L5) is a highly specific Kv1.3 blocker that exhibits 100-fold selectivity for Kv1.3 (Kd = 69 pM) over Kv1.1 and greater than 250-fold selectivity over all other channels tested. ShK(L5) suppresses the proliferation of human and rat TEM cells and inhibits interleukin-2 production at picomolar concentrations. Naive and central memory human T cells are initially 60-fold less sensitive than TEM cells to ShK(L5) and then become resistant to the peptide during activation by up-regulating the calcium-activated KCa3.1 channel. ShK(L5) does not exhibit in vitro cytotoxicity on mammalian cell lines and is negative in the Ames test. It is stable in plasma and when administered once daily by subcutaneous injection (10 μg/kg) attains “steady state” blood levels of ∼300 pM. This regimen does not cause cardiac toxicity assessed by continuous EKG monitoring and does not alter clinical chemistry and hematological parameters after 2-week therapy. ShK(L5) prevents and treats experimental autoimmune encephalomyelitis and suppresses delayed type hypersensitivity in rats. ShK(L5) might prove useful for therapy of autoimmune disorders.
Autoimmune diseases afflict millions worldwide and may have a common pathogenic mechanism. Pathogenesis may involve the “awakening” of dormant disease-specific autoreactive T cells—for instance myelin-specific T cells in patients with multiple sclerosis (MS)—by molecular mimicry or other undetermined mechanisms. Once awakened, autoreactive T cells might differentiate from a naive state into continuously activated memory T cells as a consequence of repeated autoantigen stimulation and contribute to inflammatory damage by migrating rapidly into tissues, secreting inflammatory cytokines, and exhibiting immediate effector function (Sallusto et al., 1999). Several lines of evidence support this scheme. First, a majority of myelin-specific T cells from patients with MS are costimulation-independent activated effector memory T (TEM) cells (Lovett-Racke et al., 1998; Scholz et al., 1998; Markovic-Plese et al., 2001; Wulff et al., 2003). Second, transfer of myelin-specific TEM cells into naive rat recipients induces experimental autoimmune encephalomyelitis (EAE), a model for MS (Beeton et al., 2001b). Third, T cells from patients with diabetes mellitus type 1 that are specific for the disease-associated autoantigen glutamic acid decarboxylase 65 are continuously activated memory cells (Viglietta et al., 2002). Last, a majority of T cells in the synovium of patients with rheumatoid arthritis and in skin lesions of patients with psoriasis are TEM cells, as are T cells that cause delayed type hypersensitivity (DTH) (Ezawa et al., 1997; Friedrich et al., 2000; Soler et al., 2003). Memory B cells, especially those belonging to the class-switched CD27+IgD- subset, also probably contribute to the pathogenesis of many autoimmune diseases (Iglesias et al., 2001; O’Connor et al., 2001; Corcione et al., 2004). Therapies that target TEM and class-switched memory B cells without impairing the activity of other lymphocyte subsets would therefore target the pathogenic cells in patients with autoimmune diseases but without compromising acute immune responses.
An exciting new therapeutic target for immunomodulation of TEM and class-switched memory B cells is the voltage-gated Kv1.3 K+ channel. TEM cells up-regulate Kv1.3 upon activation and their antigen-driven proliferation is exquisitely sensitive to Kv1.3 blockers (Wulff et al., 2003). Naive and TCM cells in contrast are significantly less sensitive to Kv1.3 blockers to begin with and rapidly become resistant to Kv1.3 blockade by up-regulating the calcium-activated K+ channel KCa3.1 (Wulff et al., 2003; Chandy et al., 2004). B cells, like T cells, change their potassium channel dependence from KCa3.1 to Kv1.3 as they differentiate from naive into class-switched CD27+IgD- memory B cells (Wulff et al., 2004). Kv1.3 blockers inhibit the proliferation of these cells without affecting naive and CD27+IgD+ memory B cells. By targeting TEM cells and class-switched memory B cells with Kv1.3 blockers, it might be possible to ameliorate autoimmune diseases without compromising the bulk of the immune response. The functionally restricted tissue distribution of Kv1.3 and the fact that, in vivo, Kv1.3 blockade ameliorates EAE, bone resorption in periodontal disease, and DTH in animal models without causing obvious side effects has enhanced the attractiveness of Kv1.3 as a therapeutic target (Koo et al., 1997; Beeton et al., 2001b; Valverde et al., 2004). Although Kv1.3 blockers would suppress all activated TEM (for example, TEM cells specific for vaccine antigens) and class-switched memory B cells, a Kv1.3-based therapy would be a significant improvement over current therapies that broadly and indiscriminately suppress the entire immune system. An additional advantage of Kv1.3 blockers is that they are reversible. Thus, one could titrate the therapeutic effect of Kv1.3 blockers when needed and stop therapy in the face of infection, unlike chemotherapeutic agents or targeted monoclonal antibody therapies, which take months to subside.
Despite extensive efforts, selective and potent inhibitors of the Kv1.3 channel have not been developed (Chandy et al., 2004). The most potent Kv1.3 inhibitor is the peptide ShK from the Caribbean sea anemone Stichodactyla helianthus (Pennington et al., 1995). ShK blocks Kv1.3 (Kd, ∼10 pM), suppresses proliferation of TEM cells at picomolar concentrations (Wulff et al., 2003), and ameliorates EAE (Beeton et al., 2001b). A potential drawback of ShK is its affinity (Kd, 28 pM) for the neuronal Kv1.1 channel (Kalman et al., 1998). Although no side effects were observed with ShK in EAE trials (Beeton et al., 2001b), ingress of high concentrations of ShK into the brain could lead to unwanted neurotoxicity. Other inhibitors including correolide, trans-PAC, CP-339818, UK-78282, Psora-4, margatoxin, and luteolin are less selective for Kv1.3 (Chandy et al., 2004). The development of a more selective ShK derivative is therefore necessary.
We have developed ShK(L5), a synthetic analog of ShK that blocked Kv1.3 with picomolar affinity and exhibited greater than 100-fold selectivity for Kv1.3 over Kv1.1 and other channels. Selectivity was achieved by attaching a negatively charged l-phosphotyrosine (l-pTyr) via a hydrophilic linker to ShK-Arg1. ShK(L5) suppressed TEM cell proliferation in vitro at picomolar concentrations without compromising the function of naive and TCM cells. In proof-of-concept in vivo studies, ShK(L5) ameliorated EAE caused by the transfer of myelin-specific TEM cells into naive rat recipients and suppressed the DTH response also caused by TEM cells. ShK(L5) may have use as a therapy for multiple sclerosis and other T and B cell-mediated autoimmune diseases.
Materials and Methods
Synthesis of ShK Analogs
For the Fmoc-Pmp analogs, the ethyl protecting groups were removed by treating the Fmoc-Pmp-(Ethyl)2-OH with aqueous 6 N HCl at reflux. After 16 h, a white solid precipitated out that was then isolated by filtration and washed with water until the washings were neutral (Hammerschmidt and Hanbauer, 2000). Partial removal of the ethyl protecting groups from Pmp-phosphonate resulted in either Pmp, Pmp-Et, or Pmp(Ethyl)2. All Fmoc-amino acid derivatives were obtained from Bachem AG (Bubendorf, Switzerland), except for Fmoc-d-TyrPO3(benzyl)-OH, which was obtained from Nova Biochem (San Diego, CA), and Fmoc-Pmp(Ethyl)-OH and Fmoc-d-Pmp(Ethyl)2-OH, which were obtained from Chem Impex (Wood Dale, IL). Solid-phase assembly was initiated with Fmoc-Cys(Trt)-2-chlorotrityl resin to minimize potential racemization of the C-terminal Cys residue (Fujiwara et al., 1994). Automated assembly was carried out on an ABI-431A peptide synthesizer (Applied Biosystems, Foster City, CA). Fmoc-Aeea-OH was coupled to the N terminus after the assembly of ShK. The resin was divided into nine aliquots. Either Fmoc-Tyr(PO3Bzl)-OH, Fmoc-d-Tyr(PO3Bzl)-OH, Fmoc-Tyr(PO3Me2)-OH, Fmoc-Pmp-OH, Fmoc-d-Pmp-OH, Fmoc-Pmp(Ethyl)-OH, Fmoc-Pmp(Et)2-OH, Fmoc-Tyr(tert-butyl)-OH, or Fmoc-p-amino-l-phenylalanine(tert-butyloxycarbonyl)-OH was coupled, using diisopropylcarbodiimide and 1-hydroxybenzotriazole, to one of the resin aliquots. The deblocked peptide resin was cleaved and deprotected with reagent K (King et al., 1990) containing 5% triisopropylsilane for 2 h at RT. Met(O) was reduced by addition of solid NH4I to the cleavage cocktail at t - 15 min. (Nicolas et al., 1995). For the peptides containing Tyr(PO3Me2)-OH, a cleavage cocktail containing 1 M trimethylsilyl bromide in TFA (trifluoroacetic acid) containing thioanisole as a scavenger for 18 h at 4°C was used (Tian et al., 1993). Incomplete removal of the methyl protecting groups is common when using this method, and two of the species [p-phosphotyrosine) and Tyr(PO3HMe)] are easily purified by RP-HPLC. The Tyr(PO4Me2) containing analog was cleaved via standard Reagent K cleavage keeping both methyl groups intact. In each case, the cleavage mixture was filtered and the crude peptide was precipitated into ice-cold diethyl ether. The precipitate was collected, yielding approximately 75 mg of peptide from 200 mg of resin. The crude product was dissolved in 20 ml of 50% aqueous AcOH and diluted into 0.75 l of H2O. The pH of the solution was adjusted with NH4OH to 8.2, and it was allowed to fold overnight with the addition of glutathione (2 mM:1 mM) (reduced:oxidized). All analogs were purified using RP-HPLC using a linear gradient of water versus acetonitrile buffered with trifluoroacetic acid as described previously (Pennington et al., 1995, 1996a,b). Pure fractions were pooled and lyophilized, resulting in a trifluoroacetate salt of each peptide. Purity of the peptides was greater than 95%. Each sample was confirmed by reversed-phase high-performance liquid chromatography, amino acid analysis, and matrix-assisted laser desorption ionization/time of flight mass spectrometry and adjusted to account for peptide content before bioassay. ShK and margatoxin were obtained from Bachem Biosciences (King of Prussia, PA). Luteolin was purchased from Sigma-Aldrich (St Louis, MO).
Ion Channels
We used the IUPHAR nomenclature for the ion channels described in this article (Gutman et al., 2003). Cells stably expressing mKv1.1, rKv1.2, mKv1.3, hKv1.5, and mKv3.1 have been described previously (Grissmer et al., 1994). Cell lines stably expressing other mammalian ion channels were gifts from several sources: mKv1.7 in CHL cells and hKCa2.3 in COS-7 cells from Aurora Biosciences Corp. (San Diego, CA); hKv1.4 in LTK cells from Michael Tamkun (University of Colorado, Boulder, CO); hKv2.1 in HEK293 cells from Jim Trimmer (University of California, Davis, CA); Kv11.1 (HERG) in HEK293 cells from Craig January (University of Wisconsin, Madison, WI); HEK293 cells expressing hKCa1.1 or rKCa2.1 or hKCa3.1 from Khaled Houamed (University of Chicago, Chicago, IL); hNav1.4 in HEK-293 cells from Frank Lehmann-Horn (University of Ulm, Germany), and Cav1.2 in HEK-293 cells from Franz Hofmann (Munich, Germany). RBL-2H3 (expressing Kir2.1) and N1E-115 neuroblastoma cells (expressing Nav1.2) were obtained from the American Type Culture Collection (Manassas, VA). hKv1.6 and rKv3.2 (both in pcDNA3) were obtained from Protinac GmbH (Hamburg, Germany) and transiently-transfected into COS-7 cells with Fugene-6 (Roche, Mannheim, Germany) according to the manufacturers’ protocol.
Lymphoid Cells and Cell Lines
Histopaque-1077 gradients (Sigma-Aldrich) were used to isolate splenocytes from Lewis rats and human peripheral blood mononuclear cells (PBMCs) from the blood of healthy volunteers. Human myelin oligodendrocyte glycoprotein- or tetanus toxoid-specific TEM cells were generated as described previously (Wulff et al., 2003). The encephalitogenic CD4+ Lewis rat T cell line PAS (Beraud et al., 1993) was a gift from Evelyne Béraud (University of Marseille, Marseille, France), and RPMI 8226 plasmacytoma cells were a gift from Shastri Gollapudi (University of California, Irvine, CA). Jurkat and Burkitt cells were purchased from the American Type Culture Collection.
Electrophysiological Analysis
Experiments were conducted in the whole-cell configuration of the patch-clamp technique. KV currents were elicited by 200-ms depolarizing pulses from a holding potential of -80 to 40 mV as described previously (Zhou et al., 1998; Wulff et al., 2000; Bardien-Kruger et al., 2002; Kolski-Andreaco et al., 2004; Vennekamp et al., 2004). Each channel blocker was tested at multiple concentrations. The measured reduction in peak current at 40 mV for each concentration was used to generate a dose-response curve, and the Kd and Hill coefficient were determined with Origin software (OriginLab Corp., Northampton, MA) as described previously (Zhou et al., 1998; Wulff et al., 2000; Bardien-Kruger et al., 2002; Kolski-Andreaco et al., 2004; Vennekamp et al., 2004). The Kd was also determined from the on (TON) and off (TOFF) rates for channel block. After stabilization of peak Kv1.3 current amplitude, 70 pM ShK(L5) was perfused onto the cell, and peak current values plotted as a function of time were fitted to a single exponential function to determine TON. After reaching equilibrium block, perfusion was switched back to blocker-free bath solution. Peak currents were plotted as described above to determine TOFF. KON, KOFF, and Kd were calculated assuming a simple bimolecular reaction between ShK(L5) and Kv1.3: KON = 1 - TON × KOFF / [TON × ShK(L5) concentration]; KOFF = 1 / TOFF; Kd = KOFF / KON (Peter et al., 2001). For Kv11.1 channels, current block was measured both at 20 and -50 mV (tail current). For KCa channels, Kir2.1, and swelling-activated chloride currents, we measured the change in slope conductance by the ShK analogs and, for Na+ and Ca2+ currents, the reduction of minimum current.
Staining for Flow Cytometry and Fluorescence Microscopy
The T cell phenotypes of human PBMCs, rat splenocytes, and PAS T cells were determined by flow cytometry. PBMCs were triplestained with anti-CD3 antibody conjugated to Cy-chrome (BD Pharmingen, San Diego, CA), anti-CD45RA antibody conjugated to phycoerythrin (BD Pharmingen), and anti-CCR7 antibody conjugated to fluorescein isothiocyanate (R&D Systems, Minneapolis, MN). Rat splenocytes and PAS-stained T cells were double-stained with anti-CD3 antibody conjugated to Cy-chrome and anti-CD45RC antibody conjugated to fluorescein isothiocyanate (BD Pharmingen). The stained cells were analyzed with a FACScan (BD Biosciences, San Jose, CA).
Two approaches were used to evaluate Kv1.3 protein expression in PAS T cells. First, PAS cells were stained with ShK-F6CA (10 nM; Bachem Bioscience Inc.), a fluorophore-tagged ShK analog, and analyzed by flow cytometry as described previously (Beeton et al., 2003). For competition experiments, PAS cells were preincubated with excess unlabeled ShK(L5) (100 nM) before addition of 10 nM ShK-F6CA. Second, PAS cells were permeabilized and stained with anti-Kv1.3 antibody (Koch et al., 1997) (gift from Hans-Gunther Knaus, Innsbruck, Austria) followed by a secondary antibody conjugated to Alexa-488 (Molecular Probes, Eugene, OR). Stained cells were visualized with a Zeiss LSM-510 META confocal microscope (Carl Zeiss GmbH, Jena, Germany), fluorescence intensities were measured for individual cells (n = 10-15), and statistical analysis carried out using the Mann-Whitney U test.
Functional Studies
Proliferation of human and rat T cells was determined with [3H]thymidine incorporation assays as described previously (Beeton et al., 2001a,b; Wulff et al., 2003). For measurements of IL2 production, PAS T cells were activated with MBP in the presence or the absence of ShK or ShK(L5) for 8 h, and the culture supernatants were collected as described previously (Beeton et al., 2001a). IL2 was detected in supernatants using the rat IL2 Quantikine kit (R&D Systems) according to manufacturer’s instructions. Effect of exogenous IL2 (20 units/ml; Sigma-Aldrich, St Louis, MO) on proliferation of PAS T cells was determined as described previously (Beeton et al., 2001a).
Circulating Half-Life Determination and Plasma Stability
Known amounts of ShK(L5) were added to Lewis rat serum, and the blocking activity on Kv1.3 channels was tested by patch-clamp to establish a standard dose-response curve. Serum samples from Lewis rats obtained at various times after single subcutaneous or intravenous injections of ShK(L5) were tested for Kv1.3 blocking activity by patch-clamp and the levels of ShK(L5) determined from the standard curve as described previously (Beeton et al., 2001b). In other experiments, Lewis rats received single daily injections of 10 μg/kg ShK(L5) and serum levels of ShK(L5) were determined 24 h after each injection on days 1 to 5. To determine plasma stability of ShK(L5), rat plasma spiked with a known amount of ShK(L5) was incubated at 37°C for varying durations and then tested for Kv1.3 blocking activity; the amount of residual ShK(L5) in these samples was determined from the standard curve.
Cytotoxicity Assays and Ames Test
Human PBMCs, PAS, Jurkat, RPMI 8226, and Burkitt cells were grown for 48 h in the presence or the absence of 100 nM ShK(L5). Cells were then stained with the LIVE/DEAD viability/cytotoxicity kit (Molecular Probes) according to the manufacturer’s instructions, and the percentage of live and dead cells was determined using a fluorescence microplate reader (CytoFluor; Applied Biosystems, Foster City, CA). Triton X-100 (0.1%) was used as a positive control for cell death. For the Ames test, the mutagenic activity of ShK(L5) was determined on the Salmonella typhimurium tester strain TA97a by Nelson Laboratories (Salt Lake City, UT).
EKG Studies to Evaluate Cardiac Toxicity
Electrocardiographic studies with implanted EKG transmitters (Data Sciences International, Arden Hills, MN) were used for heart rate variability analysis in animals that received ShK(L5) or vehicle. Experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committee of UC Davis. Six Lewis rats (9-11 weeks old; weight = 219 ± 9 g) were anesthetized with a mixture of ketamine (80 mg/kg) and xylazine (7.5 mg/kg) administered by intramuscular injection. EKG transmitters were placed in the peritoneal cavity of each rat and two EKG leads were tunneled subcutaneously to the right shoulder and to the xiphoid space caudal to the ribcage. An analgesic, carprofen (5 mg/kg, subcutaneous), was administered at the end of surgery. Two weeks after surgery, we performed baseline EKG recording on the rats for 2 h (day 1). We then injected the vehicle (PBS + 2% rat serum) subcutaneously and continued recording for another 8 h. The animals were then returned to their rooms. On day 2, we performed baseline EKG recording for 2 h after which ShK(L5) (10 μg/kg dissolved in vehicle) was injected subcutaneously and EKG recording continued for another 8 h. Data recorded from 1.5 to 3.5 h after the injections were used for standard heart rate variability parameters-analysis in both time and frequency domains using Nevokard software (Bio-Impedance Technology, Inc., Chapel Hill, NC).
Subchronic Toxicity Studies
Lewis rats (9-11 weeks old; weight, 199 ± 7 g) received subcutaneous injections of ShK(L5) 10 μg/kg/day (n = 6) or saline (n = 6) for 2 weeks. Rats were weighed daily. The Comparative Biology Laboratory at University of California, Davis performed chemical (COBAS MIRA Plus; Roche Diagnostic Systems, Branchburg, NJ) and hematological (HEMAVET® 850 Multispecies Hematology Analyzer; CDC Technologies, Oxford, CT) analysis on blood samples drawn at the end of 2 weeks. Single-cell suspensions prepared from thymuses and spleens removed from six animals given ShK(L5) and six given saline were stained with antibodies specific for various T and B cell markers (BD Pharmingen) and analyzed by flow cytometry.
Prevention and Treatment of Acute Adoptive EAE and Prevention of DTH in Lewis Rats
Female inbred Lewis rats 9 to 11 weeks old were purchased from Harlan-Sprague-Dawley (Indianapolis, IN) and housed under barrier conditions with irradiated rodent chow and acidified water ad libitum. All experiments were in accordance with National Institutes of Health guidelines and approved by the Institutional Animal Care and Use Committee at the University of California, Irvine. ShK(L5) was dissolved in PBS + 2% Lewis rat serum (saline) for subcutaneous injection. Acute adoptive EAE was induced as described previously (Beeton et al., 2001a,b) with 6 to 8 × 106 myelin basic protein (MBP)-activated PAS cells. MBP was extracted from frozen guinea pig spinal cords (Harlan Bioproducts, Indianapolis, IN) as described previously (Deibler et al., 1972). Rats were weighed daily and observed twice daily for clinical signs of EAE. For prevention trials, rats received 10 μg/kg/day ShK(L5) from days 0 to 5, whereas control rats received saline. For treatment trials, administration of ShK(L5) (10 μg/kg/day) or saline was begun after the onset of disease (rats had a limp tail, were hunched, and had lost ≥6% of their weight over 24 h) and continued for 3 days.
For DTH trials, Lewis rats were immunized with an emulsion of ovalbumin in complete Freund’s adjuvant (Difco, Detroit, MI). Seven days later, they received an injection of ovalbumin dissolved in saline in the pinna of one ear and saline in the other ear. Rats then received subcutaneous injections of ShK(L5) (10 μg/kg/day) or vehicle (PBS + 2% Lewis rat serum). Ear swelling was measured 24 and 48 h later using a spring-loaded micrometer (Mitutoyo, Spokane, WA).
Results
ShK(L5), a Novel ShK Analog that Exhibits 100-fold Selectivity for Kv1.3 Over Kv1.1
ShK blocks the neuronal Kv1.1 channel and the Kv1.3 channel with roughly equivalent potency. Neurotoxicity is therefore a concern under circumstances that compromise the blood-brain barrier and allow the entry of sufficient amounts of ShK to block Kv1.1 channels. Our strategy to design a Kv1.3-specific inhibitor was guided by our finding that ShK-F6CA containing fluorescein-6-carboxylate (F6CA), attached through a 20-Å Aeea linker to the N terminus of ShK exhibited 80-fold selectivity for Kv1.3 over Kv1.1 (Beeton et al., 2003). Because F6CA can exist as a restricted carboxylate or also as a cyclized lactone, it was not clear whether the Kv1.3 specificity of ShK-F6CA was a result of the negative charge of F6CA, the hydrophobicity created by this large bulky fluorescein nucleus, potential planar π-π electronic stacking, or a combination of all of these potential contributions. To distinguish between these possibilities and with the intention of developing a nonfluorescent Kv1.3-selective inhibitor, we generated a series of 12 novel N-terminally-substituted ShK analogs to probe some of these interactions. By attaching tyrosine, phenylalanine, or their derivatives (varying in charge, size, and hydrophobicity) through an Aeea linker to the N terminus of ShK, we could probe the effects of charge and hydrophobicity to gain insight into our selectivity enhancement seen with F6CA substitution.
In the example shown in Fig. 1A, l-phosphotyrosine (l-pTyr) a negatively charged (net charge -2) post-translationally modified aromatic amino acid, was attached via the AEEA linker to ShK-Arg1 to generate a novel analog called ShK(L5). ShK and ShK(L5) were tested on Kv1.3 and Kv1.1 channels stably expressed in L929 cells. Figure 1B shows the effects of ShK and ShK(L5) on Kv1.3 and Kv1.1 currents elicited by 200-ms depolarizing pulses from a holding potential of -80 to 40 mV. Both peptides reversibly blocked Kv1.3 and Kv1.1 in a dose-dependent manner with Hill coefficients of 1 (Fig. 1, B-D). Kd values were determined from the dose-response curves shown in Fig. 1C using Origin software. ShK blocked Kv1.3 (Kd = 10 ± 1 pM) and Kv1.1 (Kd = 28 ± 6 pM) with roughly equivalent potency, as expected (Fig. 1C). In contrast, ShK(L5) was 100-fold selective for Kv1.3 (Kd = 69 ± 5 pM) over Kv1.1 (Kd = 7.4 ± 0.8 nM) (Fig. 1, B and C). The time course of Kv1.3 current block by ShK(L5) and its washout is shown in Fig. 1D. The time constant (TON) of ShK(L5) wash-in was 131 ± 21 s (n = 7), whereas the time constant (TOFF) for peptide wash-out was 150 ± 28 s (n = 4). The Kd (57 ± 7 pM) calculated from the KON (15 × 106± 0.5 × 106 M-1s-1) and KOFF (0.0059 ± 0.0013 s-1) values is consistent with the Kd (69 ± 5 pM) determined with the use of Origin software.
Fig. 1.
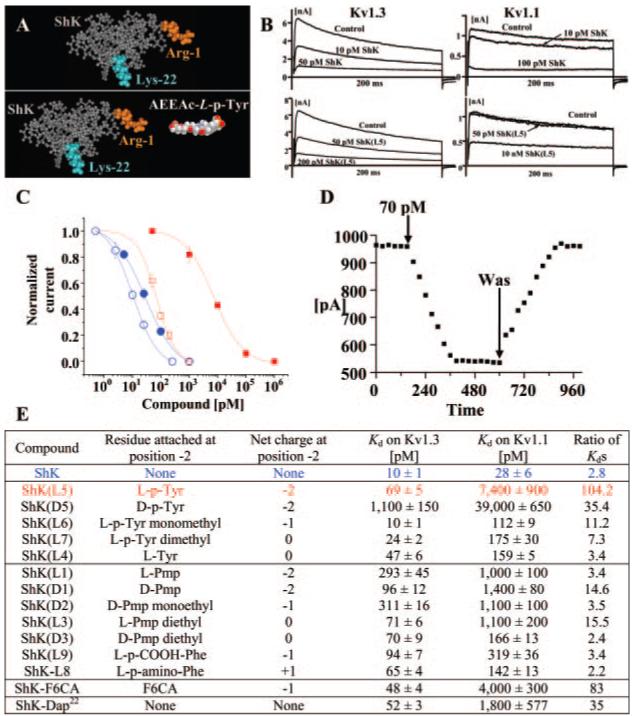
Generation of a selective Kv1.3 blocker. A, molecular model of ShK based on the published NMR structure. The Lys22, critical for channel blockade, is highlighted in orange. l-pTyr was attached to the α-amino group of Arg1 of ShK (highlighted in cyan) through an Aeea linker (right). The structures of the linker and l-pTyr were modeled with AM1 in Hyperchem. B, effect of ShK (top) and ShK(L5) (bottom) on Kv1.3 and Kv1.1 currents in stably transfected cells. C, dose-dependent inhibition of Kv1.3 (open symbols) and Kv1.1 (closed symbols) by ShK (blue) and ShK(L5) (red). Kd values on Kv1.3 = 10 ± 1 pM (ShK) and 69 ± 5 pM (ShK(L5)); Kd values on Kv1.1 = 28 ± 6 pM (ShK) and 7.4 ± 0.8 nM (ShK(L5)). D, time course of wash-in and wash-out of ShK(L5) on Kv1.3. Cells were held at a holding potential of -80 mV and depolarized for 200 ms to 40 mV every 30 s. E, Kd values shown for inhibition of Kv1.3 and Kv1.1 by ShK analogs. Kd values for ShK-F6CA and ShK-Dap22 are from published sources (Kalman et al., 1998; Beeton et al., 2003; Chandy et al., 2004).
Other ShK analogs were tested on Kv1.3 and Kv1.1 channels (Fig. 1E). ShK(D5) containing d-phosphotyrosine was 35-fold selective for Kv1.3 over Kv1.1 but was an order of magnitude less potent than ShK(L5). ShK(L6) containing l-pTyr-monomethyl showed modest (11-fold) Kv1.3 specificity, whereas ShK analogs containing l-pTyr-dimethyl or l-Tyr were not selective for Kv1.3 over Kv1.1 (Fig. 1E). Analogs that contained phenylalanine or its derivatives (varying in bulk, π electron density, and charge) were modestly specific or not specific for Kv1.3 over Kv1.1 (Fig. 1E). The 100-fold specificity of ShK(L5) for Kv1.3 over Kv1.1 is greater than that of ShK-F6CA (80-fold), ShK(D5) (35-fold), ShK-Dap22 (33-fold), or any other ShK analog tested (Fig. 1C).
ShK(L5) Is a Highly Specific Kv1.3 Inhibitor
We assessed the specificity of ShK(L5) on a panel of 20 ion channels (Table 1). ShK(L5) blocked the Kv1.3 channel in T cells with a Kd (76 pM) equivalent to its Kd on the cloned channel (69 pM). It was 100-fold selective for Kv1.3 over Kv1.1, 260-fold selective over Kv1.6, 280-fold selective over Kv3.2, 680-fold selective over Kv1.2, and >1000-fold selective over all other channels tested. It is noteworthy that it was 1600-fold Kv1.3-selective over KCa3.1, the calcium-activated K+ channel that regulates activation of human naive and TCM cells (Wulff et al., 2003). Native ShK was less selective than ShK(L5). ShK was 2.8-fold selective for Kv1.3 (Kd = 10 ± 1 pM) over Kv1.1 (Kd 28 ± 6 pM), 20-fold selective over Kv1.6 (200 ± 20 pM), 500-fold selective over Kv3.2 (Kd = 5000 ± 1000 pM), and >1000-fold selective-over Kv1.2 (10 ± 1 nM) and KCa3.1 (Kd = 28 ± 3 nM). Margatoxin, a peptide from scorpion venom that has been touted as a specific Kv1.3 inhibitor (Lin et al., 1993; Koo et al., 1997; Middleton et al., 2003) was also not specific. It was 5-fold selective for Kv1.3 (110 ± 12 pM) over Kv1.2 (Kd = 520 ± 1 pM), 9-fold selective over Kv1.1 (10 ± 1 nM), and >1000-fold selective over Kv1.6 and Kv3.2 (Kd > 100 nM). Luteolin, a nutriceutical sold for autoimmune diseases (http://www.lutimax.com) on the basis of its being a Kv1.3 inhibitor (Lahey and Rajadhyaksha, 2004), blocked Kv1.3 weakly (Kd = 65 ± 5 μM) and exhibited no selectivity over Kv1.1 (Kd = 77 ± 5 μM), Kv1.2 (Kd = 63 ± 4 μM), or Kv1.5 (Kd = 41 ± 3 μM). The exquisite specificity of ShK(L5) for Kv1.3, together with its picomolar affinity for the channel, makes it a potentially attractive immunosuppressant.
TABLE 1.
Selectivity of ShK(L5)
| Channels | Kd of ShK(L5) |
|---|---|
| pM | |
| Kv1.1 | 7000 ± 1000 |
| Kv1.2 | 48,000 ± 7000 |
| Kv1.3 (cloned) | 69 ± 5 |
| Kv1.3 (native) | 76 ± 8 |
| Kv1.4 | 137,000 ± 3000 |
| Kv1.5 | 100,000 (N.E.) |
| Kv1.6 | 18,000 ± 3000 |
| Kv1.7 | 100,000 (N.E.) |
| Kv2.1 | 100,000 (N.E.) |
| Kv3.1 | 100,000 (N.E.) |
| Kv3.2 | 20,000 ± 2000 |
| Kir2.1 | 100,000 (N.E.) |
| Kv11.1 (HERG) | 100,000 (N.E.) |
| KCa1.1 | 100,000 (N.E.) |
| KCa2.1 | 100,000 (N.E.) |
| KCa2.3 | 100,000 (N.E.) |
| KCa3.1 | 115,000 ± 5000 |
| Nav1.2 | 100,000 (N.E.) |
| Nav1.4 | 100,000 (N.E.) |
| Swelling-activated T cell Cl- channel | 100,000 (N.E.) |
| Cav1.2 | 100,000 (N.E.) |
N.E., no effect.
ShK(L5) Preferentially and Persistently Suppresses Human TEM Cell Proliferation
To assess the in vitro immunosuppressive activity of ShK(L5), we compared its ability to suppress anti-CD3 antibody-stimulated proliferation of human TEM cell lines versus human PBMCs that contain a mixture of naive and TCM cells. Flow cytometry confirmed the cell surface phenotypes of the two populations studied. The TEM lines were >90% CCR7-CD45RA- (Fig. 2A), whereas PBMCs contained 65% CCR7+CD45RA+ (naive) and 18% CCR7+CD45RA- (TCM) cells (Fig. 2B). Figure 2C shows that ShK(L5) and ShK were 60-fold more effective in suppressing the proliferation of TEM cells (IC50 = ∼80 pM) compared with PBMCs (IC50 = 5 nM, p < 0.05). The lower sensitivity of PBMCs might be explained by a rapid up-regulation of KCa3.1 channels in naive and TCM cells upon stimulation as has been reported previously (Ghanshani et al., 2000; Wulff et al., 2003). In keeping with this interpretation, PBMCs activated for 48 h to up-regulate KCa3.1 expression, then rested for 12 h and re-activated with anti-CD3 antibody, were completely resistant to ShK(L5) block (Fig. 2D, top arrow). PBMCs that had been suppressed by ShK(L5) during the first round of stimulation exhibited identical resistance to ShK(L5) when the cells were washed, rested, and re-challenged with anti-CD3 antibody. These results corroborate an earlier report showing that naive and TCM cells escape Kv1.3 inhibitors by up-regulating KCa3.1 channels (Wulff et al., 2003). Thus, ShK(L5) preferentially and persistently suppresses the proliferation of TEM cells.
Fig. 2.
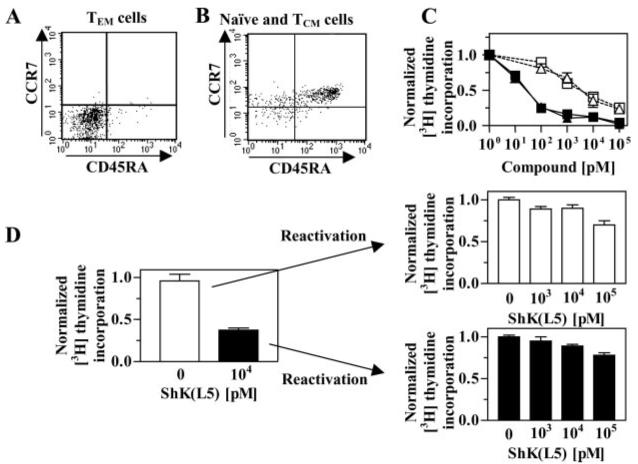
ShK(L5) preferentially suppresses the proliferation of human TEM cells. Human PBMCs (A) and a human TEM line (B) were stained with antibodies against CD3, CD45RA, and CCR7. Staining intensities of CD45RA and CCR7 were determined by flow cytometry in the CD3+-gated population. C, dose-dependent inhibition by ShK (blue) and ShK(L5) (red) of [3H] thymidine incorporation by PBMCs (open symbols, a mixture of naive/TCM cells) and TEM cells (closed symbols) stimulated for 48 h with anti-CD3 antibody. D, preactivated human PBMCs (naive/TCM cells) that up-regulate KCa3.1 expression (Ghanshani et al., 2000) become resistant to ShK(L5) inhibition when reactivated with anti-CD3 antibody. These cells have previously been shown to become sensitive to the KCa3.1-specific inhibitor TRAM-34 (Ghanshani et al., 2000).
ShK(L5) Inhibits Proliferation of and IL2 Production by Rat TEM Cells; Exogenous IL2 Partially Overrides Suppression
As a preamble to evaluating therapeutic effectiveness of ShK(L5), we examined its ability to suppress proliferation of a memory T cell line, PAS, that causes an MS-like disease in rats (Beraud et al., 1993). As a control, we used rat splenic T cells. To confirm the differentiation status of the two cell populations, we assessed the expression of CD45RC, a marker of naive T cells (Bunce and Bell, 1997). Rat splenic T cells were 76% CD45RC+ (i.e., mainly naive cells), whereas PAS cells were CD45RC-, suggesting that they are memory cells (Fig. 3A). To determine whether PAS cells are in the TEM or TCM state, we examined Kv1.3 expression before and 48 h after activation. TEM but not TCM cells are expected to significantly up-regulate Kv1.3 levels upon stimulation (Beeton et al., 2001b, 2003). Patch-clamp experiments revealed a striking increase in Kv1.3 current amplitude after MBP-stimulation of PAS cells consistent with their being TEM cells (Fig. 3B). As an independent measure of the number of Kv1.3 channels on PAS cells, we used ShK-F6CA, a fluorescently labeled ShK analog that binds specifically to Kv1.3 (Beeton et al., 2003). The intensity of ShK-F6CA staining determined by flow cytometry reflects the number of Kv1.3 tetramers expressed on the cell surface (Beeton et al., 2003). ShK-F6CA (10 nM) staining intensity increased with MBP-activation of PAS cells, and an excess of unlabeled ShK(L5) (100 nM) competitively inhibited ShK-F6CA staining (Fig. 3C). As a final test, we performed confocal microscopy on quiescent and MBP-stimulated PAS cells that had been fixed and stained with a Kv1.3-specific antibody. In keeping with data in Fig. 3, B and C, resting PAS T cells had a Kv1.3 staining intensity of 4.4 ± 0.6, and this value increased to 10.6 ± 2.3 (p < 0.005) after antigen-induced activation (Fig. 3D), showing augmentation in Kv1.3 protein expression after activation. Thus, MBP-activated PAS cells are CD45RC- Kv1.3high TEM cells, whereas rat splenic T cells used in our experiments are predominantly in the naive state.
Fig. 3.
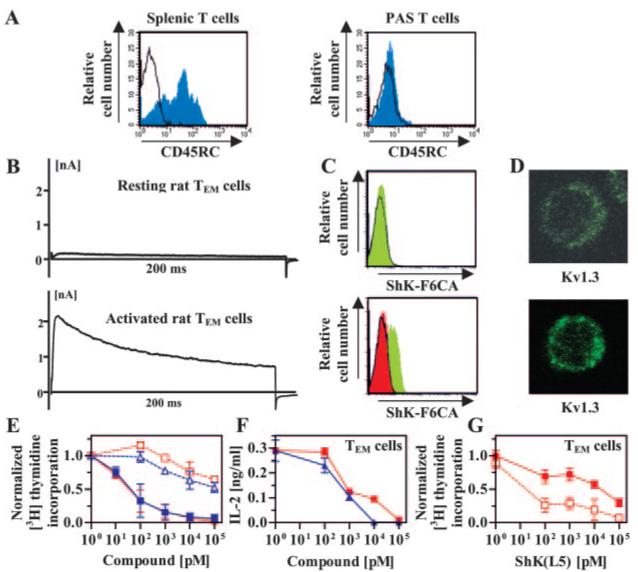
ShK(L5) preferentially suppresses the proliferation of rat TEM cells. A, CD45RC staining of rat splenic T cells (left) and PAS T cells (right) detected by flow cytometry. B, Kv1.3 currents exhibited by quiescent (top) and myelin antigen-activated (bottom) PAS T cells. C, flow cytometry profiles of ShK-F6CA-staining in quiescent (top) and myelin antigen-activated (bottom) PAS T cells. Unstained cells (black lines) and cells stained with ShK-F6CA (green filled). Competition of ShK-F6CA staining by unlabeled ShK(L5) is red-filled. D, confocal images of Kv1.3 immunostaining in quiescent (top) and myelin antigen-activated (bottom) PAS T cells. Statistical analysis was carried out using the Mann-Whitney U test. E, dose-dependent inhibition by ShK (blue) and ShK(L5) (red) of [3H]thymidine incorporation by rat naive/TCM (open symbols) and TEM (closed symbols) cells activated with Con A (1 μg/ml). F, dose-dependent inhibition by ShK (blue) and ShK(L5) (red) of IL2 secretion by PAS T cells 7 h after stimulation with MBP. G, ShK(L5)-induced inhibition of myelin-antigen triggered [3H]thymidine incorporation by PAS T cells (open symbols) is reversed by the addition of 20 units/ml IL2 (closed symbols).
MBP-triggered proliferation of PAS cells was suppressed ∼1000-fold more effectively by ShK(L5) and ShK (IC50 = ∼80 pM) than mitogen-induced proliferation of rat splenic T cells (Fig. 3E, IC50 ≈100 nM; p < 0.05). These results corroborate the findings with human T cells (Fig. 2). ShK(L5) inhibited MBP-induced IL2 production by PAS cells (Fig. 3F), and exogenous IL2 partially overrode ShK(L5) suppression of PAS cell proliferation (Fig. 3G). Earlier studies reported similar findings with less specific Kv1.3 inhibitors on human, rat and mini-pig T cells (Chandy et al., 1984; Koo et al., 1997; Beeton et al., 2001a). In summary, ShK(L5) is a powerful and selective inhibitor of human and rat TEM cells and may therefore have therapeutic use in autoimmune diseases by preferentially targeting TEM cells that contribute to the pathogenesis of these disorders (Chandy et al., 2004).
ShK(L5) Plasma Values after Subcutaneous Administration
Before embarking on in vivo studies in a rat EAE model, we used a patch-clamp bioassay to ascertain whether circulating levels of ShK(L5) after subcutaneous injection were sufficient to inhibit TEM cells. Serum samples from ShK(L5)-treated and control rats were tested for blocking activity on Kv1.3 channels. Control serum did not exhibit detectable blocking activity, indicating an absence of endogenous channel blockers. To standardize the assay, known amounts of ShK(L5) were added to rat serum, and these samples were tested on Kv1.3 channels. The spiked serum samples blocked Kv1.3 currents in a dose-dependent fashion (Kd, 77 ± 9 pM) that was indistinguishable from the effect of ShK(L5) effect in the absence of serum (Fig. 4A). Levels of ShK(L5) in treated animals were determined by comparison with the standard curve. ShK(L5) was detectable in serum 5 min after a single subcutaneous injection of 200 μg/kg (Fig. 4B). Peak levels (12 nM) were reached in 30 min and the level then fell to a baseline of approximately 300 pM over 420 min (Fig. 4B). The disappearance of ShK(L5) from the blood could be fit by a single exponential (Fig. 4C). The circulating half-life was estimated to be ∼50 min.
Fig. 4.
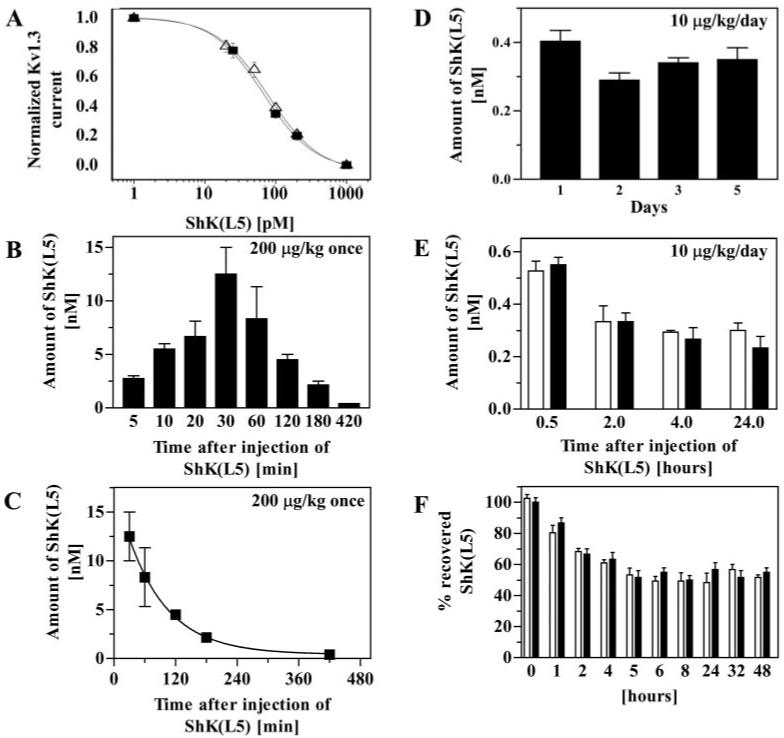
Circulating half-life and stability of ShK(L5). A, known amounts of ShK(L5) were added to rat serum (Δ) or to PBS (■) and blocking activity was determined on Kv1.3 channels stably expressed in L929 cells. B, a single dose of 200 μg/kg of ShK(L5) was injected subcutaneously into four rats. Blood was drawn at the indicated times and serum was tested by patchclamp to determine the amount of ShK(L5). C, data fit to a single exponential decay. Half-life ≈ 50 min. D, five Lewis rats received single daily subcutaneous injections of 10 μg/kg/day ShK(L5) for 5 days. Blood was drawn each morning (24 h after the previous injection) and tested for blocking activity on Kv1.3 channels by patch-clamp. E, rats received a single dose of 10 μg/kg ShK(L5) either subcutaneously (open bars; n = 4) or intravenously (closed bars; n = 4). Blood was drawn at the indicated times. Serum was tested by patch-clamp to determine the amount of ShK(L5) in blood. F, a half-blocking dose of ShK(L5) was added to rat plasma (□) or to PBS containing 2% rat plasma (■) and incubated at 37°C for varying duration. Aliquots were taken at the indicated times and blocking activity determined on Kv1.3 channels.
Because the peak serum level after 200 μg/kg (12 nM) significantly exceeds the requirement for selective blockade of Kv1.3 channels and TEM cell function, we tested lower doses. After a single injection of 10 μg/kg, the peak serum concentration of ShK(L5) reached ≈500 pM within 30 min (data not shown), a concentration sufficient to block >90% Kv1.3 but not affect Kv1.1. Repeated daily administration of this dose (10 μg/kg/day) resulted in steady-state levels of ∼300 pM (measured 24 h after injection; Fig. 4D), which is sufficient to cause 60 to 70% suppression of TEM cells with little effect on naive/TCM cells. The “steady-state” level is unexpected given the estimated circulating half-life of ∼50 min and indicates that ShK(L5) “accumulates” on repeated administration. To determine whether the “depot” was in the skin or elsewhere in the body, we measured blood levels of ShK(L5) 10 h after rats received single intravenous or subcutaneous injections of 10 μg/kg ShK(L5). The peptide disappeared with the same time course after administration by either route (Fig. 4E), indicating that the skin is not responsible for the steady-state level of 300 pM ShK(L5) reached after a single 10 μg/kg daily injection (Fig. 4D), and the depot(s) resides elsewhere.
Our success in achieving a steady-state level of 300 pM ShK(L5) after daily single injections of 10 μg/kg/day suggests that the peptide may be stable in vivo. To examine its stability, we incubated ShK(L5) in rat plasma or in PBS containing 2% rat plasma at 37°C for varying durations and then measured Kv1.3 blocking activity. In both sets of spiked samples (plasma and PBS) we observed a 50% reduction in Kv1.3-blocking activity in approximately 5 h, presumably due to peptide binding to the plastic surface of the tube, and the level then remained steady for the next 2-days (Fig. 4F). As an added test of stability, we compared the Kv1.3- versus Kv1.1-blocking activities of sera from ShK(L5)-treated rats. If ShK(L5) is modified in vivo, either by dephosphorylation of pTyr or cleavage of the Aeea-pTyr side chain, it would yield ShK(L4) and ShK, respectively, neither of which is selective for Kv1.3 over Kv1.1 (Fig. 1E). Serum samples from ShK(L5)-treated animals exhibited the same selectivity for Kv1.3 over Kv1.1 as ShK(L5), indicating that the peptide does not undergo the modifications stated above. Taken together, these results indicate that ShK(L5) is remarkably stable in plasma and attains pharmacologically relevant serum concentrations after single daily subcutaneous injections of 10 μg/kg.
Toxicity Studies
We conducted several in vitro and in vivo tests to determine whether ShK(L5) exhibits any toxicity (Table 2). Human and rat lymphoid cells incubated for 48 h with a concentration (100 nM) of ShK(L5) >1200 times greater than the Kv1.3 half-blocking dose or the IC50 for TEM suppression (70-80 pM) exhibited minimal cytotoxicity. The same high concentration of ShK(L5) was negative in the Ames test on tester strain TA97A, suggesting that it is not a mutagen. Both in vitro tests failed to detect any significant toxicity.
TABLE 2.
Toxicity study of ShK(L5)
| In vitro tests | 100 nM ShK(L5) |
|---|---|
| Cytotoxicity (% dead cells) | |
| Human PBMCs | 7.5 ± 4.3 |
| PAS T cells | 8.1 ± 0.8 |
| Jurkat cells | 5.5 ± 3.3 |
| Burkitt lymphoma | 3.1 ± 0.9 |
| RPMI 8226 myeloma | 6.5 ± 2.1 |
| Ames test | Negative |
| Acute in vivo tests | Saline | ShK(L5) 10 μg/kg |
|---|---|---|
| Electrocardiograma | ||
| Heart rate | 302 ± 13 | 311 ± 20 |
| SDNN | 13.3 ± 3.0 | 17.8 ± 4.4 |
| CV% | 6.7 ± 1.4 | 9.2 ± 2.2 |
| SDANN5 min | 5.0 ± 2.0 | 6.9 ± 2.3 |
| rMSSD | 6.8 ± 2.2 | 9.8 ± 3.5 |
| HF (n.u.) | 71 ± 21 | 79 ± 37 |
| HF (%) | 50 ± 8 | 53 ± 10 |
| LF (n.u.) | 68 ± 4 | 64 ± 10 |
| LF (%) | 50 ± 8 | 47 ± 10 |
| LF/HF | 1.1 ± 0.4 | 1.3 ± 0.7 |
| Subchronic in vivo tests | Saline | ShK(L5) 10 μg/kg/day for 2 weeks |
|---|---|---|
| Weight gain (%) | 7.2 ± 1.8 | 6.2 ± 1.7 |
| Complete blood count | ||
| Hematocrit (%) | 40.3 ± 1.4 | 39.0 ± 4.9 |
| Hemoglobin (g/dl) | 15.3 ± 0.5 | 15.0 ± 1.5 |
| MCV (fl) | 48.5 ± 0.2 | 48.3 ± 0.3 |
| MCH (pg) | 18.5 ± 0.8 | 18.5 ± 0.6 |
| MCHC (g/dl) | 38.0 ± 1.8 | 38.4 ± 1.3 |
| Total white cells (×103/mm3) | 7.1 ± 2.1 | 7.1 ± 2.5 |
| Total red cells (×106/mm3) | 8.3 ± 0.3 | 8.1 ± 1.0 |
| Total platelets (×103/mm3) | 656 ± 214 | 606 ± 106 |
| Blood chemistry | ||
| Alkaline phosphatase (U/L) | 170 ± 26 | 150 ± 18 |
| Glucose (mg/dl) | 139 ± 21 | 150 ± 18 |
| Blood urea nitrogen (mg/dl) | 17.1 ± 2.6 | 15.0 ± 1.7 |
| Creatinine (mg/dl) | 0.6 ± 0 | 0.6 ± 0.1 |
| Albumin (g/dl) | 5.0 ± 0.3 | 4.5 ± 0.4 |
| Thymic cell populations (%) | ||
| CD4-CD8- | 3.6 ± 1.1 | 4.3 ± 0.7 |
| CD4+CD8+ | 77.8 ± 6.1 | 76.8 ± 4.1 |
| CD4+CD8- | 8.5 ± 1.7 | 11.2 ± 2.0 |
| CD4-CD8+ | 10.0 ± 3.3 | 7.6 ± 1.3 |
| CD3+ | 89.5 ± 1.6 | 93.2 ± 3.5 |
| Splenic populations (%) | ||
| CD3+ | 72.4 ± 4.4 | 65.4 ± 0.1 |
| CD3+CD45RC+ | 35.6 ± 2.6 | 39.8 ± 1.1 |
| CD3+CD45RC- | 23.6 ± 2.3 | 26.5 ± 1.3 |
| CD3+CD4+ | 62.7 ± 0.1 | 66.6 ± 1.2 |
| CD3+CD8+ | 26.9 ± 0.1 | 25.0 ± 0.2 |
| IgM+ | 38.8 ± 1.5 | 33.3 ± 0.3 |
Data are expressed as mean ± S.D.
Tested with t tests, P < 0.05 on all parameters.
SDNN, standard deviation of all normal-to-normal RR intervals; CV%, 100 × SDNN/average RR interval; SDANN5 min, standard deviation of the mean of normal RR intervals for each 5-min period; rMSSD, root-mean-square of successive difference; HF (n.u.), high frequency (0.75-2.5 Hz) power in normalized unit; LF (n.u.), low frequency (0.2-0.75 Hz) power in normalized unit; MCV, mean corpuscular volume; MCH, mean corpuscular hemoglobin; MCHC, mean corpuscular hemoglobin concentration.
Drug-induced blockade of Kv11.1 (HERG) channels has contributed to major cardiac toxicity and the withdrawal of several medications from the market. ShK(L5) has no effect on Kv11.1 channels at 100 nM (>1430-fold the Kd for Kv1.3), and our chosen therapeutic regimen (10 μg/kg/day, 300 pM steady-state circulating level) should therefore not cause cardiotoxicity. As a further test, we performed heart rate variability analysis in conscious rats administered vehicle (PBS + 2% rat serum) on day 1, followed by 10 μg/kg/day ShK(L5) on day 2. ShK(L5) had no effect on heart rate or the standard HRV (heart rate variability) parameters in either the time or the frequency domain (Task Force of the European Society of Cardiology and the North American Society of Pacing Electrophysiology, 1996).
Encouraged by the acute toxicity experiments, we performed a subchronic toxicity study in which rats were administered daily subcutaneous injections of 10 μg/kg ShK(L5) or vehicle for 2 weeks (n = 6 in each group). ShK(L5)-treated animals gained weight to the same degree as rats receiving vehicle (Table 2). Hematological and blood chemistry analysis showed no difference between ShK(L5)- and vehicle-treated rats, and flow cytometric analysis revealed no differences in the proportions of thymocyte or lymphocyte subsets (Table 2). Together, these studies suggest that ShK(L5) is safe.
To determine the therapeutic safety index, we administered a 60-fold higher dose (600 μg/kg/day) of ShK(L5) to healthy rats for 5 days and observed no clinical signs of toxicity, and no toxicity was seen when healthy rats received a single injection of 1000 μg/kg ShK(L5). The situation is less sanguine when the blood-brain barrier is compromised, as happens in EAE and MS. Rats with EAE that received ShK(L5) 10 μg/kg/day for 10 days showed no signs of toxicity. In contrast, 40% of rats (5 of 12) administered 600 μg/kg/day for 5 days died on the fifth day when they developed clinical signs of EAE (extrapolated LD50 = 750 μg/kg/day). Because the peak concentration of ShK(L5) in the serum (12 nM) after administration of a single injection of 200 μg/kg is sufficient to block >50% of Kv1.1 channels, toxicity observed in EAE rats administered 600 μg/kg/day ShK(L5) is probably caused by the ingress into the brain of sufficient amounts of ShK(L5) to block Kv1.1. Thus, the effective therapeutic safety index of ShK(L5) is well in excess of 100 in situations in which the blood-brain barrier is not compromised (as seen in autoimmune diseases that do NOT affect the central nervous system), whereas the therapeutic safety index is 75 when the blood-brain barrier is breached.
ShK(L5) Prevents and Treats Acute Adoptive EAE and Prevents DTH in Lewis Rats
ShK(L5) was evaluated for immunosuppressive activity in vivo in two animal models. We tested its ability to prevent and treat acute EAE induced by the transfer of MBP-activated PAS TEM cells into Lewis rats (Beeton et al., 2001a,b; Beraud et al., 1993), as well as to suppress the DTH reaction mediated by TEM cells (Soler et al., 2003). PAS cells were activated with MBP for 48 h in vitro and then adoptively transferred (6-8 × 106 viable cells) into Lewis rats. For the prevention trial, rats then received subcutaneous injections of saline (control rats) or ShK(L5) (10 μg/kg/day) for 5 days. In the first prevention trial, control rats developed mild EAE (mean maximum clinical score 2.0 ± 1.2) with an average onset of 5.6 ± 0.6 days (not shown). ShK(L5) reduced disease severity (mean maximum clinical score, 0.7 ± 0.6, p < 0.05). In the second prevention trial, control rats developed more severe EAE (mean maximum clinical score 3.2 ± 0.4) with a mean onset of 4.8 ± 0.4 days (Fig. 5A). ShK(L5) significantly reduced disease severity (mean maximum clinical score 0.6 ± 0.4, p < 0.007) but did not significantly delay disease onset (5.5 ± 0.7 days; p = 0.07). No signs of toxicity were noted in these studies.
Fig. 5.
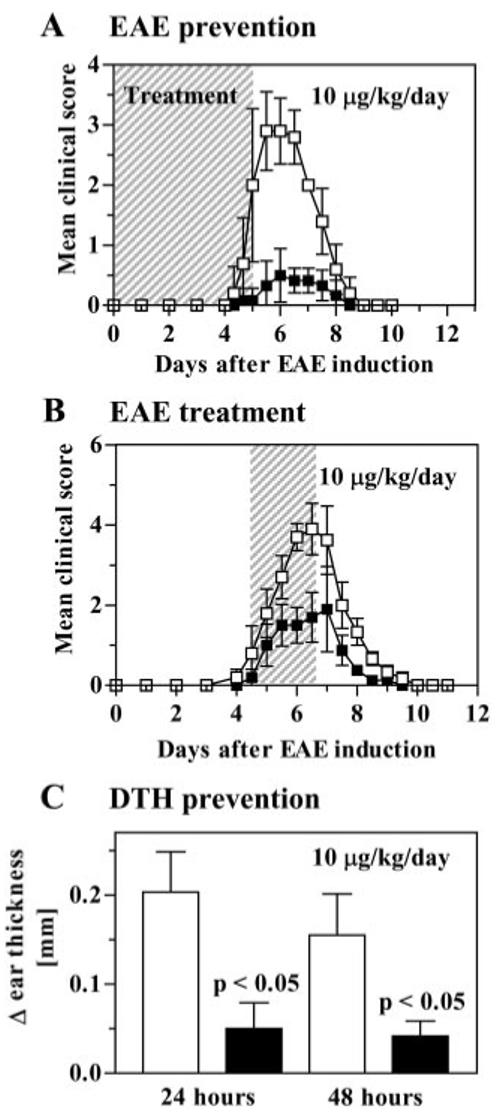
ShK-L5 prevents DTH and acute adoptive EAE in Lewis rats. A, prevention of EAE. PAS T cells were activated in vitro, washed, and injected intraperitoneally on day 0. Clinical scoring of EAE: 0 = no clinical signs, 0.5 = distal limps tail, 1 = limp tail, 2 = mild paraparesis or ataxia, 3 = moderate paraparesis, 4 = complete hind limb paralysis, 5 = 4 + incontinence, 6 = death. Rats (n = 6/group) were injected subcutaneously with vehicle (□; n = 6) or ShK(L5) (■; n = 6; 10 μg/kg/day) from day 0 to day 5. B, treatment of EAE. PAS T cells were activated in vitro, washed, and injected intraperitoneally on day 0. Treatment with ShK(L5) at 10 μg/kg/day was started when rats developed clinical signs of EAE and was continued for 3 days. C, DTH reaction was elicited against ovalbumin and rats (n = 6/group) were treated with ShK(L5) 10 μg/kg/day for 2 days, after which ear swelling was measured. Statistical analysis was carried out using the Mann-Whitney U test.
In the treatment trial (Fig. 5B) rats were injected with MBP-activated PAS cells, administered saline or 10 μg/kg/day ShK(L5) when they initially developed signs of EAE (limp tail, hunched posture, and loss of 6% or more of their weight over 24 h), and therapy was continued for 3 days. Clinical signs of EAE peaked on day 6 in the control group (score = 3.9 ± 0.7) and on day 7 in the treated group (score = 1.9 ± 0.9; p < 0.05).
As an independent assessment of the immunosuppressive activity of ShK(L5) in vivo, we also examined its effectiveness in inhibiting the DTH reaction that is mediated predominantly by skin-homing TEM cells (Soler et al., 2003). Lewis rats immunized with ovalbumin and adjuvant were challenged 7 days later with ovalbumin in one ear and saline in the other ear. Rats then received injections of saline (control rats) or ShK(L5) (10 μg/kg/day), and ear thickness was measured as an indication of DTH. All control rats developed ear swelling 24 and 48 h after ovalbumin challenge, whereas the DTH reaction was substantially milder in ShK(L5)-treated animals (Fig. 5C). Thus, ShK(L5) inhibits the TEM-mediated DTH response and prevents and ameliorates severe adoptive EAE induced by myelinactivated TEM cells.
Discussion
We have developed a highly specific Kv1.3 inhibitor by attaching the negatively charged amino acid l-pTyr to the N terminus of ShK via a 20-Å hydrophilic linker. ShK(L5) blocks Kv1.3 with a Kd of 69 pM and exhibits selectivity for Kv1.3 of 100-fold over Kv1.1, 260-fold over Kv1.6, 280-fold over Kv3.2, 680-fold over Kv1.2, and >1000-fold over all other channels tested. Other known blockers of Kv1.3 are significantly less selective than ShK(L5). Margatoxin, a peptide from Centruroides margaritatus scorpion venom, suppresses DTH in mini-pigs (Koo et al., 1997) but exhibits only 5-fold selectivity for Kv1.3 (Kd, 110 pM) over Kv1.2 (Kd, 520 pM) and 9-fold selectivity over Kv1.1 (Kd, 10 nM). Kaliotoxin from the scorpion Androctonus mauritanicus suppresses DTH in rats and ameliorates EAE (Beeton et al., 2001a) and inflammatory bone resorption in experimental periodontal disease (Valverde et al., 2004), but it is less potent (Kv1.3 Kd, 650 pM) and less selective (Grissmer et al., 1994) than ShK(L5). The first small-molecule Kv1.3 blockers with nanomolar affinity that were discovered—iminodihydroquinolines WIN-17317 and CP-339818 and the benzhydryl piperidine UK-78282—also block sodium channels (Wanner et al., 1999) and the neuronal Kv1.4 channel (Hanson et al., 1999). The small-molecule Kv1.3 inhibitors developed by Merck—correolide (Felix et al., 1999; Hanner et al., 1999; Koo et al., 1999; Bao et al., 2005), cyclohexyl-subsituted benzamides (Schmalhofer et al., 2002) and candelalides A-C (Singh et al., 2001)—are poorly selective for Kv1.3. Psora-4, the most potent small-molecule Kv1.3 blocker (Kd, 3 nM) is only 16- to 20-fold selective for Kv1.3 over Kv1.1 and Kv1.2 and 2.5-fold selective over the cardiac Kv1.5 channel (Vennekamp et al., 2004). Luteolin, a flavonoid that ameliorates EAE in rats (Hendriks et al., 2004), is sold as a nutriceutical (http://www.lutimax.com; http://www.synorx.com), ostensibly because of its ability to block Kv1.3 channels (Lahey and Rajadhyaksha, 2004). However, luteolin is a weak Kv1.3 inhibitor (Kd, 65 μM) and it is not selective for Kv1.3 over Kv1.1, Kv1.2, or Kv1.5. Other known Kv1.3 small-molecule blockers—sulfamidebenz-amidoindanes (Castle et al., 2000), dichlorophenylpyrazolopyrimidines (Atwal et al., 2001), furoquinoline Ibu-8 (Butenschon et al., 2001), tetraphenylporphyrins (Gradl et al., 2003), 3-alkyl- and 3-aryl-7H-furo[3,2-g]chromen-7-ones (Wernekenschnieder et al., 2004), and khellinone and chalcone derivatives (Baell et al., 2004), charybdotoxin, noxiustoxin, (Grissmer et al., 1994), agitoxin-2 (Garcia et al., 1994), BgK (Cotton et al., 1997), Pandinius imperator toxin1 (Peter et al., 2001), HsTx1—are neither as potent nor as selective as ShK(L5). Because of their lack of Kv1.3 selectivity, these blockers could potentially be neurotoxic if they entered the central nervous system at concentrations sufficient to block neuronal channels. The only Kv1.3 inhibitor with potency and Kv1.3-specificity comparable with ShK(L5) is the recently described synthetic analog (OSK1-Lys16Asp20) of the OSK1 toxin from scorpion Orthochirus scrobiculosus (Mouhat et al., 2005), but its activity as an immunomodulator remains undetermined.
The exquisite Kv1.3-specificity of ShK(L5) makes it an attractive drug prospect. ShK(L5) is remarkably stable in plasma and it reached steady-state blood levels of ∼300 pM after repeated single daily subcutaneous injections of 10 μg/kg. This blood concentration of ShK(L5) is sufficient to block >90% of Kv1.3 channels without affecting other ion channels and to cause 60 to 70% suppression of TEM cells while sparing naive/TCM cells. A concentration of ShK(L5) greater than 1200 times its pharmacological dose was not cytotoxic or mutagenic in vitro. ShK(L5) did not block the cardiac Kv11.1 (HERG) K+ channel responsible for drug-induced long QT syndrome (Recanatini et al., 2005), and in vivo administration of ShK(L5) at pharmacological concentrations (10 μg/kg/day) did not alter cardiac function in healthy rats based on continuous EKG monitoring. Repeated in vivo administration of 10 μg/kg/day ShK(L5) for 2 weeks did not change clinical chemistry or hematological profiles. Healthy animals administered a single 1000 μg/kg/day ShK(L5) injection (100 times the pharmacological dose) or repeated injections of 600 μg/kg/day for 5 days (60 times the pharmacological dose) did not exhibit any overt signs of toxicity. These results indicate that pharmacologically relevant blood levels can be attained after single daily subcutaneous injections of ShK(L5), and the effective therapeutic safety index in healthy rats exceeds 100.
ShK(L5) may have use as a therapeutic in autoimmune diseases by preferentially targeting continuously activated autoreactive TEM cells that have been implicated in MS, type-1 diabetes mellitus, rheumatoid arthritis, and psoriasis (Ezawa et al., 1997; Lovett-Racke et al., 1998; Scholz et al., 1998; Friedrich et al., 2000; Markovic-Plese et al., 2001; Viglietta et al., 2002; Soler et al., 2003; Wulff et al., 2003). ShK(L5) suppressed the proliferation of human and rat TEM cells (IC50 = ∼80 pM in both cases) and inhibited IL2 production at picomolar concentrations (Figs. 2 and 3). Exogenous IL2 partially overrode this block. Human naive/TCM cells were initially 60-fold less sensitive to ShK(L5) than human TEM cells and became completely resistant to the blocker during activation (Fig. 2), presumably by up-regulating KCa3.1 (Ghanshani et al., 2000; Wulff et al., 2003). Rat naive/TCM cells were 1000-fold less sensitive to ShK(L5) than TEM cells. This species variation in ShK(L5)-sensitivity of naive/TCM cells compared with TEM cells—60-fold lower in humans and 1000-fold lower in rats—can be explained by differences in K+ channel expression between human and rat naive/TCM cells. Quiescent human naive/TCM cells express more Kv1.3 channels per cell (250-400) than KCa3.1 channels (10-20) and are therefore more potently inhibited by Kv1.3 blockers than KCa3.1 blockers (Ghanshani et al., 2000; Wulff et al., 2003). Quiescent rat naive/TCM cells, in contrast, express more KCa3.1 channels per cell (10-20) than Kv1.3 channels (1-10) and are more sensitive to KCa3.1 than Kv1.3 blockers (Beeton et al., 2001b). The rat/human difference in channel expression may underlie the differential sensitivity of rat naive/TCM cells (IC50 = 100 nM) and human naive/TCM cells (IC50 = 5 nM) to ShK(L5). In summary, ShK(L5) preferentially suppresses TEM cells, whereas naive/TCM cells are less sensitive to the blocker to begin with and then rapidly escape suppression by up-regulating KCa3.1 channels (Ghanshani et al., 2000; Beeton et al., 2001b; Wulff et al., 2003).
Human class-switched memory B cells (e.g., CD27+IgG+IgD-) are implicated in the pathogenicity of autoimmune diseases (Iglesias et al., 2001; O’Connor et al., 2001; Corcione et al., 2004). Kv1.3 blockers preferentially suppress the proliferation of late memory B cells whereas naive and early memory B cells (CD27+IgD+) are significantly less sensitive (Wulff et al., 2004). ShK(L5) could therefore shut down the function of TEM and class-switched memory B cells that contribute to the development of autoimmune disorders. Of concern is the important role of class-switched memory B cells in humoral immunity (production of IgG antibodies) and the diminished capacity to mount viable immune responses to bacterial challenges that might ensue as a result of channel-based suppression of these cells. It is fortunate that human class-switched memory B cells are less sensitive to block by ShK (IC50 = 1-4 nM) than human TEM cells (IC50 = 80-400 pM), because they express higher numbers of Kv1.3 channels at rest (∼2000/cell) than TEM cells (250-400/cell) (Wulff et al., 2004). It might therefore be possible to titrate the dose of Kv1.3 blockers to preferentially suppress one or both groups of memory cells during therapy of autoimmune disease.
We evaluated ShK(L5) in two rat models of TEM cell-induced disease, EAE induced by adoptive transfer of myelin-specific TEM cells (Beeton et al., 2001b) and DTH caused by skin-homing T cells (Soler et al., 2003). ShK(L5) prevented EAE if administered as a single daily injection (10 μg/kg/day) from the time of adoptive cell transfer, and it significantly reduced disease severity when therapy was initiated at the onset of symptoms. No toxicity was observed in treated EAE rats, suggesting that the blood and tissue concentrations of ShK(L5) achieved with this treatment regimen are not sufficient to block neuronal channels, including heteromultimeric KV channels containing Kv1.3 subunits (Koch et al., 1997). ShK(L5) was also effective in suppressing DTH. These proof-of-concept studies demonstrate the therapeutic effectiveness of ShK(L5) in ameliorating TEM-mediated diseases in rat models. We determined the therapeutic safety index of ShK(L5) in EAE rats (when the blood-brain barrier is likely to be compromised) by administering daily injections of a dose (600 μg/kg/day) 60-fold higher than the therapeutically effective dose. Forty percent of rats with EAE that received this dose died on the fifth day (extrapolated LD50 = 750 μg/kg/day for 5 days), which corresponds to a therapeutic safety index of approximately 75.
In conclusion, ShK(L5) is a more selective Kv1.3 blocker than any other known inhibitor, and it might prove beneficial in autoimmune diseases by targeting both TEM cells and class-switched memory B cells. Its picomolar affinity for Kv1.3 and remarkable plasma stability coupled with its high therapeutic safety index in healthy rats (>100) as well as in EAE rats (∼75) bodes well for its potential use as a therapeutic immunomodulator. Single daily subcutaneous injections of ShK(L5) are effective in ameliorating EAE and preventing DTH, indicating that this route of peptide delivery would be feasible for therapy.
Acknowledgments
We thank Paul Munch, Suresh Raman, and Daniel Homerick for excellent technical assistance.
This work was supported by grants from the National Multiple Sclerosis Society (to H.W., K.G.C., P.A.C., M.W.P.), the National Institutes of Health (grant NS048252 to K.G.C.), the Arthritis National Research Foundation (to C.B.), and a Postdoctoral Fellowship from the National Multiple Sclerosis Society (to C.B.).
ABBREVIATIONS
- MS
multiple sclerosis
- TEM
effector memory T cell subset
- TCM
central memory T cell subset
- EAE
experimental autoimmune encephalomyelitis
- DTH
delayed type hypersensitivity
- KCa3.1
intermediate-conductance Ca2+-activated K+ channel
- Kv
voltage-gated K+ channel
- ShK
Stichodactyla helianthus toxin
- Fmoc
9-fluorenylmethoxycarbonyl
- Pmp
p-phosphonomethyl-phenylalanine
- Aeea
amino-ethyloxy-ethyloxy-acetic acid
- HEK
human embryonic kidney
- HERG
human ether-a-go-go-related gene
- PBMC
peripheral blood mononuclear cell
- IL2
interleukin 2
- PBS
phosphate-buffered saline
- MBP
myelin basic protein
- F6CA
fluorescein-6-carboxyl
- l-pTyr
l-phosphotyrosine
- CP-339818
1-benzyl-4-pentylimino-1,4-dihydroquinoline
- UK-78282
4-[diphenylmethoxy)methyl]-1-[3-(4-methoxyphenyl)propyl]-piperidine
- WIN-17317
1-benzyl-7-chloro-4-n-propylimino-1,4-dihydroquinoline hydrochloride.
Footnotes
C.B. and M.P. contributed equally to this work.
References
- Atwal KS, Vaccaro W, Lloyd J, Finlay H, Yan L, Bhandaru RS. inventors, Bristol-Myers Squibb, assignee. Heterocyclic dihydropyrimidines as potassium channel inhibitors. WO0140231. World patent. 2001 2001 Jun 7;
- Baell JB, Gable RW, Harvey AJ, Toovey N, Herzog T, Hansel W, Wulff H. Khellinone derivatives as blockers of the voltage-gated potassium channel Kv1.3: synthesis and immunosuppressive activity. J Med Chem. 2004;47:2326–2336. doi: 10.1021/jm030523s. [DOI] [PubMed] [Google Scholar]
- Bagdány M, Batista CVF, Valdez-Cruz NA, Somodi S, Rodriguez de la Vega RC, Licea AF, Gáspár R, Possani LD, Panyi G. Anuroctoxin, a new scorpion toxin of the α-KTx 6 subfamily, is highly selective for Kv1.3 over IKCa1 ion channels of human T lymphocytes. Mol Pharmacol. 2005;67:1–11. doi: 10.1124/mol.104.007187. [DOI] [PubMed] [Google Scholar]
- Bao J, Miao S, Kayser F, Kotliar AJ, Baker RK, Doss GA, Felix JP, Bugianesi RM, Slaughter RS, Kaczorowski GJ. Potent Kv1.3 inhibitors from correolide-modification of the C18 position. Bioorg Med Chem Lett. 2005;15:447–451. doi: 10.1016/j.bmcl.2004.10.058. [DOI] [PubMed] [Google Scholar]
- Bardien-Kruger S, Wulff H, Arieff Z, Brink P, Chandy KG, Corfield V. Characterisation of the human voltage-gated potassium channel gene, KCNA7, a candidate gene for inherited cardiac disorders and its exclusion as cause of progressive familial heart block I (PFHBI) Eur J Hum Genet. 2002;10:36–43. doi: 10.1038/sj.ejhg.5200739. [DOI] [PubMed] [Google Scholar]
- Beeton C, Barbaria J, Giraud P, Devaux J, Benoliel A, Gola M, Sabatier J, Bernard D, Crest M, Beraud E. Selective blocking of voltage-gated K+ channels improves experimental autoimmune encephalomyelitis and inhibits T cell activation. J Immunol. 2001a;166:936–944. doi: 10.4049/jimmunol.166.2.936. [DOI] [PubMed] [Google Scholar]
- Beeton C, Wulff H, Barbaria J, Clot-Faybesse O, Pennington M, Bernard D, Cahalan M, Chandy K, Beraud E. Selective blockade of T lymphocyte K+ channels ameliorates experimental autoimmune encephalomyelitis, a model for multiple sclerosis. Proc Natl Acad Sci USA. 2001b;98:13942–13947. doi: 10.1073/pnas.241497298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeton C, Wulff H, Singh S, Botsko S, Crossley G, Gutman GA, Cahalan MD, Pennington MW, Chandy KG. A novel fluorescent toxin to detect and investigate Kv1.3 channel up-regulation in chronically activated T lymphocytes. J Biol Chem. 2003;278:9928–9937. doi: 10.1074/jbc.M212868200. [DOI] [PubMed] [Google Scholar]
- Beraud E, Balzano C, Zamora AJ, Varriale S, Bernard D, Ben-Nun A. Pathogenic and non-pathogenic T lymphocytes specific for the encephalitogenic epitope of myelin basic protein: functional characteristics and vaccination properties. J Neuroimmunol. 1993;47:41–53. doi: 10.1016/0165-5728(93)90283-5. [DOI] [PubMed] [Google Scholar]
- Bunce C, Bell EB. CD45RC isoforms define two types of CD4 memory T cells, one of which depends on persisting antigen. J Exp Med. 1997;185:767–776. doi: 10.1084/jem.185.4.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butenschon I, Moller K, Hansel W. Angular methoxy-substituted furoand pyranoquinolinones as blockers of the voltage-gated potassium channel Kv1.3. J Med Chem. 2001;44:1249–1256. doi: 10.1021/jm001007u. [DOI] [PubMed] [Google Scholar]
- Castle NA, Hollinshead SP, Hughes PF, Mendoza GS, Searafin J, Wilson JW, Amato GS, Beaudoin S, Gross M, McNaughton-Smith G. inventors, ICAgen and Eli Lilly & Company, assignee. Potassium channel inhibitors. 6,083,986. U.S. patent. 2000 2000 Jul 4;
- Chandy KG, DeCoursey TE, Cahalan MD, McLaughlin C, Gupta S. Voltage-gated potassium channels are required for human T lymphocyte activation. J Exp Med. 1984;160:369–385. doi: 10.1084/jem.160.2.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandy KG, Wulff H, Beeton C, Pennington M, Gutman GA, Cahalan MD. K+ channels as targets for specific immunomodulation. Trends Pharmacol Sci. 2004;25:280–289. doi: 10.1016/j.tips.2004.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcione A, Casazza S, Ferretti E, Giunti D, Zappia E, Pistorio A, Gambini C, Mancardi GL, Uccelli A, Pistoria V. Recapitulation of B cell differentiation in the central nervous system of patients with multiple sclerosis. Proc Natl Acad Sci USA. 2004;101:11064–11069. doi: 10.1073/pnas.0402455101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotton J, Crest M, Bouet F, Alessandri N, Gola M, Forest E, Karlsson E, Castaneda O, Harvey AL, Vita C, et al. A potassium-channel toxin from the sea anemone Bunodosoma granulifera, an inhibitor for Kv1 channels. Revision of the amino acid sequence, disulfide-bridge assignment, chemical synthesis, and biological activity. Eur J Biochem. 1997;244:192–202. doi: 10.1111/j.1432-1033.1997.00192.x. [DOI] [PubMed] [Google Scholar]
- Deibler GE, Martenson RE, Kies MW. Large scale preparation of myelin basic protein from central nervous tissue of several mammalian species. Prep Biochem. 1972;2:139–165. doi: 10.1080/00327487208061467. [DOI] [PubMed] [Google Scholar]
- Ezawa K, Yamamura M, Matsui H, Ota Z, Makino H. Comparative analysis of CD45RA- and CD45RO-positive CD4+ T cells in peripheral blood, synovial fluid and synovial tissue in patients with rheumatoid arthritis and osteoarthritis. Acta Med Okayama. 1997;51:25–31. doi: 10.18926/AMO/30810. [DOI] [PubMed] [Google Scholar]
- Felix JP, Bugianesi RM, Schmalhofer WA, Borris R, Goetz MA, Hensens OD, Bao JM, Kayser F, Parsons WH, Rupprecht K, et al. Identification and biochemical characterization of a novel nortriterpene inhibitor of the human lymphocyte voltage-gated potassium channel, Kv1.3. Biochemistry. 1999;38:4922–4930. doi: 10.1021/bi982954w. [DOI] [PubMed] [Google Scholar]
- Friedrich M, Krammig S, Henze M, Docke WD, Sterry W, Asadullah K. Flow cytometric characterization of lesional T cells in psoriasis: intracellular cytokine and surface antigen expression indicates an activated, memory/effector type 1 immunophenotype. Arch Dermatol Res. 2000;292:519–521. doi: 10.1007/s004030000167. [DOI] [PubMed] [Google Scholar]
- Fujiwara Y, Akaji K, Kiso Y. Racemization-free synthesis of C-terminal cysteine-peptide using 2-chlorotrityl resin. Chem Pharm Bull (Tokyo) 1994;42:724–726. doi: 10.1248/cpb.42.724. [DOI] [PubMed] [Google Scholar]
- Garcia ML, Garcia-Calvo M, Hidalgo P, Lee A, MacKinnon R. Purification and characterization of the three inhibitors of voltage-dependent K+ channels from Leiurus quinquestriatus var. hebraeus venom. Biochemistry. 1994;33:6834–6839. doi: 10.1021/bi00188a012. [DOI] [PubMed] [Google Scholar]
- Ghanshani S, Wulff H, Miller MJ, Rohm H, Neben A, Gutman GA, Cahalan MD, Chandy KG. Up-regulation of the IKCa1 potassium channel during T-cell activation: molecular mechanism and functional consequences. J Biol Chem. 2000;275:37137–37149. doi: 10.1074/jbc.M003941200. [DOI] [PubMed] [Google Scholar]
- Gradl SN, Felix JP, Isacoff EY, Garcia ML, Trauner D. Protein surface recognition by rational design: nanomolar ligands for potassium channels. J Am Chem Soc. 2003;125:12668–12669. doi: 10.1021/ja036155z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grissmer S, Nguyen AN, Aiyar J, Hanson DC, Mather RJ, Gutman GA, Karmilowicz MJ, Auperin DD, Chandy KG. Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol Pharmacol. 1994;45:1227–1234. [PubMed] [Google Scholar]
- Gutman GA, Chandy KG, Adelman JP, Aiyar J, Bayliss DA, Clapham DE, Covarriubias M, Desir GV, Furuichi K, Ganetzky B, et al. International Union of Pharmacology. XLI. compendium of voltage-gated ion channels: potassium channels. Pharmacol Rev. 2003;55:583–586. doi: 10.1124/pr.55.4.9. [DOI] [PubMed] [Google Scholar]
- Hammerschmidt F, Hanbauer M. Transformation of arylmethylamines into alpha-aminophosphonic acids via metalated phosphoramidates: rearrangements of partly configurationally stable N-phosphorylated alpha-aminocarbanions. J Org Chem. 2000;65:6121–6131. doi: 10.1021/jo000585f. [DOI] [PubMed] [Google Scholar]
- Hanner M, Schmalhofer WA, Green B, Bordallo C, Liu J, Slaughter RS, Kaczorowski GJ, Garcia ML. Binding of correolide to Kv1 family potassium channels. J Biol Chem. 1999;274:25237–25244. doi: 10.1074/jbc.274.36.25237. [DOI] [PubMed] [Google Scholar]
- Hanson DC, Nguyen A, Mather RJ, Rauer H, Koch K, Burgess LE, Rizzi JP, Donovan CB, Bruns MJ, Canniff PC, et al. UK-78,282, a novel piperidine compound that potently blocks the Kv1.3 voltage-gated potassium channel and inhibits human T cell activation. Br J Pharmacol. 1999;126:1707–1716. doi: 10.1038/sj.bjp.0702480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriks JJA, Alblas J, van der Pol SMA, van Tol EAF, Dijkstra CD, de Vries HE. Flavonoids influence monocytic GTPase activity and are protective in experimental allergic encephalitis. J Exp Med. 2004;200:1667–1672. doi: 10.1084/jem.20040819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias A, Bauer J, Litzenburger T, Schubart A, Linington C. T- and B-cell responses to myelin oligodendrocyte glycoprotein in experimental autoimmune encephalomyelitis and multiple sclerosis. Glia. 2001;36:220–234. doi: 10.1002/glia.1111. [DOI] [PubMed] [Google Scholar]
- Kalman K, Pennington MW, Lanigan MD, Nguyen A, Rauer H, Mahnir V, Paschetto K, Kem WR, Grissmer S, Gutman GA, et al. ShK-Dap22, a potent Kv1.3-specific immunosuppressive polypeptide. J Biol Chem. 1998;273:32697–32707. doi: 10.1074/jbc.273.49.32697. [DOI] [PubMed] [Google Scholar]
- King DS, Fields CG, Fields GB. A cleavage method which minimizes side reactions following Fmoc solid phase peptide synthesis. Int J Pept Protein Res. 1990;36:255–266. doi: 10.1111/j.1399-3011.1990.tb00976.x. [DOI] [PubMed] [Google Scholar]
- Koch RO, Wanner SG, Koschak A, Hanner M, Schwarzer C, Kaczorowski GJ, Slaughter RS, Garcia ML, Knaus HG. Complex subunit assembly of neuronal voltage-gated K+ channels. Basis for high-affinity toxin interactions and pharmacology. J Biol Chem. 1997;272:27577–27581. doi: 10.1074/jbc.272.44.27577. [DOI] [PubMed] [Google Scholar]
- Kolski-Andreaco A, Tomita H, Shakkottai VG, Gutman GA, Cahalan MD, Gargus JJ, Chandy KG. SK3-1C, a dominant-negative suppressor of SKCa and IKCa channels. J Biol Chem. 2004;279:6893–6904. doi: 10.1074/jbc.M311725200. [DOI] [PubMed] [Google Scholar]
- Koo GC, Blake JT, Shah K, Staruch MJ, Dumont F, Wunderler D, Sanchez M, McManus OB, Sirotina-Meisher A, Fischer P, et al. Correolide and derivatives are novel immunosuppressants blocking the lymphocyte Kv1.3 potassium channels. Cell Immunol. 1999;197:99–107. doi: 10.1006/cimm.1999.1569. [DOI] [PubMed] [Google Scholar]
- Koo GC, Blake JT, Talento A, Nguyen M, Lin S, Sirotina A, Shah K, Mulvany K, Hora D, Jr, Cunningham P, et al. Blockade of the voltage-gated potassium channel Kv1.3 inhibits immune responses in vivo. J Immunol. 1997;158:5120–5128. [PubMed] [Google Scholar]
- Koschak A, Bugianesi RM, Mitterdorfer J, Kaczorowski GJ, Garcia ML, Knaus HG. Subunit composition of brain voltage-gated potassium channels determined by hongotoxin-1, a novel peptide derived from Centruroides limbatus venom. J Biol Chem. 1998;273:2639–2644. doi: 10.1074/jbc.273.5.2639. [DOI] [PubMed] [Google Scholar]
- Lahey T, Rajadhyaksha VJ. 3-Deoxyflavonoid inhibition of T-lymphocyte activation and therapeutic use. 2004102386. US patent. 2004:19.
- Lin CS, Boltz RC, Blake JT, Nguyen M, Talento A, Fischer PA, Springer MS, Sigal NH, Slaughter RS, Garcia ML, et al. Voltage-gated potassium channels regulate calcium-dependent pathways involved in human T lymphocyte activation. J Exp Med. 1993;177:637–645. doi: 10.1084/jem.177.3.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett-Racke AE, Trotter JL, Lauber J, Perrin PJ, June CH, Racke MK. Decreased dependence of myelin basic protein-reactive T cells on CD28-mediated costimulation in multiple sclerosis patients: a marker of activated/memory T cells. J Clin Investig. 1998;101:725–730. doi: 10.1172/JCI1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovic-Plese S, Cortese I, Wandinger KP, McFarland HF, Martin R. CD4+CD28- costimulation-independent T cells in multiple sclerosis. J Clin Investig. 2001;108:1185–1194. doi: 10.1172/JCI12516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton RE, Sanchez M, Linde AR, Bugianesi RM, Dai G, Felix JP, Koprak SL, Staruch MJ, Bruguera M, Cox R, et al. Substitution of a single residue in Stichodactyla helianthus peptide, ShK-Dap22, reveals a novel pharmacological profile. Biochemistry. 2003;42:13698–13707. doi: 10.1021/bi035209e. [DOI] [PubMed] [Google Scholar]
- Mouhat S, Visan V, Ananthakrishnan S, Wulff H, Andreotti N, Grissmer S, Darbon H, De Waard M, Sabatier JM. K+ channel types targeted by synthetic OSK1, a toxin from Orthochirus scrobiculosus scorpion venom. Biochem J. 2005;385:95–104. doi: 10.1042/BJ20041379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas E, Vilaseca M, Giralt E. A study of the use of NH4I for the reduction of methionine sulphoxide in peptides containing cysteine and cystine. Tetrahedron. 1995;51:5701–5710. [Google Scholar]
- O’Connor K, Bar-Or A, Hafler DA. The neuroimmunology of multiple sclerosis: possible roles of T and B lymphocytes in immunopathogenesis. J Clin Immunol. 2001;21:81–92. doi: 10.1023/a:1011064007686. [DOI] [PubMed] [Google Scholar]
- Pennington M, Byrnes M, Zaydenberg I, Khaytin I, de Chastonay J, Krafte D, Hill R, Mahnir V, Volberg W, Gorczyca W, et al. Chemical synthesis and characterization of ShK toxin: a potent potassium channel inhibitor from a sea anemone. Int J Pept Protein Res. 1995;346:354–358. doi: 10.1111/j.1399-3011.1995.tb01068.x. [DOI] [PubMed] [Google Scholar]
- Pennington M, Mahnir V, Khaytin I, Zaydenberg I, Byrnes M, Kem W. An essential binding surface for ShK toxin interaction with rat brain potassium channels. Biochemistry. 1996a;35:16407–16411. doi: 10.1021/bi962463g. [DOI] [PubMed] [Google Scholar]
- Pennington M, Mahnir V, Krafte D, Zaydenberg I, Byrnes M, Khaytin I, Crowley K, Kem W. Identification of three separate binding sites on ShK toxin, a potent inhibitor of voltage-dependent potassium channels in human T-lymphocytes and rat brain. Biochem Biophys Res Commun. 1996b;219:696–701. doi: 10.1006/bbrc.1996.0297. [DOI] [PubMed] [Google Scholar]
- Peter MJ, Varga Z, Hajdu P, Gaspar RJ, Damjanovich S, Horjales E, Possani LD, Panyi G. Effects of toxins Pi2 and Pi3 on human T lymphocyte Kv1.3 channels: the role of Glu7 and Lys24. J Membr Biol. 2001;179:13–25. doi: 10.1007/s002320010033. [DOI] [PubMed] [Google Scholar]
- Recanatini M, Poluzzi E, Masetti M, Cavalli A, De Ponti F. QT prolongation through hERG K+ channel blockade: current knowledge and strategies for the early prediction during drug development. Med Res Rev. 2005;25:133–166. doi: 10.1002/med.20019. [DOI] [PubMed] [Google Scholar]
- Regaya I, Beeton C, Ferrat G, Andreotti N, Darbon H, De Waard M, Sabatier JM. Evidence for domain-specific recognition of SK and Kv channels by MTX and HsTX1 scorpion toxins. J Biol Chem. 2004;279:55690–55696. doi: 10.1074/jbc.M410055200. [DOI] [PubMed] [Google Scholar]
- Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature (Lond) 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- Schmalhofer WA, Bao J, McManus OB, Green B, Matyskiela M, Wunderler D, Bugianesi RM, Felix JP, Hanner M, Linde-Arias AR, et al. Identification of a new class of inhibitors of the voltage-gated potassium channel, Kv1.3, with immunosuppressant properties. Biochemistry. 2002;41:7781–7794. doi: 10.1021/bi025722c. [DOI] [PubMed] [Google Scholar]
- Scholz C, Patton KT, Anderson DE, Freeman GJ, Hafler DA. Expansion of autoreactive T cells in multiple sclerosis is independent of exogenous B7 costimulation. J Immunol. 1998;160:1532–1538. [PubMed] [Google Scholar]
- Singh S, Zink DL, Dombrowski AW, Dezeny G, Bills GF, Felix J, Slaughter RS, Goetz MA. Candelalides A-C: novel diterpenoid pyrones from fermentations of Sesquicillium candelabrum as blockers of the voltage-gated potassium channel Kv1.3. Org Lett. 2001;3:247–250. doi: 10.1021/ol006891x. [DOI] [PubMed] [Google Scholar]
- Soler D, Humphreys TL, Spinola SM, Campbell JJ. CCR4 versus CCR10 in human cutaneous TH lymphocyte trafficking. Blood. 2003;101:1677–1682. doi: 10.1182/blood-2002-07-2348. [DOI] [PubMed] [Google Scholar]
- Task Force of the European Society of Cardiology. the North American Society of Pacing Electrophysiology Heart Rate variability: standards of measurement, physiological interpretation and clinical use. Circulation. 1996;93:1043–1065. [PubMed] [Google Scholar]
- Tian Z, Gu C, Roeske RW, Zhou M, Van Etten RL. Synthesis of phosphotyrosine-containing peptides by solid-phase method. Int J Peptide Protein Res. 1993;42:155–158. doi: 10.1111/j.1399-3011.1993.tb00491.x. [DOI] [PubMed] [Google Scholar]
- Valverde P, Kawai T, Taubman M. Selective blockade of voltage-gated potassium channels reduces inflammatory bone resorption in experimental periodontal disease. J Bone Miner Res. 2004;19:155–164. doi: 10.1359/JBMR.0301213. [DOI] [PubMed] [Google Scholar]
- Vennekamp J, Wulff H, Beeton C, Calabresi PA, Grissmer S, Hansel W, Chandy KG. Kv1.3 blocking 5-phenylalkoxypsoralens: a new class of immunomodulators. Mol Pharmacol. 2004;65:1364–1374. doi: 10.1124/mol.65.6.1364. [DOI] [PubMed] [Google Scholar]
- Viglietta V, Kent SC, Orban T, Hafler DA. GAD65-reactive T cells are activated in patients with autoimmune type 1a diabetes. J Clin Investig. 2002;109:895–903. doi: 10.1172/JCI14114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanner SG, Glossmann H, Knaus HG, Baker R, Parsons W, Rupprecht KM, Brochu R, Cohen CJ, Schmalhofer W, Smith M, et al. WIN 17317-3, a new high affinity probe for voltage-gated sodium channels. Biochemistry. 1999;38:11137–11146. doi: 10.1021/bi990336p. [DOI] [PubMed] [Google Scholar]
- Wernekenschnieder A, Korner P, Hansel W. 3-Alkyl- and 3-aryl-7H-furo[3,2-g]chromen-7-ones as blockers of the voltage-gated potassium channel Kv1.3. Pharmazie. 2004;59:319–320. [PubMed] [Google Scholar]
- Wulff H, Calabresi P, Allie R, Yun S, Pennington MW, Beeton C, Chandy KG. The voltage-gated Kv1.3 K+ channel in effector memory T cells as new target for MS. J Clin Investig. 2003;111:1703–1713. doi: 10.1172/JCI16921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff H, Knaus H, Pennington M, Chandy KG. K+ channel expression during B cell differentiation: implications for immunomodulation and autoimmunity. J Immunol. 2004;173:776–786. doi: 10.4049/jimmunol.173.2.776. [DOI] [PubMed] [Google Scholar]
- Wulff H, Miller MJ, Haensel W, Grissmer S, Cahalan MD, Chandy KG. Design of a potent and selective inhibitor of the intermediate-comductance Ca2+-activated K+ channel, IKCa1: a potential immunosuppressant. Proc Natl Acad Sci USA. 2000;97:8151–8156. doi: 10.1073/pnas.97.14.8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Cayabyab FS, Pennefather PS, Schlichter LC, DeCoursey TE. HERG-like K+ channels in microglia. J Gen Physiol. 1998;111:781–794. doi: 10.1085/jgp.111.6.781. [DOI] [PMC free article] [PubMed] [Google Scholar]