

Abstract
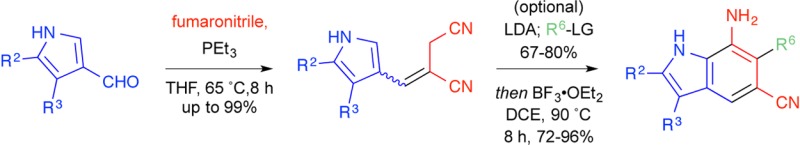
Among privileged structures, indoles occupy a central place in medicinal chemistry and alkaloid research. Here we report a flexible and efficient conversion of pyrrole-3-carboxaldehydes to substituted 7-amino-5-cyanoindoles. Phosphine addition to fumaronitrile proceeds with prototropic rearrangement of the initially formed zwitterion to the thermodynamically favored phosphonium ylide, which is poised for in situ Wittig olefination. The predominantly E-alkene product positions the allylic nitrile for facile intramolecular Hoeben–Hoesch reaction in the presence of BF3·OEt2. Syntheses of 2,5- and 3,5-disubstituted 7-aminoindoles are illustrated. Additionally, dianion alkylation of the allylic nitrile is demonstrated to furnish, after cyclization, 5,6-disubstituted 7-aminoindoles to further exemplify this scalable and high-yielding method.
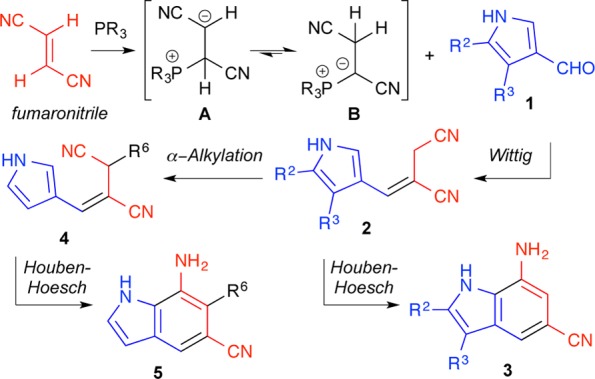
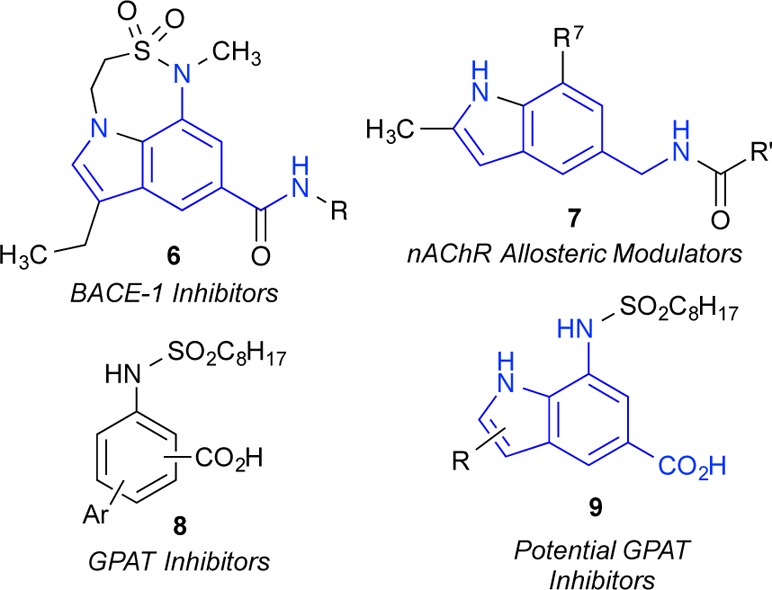
Synthetic routes to indoles have long been prized owing to the prevalence of the aromatic heterocycles in natural products as well as synthetic medicinal agents.1 Classical indole syntheses, including the work of Fischer,2 Bartoli,3 Larock,4 Reissert,5 and many others,6 remain benchmarks in the development of heterocyclic methodology. The majority of these approaches utilize a functionalized benzene ring to annulate the 5-membered pyrrolic portion of the indole. In contrast, few synthetic strategies exist that originate from a substituted pyrrole to build up the benzenoid portion of the indole.7 The latter strategy can benefit from the unique chemistry of pyrroles, which react with electrophiles preferentially at C-2 but exclusively at C-3 when N-protected with the sterically cumbersome triisopropylsilyl group.8 This tunable reactivity allows for ready introduction of the would be C-2 and C-3 substituents of the indole prior to annulation. Further, the intrinsic reactivity of pyrroles at C-2 introduces a disconnection optimal for the indole-forming cyclization itself. Tactical issues of symmetry and regiochemistry inherent to routes from substituted benzene rings are avoided. This strategy, shown in Figure 1, utilizes a three-component Wittig reaction of pyrrole-3-carboxaldehydes with fumaronitrile and a trialkylphosphine to generate predominantly E-alkenes. These allylic nitriles are positioned for intramolecular cyclization under Houben–Hoesch conditions to furnish, after aromatization, substituted 7-aminoindoles. An optional tailoring step to install C-6 substituents prior to ring closure can also be envisioned. This scheme would allow for control of the C-2, C-3, and C-6 indole substituents on the resulting 7-amino-5-cyanoindoles that are not readily accessible using existing methods. The amine (e.g., by diazotization, acylation) and the nitrile (e.g., by hydration, hydrolysis, reduction, etc.) can be differentially modified for rapid structural diversification. Potential targets (Figure 2) include a class of potent β-secretase 1 (BACE1) inhibitors,9 a class of nicotinic acetylcholine receptor (nAChR) allosteric modulators,10 and analogues of recently reported inhibitors of glycerol-3-phosphate acyltransferase (GPAT).11
Figure 1.
Proposed route to substituted 7-aminoindoles.
Figure 2.
Representative medicinal chemistry targets.
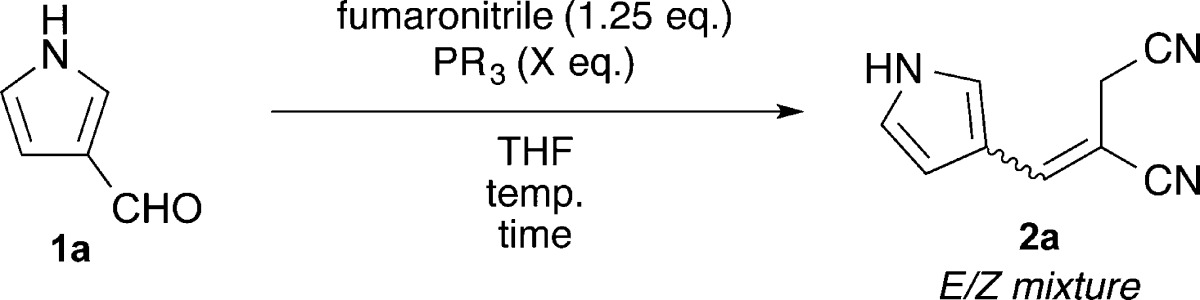
Of fundamental importance to the planned synthesis was the acquisition of E-alkenes such as 2 (Figure 1). The E-selective olefination of aldehydes with esters of fumarate and maleate, maleic anhydride, or maleimide and tributyl- or triphenylphosphine has been previously described.12 We envisioned a similar 1,4-addition of a phosphine to fumaronitrile proceeded by rapid prototropic rearrangement of the initially formed zwitterion A to the thermodynamically favored phosphonium ylide B, which is poised for in situ Wittig olefination with pyrrole-3-carboxaldehydes such as 1. Trimethyl-, triethyl-, tributyl-, and triphenylphosphine, as well as trimethyl phosphite, were evaluated for the efficiency of their reactions with fumaronitrile and aldehyde 1a as shown in Table 1. PPh3 and P(OMe)3 were unreactive under the reaction conditions. PMe3, PEt3, and PBu3, when used in excess, exhibited understandably similar reactivity, converting aldehyde 1a to alkene 2a in moderate yield (72–80%) but with only slight E-selectivity (4:3) after extended reaction time (48 h). Concerned that the excess phosphine was adding into the product, itself an α,β-unsaturated nitrile, and causing isomerization of the alkene, we next attempted the reaction with limiting phosphine. At room temperature, these reactions gave the desired alkene 2a in high yield (91–99%) and good E/Z ratio (3:1), suggesting that the excess phosphine was indeed causing alkene isomerization. To ameliorate the sluggish reaction rate, we heated the reaction with PEt3 to 65 °C. These improved conditions cut the reaction time 6-fold while maintaining the favorable yield (97%) and E/Z ratio (3:1) of the room temperature reaction. There are distinct advantages to each of the phosphines tested. The volatility of PMe3 (bp 38 °C) allows for easy removal upon workup but prohibits heating of the reaction without a sealed vessel. PBu3 (bp 240 °C) requires an additional oxidative workup step but is the most economical choice. Finally, PEt3 (bp 126 °C) afforded the highest yield and allows for heating to reflux while maintaining the option of low-pressure removal.
Table 1. Optimization of Wittig Reaction with Aldehyde 1a.
| entry | PR3 | X | temp (°C) | time (h) | yield of 2aa (%) | E/Zb |
|---|---|---|---|---|---|---|
| 1 | PMe3 | 1.4 | 23 | 48 | 76 | 4:3 |
| 2 | PEt3 | 1.4 | 23 | 48 | 80 | 4:3 |
| 3 | PBu3 | 1.4 | 23 | 48 | 72 | 4:3 |
| 4 | PPh3 | 1.4 | 23 | 48 | 0 | |
| 5 | P(OMe)3 | 1.4 | 23 | 48 | 0 | |
| 6 | PMe3 | 1.2 | 23 | 48 | 95 | 3:1 |
| 7 | PEt3 | 1.2 | 23 | 48 | 99 | 3:1 |
| 8 | PBu3 | 1.2 | 23 | 48 | 91 | 3:1 |
| 9 | PEt3 | 1.2 | 65 | 8 | 97 | 3:1 |
Isolated yield.
E/Z values were calculated from the ratio of the integrals of the allylic methylenes in the 1H NMR spectra.
To demonstrate scope, the optimized conditions using PEt3 at 65 °C were applied to various 4- and 5-substituted pyrrole-3-carboxaldehydes 1a–g. Alkenes 2a–g were obtained in generally good to excellent yield and moderate diastereoselectivity (Table 2). The reaction conditions were tolerant of a wide range of substituents including alkyl, aryl, halogen, and ester moieties. Electron-withdrawing substituents at the pyrrolic α-position led to slower conversion. Additionally, minor substituent effects were observed for the diastereoselectivity of the reactions.
Table 2. Scope of Wittig Reaction.
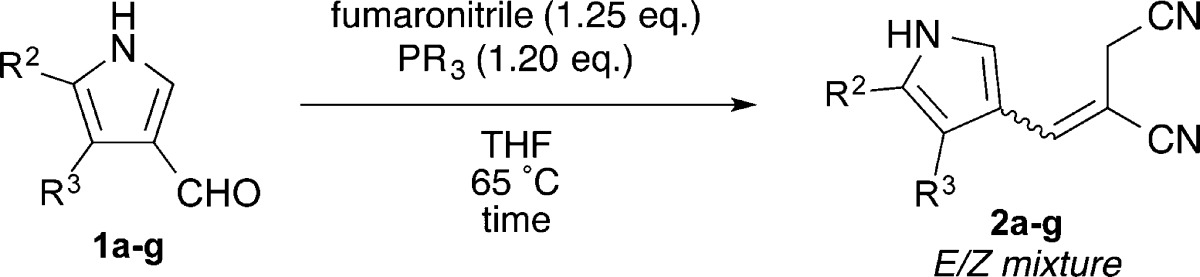
| entry | 1 | R2 | R3 | time (h) | product | yield (%) | E/Za |
|---|---|---|---|---|---|---|---|
| 1 | 1a | –H | –H | 8 | 2a | 97 | 3:1 |
| 2 | 1b | –Br | –H | 10 | 2b | 97 | 3:1 |
| 3 | 1c | –CO2Et | –H | 10 | 2c | 96 | 5:1 |
| 4 | 1d | –Ph | –H | 8 | 2d | 88 | 3:1 |
| 5 | 1e | –H | –Me | 8 | 2e | 85 | 4:1 |
| 6 | 1f | –H | –Et | 8 | 2f | 85 | 4:1 |
| 7 | 1g | –H | –Br | 8 | 2g | 93 | 5:3 |
E/Z values were calculated from the ratio of the integrals of the allylic methylenes in the 1H NMR spectra.
To further diversify the library of E-alkenes, we next sought to functionalize the allylic nitrile. To accomplish this task without the use of protecting groups, we elected to exploit the relatively more acidic pyrrole N–H and developed a dianion approach to alkylate the allylic position chemoselectively (Table 3). Addition of 2.1 equiv of LDA followed by the addition of various electrophiles effected selective α-alkylation of nitrile 2a to afford 4a–f without detection of N-alkylated pyrrole side products 10a–f. Analogous attempts with the electrophiles ethyl chloroformate, methyl acrylate, Br2, and NBS showed no desired reaction.
Table 3. Alkylation of Allylic Nitrile 2a.
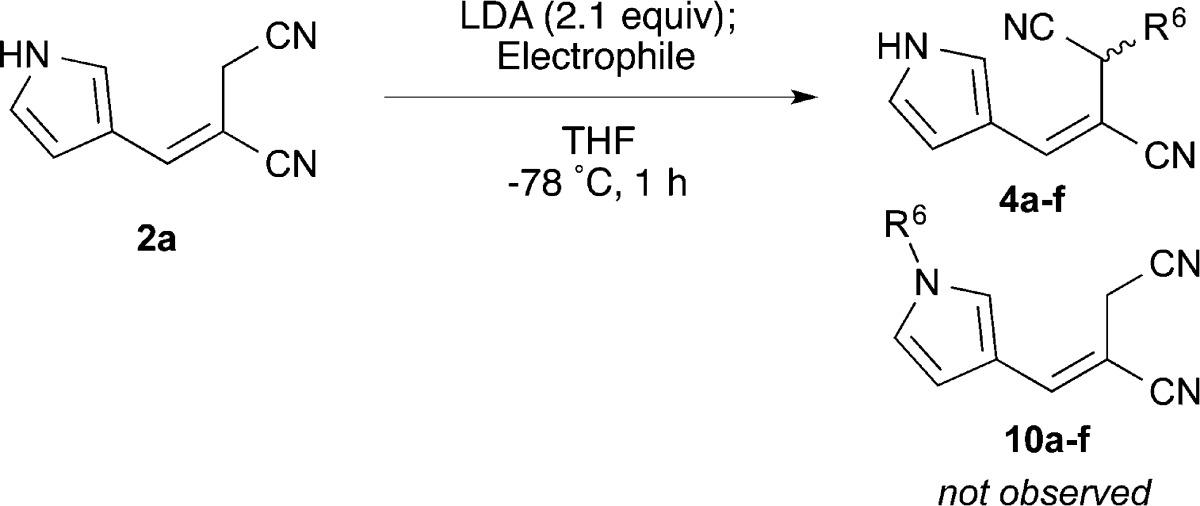
| entry | electrophile | product | R6 | yield (%) |
|---|---|---|---|---|
| 1 | MeI | 4a | –CH3 | 77 |
| 2 | EtI | 4b | –CH2CH3 | 80 |
| 3 | BnBr | 4c | –CH2Ph | 75 |
| 4 | allyl-Br | 4d | –CH2CH=CH2 | 67 |
| 5 | propargyl-Br | 4e | –CH2C≡CH | 72 |
| 6 | BrCH2CO2Et | 4f | –CH2CO2Et | 80 |
With reaction conditions established to E-olefinic precursors 2a–g and 4a–f, we next sought to optimize indole cyclization conditions as shown in Table 4. Of the Lewis acids tested, only BF3·OEt2 successfully effected cyclization. Initial attempts to use a catalytic amount of BF3·OEt2 gave only stoichiometric yield. Presumably, this is due to chelation of the Lewis acid by the 7-aminoindole product thereby preventing catalytic turnover. Similarly, using THF as the solvent afforded no reaction, likely owing to association of the Lewis acid with the ethereal solvent. The optimized conditions used 2.5 equiv of BF3·OEt2 in 1,2-dichloroethane at 90 °C for 12 h, which gave efficient annulation of alkene 2a to indole 3a.
Table 4. Optimization of Cyclization to indole 3a.
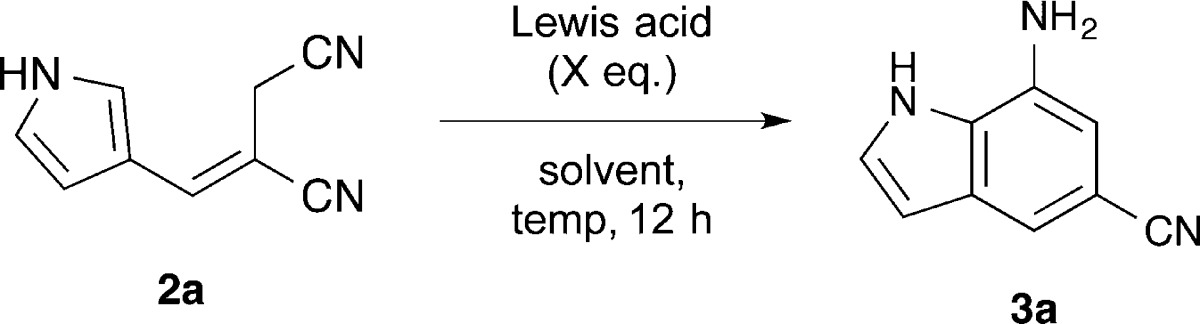
| entry | Lewis acid | X | solvent | temp (°C) | yield (%) |
|---|---|---|---|---|---|
| 1 | none | CH2Cl2 | 45 | 0 | |
| 2 | AlCl3 | 0.2 | CH2Cl2 | 45 | 0 |
| 3 | Sc(OTf)3 | 0.2 | CH2Cl2 | 45 | 0 |
| 4 | TiCl4 | 0.2 | CH2Cl2 | 45 | 0 |
| 5 | BF3·OEt2 | 0.2 | CH2Cl2 | 45 | 19 |
| 6 | BF3·OEt2 | 1.1 | THF | 70 | 0 |
| 7 | BF3·OEt2 | 1.1 | PhCH3 | 85 | 61 |
| 8 | BF3·OEt2 | 2.5 | DCE | 90 | 91 |
Application of the annulation conditions to the E-alkenes 2a–g and 4a–f gave indoles 3a–g and 5a–f in good to excellent yields (Table 5). The reaction conditions were amenable to a wide scope of substituents. Of particular interest to us, the bromo- and propargyl-substituted indoles can be further functionalized through coupling reactions or click chemistry, respectively, to rapidly generate large libraries of compounds. The more electron-deficient pyrroles (e.g., 2c, 2g) demonstrated lower yields, typical of electrophilic aromatic substitution with an electron-deficient arene. The reaction times for the 6-substituted alkenes were slightly shorter than for cyclization of the 2- and 3-substituted examples. This behavior points to a possible Thorpe–Ingold effect, whereby the C-6 substituent increases the population of the reactive rotamer resulting in an increased reaction rate. It is likely that this tactic can also be applied to pyrroles additionally substituted at the 2- and 3-positions.
Table 5. Scope of Indole Annulation.
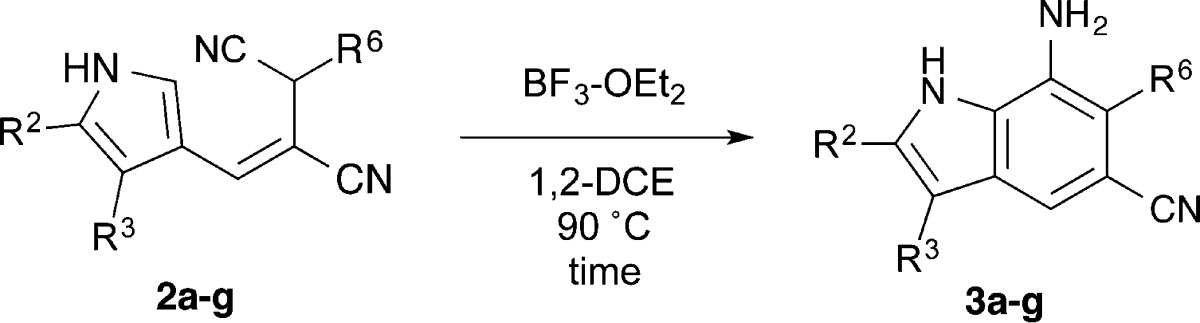
| entry | pyrrole | R2 | R3 | R6 | indole | time (h) | yield (%) |
|---|---|---|---|---|---|---|---|
| 1 | 2a | –H | –H | –H | 3a | 12 | 91 |
| 2 | 2b | –Br | –H | –H | 3b | 12 | 67 |
| 3 | 2c | –CO2Et | –H | –H | 3c | 12 | 62 |
| 4 | 2d | –Ph | –H | –H | 3d | 12 | 87 |
| 5 | 2e | –H | –CH3 | –H | 3e | 12 | 94 |
| 6 | 2f | –H | –CH2CH3 | –H | 3f | 12 | 92 |
| 7 | 2g | –H | –Br | –H | 3g | 12 | 75 |
| 8 | 4a | –H | –H | –CH3 | 5a | 8 | 88 |
| 9 | 4b | –H | –H | –CH2CH3 | 5b | 8 | 95 |
| 10 | 4c | –H | –H | –CH2Ph | 5c | 8 | 96 |
| 11 | 4d | –H | –H | –CH2CH=CH2 | 5d | 8 | 93 |
| 12 | 4e | –H | –H | –CH2C≡CH | 5e | 8 | 92 |
| 13 | 4f | –H | –H | –CH2CO2Et | 5f | 8 | 72 |
In addition, a functionally one-pot reaction was run to indole 3a, shown in Scheme 1, in which pyrrole-3-carboxaldehyde and fumaronitrile were reacted in dry THF with PMe3 as before (Table 1). Taking advantage of the relatively low boiling point of PMe3, the solvent and any unreacted phosphine were removed by rotary evaporation and the residue taken up in 1,2-DCE. Treatment of this mixture as above with BF3·OEt2 gave the desired indole in 77% yield, approximately the theoretical limit from the E-isomer, after crystallization of the product from the crude mixture containing unreacted Z-isomer.
Scheme 1. One-Pot Synthesis of indole 3a.

Encouraged by the success of this approach, we next tested whether the corresponding furan- and thiophene-3-carboxaldehydes could undergo a similar two-step sequence to the corresponding 7-amino-5-cyanobenzofurans and benzothiophenes, respectively (Scheme 2). We hypothesized that, with decreased C-2 nucleophilicity and increased heteroatomic interaction with the BF3·OEt2, the annulation of these heterocycles would be less facile. First, Wittig reaction using the optimized conditions afforded the olefins 12a,b in excellent yield and better stereoselectivity than for the pyrrolic reactions. Treatment with BF3·OEt2 effected annulation to benzofuran 13a and benzothiophene 13b in low to moderate yield. As expected, the reaction rates were much slower with conversion stalling around 24 h. Neither additional equivalents of Lewis acid nor longer reaction times improved the yields. It is possible that use of a less oxophilic Lewis acid could improve the yields for these classes of heterocycles.
Scheme 2. Application to the Synthesis of Benzofuran and Benzothiophene Derivatives.

Finally, to illustrate practical applications of this method we chose to demonstrate functional group conversions from the 7-amino-5-cyanoindole intermediates to readily identifiable precursors of the medicinal chemistry targets noted above (Figure 2). First, we utilized Sandmeyer chemistry to substitute the amino group of indole 3a with chloro or bromo as shown in Scheme 3. These aryl halides allow access to a variety of C-7 substituted indoles, such as the nAChR allosteric modulators represented by 7 (Figure 2), through metal-catalyzed coupling reactions. Second, we demonstrated sulfonamide coupling of the aniline with octanesulfonyl chloride, followed by alkaline nitrile hydrolysis to give the GPAT inhibitor analogue 9a. A similar sulfonamide coupling and hydrolysis sequence can be envisioned to access the diversifiable precursor of the BACE1 inhibitors represented by 6 (Figure 2).
Scheme 3. Functional Group Conversion of 3a to Precursors of Medicinal Chemistry Targets.

In conclusion, we have developed a flexible and step-efficient method to obtain highly functionalized 7-aminoindoles from pyrrole-3-carboxaldehydes. This method utilizes a three-component Wittig reaction of pyrrole-3-carboxaldehydes with fumaronitrile and PEt3 to generate predominantly cis-allylic nitriles, which can be optionally elaborated further by a chemoselective alkylation at C-6. These cis-allylic nitriles undergo cyclization through an intramolecular Houben–Hoesch reaction to afford highly substituted indoles. A one-pot procedure combining the Wittig and Houben–Hoesch reactions was also demonstrated. While the method is substantially linear, the steps are typically high-yielding with chromatographic purification minimized or eliminated, and intermediates and products isolated by crystallization. The reactions are scalable and require no special precautions or protecting groups—in sum, attributes favorable for large-scale applications. Further studies will aim to expand the scope of this method and to demonstrate its potential for the synthesis of bioactive targets of interest.
Acknowledgments
We thank Dr. I. P. Mortimer for mass spectral analysis and Dr. C. Moore for NMR assistance. This work was supported by the National Institutes of Health research grant ES001670.
Supporting Information Available
Detailed synthetic procedures and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Sharma V.; Kumar P.; Pathak D. J. Heterocycl. Chem. 2010, 41. [Google Scholar]
- Fischer E.; Hess O. Ber. Chem. Ges. 1884, 17, 559. [Google Scholar]
- Bartoli G.; Palmieri G.; Bosco M.; Dalpozzo R. Tetrahedron Lett. 1989, 30, 2129. [Google Scholar]
- Larock R. C.; Yum E. K. J. Am. Chem. Soc. 1991, 113, 6689. [Google Scholar]
- Reissert A. Ber. Chem. Ges. 1897, 30, 1030. [Google Scholar]
- For recent reviews, see:; a Taber D. F.; Tirunahari P. K. Tetrahedron 2011, 67, 7195–7210. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cacchi S.; Fabrizi G. Chem. Rev. 2005, 105, 2873–2920. [DOI] [PubMed] [Google Scholar]
- For pyrrole-based indole synthesis, see:; a Moskal J.; van Leusen A. M. J. Org. Chem. 1986, 51, 4131. [Google Scholar]; b Ishibashi H.; Tabata T.; Hanaoka K.; Iriyama H.; Akamatsu S.; Ikeda M. Tetrahedron Lett. 1993, 34, 489. [Google Scholar]; c Della Rossa C.; Kneeteman M.; Mancini P. Tetrahedron Lett. 2007, 48, 1435. [Google Scholar]; d Kim M.; Vedejs E. J. Org. Chem. 2004, 69, 6945. [DOI] [PubMed] [Google Scholar]; e Iwasaki M.; Kobayashi Y.; Li J.-P.; Matsuzaka H.; Ishii Y.; Hidai M. J. Org. Chem. 1991, 56, 1922. [Google Scholar]; f Katritzky A. R.; Ledoux S.; Nair S. K. J. Org. Chem. 2003, 68, 5728. [DOI] [PubMed] [Google Scholar]; g Asao N.; Aikawa H. J. Org. Chem. 2006, 71, 5249. [DOI] [PubMed] [Google Scholar]; h Zhao J.; Hughes C. O.; Toste F. D. J. Am. Chem. Soc. 2006, 128, 7436. [DOI] [PubMed] [Google Scholar]
- Muchowski J. M.; Naef R. Helv. Chim. Acta 1984, 67, 1168–1172. [Google Scholar]
- Charrier N.; Clarke B.; Cutler L.; Demont E.; Dingwall C.; Dunsdon R.; East P.; Hawkins J.; Howes C.; Hussain I.; Jeffrey P.; Maile G.; Matico R.; Mosley J.; Naylor A.; O’Brien A.; Redshaw S.; Rowland P.; Soleil V.; Smith K. J.; Sweitzer S.; Theobald P.; Vesey D.; Walter D. S.; Wayne G. J. Med. Chem. 2008, 51, 3313–3317. [DOI] [PubMed] [Google Scholar]
- Sams A. G.; Eskildsen J.; Bastlund J. F. Int. Patent WO 2012/131031 A1, Oct 4, 2012.
- a Outlaw V. K.; Wydysh E. A.; Vadlamudi A.; Medghalchi S. M.; Townsend C. A. Med. Chem. Commun. 2014, 5, 826–830. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wydysh E. A.; Medghalchi S. M.; Vadlamudi A.; Townsend C. A. J. Med. Chem. 2009, 52, 3317–3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hedaya E.; Theodoropulos S. Tetrahedron 1968, 24, 2241–2254. [Google Scholar]; b Eyjólfsson R. Acta Chem. Scand. 1970, 24, 3075–3078. [DOI] [PubMed] [Google Scholar]; c Balasubramaniyan V.; Tongare D. B.; Gosavi S. S.; Babar S. M. Proc. Indian Acad. Sci., Chem. Sci. 1993, 105, 265–271. [Google Scholar]; d McCombie S. W.; Luchaco C. A. Tetrahedron Lett. 1997, 38, 5775–5776. [Google Scholar]; e Marcq V.; Mirand C.; Decarme M.; Emonard H.; Hornebeck W. Bioorg. Med. Chem. Lett. 2003, 13, 2843–2846. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.